Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier

{kind=link}
{kind=link}
{kind=link}
Abstract
: In past two decades poly lactic-co-glycolic acid (PLGA) has been among the most attractive polymeric candidates used to fabricate devices for drug delivery and tissue engineering applications. PLGA is biocompatible and biodegradable, exhibits a wide range of erosion times, has tunable mechanical properties and most importantly, is a FDA approved polymer. In particular, PLGA has been extensively studied for the development of devices for controlled delivery of small molecule drugs, proteins and other macromolecules in commercial use and in research. This manuscript describes the various fabrication techniques for these devices and the factors affecting their degradation and drug release.1. Introduction
A considerable amount of research has been conducted on drug delivery by biodegradable polymers since their introduction as bioresorbable surgical devices about three decades ago. Amongst all the biomaterials, application of the biodegradable polymer poly lactic-co-glycolic acid (PLGA) has shown immense potential as a drug delivery carrier and as scaffolds for tissue engineering. PLGA are a family of FDA-approved biodegradable polymers that are physically strong and highly biocompatible and have been extensively studied as delivery vehicles for drugs, proteins and various other macromolecules such as DNA, RNA and peptides [1–3]. PLGA is most popular among the various available biodegradable polymers because of its long clinical experience, favorable degradation characteristics and possibilities for sustained drug delivery. Recent literature has shown that degradation of PLGA can be employed for sustained drug release at desirable doses by implantation without surgical procedures. Additionally, it is possible to tune the overall physical properties of the polymer-drug matrix by controlling the relevant parameters such as polymer molecular weight, ratio of lactide to glycolide and drug concentration to achieve a desired dosage and release interval depending upon the drug type [4–6]. However the potential toxicity from dose dumping, inconsistent release and drug-polymer interactions require detailed evaluation. Here we present a review on the PLGA primarily as a delivery vehicle for various drugs, proteins and other macromolecules in commercial use and in research. We also present possible directions for future uses of PLGA in drug delivery applications.
2. Biodegradable Polymers
Biodegradable materials are natural or synthetic in origin and are degraded in vivo, either enzymatically or non-enzymatically or both, to produce biocompatible, toxicologically safe by-products which are further eliminated by the normal metabolic pathways. The number of such materials that are used in or as adjuncts in controlled drug delivery has increased dramatically over the past decade. The basic category of biomaterials used in drug delivery can be broadly classified as (1) synthetic biodegradable polymers, which includes relatively hydrophobic materials such as the α-hydroxy acids (a family that includes poly lactic-co-glycolic acid, PLGA), polyanhydrides, and others, and (2) naturally occurring polymers, such as complex sugars (hyaluronan, chitosan) and inorganics (hydroxyapatite) [7–9]. The breath of materials used in drug delivery arises from the multiplicity of diseases, dosage range and special requirements that may apply. Biocompatibility is clearly important, although it is important to note that biocompatibility is not an intrinsic property of a material, but depends on the biological environment and the tolerability that exists with respect to specific drug-polymer-tissue interactions [9].
2.1. Poly Lactic-co-Glycolic Acid (PLGA)
Polyester PLGA is a copolymer of poly lactic acid (PLA) and poly glycolic acid (PGA). It is the best defined biomaterial available for drug delivery with respect to design and performance. Poly lactic acid contains an asymmetric α-carbon which is typically described as the D or L form in classical stereochemical terms and sometimes as R and S form, respectively. The enantiomeric forms of the polymer PLA are poly D-lactic acid (PDLA) and poly L-lactic acid (PLLA). PLGA is generally an acronym for poly D,L-lactic-co-glycolic acid where D- and L- lactic acid forms are in equal ratio.
2.1.1. Physico-Chemical Properties
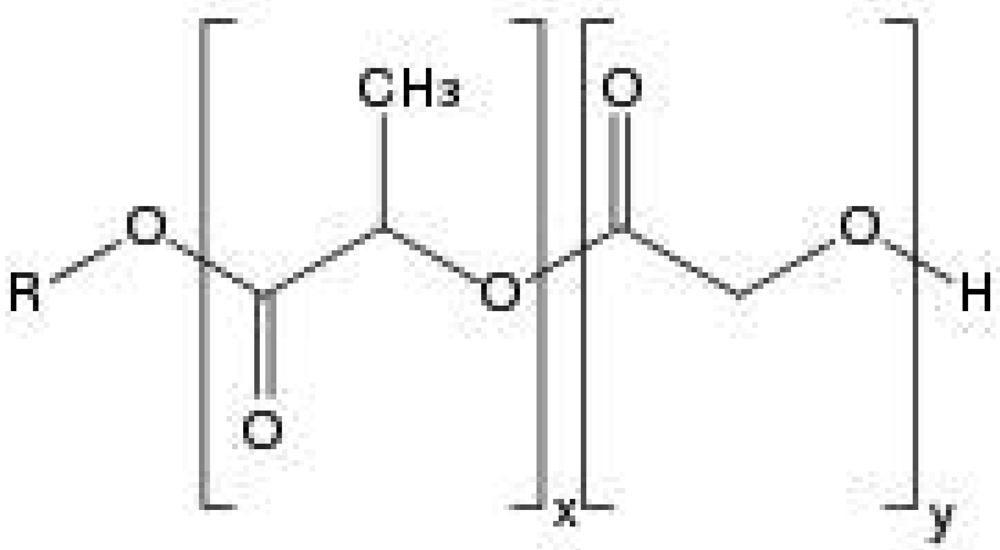
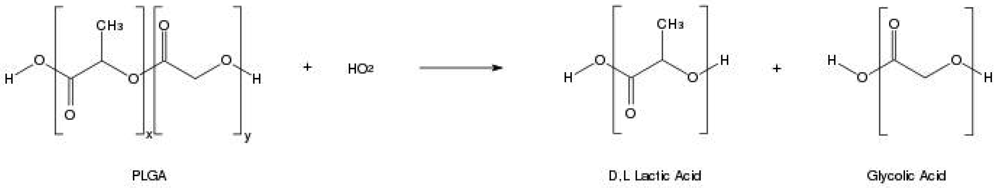
In order to design a better controlled drug delivery device, it is essential to understand the physical, chemical and biological properties of PLGA. The physicochemical properties of optically active PDLA and PLLA are nearly the same. In general, the polymer PLA can be made in highly crystalline form (PLLA) or completely amorphous (PDLA) due to disordered polymer chains. PGA is void of any methyl side groups and shows highly crystalline structure in contrast to PLA as shown in Figure 1. PLGA can be processed into almost any shape and size, and can encapsulate molecules of virtually any size. It is soluble in wide range of common solvents including chlorinated solvents, tetrahydofuran, acetone or ethyl acetate [7,10]. In water, PLGA biodegrades by hydrolysis of its ester linkages (Figure 2). Presence of methyl side groups in PLA makes it more hydrophobic than PGA and hence lactide rich PLGA copolymers are less hydrophilic, absorb less water and subsequently degrade more slowly. Due to the hydrolysis of PLGA, parameters that are typically considered invariant descriptions of a solid formulation can change with time, such as the glass transition temperature (Tg), moisture content and molecular weight. The effect of these polymer properties on the rate of drug release from biodegradable polymeric matrices has been widely studied. The change in PLGA properties during polymer biodegradation influences the release and degradation rates of incorporated drug molecules. PLGA physical properties themselves have been shown to depend upon multiple factors, including the initial molecular weight, the ratio of lactide to glycolide, the size of the device, exposure to water (surface shape) and storage temperature [11]. Mechanical strength of the PLGA is affected by physical properties such as molecular weight and polydispersity index. These properties also affect the ability to be formulated as a drug delivery device and may control the device degradation rate and hydrolysis. Recent studies have found, however, that the type of drug also plays a role in setting the release rate [12]. Mechanical strength, swelling behavior, capacity to undergo hydrolysis and subsequently biodegradation rate of the polymer are directly influenced by the degree of crystallinity of the PLGA, which is further dependent on the type and molar ratio of the individual monomer components in the copolymer chain. Crystalline PGA, when co-polymerized with PLA, reduces the degree of crystallinity of PLGA and as a result increase the rate of hydration and hydrolysis. As a rule, higher content of PGA leads to quicker rates of degradation with an exception of 50:50 ratio of PLA/PGA, which exhibits the fastest degradation, with higher PGA content leading to increased degradation interval below 50%. Degree of crystallinity and melting point of the polymers are directly related to the molecular weight of the polymer. The Tg (glass transition temperature) of the PLGA copolymers are reported to be above the physiological temperature of 37 °C and hence are glassy in nature, thus exhibiting fairly rigid chain structure. It has been further reported that Tg of PLGAs decrease with a decrease of lactide content in the copolymer composition and with a decrease in molecular weight [13]. Commercially available PLGA polymers are usually characterized in terms of intrinsic viscosity, which is directly related to their molecular weights.
