
C-C Motif Chemokine Ligand-17 as a Novel Biomarker and Regulator of Epithelial Mesenchymal Transition in Renal Fibrogenesis

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Biochemical Analysis
2.2. Chemical Reagent and Antibody
2.3. Unilateral Ureteral Obstruction (UUO) and Immunohistochemistry
2.4. Human Cytokine Assay
2.5. Cell Culture
2.6. Cell Cycle Assay
2.7. DNA Transient Transfection
2.8. Assessment of Cell Growth by MTT Assay
2.9. Assessment of Gene Expression by qRT-PCR
2.10. In Vitro Migration Assay
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
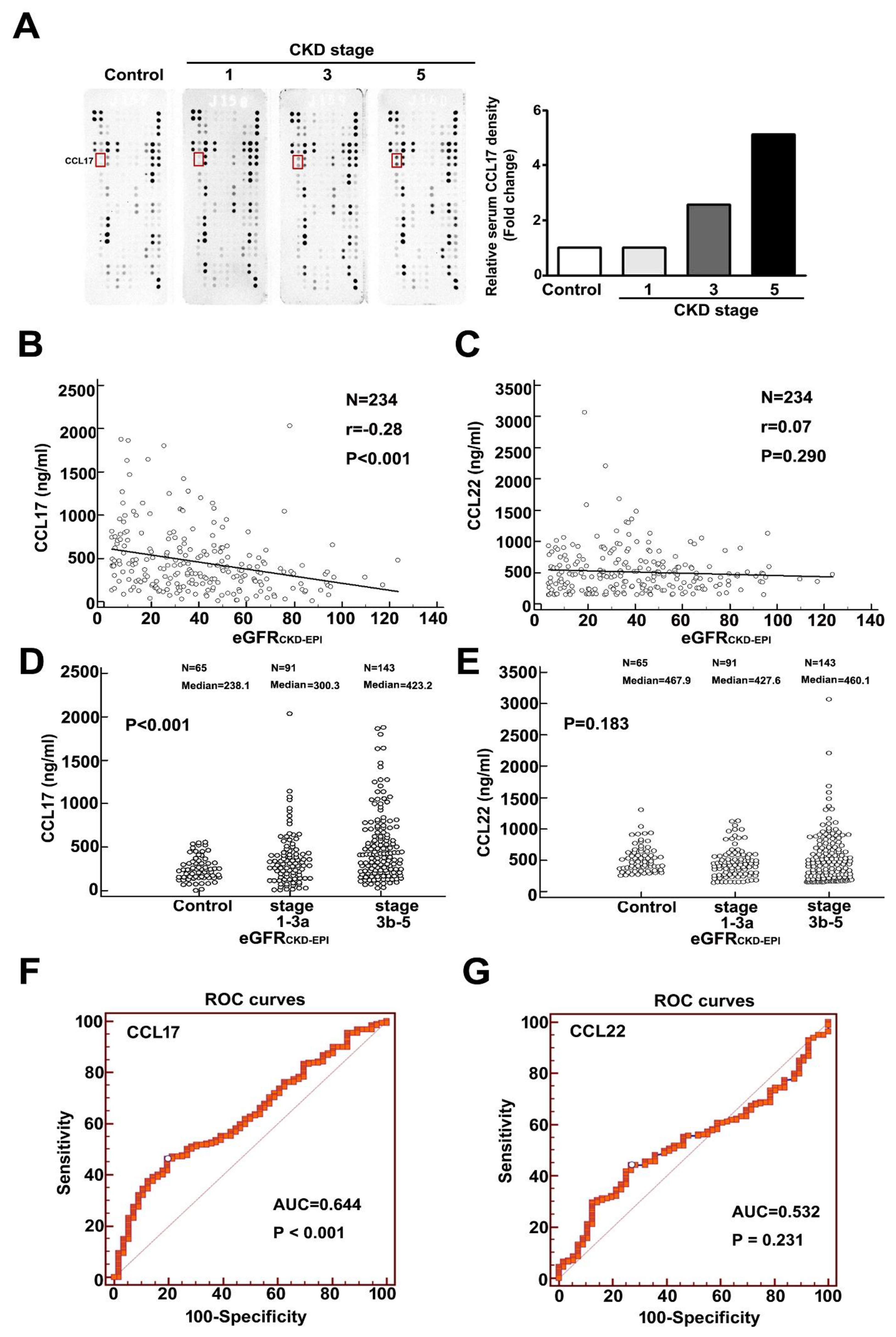
3.1. Upregulation of CCL17 in Patients with Advanced Chronic Kidney Disease
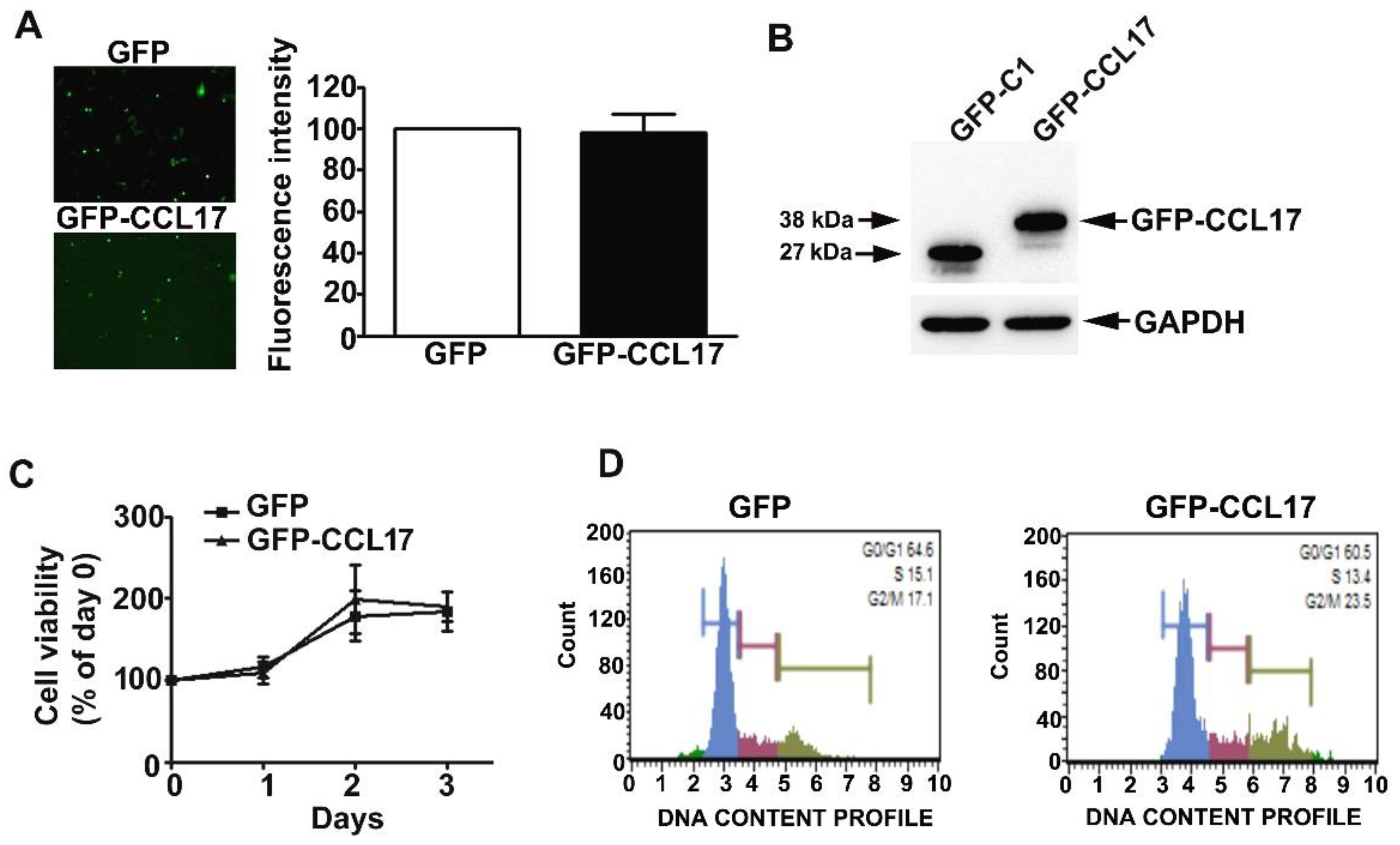
3.2. Overexpression of CCL17 in HK2 Cells Does Not Affect Their Viability or Cell Cycle Phase Distribution
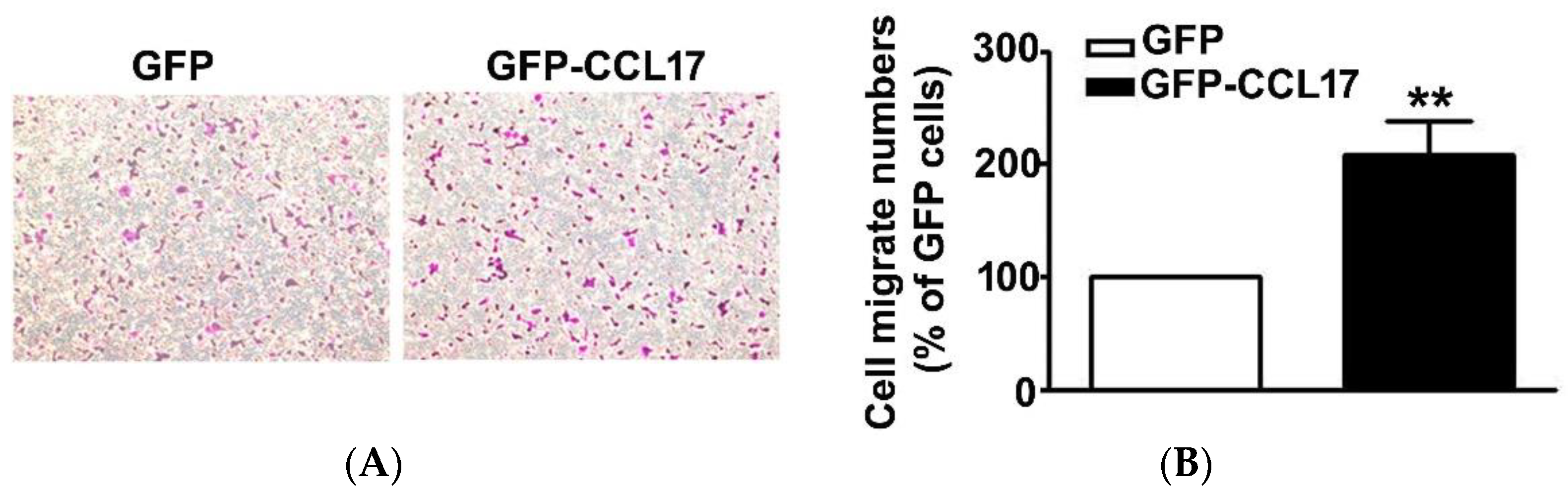
3.3. CCL17 Overexpression Increased HK2 Cell Migration Ability
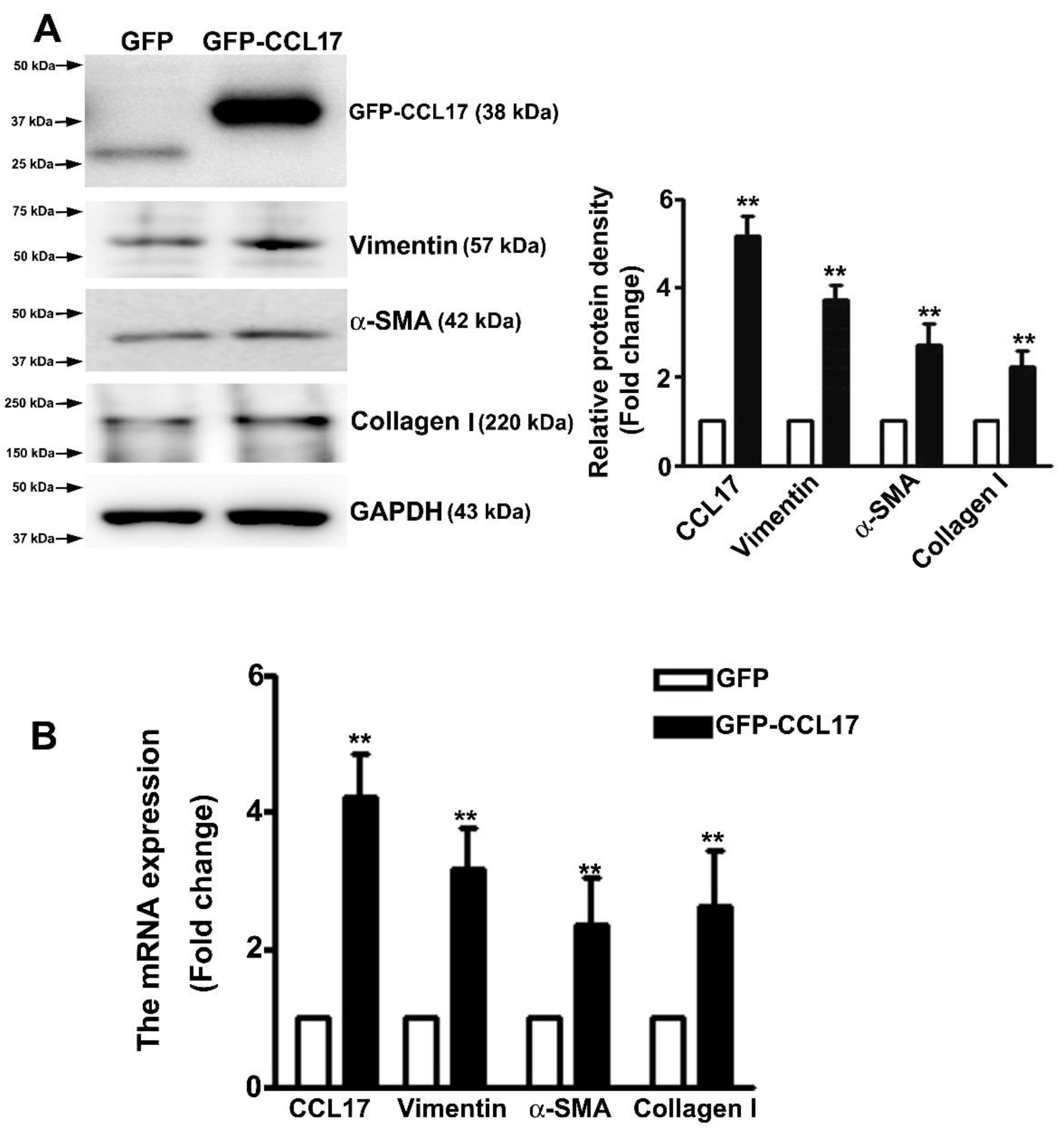
3.4. Overexpressed CCL17 Involved in Epithelial–Mesenchymal Transition (EMT) and Fibrosis in HK2 Cells
3.5. Increased Expression of CCL17 and EMT-Related Proteins in Obstructed Kidneys
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. Mechanisms and Treatment of CKD. J. Am. Soc. Nephrol. 2012, 23, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-R.; Yang, Y.; Wang, S.-C.; Chiu, P.-F.; Chou, W.-Y.; Lin, C.-Y.; Chang, J.-M.; Chen, T.-W.; Ferng, S.-H.; Lin, C.-L. Effectiveness of multidisciplinary care for chronic kidney disease in Taiwan: A 3-year prospective cohort study. Nephrol. Dial. Transplant. 2012, 28, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.-J.; Tsai, J.-C.; Chen, H.