
Key Cannabis Salt-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Varieties



 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material and Growth Conditions
2.2. Physiological Analysis
2.3. RNA-Seq Library Construction and Sequencing
2.4. Data Analysis
2.5. Alignment and Annotations of New Transcripts
2.6. Expression Analysis
2.7. qRT-PCR Analysis
3. Results
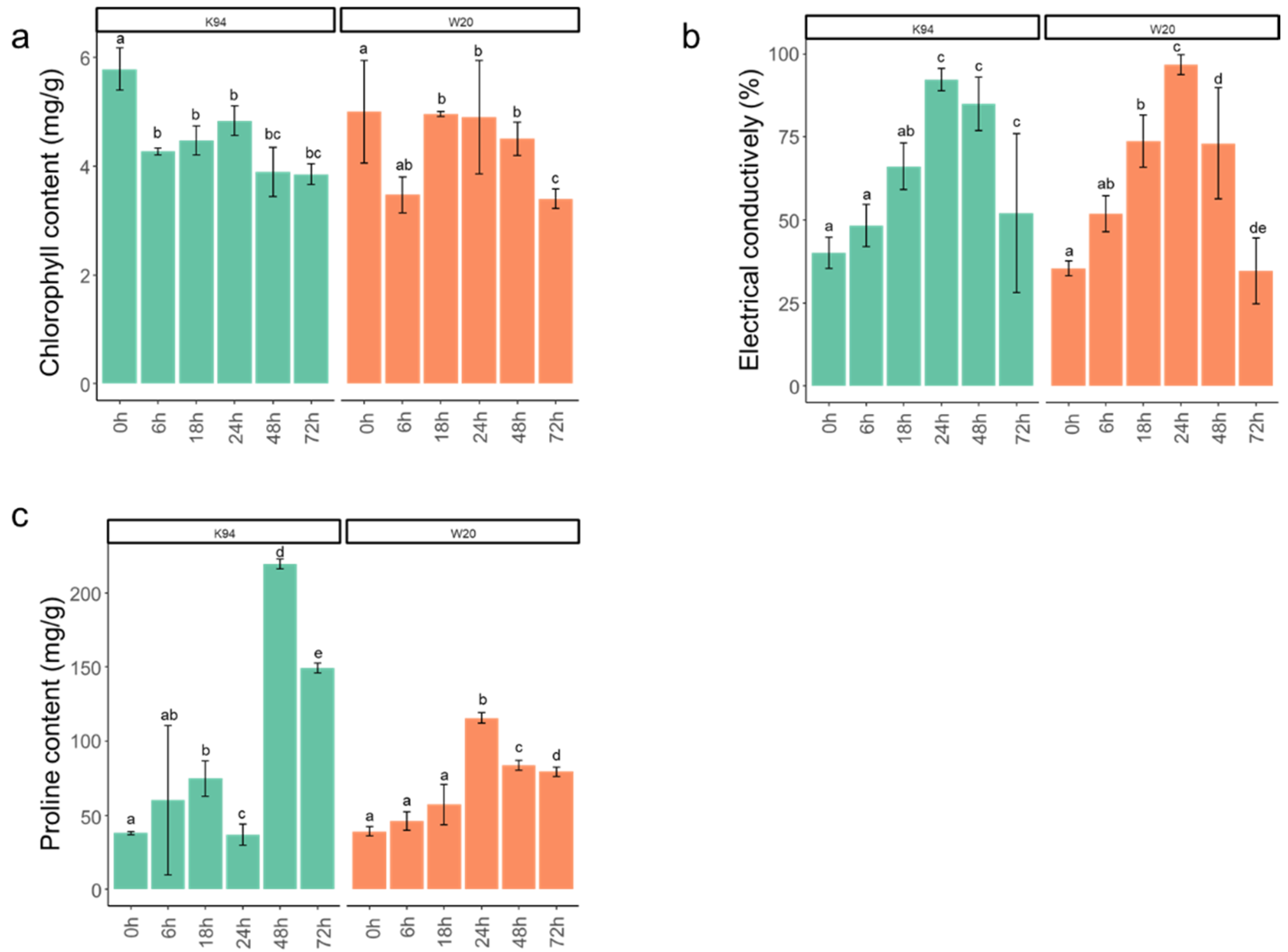
3.1. Physiological Analysis of Two Cannabis Seedling Varieties’ Responses to Salt Stress
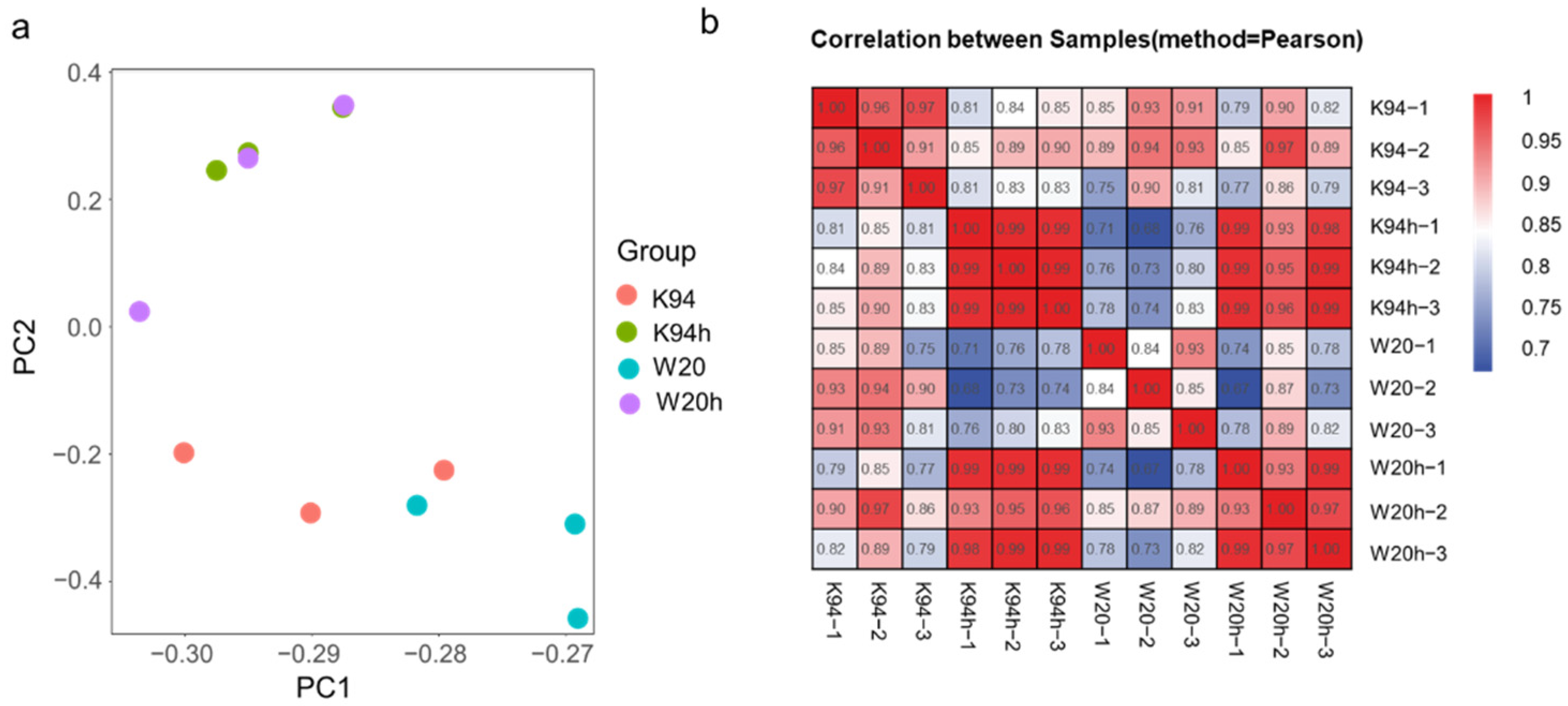
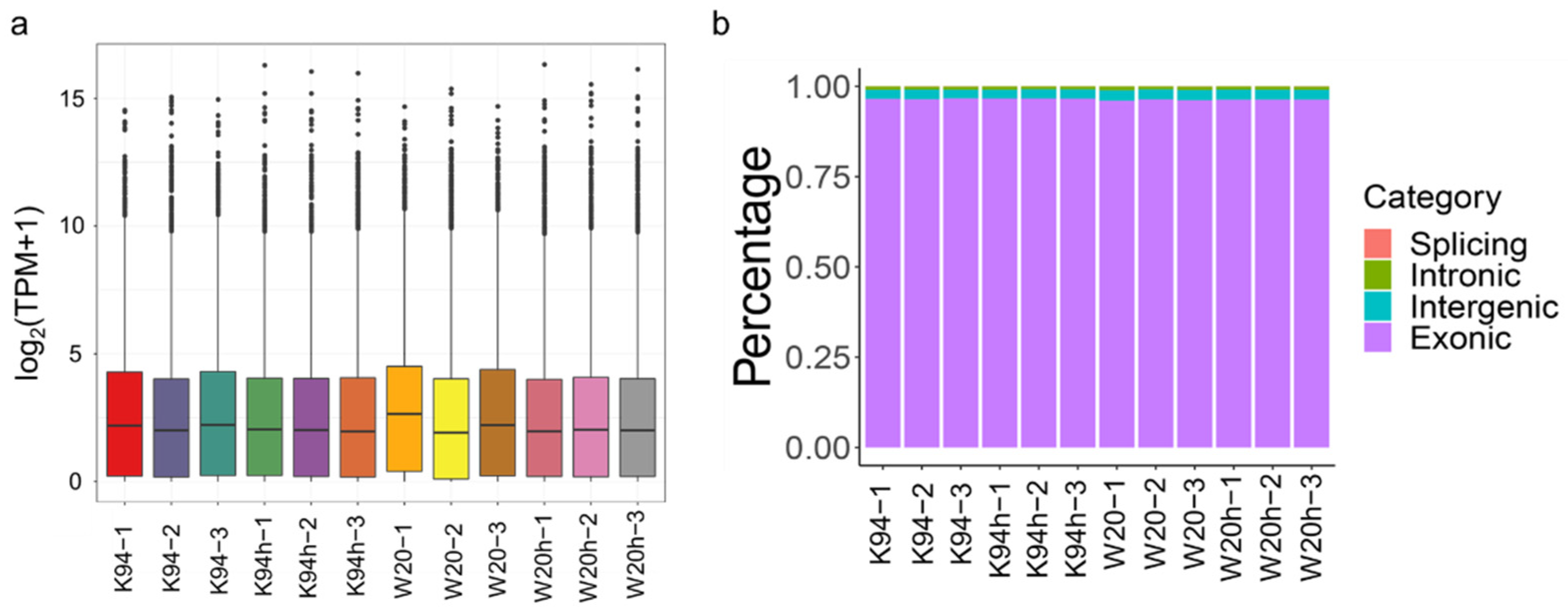
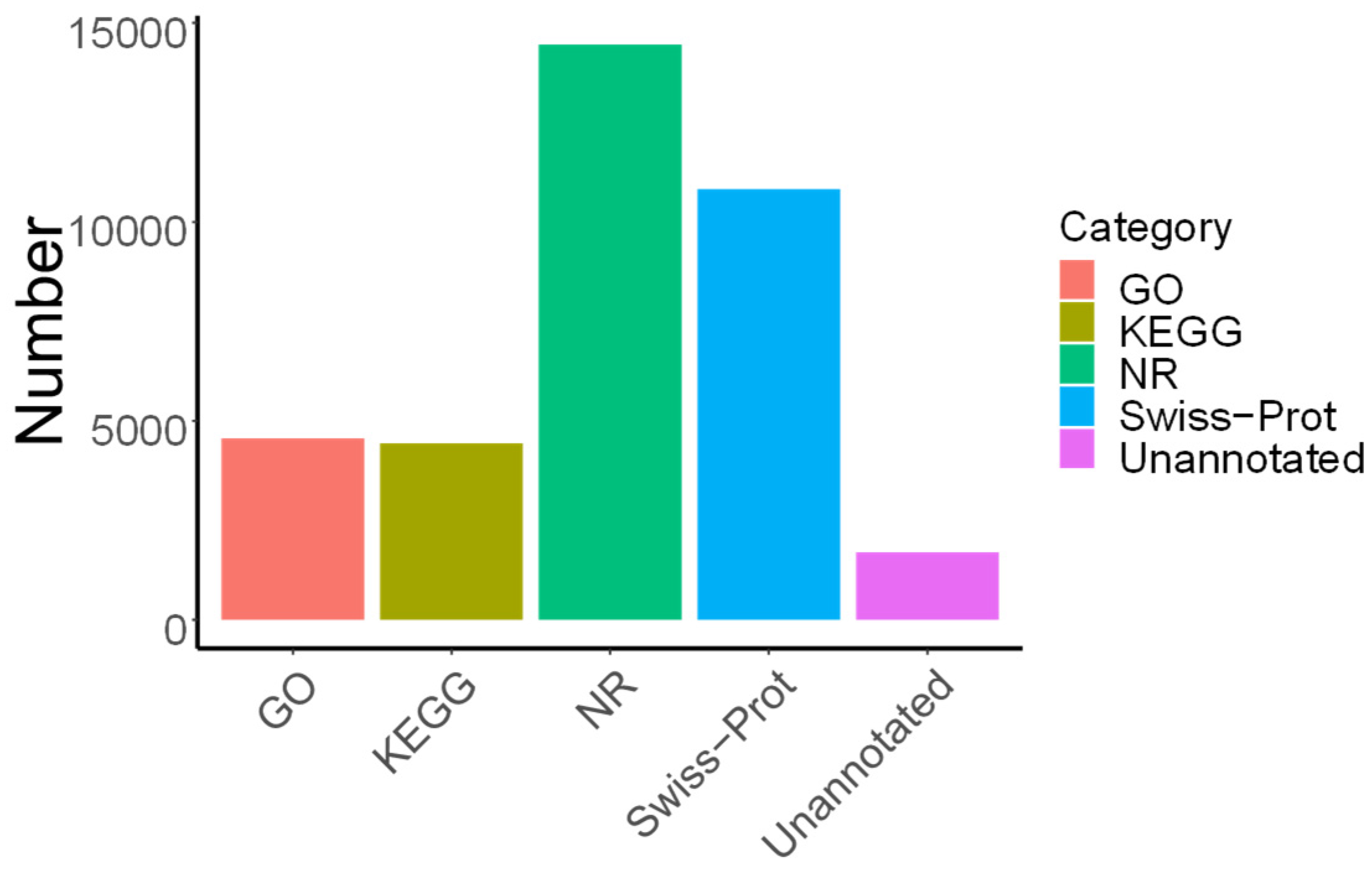
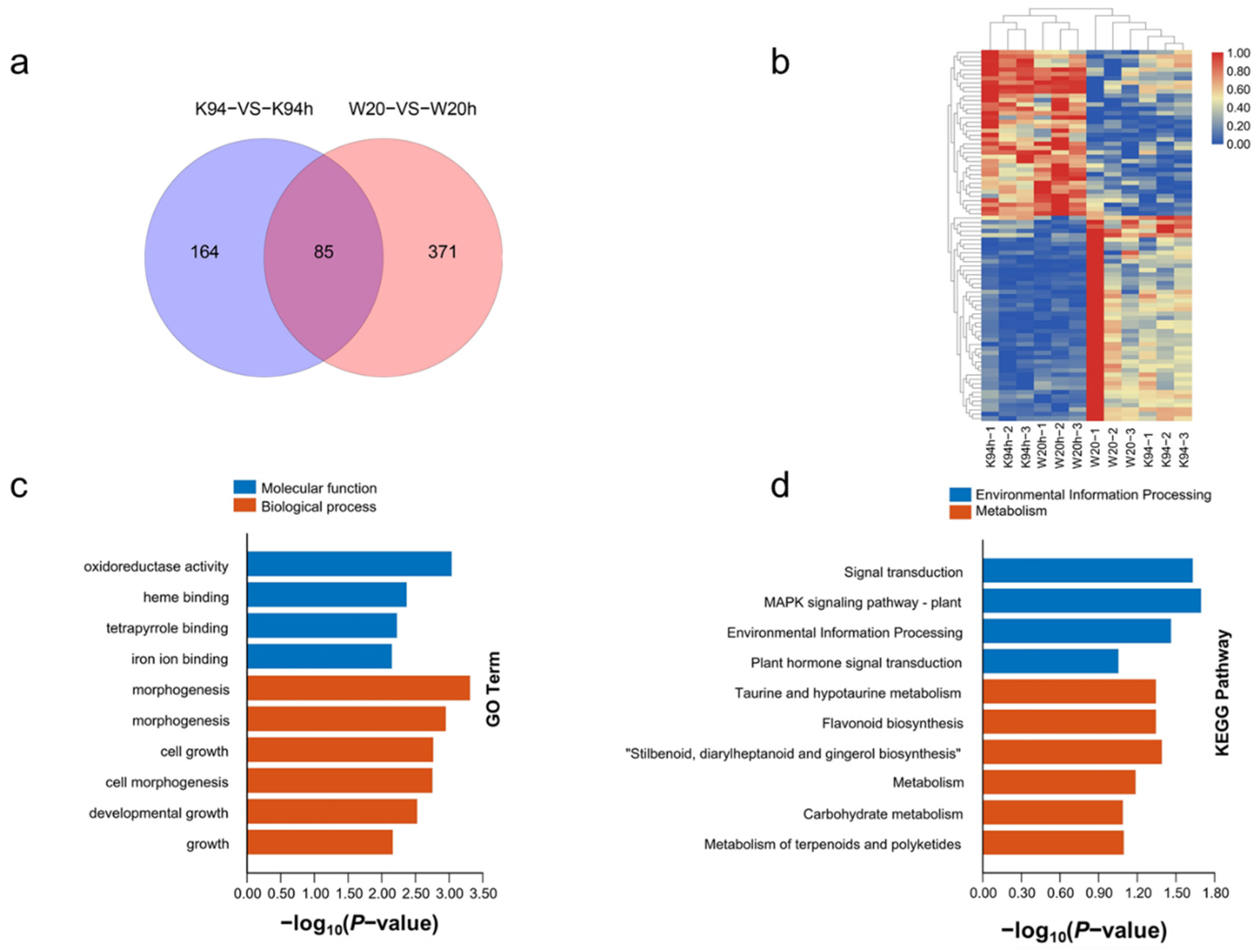
3.2. Transcriptome Sequencing Analysis
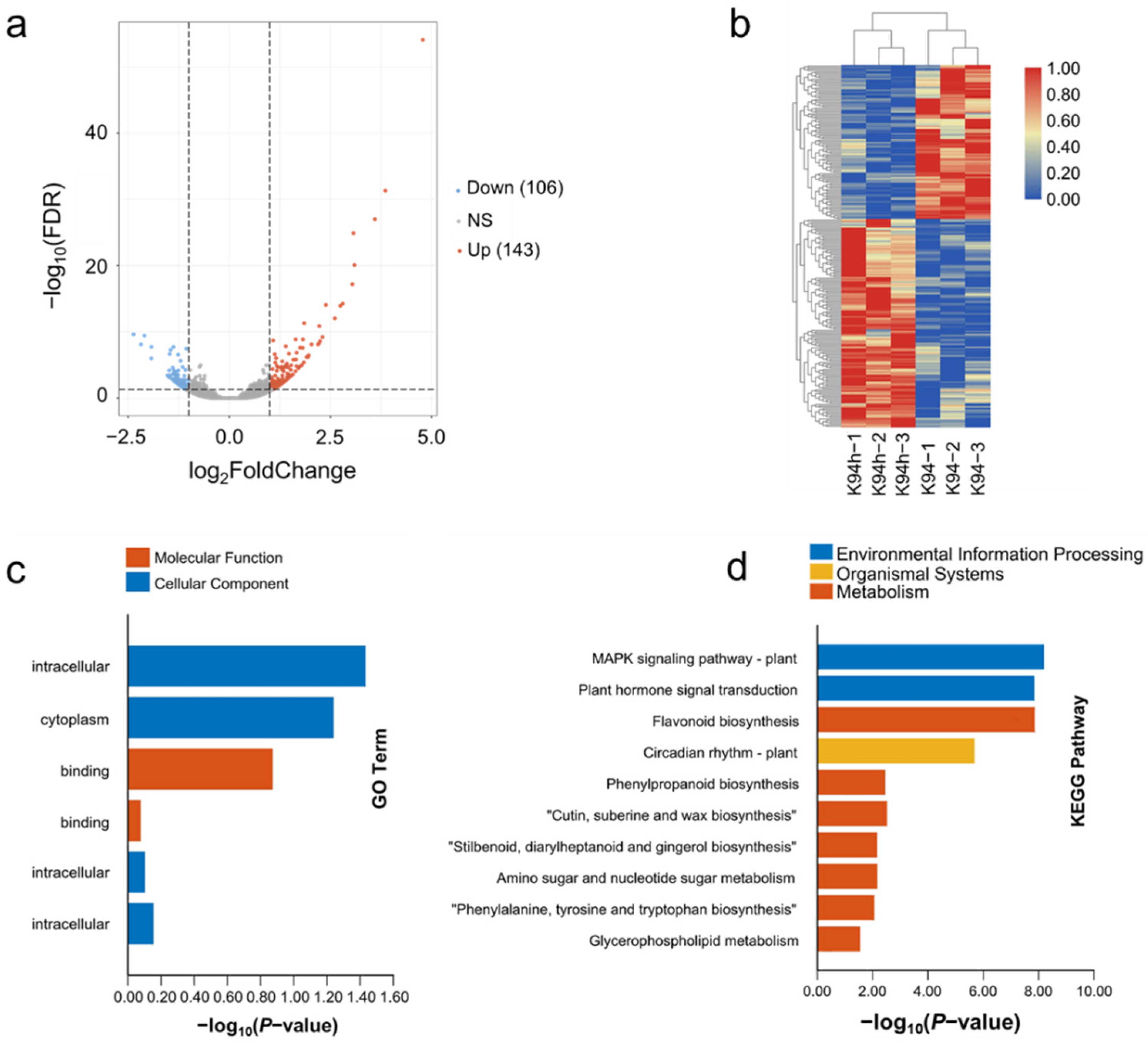
3.3. The Expression Pattern of the Differential Expression Genes Analyzed in K94
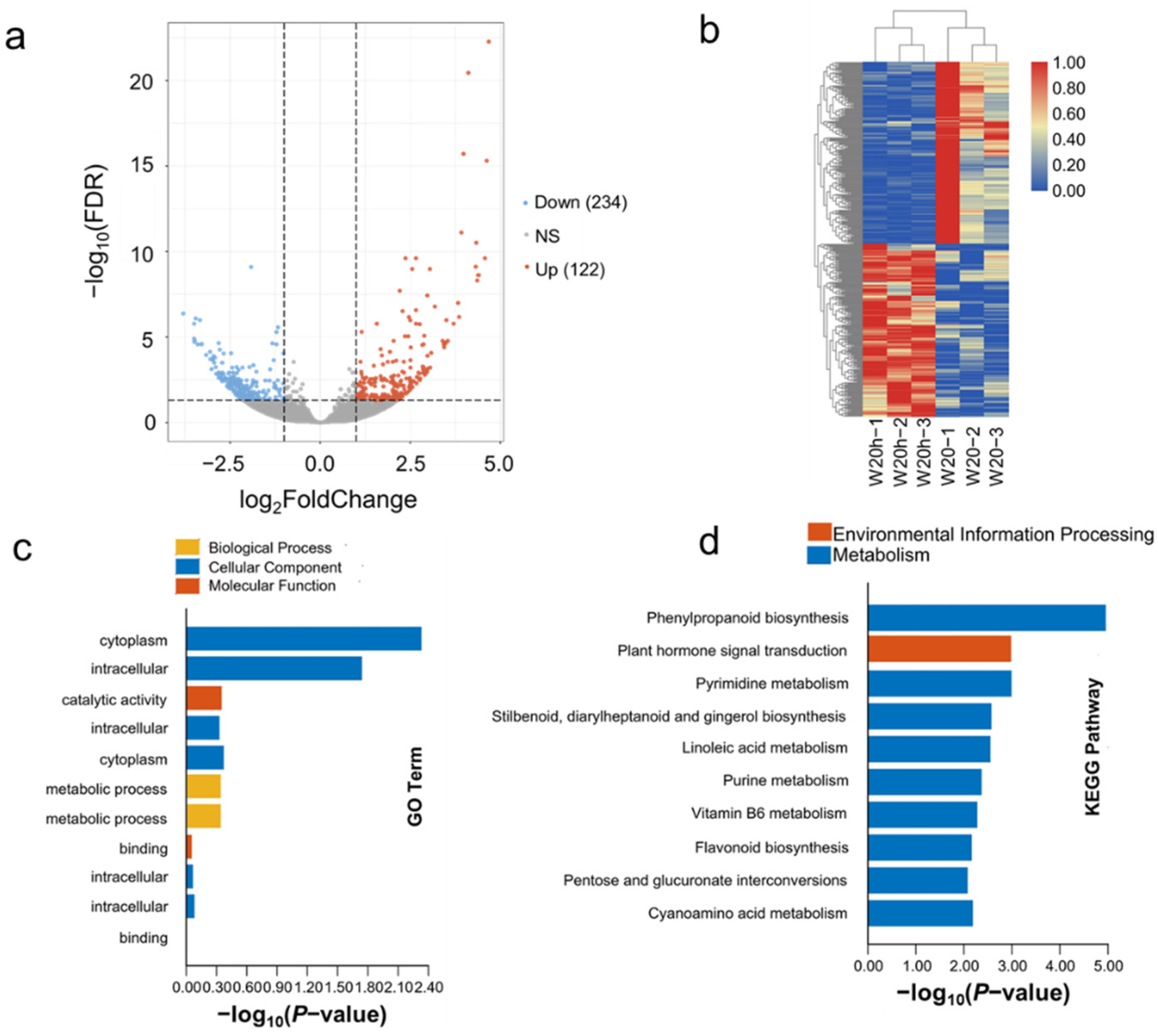
3.4. The Expression Pattern of DEGs Analyzed in W20
3.5. Identification of DEGs between K94 and W20
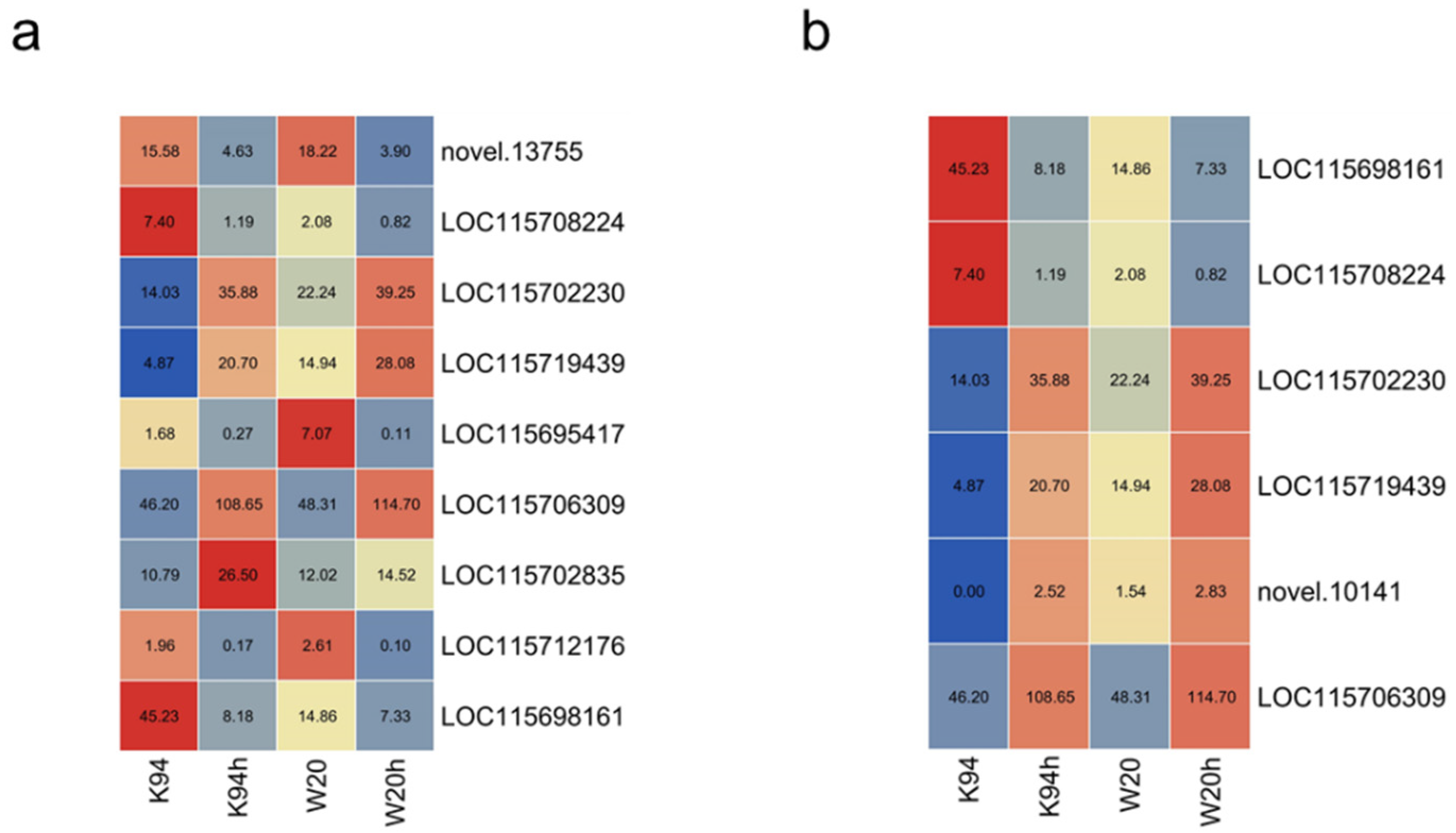
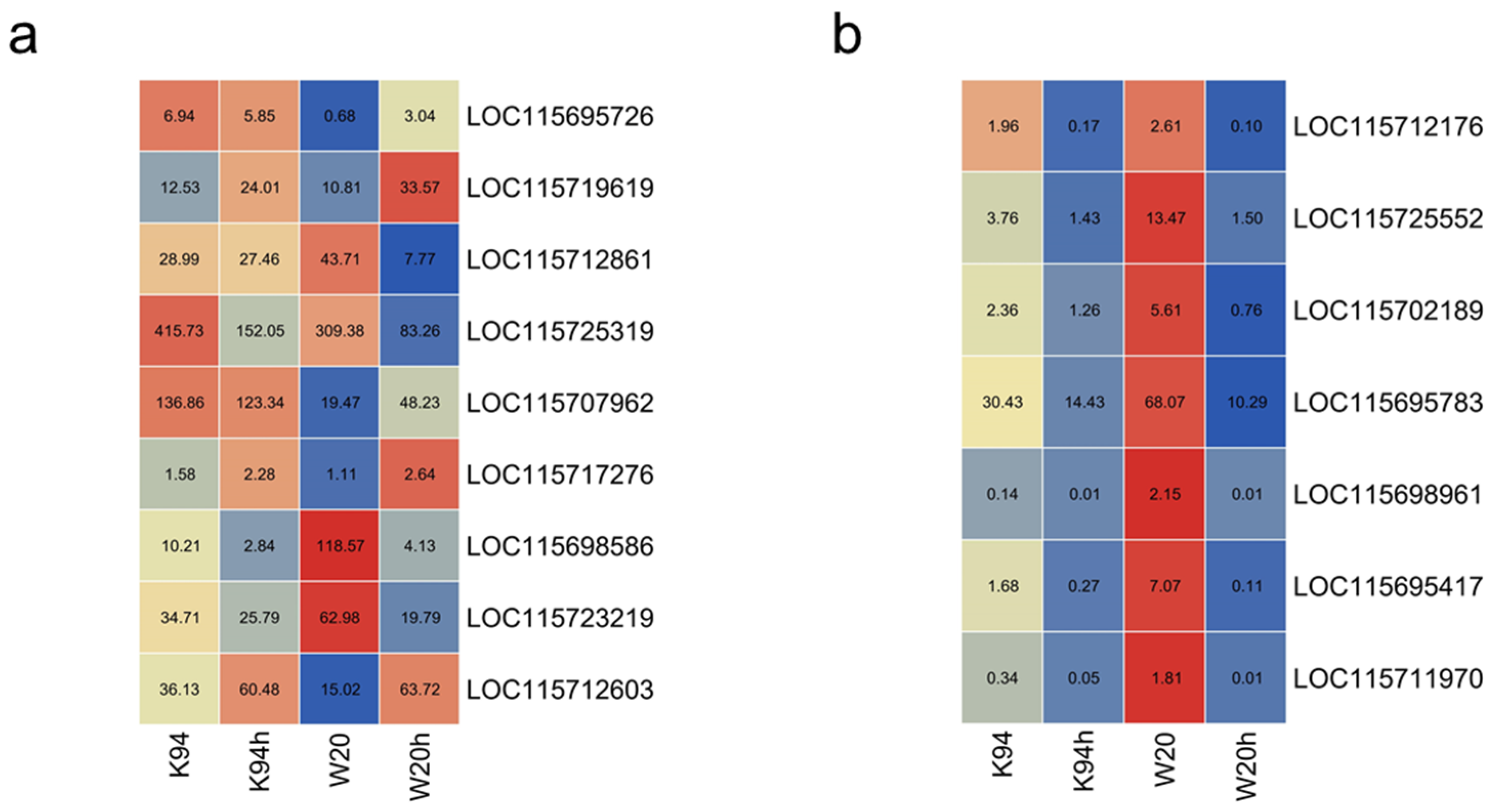
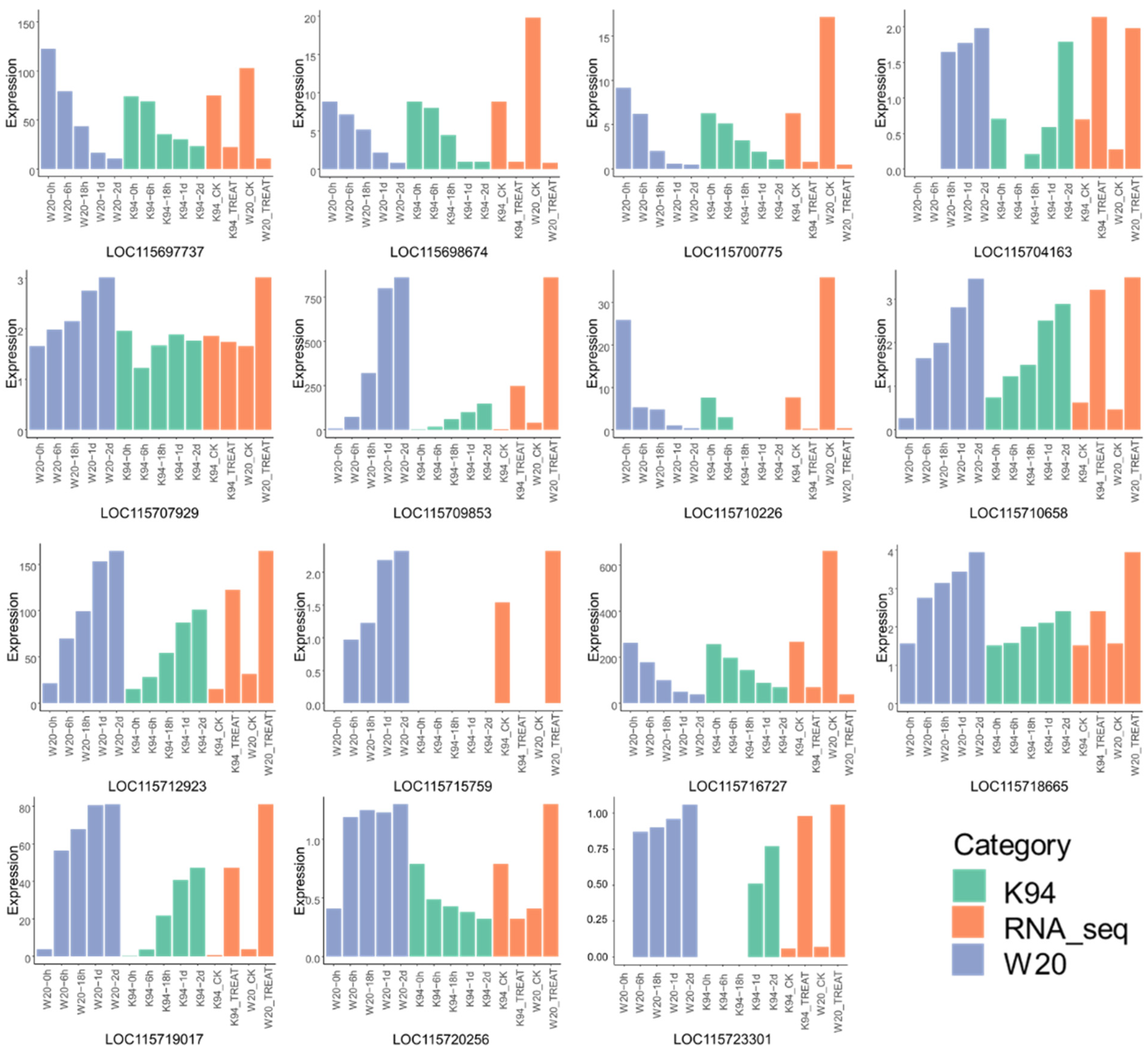
3.6. Comparison of DEG Tag Data with qRT-PCR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herbig, C.; Sirocko, F. Palaeobotanical evidence for agricultural activities in the Eifel region during the Holocene: Plant macro-remain and pollen analyses from sediments of three maar lakes in the Quaternary Westeifel Volcanic Field (Germany, Rheinland-Pfalz). Veg. Hist. Archaeobot. 2013, 22, 447–462. [Google Scholar] [CrossRef]
- Li, H.L. An archaeological and historical account of cannabis in China. Econ. Bot. 1973, 28, 437–448. [Google Scholar] [CrossRef]
- Amaducci, S.; Scordia, D.; Liu, F.H.; Zhang, Q.; Guo, H.; Testa, G.; Cosentino, S.L. Key cultivation techniques for hemp in Europe and China. Ind. Crops Prod. 2015, 68, 2–16. [Google Scholar] [CrossRef]
- Duvall, C.S. Drug laws, bioprospecting and the agricultural heritage of Cannabis in Africa. Space Polity 2016, 20, 10–25. [Google Scholar] [CrossRef]
- Obradovic, I. From prohibition to regulation: A comparative analysis of the emergence and related outcomes of new legal cannabis policy models (Colorado, Washington State and Uruguay). Int. J. Drug Policy 2019, 91, 102590. [Google Scholar] [CrossRef]
- Aliferis, K.A.; Bernard-Perron, D. Cannabinomics: Application of Metabolomics in Cannabis (Cannabis sativa L.) Research and Development. Front. Plant Sci. 2020, 11, 554. [Google Scholar] [CrossRef]
- Hall, W.; Degenhardt, L. Prevalence and correlates of cannabis use in developed and developing countries. Curr. Opin. Psychiatry 2007, 20, 393–397. [Google Scholar] [CrossRef]
- Cromack, H.T.H. The effect of cultivar and seed density on the production and fiber content of Cannabis sativa: In southern England. Ind. Crops Prod. 1998, 7, 205–210. [Google Scholar] [CrossRef]
- Salentijn, E.M.J.; Zhang, Q.; Amaducci, S.; Yang, M.; Trindade, L.M. New developments in fiber hemp (Cannabis sativa L.) breeding. Ind. Crops Prod. 2015, 68, 32–41. [Google Scholar] [CrossRef]