2.1.2. Pharmacokinectic and Biodistribution Profile
The drug delivery specific vehicle, i.e., PLGA, must be able to deliver its payload with appropriate duration, biodistribution and concentration for the intended therapeutic effect. Therefore, design essentials, including material, geometry and location must incorporate mechanisms of degradation and clearance of the vehicle as well as active pharmaceutical ingredients (API). Biodistribution and pharmacokinetics of PLGA follows a non-linear and dose-dependent profile [14]. Furthermore, previous studies suggest that both blood clearance and uptake by the mononuclear phagocyte system (MPS) may depend on dose and composition of PLGA carrier systems [15]. Additionally whole-body autoradiography and quantitative distribution experiments indicate that some formulations of PLGA, such as nanoparticles, accumulate rapidly in liver, bone marrow, lymph nodes, spleen and peritoneal macrophages. The degradation of the PLGA carriers is quick on the initial stage (around 30%) and slows eventually to be cleared by respiration in the lung [16]. To address these limitations, studies have investigated the role of surface modification, suggesting that incorporation of surface modifying agents can significantly increase blood circulation half-life [17].
2.2. Copolymers of PLGA
The need for better delivery formulations that incorporate a variety in drugs and methods of administration has resulted in the development of various types of block copolymers of polyesters with poly ethylene glycol (PEG). PLGA/PEG block copolymers have been processed as diblock (PLGA-PEG) [18,19] or triblock molecules with both ABA (PLGA-PEG-PLGA) [20] and BAB (PEG-PLGA-PEG) [21] types. In diblock types, PEG chains orient themselves towards the external aqueous phase in micelles, thus surrounding the encapsulated species. This layer of PEG acts as a barrier and reduces the interactions with foreign molecules by steric and hydrated repulsion, giving enhanced shelf stability [22]. However, the addition of PEG to the system also results in reduction of encapsulation efficiency for drugs and proteins, even with the most appropriate fabrication techniques. The reduced drug incorporation may be due to steric interference of drug/protein-polymer interaction by the PEG chains. The precise mechanism for this effect is unclear. Better release kinetics from formulations of diblock copolymers have been demonstrated in comparison to PLGA alone. Various mechanisms of targeted delivery of drugs from diblock nanoparticles have also been reported [18,23,24].
Triblock copolymers of both ABA and BAB type can act as a thermogel with an A-block covalently coupled with a B-block via ester link. The copolymer is usually a free flowing solution at low temperature and can form a high viscosity gel at body temperature. These temperature-responsive copolymers, PLGA-PEG-PLGA or PEG-PLGA-PEG, are a kind of block copolymers composed of hydrophobic PLGA segments and hydrophilic PEG segments. The hydrophobic PLGA segments form associative crosslinks and the hydrophilic PEG segments allow the copolymer molecules to stay in solution. At lower temperatures, hydrogen bonding between hydrophilic PEG segments and water molecules dominates the aqueous solution, resulting in their dissolution in water. As the temperature increases, the hydrogen bonding becomes weaker, while hydrophobic forces among the PLGA segments are strengthened, leading to solution-gel transition. The ease of handling during fabrication, formulation, filtration and filling makes such thermoresponsive polymers attractive candidates. Drug and/or protein release from both ABA and BAB copolymers occurs by two principal mechanisms: (i) drug diffusion from the hydrogel during the initial release phase; and (ii) release of drug by the erosion of the hydrogel matrix during the later phase. During the degradation of a PEG-PLGA-PEG gel, there is a preferential mass loss of PEG-rich components. Therefore, the remaining gel becomes more hydrophobic in an aqueous environment, resulting in less water content [20,25–28]. This motif can also be applied to other co-polymer combinations, including but not limited to various copolymers of PLGA and polycaprolactone [29,30].
3. Fabrication Techniques for PLGA Carriers
Drugs and proteins are the most rapidly growing class of pharmaceuticals for which controlled or targeted release is used to increase specificity, lower toxicity and decrease the risk associated with treatment. However, the stability and delivery challenges associated with these agents have limited the number of marketed products. Maintaining adequate shelf-life of peptide and protein drugs often requires solid-state formulation to limit hydrolytic degradation reactions [31]. Drug delivery of peptides and proteins may also require parenteral formulations to avoid degradation in the digestive tract and first pass metabolism, while the short circulating half-lives of peptides and proteins contribute to the need for parenteral formulations that will reduce dosing frequency. In order to avoid the inconvenient surgical insertion of large implants, injectable biodegradable and biocompatible PLGA particles (microspheres, microcapsules, nanocapsules, nanospheres) could be employed for controlled-release dosage forms. Drugs formulated in such polymeric devices are released either by diffusion through the polymer barrier, or by erosion of the polymer material, or by a combination of both diffusion and erosion mechanisms. In addition to its biocompatibility, drug compatibility, suitable biodegradation kinetics and mechanical properties, PLGA can be easily processed and fabricated in various forms and sizes. This section describes various fabrication techniques of PLGA controlled drug delivery devices [9].
3.1. Microparticle Preparation Techniques
3.1.1. Solvent Evaporation Method
Single emulsion process
Oil-in-water emulsification processes are examples of single emulsion processes. Polymer in the appropriate amount is first dissolved in a water immiscible, volatile organic solvent (e.g., dichloromethane (DCM)) in order to prepare a single phase solution. The drug of particle size around 20–30 μm is added to the solution to produce a dispersion in the solution. This polymer dissolved drug dispersed solution is then emulsified in large volume of water in presence of emulsifier (polyvinyl alcohol (PVA) etc.) in appropriate temperature with stirring. The organic solvent is then allowed to evaporate or extracted to harden the oil droplets under applicable conditions. In former case, the emulsion is maintained at reduced or atmospheric pressure with controlling the stir rate as solvent evaporates. In the latter case, the emulsion is transferred to a large quantity of water (with or without surfactant) or other quench medium to diffuse out the solvent associated with the oil droplets. The resultant solid microspheres are then washed and dried under appropriate conditions to give a final injectable microsphere formulation [32–35].