-C. Epidemiology, impact and preventive care of chronic kidney disease in Taiwan. Nephrology 2010, 15 (Suppl. S2), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Essawy, M.; Soylemezoglu, O.; Muchaneta-Kubara, E.C.; Shortland, J.; Brown, C.B.; El Nahas, A.M. Myofibroblasts and the progression of diabetic nephropathy. Nephrol. Dial. Transplant. 1997, 12, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, I.S.; Burrows, C.; Shanks, J.H.; Venning, M.; McWilliam, L.J. Interstitial myofibroblasts: Predictors of progression in membranous nephropathy. J. Clin. Pathol. 1997, 50, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshie, O.; Imai, T.; Nomiyama, H. Chemokines in Immunity. Adv. Immunol. 2001, 78, 57–110. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ma, Z.; Peng, H.; He, L.; Hu, Z.; Wang, Y. CXCL16 Deficiency Attenuates Renal Injury and Fibrosis in Salt-Sensitive Hypertension. Sci. Rep. 2016, 6, 28715. [Google Scholar] [CrossRef]
- Ma, Z.; Jin, X.; He, L.; Wang, Y. CXCL16 regulates renal injury and fibrosis in experimental renal artery stenosis. Am. J. Physiol. Circ. Physiol. 2016, 311, H815–H821. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Entman, M.L.; Wang, Y. CCR2 Regulates the Uptake of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. PLoS ONE 2013, 8, e77493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, C.-H.; Lin, S.-R.; Liu, C.-H.; Pan, S.-Y.; Hsu, H.; Chen, Y.-T.; Yen, C.-T.; Yu, I.-S.; Wu, H.-L.; Lin, S.-L. Targeting fibroblast CD248 attenuates CCL17-expressing macrophages and tissue fibrosis. Sci. Rep. 2020, 10, 16772. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, S.; Ikeda, E.; Yoshie, O.; Tsukamoto, N.; Aiso, S.; Aikawa, N.; Kubo, A.; Matsushima, K.; Yamaguchi, K. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur. Respir. J. 2004, 24, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Yogo, Y.; Fujishima, S.; Inoue, T.; Saito, F.; Shiomi, T.; Yamaguchi, K.; Ishizaka, A. Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis. Respir. Res. 2009, 10, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, Y. Thymus and activation-regulated chemokine as a clinical biomarker in atopic dermatitis. J. Dermatol. 2014, 41, 221–229. [Google Scholar] [CrossRef]
- Chen, Y.; Hsu, H.; Lin, C.; Pan, S.; Liu, S.; Wu, C.; Tsai, P.; Liao, C.; Cheng, H.-T.; Chiang, W.; et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J. Pathol. 2020, 250, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Wohlfahrtova, M.; Tycova, I.; Honsova, E.; Lodererova, A.; Viklicky, O. Molecular Patterns of Subclinical and Clinical Rejection of Kidney Allograft: Quantity Matters. Kidney Blood Press. Res. 2015, 40, 244–257. [Google Scholar] [CrossRef]
- Lebherz-Eichinger, D.; Klaus, D.A.; Reiter, T.; Hörl, W.H.; Haas, M.; Ankersmit, H.J.; Krenn, C.G.; Roth, G.A. Increased chemokine excretion in patients suffering from chronic kidney disease. Transl. Res. 2014, 164, 433–443. [Google Scholar] [CrossRef]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Ozols, E.; Kanellis, J.; Badal, S.S.; Liles, J.T.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin Inhibition Protects Against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis. Int. J. Mol. Sci. 2020, 22, 271. [Google Scholar] [CrossRef]
- Hung, T.-W.; Chu, C.-Y.; Yu, C.-L.; Lee, C.-C.; Hsu, L.-S.; Chen, Y.-S.; Hsieh, Y.-H.; Tsai, J.-P. Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins 2020, 12, 506. [Google Scholar] [CrossRef]