- Callaway, J.C. Hempseed as a nutritional resource: An overview. Euphytica 2004, 140, 65–72. [Google Scholar] [CrossRef]
- House, J.D.; Neufeld, J.; Leson, G. Evaluating the quality of protein from hemp seed (Cannabis sativa L.) products through the use of the protein digestibility-corrected amino acid score method. J. Agric. Food Chem. 2010, 58, 11801–11807. [Google Scholar] [CrossRef] [PubMed]
- Galasso, I.; Russo, R.; Mapelli, S.; Ponzoni, E.; Brambilla, I.M.; Battelli, G.; Reggiani, R. Variability in seed traits in a collection of cannabis sativa L. genotypes. Front. Plant Sci. 2016, 7, 688. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.H.; Ten, Z.; Wang, X.S.; Yang, X.Q. Physicochemical and functional properties of hemp (Cannabis sativa L.) protein isolate. J. Agric. Food Chem. 2006, 54, 8945–8950. [Google Scholar] [CrossRef]
- Deferne, J.; Pate, D.W. Hemp seed oil: A source of valuable essential fatty acids. J. Int. Hemp Assoc. 1996, 3, 4–7. [Google Scholar]
- Crescente, G.; Piccolella, S.; Esposito, A.; Scognamiglio, M.; Fiorentino, A.; Pacifico, S. Chemical composition and nutraceutical properties of hempseed: An ancient food with actual functional value. Phytochem. Rev. 2018, 17, 733–749. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Blumwald, E. Developing salt-tolerant crop plants: Challenges and opportunities. Trends Plant Sci. 2005, 10, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, K.; Tester, M.; Roy, S.J. Quantifying the three main components of salinity tolerance in cereals. Plant Cell Environ. 2009, 33, 237–249. [Google Scholar] [CrossRef]
- Sanower, H.M.; Sultan, A.S.J. Present Scenario of Global Salt Affected Soils, its Management and Importance of Salinity Research. Int. Res. J. Biol. Sci. 2019, 1, 1–3. [Google Scholar]
- Munns, R. Genes and salt tolerance: Bringing them together. New Phytol. 2005, 167, 645–663. [Google Scholar] [CrossRef]
- Oliveira, M.; Hoste, H.; Custódio, L. A systematic review on the ethnoveterinary uses of mediterranean salt-tolerant plants: Exploring its potential use as fodder, nutraceuticals or phytotherapeutics in ruminant production. J. Ethnopharmacol. 2021, 267, 113464. [Google Scholar] [CrossRef]
- Passaia, G.; Spagnolo Fonini, L.; Caverzan, A.; Jardim-Messeder, D.; Christoff, A.P.; Gaeta, M.L.; de Araujo Mariath, J.E.; Margis, R.; Margis-Pinheiro, M. The mitochondrial glutathione peroxidase GPX3 is essential for H2O2 homeostasis and root and shoot development in rice. Plant Sci. 2013, 208, 93–101. [Google Scholar] [CrossRef]
- Singh, V.; Singh, A.P.; Bhadoria, J.; Giri, J.; Singh, J.; Vineeth, T.V.; Sharma, P.C. Differential expression of salt-responsive genes to salinity stress in salt-tolerant and salt-sensitive rice (Oryza sativa L.) at seedling stage. Protoplasma 2018, 255, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Deyholos, M.K. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Wu, R.; Wee, C.W.; Xie, F.; Wei, X.; Chan, P.M.Y.; Tham, C.; Duan, L.; Dinneny, J.R. A spatio-temporal understanding of growth regulation during the salt stress response in Arabidopsis. Plant Cell 2013, 25, 2132–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fang, Z.; Yang, L.; Chan, Z. Transcriptional variation analysis of Arabidopsis ecotypes in response to drought and salt stresses dissects commonly regulated networks. Physiol. Plant. 