Double (Multiple) emulsion process
Water-in-oil-in-water emulsion methods are best suited to encapsulate water-soluble drugs like peptides, proteins, and vaccines, unlike single emulsion methods which is ideal for water-insoluble drugs like steroids. First, an appropriate amount of drug is dissolved in aqueous phase (deionised water) and then this drug solution is added to organic phase consisting of PLGA and/or PLA solution in DCM or chloroform with vigorous stirring to yield a water-in-oil emulsion. Next, the water-in-oil primary emulsion is added to PVA aqueous solution and further emulsified for around a minute at appropriate stress mixing conditions. The organic solvent is then allowed to evaporate or is extracted in the same manner as oil-in-water emulsion techniques. In double emulsion processes, choice of solvents and stirring rate predominantly affects the encapsulation efficiency and final particle size [32,36,37].
3.1.2. Phase Separation (Coacervation)
Coacervation is a process focused on preparation of micrometer sized biodegradable polymer encapsulation formulations via liquid-liquid phase separation techniques. The process yields two liquid phases (phase separation) including the polymer containing coacervate phase and the supernatant phase depleted in polymer. The drug which is dispersed/dissolved in the polymer solution is coated by the coacervate. Thus, the coacervation process includes the following three steps as reported in literature [38–40]
Phase separation of the coating polymer solution,
Adsorption of the coacervate around the drug particles, and
Quenching of the microspheres.
Solutions are prepared by mixing polymer and solvent in appropriate ratios. Hydrophilic drugs like peptides and proteins are dissolved in water and dispersed in polymer solution (water-in-oil emulsion). Hydrophobic drugs like steroids are either solubilized or dispersed in the polymer solution (oil-in-water emulsion). Gradual addition of organic medium to the polymer-drug-solvent phase while stirring, extracts the polymer solvent resulting in phase separation of polymer by forming a soft coacervate of drug containing droplets. The size of these droplets can be controlled by varying stirring rate and temperature of the system. The system is then quickly dipped into a medium in which it is not soluble (both organic or aqueous) to quench these microdroplets. The soaking time in the quenching bath controls the coarsening and hardness of the droplets. The final form of the microspheres is collected by washing, sieving, filtration, centrifugation or freeze drying. The processing parameters including polymer concentration, quenching temperature, quenching time and solvent composition affect the morphology and size of the microspheres [41–43].
3.1.3. Spray Drying
Emulsion techniques require precise control of processing parameters for higher encapsulation efficiency, and phase separation techniques tend to produce agglomerated particles and also require removal of large quantities of the organic phase from the microspheres. This makes the process difficult for mass production. Alternatively, spray drying is very rapid, convenient and has very few processing parameters, making it suitable for industrial scalable processing. In this process, drug/protein/peptide loaded microspheres are prepared by spraying a solid-in-oil dispersion or water-in-oil emulsion in a stream of heated air. The type of drug (hydrophobic or hydrophilic) decides the choice of solvent to be used in the process. The nature of solvent used, temperature of the solvent evaporation and feed rate affects the morphology of the microspheres. The main disadvantage of this process is the adhesion of the microparticles to the inner walls of the spray-dryer. Various spray drying techniques have been reported [44–49]. This method is known to encapsulate all kinds of drugs/peptides/proteins into microparticles without significant loss in their biological activity. Recently, coaxial capillary flows have become a preferable technique to produce monodispersed micro/nanoparticles with either simple or core-shell structure because of their precise control on mean particle size [50,51]. Using these techniques, processing parameters such as orientation of jets, material flow rates, and rate of solvent extraction can be controlled to create uniform and well-centered double-walled microspheres exhibiting a controllable shell thickness [52]. Additionally, microfluidic devices can incorporate the use of electrostatic forces to control the size and shape of particles for increased tuning of release characteristics [53].
3.2. Nanoparticle Preparation Techniques
Various groups have also reported successful preparation of PLGA nanoparticles. All the above described microparticle techniques can be employed for manufacturing PLGA nanoparticles (nanospheres and nanocapsules) by adjusting the processing parameters. These parameters usually use a small dispersed phase ratio and rate of stirring. The most common method used for the preparation of solid, polymeric nanoparticles is the emulsification-solvent evaporation technique. However, this method is primarily used in encapsulation of hydrophobic drugs. A modification on this procedure called the double or multiple emulsion technique has become the favored protocol for encapsulating hydrophilic compounds and proteins [37]. Nanoparticles can also be synthesized by nanoprecipitation methods. Polymer and drug are dissolved in acetone and added to an aqueous solution containing Pluronic F68. The acetone is evaporated at appropriate temperatures and reduced pressures leaving behind the polymer encapsulated nanoparticles with drug [54]. Salting out is another method in which a water-in-oil emulsion is first formed containing polymer, solvent (usually non chlorinated like acetone), salt (e.g., magnesium acetate tetrahydrate) and stabilizer. Water is then added to the solution until the volume is sufficient to diffuse acetone into the water, resulting in nanoparticle formulations [55–58].
3.3. Implant Preparation Techniques
3.3.1. Solvent-Casting and Compression Molding
Solvent casting is a method to fabricate a macroscopic millimeter size formulation which can be implanted or inserted for long term medication [59]. Large size, macroscopic formulations act as a reservoir for drug that can be delivered over a longer interval. In this method, a polymer and drug mixture is dissolved in a common solvent (e.g., acetone) in the desirable proportion, and the solvent is cast at around 60 °C until complete evaporation. Their resultant structure is a composite material of the drug together with the polymer. The solvent cast material is then compression molded into its desired geometry at around 80 °C and 25,000 psi to final density of 1 g/cc. This implant can be subcutaneously delivered in the body. The main advantage of this approach over micro/nanospheres is related to the ability to manage adverse events, since implants retain a degree of reversibility which is not available in depot mechanisms [12,59].
3.3.2. Extrusion
Solvent-casting methods are not ideal for industrial scale-up for many reasons. First, the process requires large amounts of organic solvent to dissolve PLGA and the active pharmaceutical agent (API) to combinethe drug and polymer for pellet fabrication. Such systems are also open to the risk of denaturation of drugs and/or proteins during encapsulation because of the use of organic solvents. Denatured species are therapeutically inactive and can cause unpredictable side effects, such as immunogenicity or other toxicity. Second, this process requires a very long time to completely remove solvents from the resulting material. Third, solvent-casting and compression molding are not continuous processes, which may increase batch-to-batch variation in the composition of implants as well as cost of manufacturing [60].
Unlike solvent-casting, extrusion is a continuous process of drawing polymer-drug mixture through a die to create implants of fixed cross-sectional profile without any use of solvent. The process requires an extruder and polymer-drug mixture with required micron size feed material. During the process, the polymer-drug mixture is heated to semi-liquid state by a combination of heating elements and shear stress from the extrusion screw. The screw pushes the mixture through the die. The resulting extrudate is then cooled and solidified before cutting into desired lengths for implants or other applications [61]. Exposure of drug to high temperature can be disadvantageous as denaturation can take place. Therefore, the extrusion process possess a limitation on the drugs that can be used based on their melting point, polymorph stability and chemical interactions with PLGA.