- Wen, Y.; Lin, Y.; Chu, C.; Yang, Y.; Yang, S.; Liu, Y.; Hsiao, M.; Lee, W.; Chien, M. Melatonin-triggered post-transcriptional and post-translational modifications of ADAMTS1 coordinately retard tumorigenesis and metastasis of renal cell carcinoma. J. Pineal Res. 2020, 69, e12668. [Google Scholar] [CrossRef] [PubMed]
- Nagavally, R.R.; Sunilkumar, S.; Akhtar, M.; Trombetta, L.D.; Ford, S.M. Chrysin Ameliorates Cyclosporine-A-Induced Renal Fibrosis by Inhibiting TGF-β1-Induced Epithelial–Mesenchymal Transition. Int. J. Mol. Sci. 2021, 22, 10252. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sanchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A. Tubulointerstitial Changes as a Major Determinant in the Progression of Renal Damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Beshay, O.N.; Ewees, M.G.; Abdel-Bakky, M.S.; Hafez, S.M.N.A.; Abdelrehim, A.B.; Bayoumi, A.M. Resveratrol reduces gentamicin-induced EMT in the kidney via inhibition of reactive oxygen species and involving TGF-β/Smad pathway. Life Sci. 2020, 258, 118178. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, W.; Yin, P.; Gao, J.; Na, L.; Sun, Y.; Wang, Z.; Zhang, Z.; Zhao, C. Ruxolitinib Alleviates Renal Interstitial Fibrosis in UUO Mice. Int. J. Biol. Sci. 2020, 16, 194–203. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase Protects against Renal Fibrosis by Inhibiting the Activation of the ERK Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 855. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Olbromski, M.; Dzięgiel, P. CCL18 in the Progression of Cancer. Int. J. Mol. Sci. 2020, 21, 7955. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, X.; Deng, H.; Xu, B.; Chen, L.; Wang, Q.; Zhou, Q.; Zhang, D.; Wu, C.; Jiang, J. Expression of CCR6 in esophageal squamous cell carcinoma and its effects on epithelial-to-mesenchymal transition. Oncotarget 2017, 8, 115244–115253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Fu, R.; Chen, C.; Cheng, X.; Guo, T.; Huangfu, L.; Li, X.; Du, H.; Xing, X.; Ji, J. CXCL16 Promotes Gastric Cancer Tumorigenesis via ADAM10-Dependent CXCL16/CXCR6 Axis and Activates Akt and MAPK Signaling Pathways. Int. J. Biol. Sci. 2021, 17, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lin, Z.; Lu, J.; Lin, X.; Xu, W.; Wang, N.; Huang, S.; Wang, Y.; Zhu, Y.; Chen, Z.; et al. CCL2-CCL5/CCR4 con-tributed to radiation-induced epithelial-mesenchymal transition of HPAEpiC cells via the ERK signaling pathways. Am. J. Transl. Res. 2019, 11, 733–743. [Google Scholar] [PubMed]
- Han, G.; Wu, D.; Yang, Y.; Li, Z.; Zhang, J.; Li, C. CrkL meditates CCL20/CCR6-induced EMT in gastric cancer. Cytokine 2015, 76, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shao, Z.; Jiang, E.; Zhou, X.; Wang, L.; Wang, H.; Luo, X.; Chen, Q.; Liu, K.; Shang, Z. CCL21/CCR7 interaction promotes EMT and enhances the stemness of OSCC via a JAK2/STAT3 signaling pathway. J. Cell. Physiol. 2020, 235, 5995–6009. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sun, C.; Lu, J. The Role of Chemokine Receptors in Renal Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2020, 177, 1–24. [Google Scholar] [CrossRef]
- Zhu, F.; Li, X.; Chen, S.; Zeng, Q.; Zhao, Y.; Luo, F. Tumor-associated macrophage or chemokine ligand CCL17 positively regulates the tumorigenesis of hepatocellular carcinoma. Med. Oncol. 2016, 33, 17. [Google Scholar] [CrossRef]