2021, 172, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Nakamura, A.; Hara, N.; Toki, S.; Tanaka, Y. Molecular and functional analyses of rice NHX-type Na+/H+ antiporter genes. Planta 2011, 233, 175–188. [Google Scholar] [CrossRef]
- He, S.; Tan, L.; Hu, Z.; Chen, G.; Wang, G.; Hu, T. Molecular characterization and functional analysis by heterologous expression in E. coli under diverse abiotic stresses for OsLEA5, the atypical hydrophobic LEA protein from Oryza sativa L. Mol. Genet. Genom. 2012, 287, 39–54. [Google Scholar] [CrossRef]
- Zhang, F.; Li, L.; Jiao, Z.; Chen, Y.; Liu, H.; Chen, X.; Fu, J.; Wang, G.; Zheng, J. Characterization of the calcineurin B-Like (CBL) gene family in maize and functional analysis of ZmCBL9 under abscisic acid and abiotic stress treatments. Plant Sci. 2016, 253, 118–129. [Google Scholar] [CrossRef]
- Postiglione, A.E.; Muday, G.K. The Role of ROS Homeostasis in ABA-Induced Guard Cell Signaling. Front. Plant Sci. 2020, 11, 968. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, W.; Dai, Y.; Xiao, N.; Zhang, C.; Li, H.; Lu, Y.; Wu, M.; Tao, X.; Deng, D.; et al. A maize phytochrome-interacting factor 3 improves drought and salt stress tolerance in rice. Plant Mol. Biol. 2015, 87, 413–428. [Google Scholar] [CrossRef]
- Ying, S.; Zhang, D.F.; Fu, J.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta 2012, 235, 253–266. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, S.; Pan, J.; Kong, X.; Zhou, Y.; Liu, Y.; Li, D. The overexpression of a maize mitogen-activated protein kinase gene (ZmMPK5) confers salt stress tolerance and induces defence responses in tobacco. Plant Biol. 2013, 16, 558–570. [Google Scholar]
- Riechmann, J.L. Transcriptional Regulation: A Genomic Overview. Arab. Book 2002, 1, e0085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Liu, X.; Pan, Q.; Mi, G.; Chen, F.; Yuan, L. Combined physiological, transcriptome, and genetic analysis reveals a molecular network of nitrogen remobilization in maize. J. Exp. Bot. 2020, 70, 5061–5073. [Google Scholar] [CrossRef]
- Bamsey, M.; Berinstain, A.; Dixon, M. Development of a potassium-selective optode for hydroponic nutrient solution monitoring. Anal. Chim. Acta 2012, 737, 72–82. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Firouzabadi, F.N.; Shobbar, Z.S. Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS ONE 2019, 14, e0213305. [Google Scholar] [CrossRef] [PubMed]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: Verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 1989, 975, 384–394. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Adal, A.M.; Doshi, K.; Holbrook, L.; Mahmoud, S.S. Comparative RNA-Seq analysis reveals genes associated with masculinization in female Cannabis sativa. Planta 2021, 253, 17. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Pan, Z.; Chen, L.; Dai, Y.; Wan, J.; Ye, H.; Nguyen, H.T.; Zhang, G.; Chen, H. Analysis of whole transcriptome rna-seq data reveals many alternative splicing events in soybean roots under drought stress conditions. Genes 2020, 11, 1520. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.; Quillérou, E.; Nangia, V.; Murtaza, G.; Singh, M.; Thomas, R.J.; Drechsel, P.; Noble, A.D. Economics of salt-induced land degradation and restoration. Nat. Resour. Forum. 2014, 38, 282–295. [Google Scholar] [CrossRef]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 28, 164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Blumwald, E. Transgenic salt-tolerant tomato plants accumulate salt in foliage but not in fruit. Nat. Biotechnol. 2001, 19, 765–768. [Google Scholar] [CrossRef]
- Zhang, H.X.; Hodson, J.N.; Williams, J.P.; Blumwald, E. Engineering salt-tolerant Brassica plants: Characterization of yield and seed oil quality in transgenic plants with increased vacuolar sodium accumulation. Proc. Natl. Acad. Sci. USA 2001, 98, 12832–12836. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018, 217, 523–539. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhou, X.; Tao, M.; Yuan, F.; Liu, L.; Wu, F.; Wu, X.; Xiang, Y.; Niu, Y.; Liu, F.; et al. Plant cell-surface GIPC sphingolipids sense salt to trigger Ca2+ influx. Nature 2019, 572, 341–346. [Google Scholar] [CrossRef]
- Kamran, M.; Parveen, A.; Ahmar, S.; Malik, Z.; Hussain, S.; Chattha, M.S.; Saleem, M.H.; Adil, M.; Heidari, P.; Chen, J.T. An overview of hazardous impacts of soil salinity in crops, tolerance mechanisms, and amelioration through selenium supplementation. Int. J. Mol. Sci. 2020, 21, 148. [Google Scholar] [CrossRef] [Green Version]
- Lodeyro, A.F.; Carrillo, N. Salt stress in higher plants: Mechanisms of toxicity and defensive responses. In Stress Responses in Plants: Mechanisms of Toxicity and Tolerance; Springer: Cham, Switzerland, 2015; pp. 1–33. [Google Scholar]
- Qin, H.; Huang, R. The phytohormonal regulation of Na+/K+ and reactive oxygen species homeostasis in rice salt response. Mol. Breed. 2020, 40, 47. [Google Scholar] [CrossRef]
- Hussain, M.; Ahmad, S.; Hussain, S.; Lal, R.; Ul-Allah, S.; Nawaz, A. Rice in Saline Soils: Physiology, Biochemistry, Genetics, and Management. Adv. Agron. 2018, 148, 231–287. [Google Scholar]
- Razzaq, A.; Ali, A.; Safdar, L.B.; Zafar, M.M.; Rui, Y.; Shakeel, A.; Shaukat, A.; Ashraf, M.; Gong, W.; Yuan, Y. Salt stress induces physiochemical alterations in rice grain composition and quality. J. Food Sci. 2020, 85, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Yasar, F.; Uzal, O. Effect of calcium applications on ion accumulation in different organs of pepper plant under salt stress. BIO Web Conf. 2020, 17, 00231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, Y.; Deng, C.; Deng, S.; Li, N.; Zhao, C.; Zhao, R.; Liang, S.; Chen, S. The Arabidopsis Ca2+ -dependent protein kinase CPK12 is involved in plant response to salt stress. Int. J. Mol. Sci. 2018, 19, 4062. [Google Scholar] [CrossRef] [Green Version]