3.4. Miscellaneous Systems
3.4.1. Multi-Drug Delivery Devices
A pulsated drug release profile is sometimes preferred over the continuous presence of the drug, which may lead to downregulation of receptors or the development of tolerance. Novel multi-pulsatile delivery devices have developed in which there is a predetermined off period followed by rapid and transient drug release in a cycle until the device is degraded. Such devices have also been shown to be capable of releasing multiple drugs for a sequence of cycles. PLGA is also an attractive candidate for devices with multi-drug delivery and multi-pulsed delivery applications because of its desirable and tunable properties [62–65]. Such systems can be extended to achieve programmed delivery of multiple drugs in a predetermined sequence of pulses from a single device [64]. For example, a single biodegradable polymeric microchip can be constructed of PLGA and/or PLLA in combination with multiple drugs to achieve pulsatile drug delivery over a long period of time [65].
3.4.2. Supercritical CO2
More recently, alternative methods of fabrication using supercritical CO2 as the foaming agent have also been proposed to overcome some limitations that result from conventional methods of microporous foam formation, including solvent-casting and particulate leaching techniques. Conventional methods usually require large amounts of organic solvents and thus require additional extensive purification steps to remove the residual solvent. Using supercritical CO2 as a foaming agent, organic solvents can be minimized or eliminated in production of PLGA foams [66]. Polymer encapsulated with drug/protein is usually manufactured using emulsion techniques. to apply this technique, a solution is placed into a CO2 pressure cell immediately after emulsion. Under high pressure CO2, the glass transition temperature of PLGA is reduced, resulting in a CO2 dissolved polymer liquid. After such pressurization and rapid depressurization sequence, the thermodynamic instability of CO2 molecules leads to their clustering inside the liquid polymer. As CO2 leaves, the emulsion results in a porous polymer structure [66,67]. Since micro-porous foams have higher surface-to-volume ratios, more efficient drug release has been reported [68].
3.4.3. Multifunctional PLGA Micro/Nanoparticles
PLGA micro/nanoparticles have also been used for multiple applications in a single formulation. PLGA particles may be used to encapsulate absorption and fluorescence dyes in addition to a drug for multimodal imaging using fluorescence (FL), ultrasound (US), or photoacoustic tomography (PAT) [69]. Such multifunctional particles can also be formulated from a component material to reduce side effects of the encapsulated drug [70]. These particles can not only serve as a delivery system for the encapsulated drug but also reduce the harmful side effects through targeted drug delivery. However, the chemical reactivity among these adjutants needs to be assessed before determination of a final formulation. The fabrication of such multifunctional particles is usually achieved through emulsion techniques. However, the high rate clearance of micro/nanoparticles by the body's reticuloendothelial system (RES) and the difficultyfor such particles to penetrate many tissue types may limit their use to the vascular space [69–71].
4. Drug Release Behavior
4.1. Biphasic Release
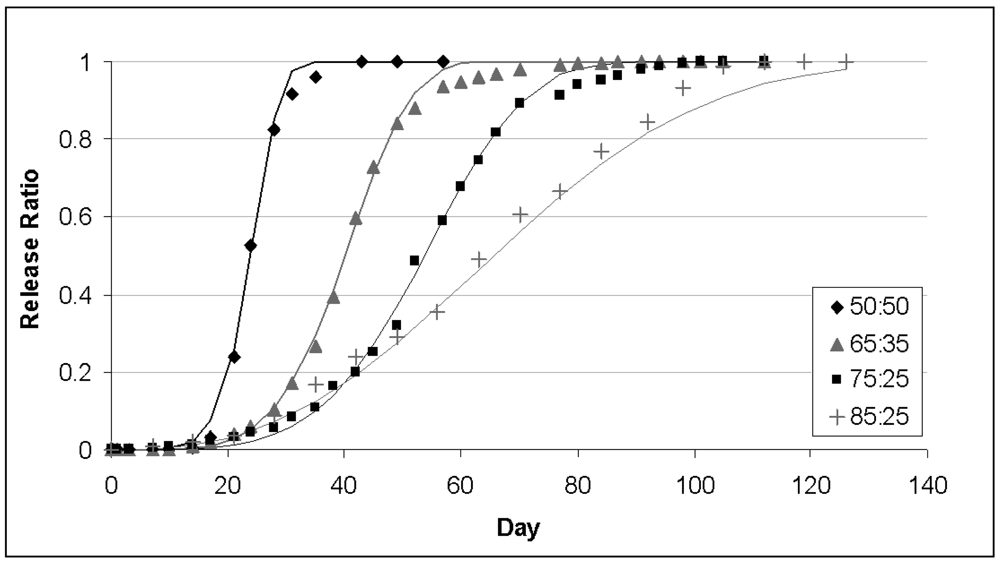
PLGA copolymer undergoes degradation by hydrolysis or biodegradation through cleavage of its backbone ester linkages into oligomers and, finally monomers. This has been demonstrated in both in vivo and in vitro for various drug types and proteins with different polymer ratios [72,73]. The degradation process for these polymers is mainly through uniform bulk degradation of the matrix where the water penetration into the matrix is higher than the rate of polymer degradation. Furthermore, the increase of carboxylic end groups as a result of biodegradation autocatalyses the process. The degradation of PLGA copolymer is the collective process of bulk diffusion, surface diffusion, bulk erosion and surface erosion. Since there are many variables that influence the degradation process, the release rate pattern is often unpredictable. The biodegradation rate of the PLGA copolymers are dependent on the molar ratio of the lactic and glycolic acids in the polymer chain, molecular weight of the polymer, the degree of crystallinity, and the Tg of the polymer. The release of drug from the homogeneously degrading matrix is more complicated. A biphasic curve for drug release as a result of PLGA biodegradation has been shown to display following pattern: (Figure 3) [72–74]
Initial burst of drug release is related to drug type, drug concentration and polymer hydrophobicity. Drug on the surface, in contact with the medium, is released as a function of solubility as well as penetration of water into polymer matrix. Random scission of PLGA decreases molecular weight of polymer significantly, but no appreciable weight loss and no soluble monomer product are formed in this phase.
In the second phase, drug is released progressively through the thicker drug depleted layer. The water inside the matrix hydrolyzes the polymer into soluble oligomeric and monomeric products. This creates a passage for drug to be released by diffusion and erosion until complete polymer solubilization. Drug type also plays an important role here in attracting the aqueous phase into the matrix.
The role of enzymes in any PLGA biodegradation is unclear. Most literature indicate that the PLGA biodegradation does not involve any enzymatic activity and is purely through hydrolysis. However, some investigators have suggested an enzymatic role in PLGA breakdown based upon the difference in the in vitro and in vivo degradation rates. The PLGA polymer biodegrades into lactic and glycolic acids. Lactic acid enters the tricarboxylic acid cycle and is metabolized and subsequently eliminated from the body as carbon dioxide and water [75]. Glycolic acid is either excreted unchanged in the kidney or it enters the tricarboxylic acid cycle and is eventually eliminated as carbon dioxide and water. Ideally PLGA polymer systems should have considerable mechanical strength, since the drug delivery devices formulated using them are subjected to significant physical stress, which can also influence mechanical breakdown of implants and alter surface area and hydration/hydrolysis [76].