- Okamura, D.M.; López-Guisa, J.M.; Koelsch, K.; Collins, S.; Eddy, A. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol. Physiol. 2007, 293, F575–F585. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Lin, S.-C.; Chen, J.; He, L.; Dong, F.; Xu, J.; Han, S.; Du, J.; Entman, M.L.; Wang, Y. CXCL16 Recruits Bone Marrow-Derived Fibroblast Precursors in Renal Fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1876–1886. [Google Scholar] [CrossRef] [Green Version]
- Segerer, S.; Cui, Y.; Hudkins, K.L.; Goodpaster, T.; Eitner, F.; Mack, M.; Schlöndorff, D.; Alpers, C.E. Expression of the Chemokine Monocyte Chemoattractant Protein-1 and Its Receptor Chemokine Receptor 2 in Human Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2000, 11, 2231–2242. [Google Scholar] [CrossRef]
- Wada, T.; Furuichi, K.; Sakai, N.; Iwata, Y.; Yoshimoto, K.; Shimizu, M.; Takeda, S.-I.; Takasawa, K.; Yoshimura, M.; Kida, H.; et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 2000, 58, 1492–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, K.; Wada, T.; Furuichi, K.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Takeya, M.; Kuziel, W.A.; Matsushima, K.; Mukaida, N.; et al. Blockade of CCR2 Ameliorates Progressive Fibrosis in Kidney. Am. J. Pathol. 2004, 165, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Nkuipou-Kenfack, E.; Zürbig, P.; Mischak, H. The long path towards implementation of clinical proteomics: Exemplified based on CKD273. Proteom.-Clin. Appl. 2017, 11, 11. [Google Scholar] [CrossRef]