- Slabu, S.; Zorb, C.; Steffens, D.; Schubert, S. Is salt stress of faba bean (Vicia faba) caused by Na+ or Cl- toxicity? J. Plant Nutr. Soil Sci. 2009, 172, 644–650. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamaji, N.; Costa, A.; Okuma, E.; Kobayashi, N.I.; Kashiwagi, T.; Katsuhara, M.; Wang, C.; Tanoi, K.; Murata, Y.; et al. OsHKT1;4-mediated Na+ transport in stems contributes to Na+ exclusion from leaf blades of rice at the reproductive growth stage upon salt stress. BMC Plant Biol. 2016, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.M.A.; Serralheiro, R.P. Soil salinity: Effect on vegetable crop growth. Management practices to prevent and mitigate soil salinization. Horticulturae 2017, 3, 30. [Google Scholar] [CrossRef]
- Yasir, M.; He, S.; Sun, G.; Geng, X.; Pan, Z.; Gong, W.; Jia, Y.; Du, X. A genome-wide association study revealed key SNPs/genes associated with salinity stress tolerance in upland cotton. Genes 2019, 10, 829. [Google Scholar] [CrossRef] [Green Version]
- Munns, R. Comparative physiology of salt and water stress. Plant Cell Environ. 2002, 25, 239–250. [Google Scholar] [CrossRef]
- Sellamuthu, G.; Jegadeeson, V.; Sajeevan, R.S.; Rajakani, R.; Parthasarathy, P.; Raju, K.; Shabala, L.; Chen, Z.H.; Zhou, M.; Sowdhamini, R.; et al. Distinct Evolutionary Origins of Intron Retention Splicing Events in NHX1 Antiporter Transcripts Relate to Sequence Specific Distinctions in Oryza Species. Front. Plant Sci. 2020, 11, 267. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Smith, S.A. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genom. 2013, 14, 328. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Zhu, P.; Qian, L.; Qian, X.; Mi, Y.; Lin, Z.; Dong, S.; Aronsson, H.; Zhang, H.; Cheng, J. Identification and fine mapping of qGR6.2, a novel locus controlling rice seed germination under salt stress. BMC Plant Biol. 2021, 21, 36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Tong, Y.; Li, Y.; Cheng, Z.M.; Zhong, Y. Genome-wide identification of the HKT genes in five Rosaceae species and expression analysis of HKT genes in response to salt-stress in Fragaria vesca. Genes Genom. 2019, 41, 325–336. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, H.; Qiu, Z.; Hu, B.; Zeng, B.; Zhong, C.; Fan, C. Comprehensive analysis of SNRK gene family and their responses to salt stress in eucalyptus grandis. Int. J. Mol. Sci. 2019, 20, 2786. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ye, Y.; Xu, M.; Feng, L.; Xu, L.A. Roles of the SPL gene family and miR156 in the salt stress responses of tamarisk (Tamarix chinensis). BMC Plant Biol. 2019, 19, 370. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Téllez, S.; Anoman, A.D.; Alcántara-Enguídanos, A.; Garza-Aguirre, R.A.; Alseekh, S.; Ros, R. PGDH family genes differentially affect Arabidopsis tolerance to salt stress. Plant Sci. 2020, 290, 110284. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, P.; Zhu, R.; Fu, J.; Su, J.; Zheng, J.; Wang, Z.; Wang, D.; Gong, Q. Autophagy is rapidly induced by salt stress and is required for salt tolerance in Arabidopsis. Front. Plant Sci. 2017, 8, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awlia, M.; Alshareef, N.; Saber, N.; Korte, A.; Oakey, H.; Panzarová, K.; Trtílek, M.; Negrão, S.; Tester, M.; Julkowska, M.M. Genetic mapping of the early responses to salt stress in Arabidopsis thaliana. Plant J. 2021, 107, 544–563. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Subramanian, S.; Smith, D.L. Plant endophytes promote growth and alleviate salt stress in Arabidopsis thaliana. Sci. Rep. 2020, 10, 12740. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Q.; Nan, H.; Li, X.; Lu, S.; Zhao, X.; Liu, B.; Guo, C.; Kong, F.; Cao, D. Overexpression of GmFDL19 enhances tolerance to drought and salt stresses in soybean. PLoS ONE 2017, 12, e0179554. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Song, G.; Guo, W.; Wang, W.; Zhao, H.; Gao, T.; Lv, Q.; Yang, X.; Xu, F.; Dong, Y.; et al. Dynamic Changes in Genome-Wide Histone3 Lysine27 Trimethylation and Gene Expression of Soybean Roots in Response to Salt Stress. Front. Plant Sci. 2019, 10, 1031. [Google Scholar] [CrossRef]
- Li, M.; Chen, R.; Jiang, Q.; Sun, X.; Zhang, H.; Hu, Z. GmNAC06, a NAC domain transcription factor enhances salt stress tolerance in soybean. Plant Mol. Biol. 2021, 105, 333–345. [Google Scholar] [CrossRef]
- Verma, P.K.; Verma, S.; Tripathi, R.D.; Pandey, N.; Chakrabarty, D. CC-type glutaredoxin, OsGrx_C7 plays a crucial role in enhancing protection against salt stress in rice. J. Biotechnol. 2021, 329, 192–203. [Google Scholar] [CrossRef]
- Yan, F.; Wei, H.; Ding, Y.; Li, W.; Liu, Z.; Chen, L.; Tang, S.; Ding, C.; Jiang, Y.; Li, G. Melatonin regulates antioxidant strategy in response to continuous salt stress in rice seedlings. Plant Physiol. Biochem. 2021, 165, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Zhang, J.; Li, W.; Ding, Y.; Zhong, Q.; Xu, X.; Wei, H.; Li, G. Exogenous melatonin alleviates salt stress by improving leaf photosynthesis in rice seedlings. Plant Physiol. Biochem. 2021, 163, 367–375. [Google Scholar] [CrossRef]
- Li, P.C.; Yang, X.Y.; Wang, H.M.; Pan, T.; Yang, J.Y.; Wang, Y.Y.; Xu, Y.; Yang, Z.F. Metabolic responses to combined water deficit and salt stress in maize primary roots. J. Integr. Agric. 2021, 20, 109–119. [Google Scholar] [CrossRef]
- Ren, J.; Ye, J.; Yin, L.; Li, G.; Deng, X.; Wang, S. Exogenous melatonin improves salt tolerance by mitigating osmotic, ion, and oxidative stresses in maize seedlings. Agronomy 2020, 10, 663. [Google Scholar] [CrossRef]
- Weng, Q.; Zhao, Y.; Yanan, Z.; Song, X.; Yuan, J.; Liu, Y. Identification of salt stress-responsive proteins in maize (Zea may) seedlings using itraq-based proteomic technique. Iran. J. Biotechnol. 2021, 19, e2512. [Google Scholar] [PubMed]