4.2. Factors Affecting Degradation
To enhance the desirable properties of PLGA, it is essential to understand the factors affecting the PLGA degradation and design a drug delivery device accommodating all these factors to make it more efficient and efficacious.
4.2.1. Effect of Composition
Polymer composition is the most important factor to determine the hydrophilicity and rate of degradation of a delivery matrix which influence the rate of degradation. A systematic study of polymer composition with its degradation has been shown by many groups [77,78]. These results show that increase in glycolic acid percentage in the oligomers accelerates the weight loss of polymer. PLGA 50:50 (PLA/PGA) exhibited a faster degradation than PLGA 65:35 due to preferential degradation of glycolic acid proportion assigned by higher hydrophilicity. Subsequently PLGA 65:35 shows faster degradation than PLGA 75:25 and PLGA 75:25 than PLGA 85:15 [79]. Thus absolute value of the degradation rate increases with the glycolic acid proportion. The amount of glycolic acid is a critical parameter in tuning the hydrophilicity of the matrix and thus the degradation and drug release rate.
4.2.2. Effect of Crystallinity (or Tg)
Copolymer composition also affects important properties such as glass transition temperature and crystallinity which have indirect effects on degradation rate. At the moment, there are conflicting reports on the effect of crystallinity on the degradation rate [80]. Few groups [81] have proposed that the crystallinity of lactic acid (PLLA) increases the degradation rate because the degradation of semi-crystalline polymer is accelerated due to an increase in hydrophilicity. In contrast, various other studies have shown a decrease of degradation rate with increase in sample crystallinity [82].
4.2.3. Effect of Weight Average Molecular Weight (Mw)
Polymers with higher molecular weight have generally exhibited lower degradation rates [83]. Molecular weight has a direct relation with the polymer chain size. Polymers having higher molecular weight have longer polymer chains, which require more time to degrade than small polymer chains. However this is opposite for PLLA due to an inversely proportional degree of crystallinity with the molecular weight [83,84].
4.2.4. Effect of Drug Type
The mechanism of polymer-drug matrix degradation and the parameters of drug release rate vary as a function of drug type [85]. The presence of drug may change the degradation mechanism from bulk erosion to surface degradation, as well as affect the rate of matrix degradation [12]. The drug release profile, as defined by the time required for 100% release and the steady-state rate also varies significantly. However, efforts to correlate the release rate parameters to the drug chemistry (as defined by the density of OH groups) or hydrophilicity (as given by solubility in water) do not yield a strong relationship. However, it is clear that one must seriously consider the effect of the chemical properties of the drug to explain the drug-release mechanisms of a particular system using biodegradable polymers.
4.2.5. Effect of Size and Shape of the Matrix
The ratio of surface area to volume has shown to be a significant factor for degradation of large devices. Higher surface area ratio leads to higher degradation of the matrix. It has also been reported that bulk degradation is faster than pure surface degradation for PLGA, which makes the release of the drug faster from the devices with higher surface area to volume [82,86,87].
4.2.6. Effect of pH
The in vitro biodegradation/hydrolysis of PLGA showed that both alkaline and strongly acidic media accelerate polymer degradation [88]. However, the difference between the slightly acidic and neutral media is less pronounced due to autocatalysis by the carboxylic end groups [89].
4.2.7. Effect of Enzymes
There are conflicting results published on the effect of enzymes on degradation mechanisms (hydrolytic versus enzymatic cleavage) partially due to observations that degradation in vivo cannot be entirely correlated to in vitro assessment [80]. It has been proposed that PLGA degrades primarily through hydrolytic degradation but it has also been suggested that enzymatic degradation may play a role in the process. Due to a lack of uniformity in in vivo tests, there is difficulty in comparing and demonstrating the choice of proposed enzymes and their contribution in the degradation process [90,91].
4.2.8. Effect of Drug Load
Amount of drug loading in the drug delivery matrix plays a significant role on the rate and duration of drug release. Matrices having higher drug content possess a larger initial burst release than those having lower content because of their smaller polymer to drug ratio. However, this drug content effect is attenuated when the drug content reaches a certain level depending upon drug type [92].
4.3. Toxicology
Toxicological studies with PLGA devices suggest that local tissue reactions at the site of application may occur [93,94]. Although these reactions are generally mild and PLGA has been shown to be extremely safe as a material for macroscopic and microparticle systems, unique considerations may arise when using nanoscale applications. Several studies suggest that nanoparticles of any material may create specific biodistribution and toxicological profiles [95].
4.4. Modeling of PLGA Drug Release Profiles
PLGA degradation and drug release from a matrix is a combination of surface diffusion, bulk diffusion, and erosion of the matrix which is attributed to a variety of physical, chemical and processing parameters of that corresponding system. However, underlying mechanisms of this complex process are not clearly understood. The first stage of drug release is through random scission of the polymer without any polymer weight loss and is mainly through diffusion, while the second stage is characterized by the onset of weight loss. One proposed diffusion model of polymer degradation incorporates the effect of polymer degradation on the drug diffusivity in the polymer and the non-uniform distribution of that drug inside the formulation. The equation describing the release is the common diffusion equation: [96]
The initial drug distribution within the structure f(r) is usually obtained empirically. The first stage of hydrolytic degradation has been widely investigated and molecular weight as a function of time is given by Equation (4)
If micro/nanoparticles are used, the effect on the drug release from the population size distribution has to be considered in the cumulative equation. From previous studies, microparticle size distribution is best modeled as a Weibull distribution or Rosin–Rammler mathematical distribution
5. Conclusions and Future Prospects
PLGA polymers have been shown to be excellent delivery carriers for controlled administration of drugs, peptides and proteins due to their biocompatibility and biodegradability. In general, the PLGA degradation and the drug release rate can be accelerated by greater hydrophilicity, increase in chemical interactions among the hydrolytic groups, less crystallinity and larger volume to surface ratio of the device. All of the these factors should be taken into consideration in order to tune the degradation and drug release mechanism for desired application. Thus, for a short-term release requirement (up to one month), an amorphous polymer with high hydrophilicity is recommended. For a longer-term release requirement (one to six months), the choice of an amorphous polymer with high molecular weight would be appropriate. Also, for very long-term release (more than six months), semi-crystalline polymer with a high degree of crystallinity can be considered. Furthermore, multiple studies demonstrate that PLGA can easily be formulated into the drug carrying devices at all scales, i.e., as nanospheres, as microspheres and even as millimeter sized implants, can encapsulate a wide range of drugs, peptides or proteins and can be delivered over different periods of time with diverse routes of delivery.



Acknowledgments
This study was supported by R01MH074672.