- Good, D.M.; Zürbig, P.; Argilés, À.; Bauer, H.W.; Behrens, G.; Coon, J.J.; Dakna, M.; Decramer, S.; Delles, C.; Dominiczak, A.; et al. Naturally Occurring Human Urinary Peptides for Use in Diagnosis of Chronic Kidney Disease. Mol. Cell. Proteom. 2010, 9, 2424–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belperio, J.A.; Dy, M.; Murray, L.; Burdick, M.D.; Xue, Y.Y.; Strieter, R.M.; Keane, M.P. The role of the Th2 CC chemokine ligand CCL17 in pulmonary fibrosis. J. Immunol. 2004, 173, 4692–4698. [Google Scholar] [CrossRef] [Green Version]
- Sumiyoshi, K.; Nakao, A.; Setoguchi, Y.; Tsuboi, R.; Okumura, K.; Ogawa, H. TGF-β/Smad signaling inhibits IFN-γ and TNF-α-induced TARC (CCL17) production in HaCaT cells. J. Dermatol. Sci. 2003, 31, 53–58. [Google Scholar] [CrossRef]
- Kwon, D.-J.; Bae, Y.-S.; Ju, S.M.; Goh, A.R.; Youn, G.S.; Choi, S.Y.; Park, J. Casuarinin suppresses TARC/CCL17 and MDC/CCL22 production via blockade of NF-κB and STAT1 activation in HaCaT cells. Biochem. Biophys. Res. Commun. 2012, 417, 1254–1259. [Google Scholar] [CrossRef]
- Qi, X.-F.; Kim, D.-H.; Yoon, Y.-S.; Li, J.-H.; Song, S.-B.; Jin, D.; Huang, X.-Z.; Teng, Y.-C.; Lee, K.-J. The adenylyl cyclase-cAMP system suppresses TARC/CCL17 and MDC/CCL22 production through p38 MAPK and NF-κB in HaCaT keratinocytes. Mol. Immunol. 2009, 46, 1925–1934. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Early CKD | Advanced CKD | ||||||
|---|---|---|---|---|---|---|---|
| CKD Stage | Normal | 1 | 2 | 3a | 3b | 4 | 5 |
| Number | 65 | 12 | 38 | 41 | 52 | 41 | 50 |
| Age (year) | 50.17 ± 10.70 | 52.42 ± 12.43 | 64.29 ± 10.28 | 71.22 ± 9.73 | 72.02 ± 10.52 | 75.49 ± 9.59 | 72.22 ± 13.29 |
| Female/Male | 35/30 | 4/8 | 11/27 | 13/28 | 19/33 | 20/21 | 27/23 |
| BUN (mg/dL) | 9.09 ± 2.73 | 10.91 ± 3.34 | 15.76 ± 5.35 | 19.34 ± 5.97 | 25.00 ± 7.16 | 37.22 ± 112.53 | 68.60 ± 24.15 |