- Li, J.; Liu, J.; Zhu, T.; Zhao, C.; Li, L.; Chen, M. The role of melatonin in salt stress responses. Int. J. Mol. Sci. 2019, 20, 1735. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Zhao, Y.; Wang, Y.; Shi, Z.; Zhang, P.; Zhang, Y.; Song, W.; Zhao, J. Comparative Proteomics of Contrasting Maize Genotypes Provides Insights into Salt-Stress Tolerance Mechanisms. J. Proteome Res. 2018, 17, 141–153. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, Y.; Chen, K.; Kong, M.; Song, W.; Lu, B.; Shi, Y.; Zhao, Y.; Zhao, J. Mapping of quantitative trait loci for seedling salt tolerance in maize. Mol. Breed. 2019, 39, 64. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, X.; Wang, L.; Cao, Y.; Song, W.; Shi, J.; Lai, J.; Jiang, C. A HAK family Na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants 2019, 5, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, D.; Pudussery, M.V.; Kumar, R.; Pallete, A.; Markley, P.; Bridges, W.C.; Sekhon, R.S. Characterization of natural genetic variation identifies multiple genes involved in salt tolerance in maize. Funct. Integr. Genom. 2020, 20, 261–275. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, Y.; Li, J.; Zhang, P.; Chen, K.; Song, W.; Wang, X.; Yang, J.; Lu, X.; Lu, B.; et al. Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method. Plant Biotechnol. J. 2021, 19, 1937–1951. [Google Scholar] [CrossRef]
- Mehlmer, N.; Wurzinger, B.; Stael, S.; Hofmann-Rodrigues, D.; Csaszar, E.; Pfister, B.; Bayer, R.; Teige, M. The Ca2+-dependent protein kinase CPK3 is required for MAPK-independent salt-stress acclimation in Arabidopsis. Plant J. 2010, 63, 484–498. [Google Scholar] [CrossRef] [Green Version]
- Abulfaraj, A.A. Stepwise signal transduction cascades under salt stress in leaves of wild barley (Hordeum spontaneum). Biotechnol. Biotechnol. Equip. 2020, 34, 860–872. [Google Scholar] [CrossRef]
- Su, Y.; Guo, A.; Huang, Y.; Wang, Y.; Hua, J. GhCIPK6a increases salt tolerance in transgenic upland cotton by involving in ROS scavenging and MAPK signaling pathways. BMC Plant Biol. 2020, 20, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, L.; Pan, F.; Guo, W.; Chen, B.; Yang, H.; Wang, G.; Li, X. Transcriptomic analysis reveals ethylene signal transduction genes involved in pistil development of pumpkin. PeerJ 2020, 8, e9677. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Q.; Gao, Z.; Wang, Y.; Liu, Y.; Ma, Z.; Chen, Y.; Zhang, Y.; Yan, F.; Li, J. Analysis of phytohormone signal transduction in sophora alopecuroides under salt stress. Int. J. Mol. Sci. 2021, 22, 7313. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lu, B.; Liu, L.; Duan, W.; Jiang, D.; Li, J.; Zhang, K.; Sun, H.; Zhang, Y.; Li, C.; et al. Melatonin promotes seed germination under salt stress by regulating ABA and GA3 in cotton (Gossypium hirsutum L.). Plant Physiol. Biochem. 2021, 162, 506–516. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer (5′->3′) |
|---|---|
| LOC115697737 | GCTGTTGAGATTTCAGTACTTGCC//TACCTCCTCAAACCATGCAC |
| LOC115698674 | ACTGTTTTATACACTTGGTTCCTTT//AAGTGGAGCGTGAAGAGTGG |
| LOC115700775 | TGATTGAGCGTAGTGGGAAGC//TCCCTGCCAAAAACCTCCAA |
| LOC115704163 | ACCTGCATTCTACATTAGGCA//TGCGTGCATGTAACAAGATTAAA |
| LOC115707929 | ACTAATCAAGGCACAACACACC//TTCTCCCAGCCCAGGTGTAA |
| LOC115709853 | GCAGCTCTATGCGAGTATGC//AGTGTCAACCCTAAAATGAGTTGAT |
| LOC115710226 | AGCAAGTGAAACAGCAACCAA//TCTTCTGTGTTCATTCCTACTTCT |