References
- Bouissou, C.; Rouse, J.J.; Price, R.; van der Walle, C.F. The influence of surfactant on PLGA microsphere glass transition and water sorption: Remodeling the surface morphology to attenuate the burst release. Pharm. Res. 2006, 23, 1295–1305. [Google Scholar]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar]
- Ruhe, P.Q.; Hedberg, E.L.; Padron, N.T.; Spauwen, P.H.; Jansen, J.A.; Mikos, A.G. rhBMP-2 release from injectable poly (DL-lactic-co-glycolic acid)/calcium-phosphate cement composites. J. Bone Jt. Surg. 2003, 85, 75–81. [Google Scholar]
- Allison, S.D. Effect of structural relaxation on the preparation and drug release behavior of poly(lactic-co-glycolic)acid microparticle drug delivery systems. J. Pharm. Sci. 2008, 97, 2022–2035. [Google Scholar]
- Mundargi, R.; Babu, V.; Rangaswamy, V.; Patel, P.; Aminabhavi, T. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar]
- Mohamed, F.; van der Walle, C.F. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J. Pharm. Sci. 2008, 97, 71–87. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar]
- Wu, X.S.; Wang, N. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers. Part II: Biodegradation. J. Biomater. Sci. Polym. Ed. 2001, 12, 21–34. [Google Scholar]
- Houchin, M.L.; Topp, E.M. Physical properties of PLGA films during polymer degradation. J. Appl. Polym. Sci. 2009, 114, 2848–2854. [Google Scholar]
- Siegel, S.J.; Kahn, J.B.; Metzger, K.; Winey, K.I.; Werner, K.; Dan, N. Effect of drug type on the degradation rate of PLGA matrices. Eur. J. Pharm. Biopharm. 2006, 64, 287–293. [Google Scholar]
- Passerini, N.; Craig, D.Q.M. An investigation into the effects of residual water on the glass transition temperature of polylactide microspheres using modulated temperature DSC. J. Control. Release 2001, 73, 111–115. [Google Scholar]
- Yang, Y.Y.; Chung, T.S.; Ng, N.P. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials 2001, 22, 231–241. [Google Scholar]
- Panagi, Z.; Beletsi, A.; Evangelatos, G.; Livaniou, E.; Ithakissios, D.S.; Avgoustakis, K. Effect of dose on the biodistribution and pharmacokinetics of PLGA and PLGA-mPEG nanoparticles. Int. J. Pharm. 2001, 221, 143–152. [Google Scholar]
- Bazile, D.V.; Ropert, C.; Huve, P.; Verrecchia, T.; Marlard, M.; Frydman, A.; Veillard, M.; Spenlehauer, G. Body distribution of fully biodegradable [14C]-poly(lactic acid) nanoparticles coated with albumin after parenteral administration to rats. Biomaterials 1992, 13, 1093–1102. [Google Scholar]
- Esmaeili, F.; Ghahremani, M.H.; Esmaeili, B.; Khoshayand, M.R.; Atyabi, F.; Dinarvand, R. PLGA nanoparticles of different surface properties: Preparation and evaluation of their body distribution. Int. J. Pharm. 2008, 349, 249–255. [Google Scholar]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG Nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar]
- Li, Y.; Pei, Y.; Zhang, X.; Gu, Z.; Zhou, Z.; Yuan, W.; Zhou, J.; Zhu, J.; Gao, X. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar]
- Ghahremankhani, A.A.; Dorkoosh, F.; Dinarvand, R. PLGA-PEG-PLGA tri-block copolymers as an in-situ gel forming system for calcitonin delivery. Polym. Bull. 2007, 59, 637–646. [Google Scholar]
- Jeong, B.; Bae, Y.H.; Kim, S.W. In situ gelation of PEGPLGAPEG triblock copolymer aqueous solutions and degradation thereof. J. Biomed. Mater. Res. 2000, 50, 171–177. [Google Scholar]
- Tobio, M.; Gref, R.; Sanchez, A.; Langer, R.; Alonso, M.J. Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm. Res. 1998, 15, 270–275. [Google Scholar]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA–PEG nanoparticles. Proc. Natl. Acad. Sci USA 2008, 105, 17356–17361. [Google Scholar]
- Yoo, H.S.; Park, T.G. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J. Control. Release 2004, 96, 273–283. [Google Scholar]
- Duvvuri, S.; Janoria, K.G.; Mitra, A.K. Development of a novel formulation containing poly (D,L-lactide-co-glycolide) microspheres dispersed in PLGA-PEG-PLGA gel for sustained delivery of ganciclovir. J. Control. Release 2005, 108, 282–293. [Google Scholar]
- Garinot, M.; Fievez, V.; Pourcelle, V.; Stoffelbach, F.; des Rieux, A.; Plapied, L.; Theate, I.; Freichels, H.; Jerome, C.; Marchand-Brynaert, J.; Schneider, Y.; Preat, V. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J. Control. Release 2007, 120, 195–204. [Google Scholar]
- Danhier, F.; Lecouturier, N.; Vroman, B.; Jerome, C.; Marchand-Brynaert, J.; Feron, O.; Preat, V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J. Control. Release 2009, 133, 11–17. [Google Scholar]
- Pai, S.S.; Tilton, R.D.; Przybycien, T.M. Poly(ethylene glycol)-modified proteins: Implications for poly(lactide-co-glycolide)-based microsphere delivery. AAPS J. 2009, 11, 88–98. [Google Scholar]
- Choi, S.H.; Park, T.G. Synthesis and characterization of elastic PLGA/PCL/PLGA tri-block copolymers. J. Biomater. Sci. Polym. Ed. 2002, 13, 1163–1173. [Google Scholar]
- Dong, C.M.; Guo, Y.Z.; Qiu, K.Y.; Gu, Z.W.; Feng, X.D. In vitro degradation and controlled release behavior of D, L-PLGA50 and PCL-bD, L-PLGA50 copolymer microspheres. J. Control. Release 2005, 107, 53–64. [Google Scholar]
- Houchin, M.L.; Topp, E.M. Chemical degradation of peptides and proteins in PLGA: A review of reactions and mechanisms. J. Pharm. Sci. 2008, 97, 2395–2404. [Google Scholar]
- Arshady, R. Preparation of biodegradable microspheres and microcapsules: 2. Polyactides and related polyesters. J. Control. Release 1991, 17, 1–21. [Google Scholar]
- King, T.W.; Patrick, C.W., Jr. Development and in vitro characterization of vascular endothelial growth factor (VEGF)-loaded poly (D,L-lactic-co-glycolic acid)/poly (ethylene glycol) microspheres using a solid encapsulation/single emulsion/solvent extraction technique. J. Biomed. Mater. Res. Part A 2000, 51, 383–390. [Google Scholar]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar]
- Sah, H. Microencapsulation techniques using ethyl acetate as a dispersed solvent: Effects of its extraction rate on the characteristics of PLGA microspheres. J. Control. Release 1997, 47, 233–245. [Google Scholar]
- Chaisri, W.; Hennink, W.E.; Okonogi, S. Preparation and characterization of cephalexin loaded PLGA microspheres. Curr. Drug Deliv. 2009, 6, 69–75. [Google Scholar]
- Mao, S.; Xu, J.; Cai, C.; Germershaus, O.; Schaper, A.; Kissel, T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int. J. Pharm. 2007, 334, 137–148. [Google Scholar]