| Cre (mg/dL) | 0.75 ± 0.13 | 0.77 ± 0.12 | 1.03 ± 0.18 | 1.30 ± 0.20 | 1.69 ± 0.29 | 2.40 ± 0.46 | 6.37 ± 2.66 |
| eGFRCKD-EPI (mL/min) | 101.58 ± 7.42 | 99.98 ± 10.67 | 71.43 ± 8.09 | 51.26 ± 4.40 | 36.54 ± 4.34 | 22.95 ± 4.26 | 8.27 ± 3.44 |
| CCL17 (ng/mL) | 250.01 ± 138.08 | 275.26 ± 160.47 | 333.63 ± 356.98 | 408.15 ± 291.38 | 431.22 ± 330.61 | 485.14 ± 407.62 | 638.84 ± 429.59 |
| 238.10 (149.56–321.72) | 248.05 (174.65–328.24) | 258.80 (123.20–403.20) | 355.50 (205.05–590.53) | 323.85 (203.60–554.48) | 373.68 (195.85–633.03) | 486.95 (348.60–808.60) | |
| CCL22 (ng/mL) | 519.03 ± 217.67 | 465.55 ± 235.42 | 430.75 ± 241.08 | 475.77 ± 239.78 | 584.84 ± 387.71 | 590.36 ± 572.84 | 457.72 ± 275.97 |
| 467.90 (368.60–634.10) | 427.90 (351.20–482.45) | 389.40 (252.20–507.60) | 475.50 (246.98–595.28) | 509.20 (271.60–841.85) | 449.30 (209.75–737.10) | 380.30 (221.10–626.00) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-H.; Wang, W.-C.; Hung, T.-W.; Lee, C.-C.; Tsai, J.-P. C-C Motif Chemokine Ligand-17 as a Novel Biomarker and Regulator of Epithelial Mesenchymal Transition in Renal Fibrogenesis. Cells 2021, 10, 3345. https://doi.org/10.3390/cells10123345
Hsieh Y-H, Wang W-C, Hung T-W, Lee C-C, Tsai J-P. C-C Motif Chemokine Ligand-17 as a Novel Biomarker and Regulator of Epithelial Mesenchymal Transition in Renal Fibrogenesis. Cells. 2021; 10(12):3345. https://doi.org/10.3390/cells10123345
Chicago/Turabian StyleHsieh, Yi-Hsien, Wen-Chien Wang, Tung-Wei Hung, Chu-Che Lee, and Jen-Pi Tsai. 2021. "C-C Motif Chemokine Ligand-17 as a Novel Biomarker and Regulator of Epithelial Mesenchymal Transition in Renal Fibrogenesis" Cells 10, no. 12: 3345. https://doi.org/10.3390/cells10123345