| LOC115710658 | AGTCTTTAAAATCTCATTGCGCGT//CTTGGCCAGTAAAACGCTCG |
| LOC115712923 | GGATGGGTTGTGTGGAGGAG//GTGCAAATTTAAAAGCACCTTTGGA |
| LOC115715759 | TGTGAGTGACTTGGACTGTGT//AAACTACGAAGCACCTGCAA |
| LOC115716727 | TGGCTCAGTTGCTCTTGATGA//GGGTATCATGGTCGGTGCC |
| LOC115718665 | CGGAACTTCTATAAATATTCGGTGT//GGCTTAGAAGCCTTACAACCAT |
| LOC115719017 | ATGAGTGGGTCACACAGTGG//ATGAGTGGGTCACACAGTGG |
| LOC115720256 | GGGCACAACAGAAGCACTTG//ACAACACATGTTCATGCCACAA |
| LOC115723301 | ACAAAGCTCTTGCGTTTGGTT//TTGGGAGCTAGTTGTGACCT |
| Sample | Clean Base (bp) | Total Read Pairs | GC (%) | Q30 (%) | Total Mapped Reads | Unique Mapped Reads | Multiple Mapped Reads |
|---|---|---|---|---|---|---|---|
| K94-1 | 8,004,702,300 | 26,682,341 | 43.86; 43.83 | 93.21; 92.33 | 22,619,678 (84.77%) | 21,793,155 (81.68%) | 826,523 (3.10%) |
| K94-2 | 8,138,507,400 | 27,128,358 | 44.05; 44.03 | 93.39; 93.22 | 22,827,417 (84.15%) | 21,999,162 (81.09%) | 828,255 (3.05%) |
| K94-3 | 7,414,971,000 | 24,716,570 | 43.88; 43.84 | 93.33; 90.79 | 20,871,006 (84.44%) | 20,066,538 (81.19%) | 804,468 (3.25%) |
| K94h-1 | 6,796,399,800 | 22,654,666 | 43.86; 43.83 | 92.79; 90.57 | 19,084,286 (84.24%) | 18,423,842 (81.32%) | 660,444 (2.92%) |
| K94h-2 | 7,230,943,800 | 24,103,146 | 43.91; 43.89 | 92.77; 90.54 | 20,036,029 (83.13%) | 19,334,787 (80.22%) | 701,242 (2.91%) |
| K94h-3 | 7,273,562,700 | 24,245,209 | 43.82; 43.79 | 92.69; 90.82 | 20,235,640 (83.46%) | 19,532,476 (80.56%) | 703,164 (2.90%) |
| W20-1 | 7,413,545,700 | 24,711,819 | 43.54; 43.52 | 93.68; 93.35 | 20,677,010 (83.67%) | 19,893,900 (80.50%) | 783,110 (3.17%) |
| W20-2 | 8,590,370,100 | 28,634,567 | 44.14; 44.09 | 93.95; 92.41 | 24,162,827 (84.38%) | 23,211,655 (81.06%) | 951,172 (3.32%) |
| W20-3 | 8,981,841,900 | 29,939,473 | 43.74; 43.72 | 93.85; 93.32 | 25,241,745 (84.31%) | 24,270,435 (81.07%) | 971,310 (3.24%) |
| W20h-1 | 7,666,976,100 | 25,556,587 | 43.9; 43.88 | 93.24; 92.73 | 21,712,432 (84.96%) | 20,915,160 (81.84%) | 797,272 (3.12%) |
| W20h-2 | 6,863,257,500 | 22,877,525 | 44.1; 44.08 | 93.56; 93.43 | 19,593,613 (85.65%) | 18,902,678 (82.63%) | 690,935 (3.02%) |
| W20h-3 | 7,034,013,300 | 23,446,711 | 43.86; 43.81 | 93.49; 92.22 | 19,928,289 (84.99%) | 19,227,153 (82.00%) | 701,136 (2.99%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Zhang, C.; Huang, S.; Chang, L.; Li, J.; Tang, H.; Dey, S.; Biswas, A.; Du, D.; Li, D.; et al. Key Cannabis Salt-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Varieties. Agronomy 2021, 11, 2338. https://doi.org/10.3390/agronomy11112338
Zhang J, Zhang C, Huang S, Chang L, Li J, Tang H, Dey S, Biswas A, Du D, Li D, et al. Key Cannabis Salt-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Varieties. Agronomy. 2021; 11(11):2338. https://doi.org/10.3390/agronomy11112338
Chicago/Turabian StyleZhang, Jiangjiang, Cuiping Zhang, Siqi Huang, Li Chang, Jianjun Li, Huijuan Tang, Susmita Dey, Ashok Biswas, Dengxiang Du, Defang Li, and et al. 2021. "Key Cannabis Salt-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Varieties" Agronomy 11, no. 11: 2338. https://doi.org/10.3390/agronomy11112338