- Thomasin, C.; Nam-Trân, H.; Merkle, H.P.; Gander, B. Drug microencapsulation by PLA/PLGA coacervation in the light of thermodynamics. 1. Overview and theoretical considerations. J. Pharm. Sci. 1998, 87, 259–268. [Google Scholar]
- Thomasin, C.; Merkle, H.P.; Gander, B. Drug microencapsulation by PLA/PLGA coacervation in the light of thermodynamics. 2. Parameters determining microsphere formation. J. Pharm. Sci. 1998, 87, 269–275. [Google Scholar]
- Edelman, R.; Russell, R.G.; Losonsky, G.; Tall, B.D.; Tacket, CO.; Levine, M.M.; Lewis, D.H. Immunization of rabbits with enterotoxigenic E. coli colonization factor antigen (CFA/I) encapsulated in biodegradable microspheres of poly (lactide-co-glycolide). Vaccine 1993, 11, 155–158. [Google Scholar]
- Hua, F.J.; Kim, G.E.; Lee, J.D.; Son, Y.K.; Lee, D.S. Macroporous poly(L-lactide) scaffold 1. Preparation of a macroporous scaffold by liquid-liquid phase separation of a PLLA-dioxane-water system. J. Biomed. Mater. Res. 2002, 63, 161–167. [Google Scholar]
- Hua, F.J.; Park, T.G.; Lee, D.S. A facile preparation of highly interconnected macroporous poly(D,L-lactic acid-co-glycolic acid) (PLGA) scaffolds by liquid-liquid phase separation of a PLGA-dioxane-water ternary system. Polymer 2003, 44, 1911–1920. [Google Scholar]
- Graham, P.D.; Brodbeck, K.J.; McHugh, AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. J. Control. Release 1999, 58, 233–245. [Google Scholar]
- Mu, L.; Feng, S.S. Fabrication, characterization and in vitro release of paclitaxel (Taxol®) loaded poly (lactic-co-glycolic acid) microspheres prepared by spray drying technique with lipid/cholesterol emulsifiers. J. Control. Release 2001, 76, 239–254. [Google Scholar]
- Gavini, E.; Chetoni, P.; Cossu, M.; Alvarez, M.G.; Saettone, M.F.; Giunchedi, P. PLGA microspheres for the ocular delivery of a peptide drug, vancomycin using emulsification/spray-drying as the preparation method: In vitro/in vivo studies. Eur. J. Pharm. Biopharm. 2004, 57, 207–212. [Google Scholar]
- Nie, H.; Lee, L.Y.; Tong, H.; Wang, C. PLGA/chitosan composites from a combination of spray drying and supercritical fluid foaming techniques: New carriers for DNA delivery. J. Control. Release 2008, 129, 207–214. [Google Scholar]
- Wagenaar, B.; Muller, B. Piroxicam release from spray-dried biodegradable microspheres. Biomaterials 1994, 15, 49–54. [Google Scholar]
- Sastre, R.L.; Olmo, R.; Teijoón, C.; Munñíz, E.; Teijoón, J.M.; Blanco, M.D. 5-Fluorouracil plasma levels and biodegradation of subcutaneously injected drug-loaded microspheres prepared by spray-drying poly(D,L-lactide) and poly(D,L-lactide-co-glycolide) polymers. Int. J. Pharm. 2007, 338, 180–190. [Google Scholar]
- Castelli, F.; Conti, B.; Conte, U.; Puglisi, G. Effect of molecular weight and storage times on tolmetin release from poly-lactide microspheres to lipid model membrane. A calorimetric study. J. Control. Release 1996, 40, 277–284. [Google Scholar]
- Barrero, A.; Loscertales, I.G. Micro- and nanoparticles via capillary flows. Annu. Rev. Fluid Mech. 2007, 39, 89–106. [Google Scholar]
- Freitas, S.; Merkle, H.P.; Gander, B. Microencapsulation by solvent extraction/evaporation: Reviewing the state of the art of microsphere preparation process technology. J. Control. Release 2005, 102, 313–332. [Google Scholar]
- Berkland, C.; Pollauf, E.; Pack, D.W.; Kim, K.K. Uniform double-walled polymer microspheres of controllable shell thickness. J. Control. Release 2004, 96, 101–111. [Google Scholar]
- Loscertales, I.G.; Barrero, A.; Guerrero, I.; Cortijo, R.; Marquez, M.; Ganan-Calvo, A.M. Micro/Nano encapsulation via electrified coaxial liquid jets. Science 2002, 295, 1695–1698. [Google Scholar]
- Hans, M.L.; Lowman, A.M. Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci. 2002, 6, 319–327. [Google Scholar]
- Lamprecht, A.; Ubrich, N.; Perez, M.H.; Lehr, C.M.; Hoffman, M.; Maincent, P. Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int. J. Pharm. 2000, 196, 177–182. [Google Scholar]
- Murakami, H.; Kobayashi, M.; Takeuchi, H.; Kawashima, Y. Preparation of poly(D,L-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int. J. Pharm. 1999, 187, 143–152. [Google Scholar]
- Konan, Y.N.; Cerny, R.; Favet, J.; Berton, M.; Gurny, R.; Allemann, E. Preparation and characterization of sterile sub-200 nm meso-tetra(4-hydroxylphenyl)porphyrin-loaded nanoparticles for photodynamic therapy. Eur. J. Pharm. Biopharm. 2003, 55, 115–124. [Google Scholar]
- Kwon, H.Y.; Lee, J.Y.; Choi, S.W.; Jang, Y.S.; Kim, J.H. Preparation of PLGA nanoparticles containing estrogen by emulsification-diffusion method. Colloids Surf. A Physicochem. Eng. Asp. 2001, 182, 123–130. [Google Scholar]
- Rabin, C.; Liang, Y.; Ehrlichman, R.S.; Budhian, A.; Metzger, K.L.; Majewski-Tiedeken, C.; Winey, K.I.; Siegel, S.J. In vitro and in vivo demonstration of risperidone implants in mice. Schizophr. Res. 2008, 98, 66–78. [Google Scholar]
- Widmer, M. Manufacture of porous biodegradable polymer conduits by an extrusion process for guided tissue regeneration. Biomaterials 1998, 19, 1945–1955. [Google Scholar]
- Wang, C.; Wang, W.; Meyer, R.F.; Liang, Y.; Winey, K.I.; Siegel, S.J. A rapid method for creating drug implants: Translating laboratory-based methods into a scalable manufacturing process. J. Biomed. Mater. Res. Part B Appl. Biomater. 2010, 93, 562–572. [Google Scholar]
- Yang, R.; Chen, T.; Chen, H.; Wang, W. Microfabrication of biodegradable (PLGA) honeycomb-structures and potential applications in implantable drug delivery. Sens. Actuat. B Chem. 2005, 106, 506–511. [Google Scholar]
- Grayson, A.C.R.; Cima, M.J.; Langer, R. Size and temperature effects on poly(lactic-co-glycolic acid) degradation and microreservoir device performance. Biomaterials 2005, 26, 2137–2145. [Google Scholar]
- Stubbe, B.G.; Smedt, S.C.D.; Demeester, J. “Programmed polymeric devices” for pulsed drug delivery. Pharm. Res. 2004, 21, 1732–1740. [Google Scholar]
- Grayson, A.C.R.; Choi, I.S.; Tyler, B.M.; Wang, P.P.; Brem, H.; Cima, M.J.; Langer, R. Multi-pulse drug delivery from a resorbable polymeric microchip device. Nat. Mater. 2003, 2, 767–772. [Google Scholar]
- Koushik, K.; Kompella, U.B. Preparation of large porous Deslorelin-PLGA microparticles with reduced residual solvent and cellular uptake using a supercritical carbon dioxide process. Pharm. Res. 2004, 21, 524–535. [Google Scholar]
- Davies, O.R.; Lewis, A.L.; Whitaker, M.J.; Tai, H.; Shakesheff, K.M.; Howdle, S.M. Applications of supercritical CO2 in the fabrication of polymer systems for drug delivery and tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 373–387. [Google Scholar]
- Hile, D.D.; Amirpour, M.L.; Akgerman, A.; Pishko, M.V. Active growth factor delivery from poly (D,L-lactide-co-glycolide) foams prepared in supercritical CO2. J. Control. Release 2000, 66, 177–185. [Google Scholar]
- Xu, J.S.; Huang, J.; Qin, R.; Hinkle, G.H.; Povoski, S.P.; Martin, E.W.; Xu, R.X. Synthesizing and binding dual-mode poly (lactic-co-glycolic acid) (PLGA) nanobubbles for cancer targeting and imaging. Biomaterials 2010, 31, 1716–1722. [Google Scholar]
- Zhang, L.; Xu, J.S.; Sanders, V.M.; Letson, A.D.; Roberts, C.J.; Xu, R.X. Multifunctional microbubbles for image-guided antivascular endothelial growth factor therapy. J. Biomed. Opt. 2010, 15, 030515. [Google Scholar]
- Sun, B.; Ranganathan, B.; Feng, S. Multifunctional poly(D,L-lactide-co-glycolide)/ montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials 2008, 29, 475–486. [Google Scholar]
- Ramchandani, M.; Robinson, D. In vitro and in vivo release of ciprofloxacin from PLGA 50:50 implants. J. Control Release 1998, 54, 167–175. [Google Scholar]
- Amann, L.C.; Gandal, M.J.; Lin, R.; Liang, Y.; Siegel, S.J. In vitro-in vivo correlations of scalable PLGA-risperidone implants for the treatment of schizophrenia. Pharm. Res. 2010, 27, 1730–1737. [Google Scholar]
- Faisant, N.; Siepmann, J.; Benoit, J.P. PLGA-based microparticles: Elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur. J. Pharm. Sci. 2002, 15, 355–366. [Google Scholar]
- Crotts, G.; Park, T.G. Protein delivery from poly (lactic-co-glycolic acid) biodegradable microspheres: Release kinetics and stability issues. J. Microencapsul. 1998, 15, 699–713. [Google Scholar]
- Kranz, H.; Ubrich, N.; Maincent, P.; Bodmeier, R. Physicomechanical properties of biodegradable poly(D,L-lactide) and poly(D,L-lactide-co-glycolide) films in the dry and wet states. J. Pharm. Sci. 2000, 89, 1558–1566. [Google Scholar]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly(-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar]
- Lu, L.; Garcia, C.A.; Mikos, A.G. In vitro degradation of thin poly(D,L-lactic-co-glycolic acid) films. J. Biomed. Mater. Res. 1999, 46, 236–244. [Google Scholar]
- Park, T.G. Degradation of poly(lactic-co-glycolic acid) microspheres: Effect of copolymer composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar]
- Alexis, F. Factors affecting the degradation and drug-release mechanism of poly (lactic acid) and poly [(lactic acid)-co-(glycolic acid)]. Polym. Int. 2005, 54, 36–46. [Google Scholar]
- Tsuji, H.; Mizuno, A.; Ikada, Y. Properties and morphology of poly(L-lactide). III. Effects of initial crystallinity on long-term in vitro hydrolysis of high molecular weight poly(L-lactide) film in phosphate-buffered solution. J. Appl. Polym. Sci. 2000, 77, 1452–1464. [Google Scholar]
- Schliecker, G.; Schmidt, C.; Fuchs, S.; Wombacher, R.; Kissel, T. Hydrolytic degradation of poly(lactide-co-glycolide) films: Effect of oligomers on degradation rate and crystallinity. Int. J. Pharm. 2003, 266, 39–49. [Google Scholar]
- Park, T.G. Degradation of poly (D,L-lactic acid) microspheres: Effect of molecular weight. J. Control. Release 1994, 30, 161–173. [Google Scholar]
- Liggins, R. Paclitaxel loaded poly(L-lactic acid) microspheres: Properties of microspheres made with low molecular weight polymers. Int. J. Pharm. 2001, 222, 19–33. [Google Scholar]
- Frank, A.; Rath, S.K.; Venkatraman, S.S. Controlled release from bioerodible polymers: Effect of drug type and polymer composition. J. Control. Release 2005, 102, 333–344. [Google Scholar]
- Witt, C. Morphological characterization of microspheres, films and implants prepared from poly(lactide-co-glycolide) and ABA triblock copolymers: Is the erosion controlled by degradation, swelling or diffusion? Eur. J. Pharm. Biopharm. 2001, 51, 171–181. [Google Scholar]
- Grizzi, I.; Garreau, H.; Li, S.; Vert, M. Hydrolytic degradation of devices based on poly(DL-lactic acid) size-dependence. Biomaterials 1995, 16, 305–311. [Google Scholar]
- Holy, C. In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam. Biomaterials 1999, 20, 1177–1185. [Google Scholar]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar]
- Cai, Q.; Shi, G.; Bei, J.; Wang, S. Enzymatic degradation behavior and mechanism of poly(lactide-co-glycolide) foams by trypsin. Biomaterials 2003, 24, 629–638. [Google Scholar]
- Li, S.; Girard, A.; Garreau, H.; Vert, M. Enzymatic degradation of polylactide stereocopolymers with predominant -lactyl contents. Polym. Degrad. Stab. 2000, 71, 61–67. [Google Scholar]
- Eniola, A.O.; Hammer, D.A. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: Effect of degradation on targeting activity. Biomaterials 2005, 26, 661–670. [Google Scholar]
- Dailey, L.; Jekel, N.; Fink, L.; Gessler, T.; Schmehl, T.; Wittmar, M.; Kissel, T.; Seeger, W. Investigation of the proinflammatory potential of biodegradable nanoparticle drug delivery systems in the lung. Toxicol. Appl. Pharmacol. 2006, 215, 100–108. [Google Scholar]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar]
- Olivier, J.C.; Fenart, L.; Chauvet, R.; Pariat, C.; Cecchelli, R.; Couet, W. Indirect evidence that drug brain targeting using polysorbate 80-coated polybutylcyanoacrylate nanoparticles is related to toxicity. Pharm. Res. 1999, 16, 1836–1842. [Google Scholar]
- Berchane, N.S.; Carson, K.H.; Rice-Ficht, A.C.; Andrews, M.J. Effect of mean diameter and polydispersity of PLG microspheres on drug release: Experiment and theory. Int. J. Pharm. 2007, 337, 118–126. [Google Scholar]
- Raman, C.; Berkland, C.; Kim, K.; Pack, D.W. Modeling small-molecule release from PLG microspheres: Effects of polymer degradation and nonuniform drug distribution. J. Control. Release 2005, 103, 149–158. [Google Scholar]
- Siepmann, J.; Goöpferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377-1397. https://doi.org/10.3390/polym3031377
Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers. 2011; 3(3):1377-1397. https://doi.org/10.3390/polym3031377
Chicago/Turabian StyleMakadia, Hirenkumar K., and Steven J. Siegel. 2011. "Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier" Polymers 3, no. 3: 1377-1397. https://doi.org/10.3390/polym3031377