1. Introduction
The molecular weight distribution (MWD) and architecture are two important characteristics of a polymer [
1]. They strongly affect material properties such as dynamic moduli, fracture toughness, glass transition temperature, and viscosity [
1,
2,
3]. Experimental methods for an accurate determination of MWD are thus of great interest [
3,
4,
5]. Theoretically, it is also highly desirable if the MWD of a polymer can be predicted a priori based on the knowledge of the polymerization reaction involved without even synthesizing the polymer. Such a theoretical method will be a valuable tool that is not only useful for understanding experimental measurements, but also beneficial for other theories and models aiming to predict polymer properties. For example, Nichetti and Manas-Zloczower [
6] proposed a theoretical model to predict the viscosity of a polymer melt based on its MWD that was determined by fitting the data from gel permeation chromatography to statistical distribution functions. A method to quickly generate its MWD without the necessity of experimental input thus can advance the predictive capability of such theories [
7].
A theory on the constitution and molecular size distribution of a step-growth polymer was proposed by Flory and Stockmayer several decades ago [
8,
9,
10,
11,
12,
13], and has been frequently used to determine the gel point. Flory studied the polymerization of bifunctional monomers mixed with trifunctional and tetrafunctional branching units, and made two fundamental assumptions [
8,
9,
10]. First, the same functional group has the same probability to react with another group, and this probability is not affected by the length of the polymer to which the functional group belongs, as well as the position of the functional group on that polymer. Secondly, ring polymers are not formed. Stockmayer extended the theory to branching units with arbitrary functionalities and derived the Stockmayer formula for the number of polymer chains with a given composition, though ring structures were still excluded [
11,
12,
13]. The predictions of the Flory-Stockmayer theory [
8,
9,
10,
11,
12,
13], including the gel point and the average molecular weight, have been tested experimentally [
14,
15,
16,
17,
18,
19,
20]. However, the entire MWD is hard to probe experimentally, and often only the average molecular weight is measured. Practically, it is also difficult to directly predict MWD using the Flory-Stockmayer theory because of the mathematical complexity involved in computing the amount of all possible molecules in a branched polymer system. Furthermore, the Flory-Stockmayer theory is expected to be only valid below the gel point. Schamboeck et al. [
21] suggested a new theoretical method based on directed random graphs to overcome the complexity of the Flory-Stockmayer theory, but the distribution beyond the gel point was not presented. Beyond the gel point, the formation of cyclics and closed loops in a branched structure becomes significant, and theories ignoring their presence may fail [
22,
23].
Many computational methods, such as molecular dynamics simulations and dissipative particle dynamics based on either all-atom [
24] or coarse-grained models [
25,
26,
27], can be used for simulating chemical reactions including polymerization. However, it can be challenging for these methods to treat a nearly fully reacted system because of the limitation on their accessible time scales. Furthermore, the formation of branched polymers is even more difficult for these methods to handle, as when the extent of the reaction grows larger, it becomes harder to visit monomers already incorporated into a polymer, though they may still possess active functional groups. On the other hand, a polymerization process is much more amenable to Monte Carlo (MC) simulations [
28,
29,
30].
MC methods are a class of techniques based on random sampling to numerically solve problems that have a probabilistic interpretation [
31]. MC methods have broad applications in polymer science [
32,
33,
34], especially in polymer reaction engineering [
28,
33]. Johnson and O’Driscoll [
35] used an MC simulation to study sequence distributions in step-growth copolymerization. Tobita applied MC simulation to a wide range of polymerization problems, including free-radical polymerization [
36,
37,
38], emulsion polymerization [
39,
40,
41], long-chain branching and random scission [
42,
43], and living radical polymerization [
44,
45]. Hadicke and Stutz [
46] used an amine-cured epoxy as an example to compare the structure of step-growth networks obtained via MC simulation to that predicted by the branching theory. He et al. [
47,
48] applied an MC method to simulate self-condensing vinyl polymerization in the presence of multifunctional initiators, and probed the role of reactive rate constants. Rouault and Milchev [
49], and He et al. [
50] performed MC simulations to study the kinetics and chain length distributions in living polymerization. Prescott [
51] used an MC model to show that chain-length-dependent termination plays a significant role in living/controlled free-radical polymerization systems containing reversible transfer agents. In a series of papers, Al-Harthi et al. [
52,
53,
54,
55] used dynamic MC simulations to study atom transfer radical polymerization. Polanowski et al. [
56,
57] and Bannister et al. [
58] used MC methods to study branching and gelation in the living copolymerization of vinyl and divinyl monomers. Recently, Polanowski et al. [
59] used MC simulations to investigate the polymerization of star and dendritic polymers. Lyu et al. [
22,
23] used a similar model to study the atom transfer radical polymerization and the conventional free-radical polymerization of divinyl monomers, and checked the applicability of the Flory-Stockmayer theory in such systems. Gao et al. [
60,
61] used kinetic MC methods to simulate free-radical copolymerization processes, and discussed how to accelerate such simulations using scaling approaches. Meimaroglou et al. [
62] proposed an MC algorithm to calculate the MWD for linear polymers, and the bivariate molecular weight - long chain branching distribution for highly branched polymers. They also used MC simulation to investigate the molecular, topological, and solution properties of highly branched low-density polyethylene [
63], and the ring-opening homopolymerization of L,L-Lactide [
64]. Jin et al. [
65] employed kinetic MC simulations to study the cross-linked network formation between hydroxyl-terminated poly(dimethylsiloxane) and triisocyanate. Iedema et al. [
66] developed an MC simulation model including both branching and random scission to calculate the molecular weight and branching distributions, and compared their calculations to experimental measurements on high-density polyethylene. Yaghini and Iedema [
67] compared the results on low-density polyethylene from such MC simulations to the predictions of a multiradical model based on a Galerkin finite element approach. De Keer et al. [
68] used a matrix-based kinetic MC framework to reveal the effects of intramolecular reactions on the variation of the chain length distribution and its averages for a network polymer formed via a step-growth mechanism, with the intention to go beyond the equation derived by Carothers [
69], Flory [
8,
70,
71], and Stockmayer [
11,
12,
13].
One important application of MC simulations is to quickly compute MWD [
72,
73,
74,
75,
76]. In MC simulations, all structures, including rings and networks allowed by a polymerization reaction, can be produced [
77], no matter whether the system is below or beyond the gel point [
78]. Various schemes can also be implemented to capture different polymerization kinetics, which thus allows us to test the specific assumptions made by a theory. The Gillespie algorithm can be used to speed up the kinetics of a reaction in silico and enable a reactive system to quickly reach a steady state [
79,
80]. In this paper, we develop an MC simulation model based on the Gillespie algorithm to study the formation of branched chains via step-growth polymerization. This model is applied to the polymerization of polyetherimides (PEIs) in the presence of chain terminators and branching agents. The results from the MC simulations are used to test the Flory-Stockmayer theory [
8,
9,
10,
11,
12,
13], including its assumption on the reaction rates.
This paper is organized as follows. In
Section 2, the Flory-Stockmayer theory is introduced, the technical challenge of computing MWD with this theory is discussed, and an approximation method is proposed. In
Section 3, the MC model of the polymerization process of the branched PEIs being studied is described in detail. This particular polymerization system serves as an example to facilitate understanding. However, the MC model described can be applied to other branched polymers as well. In
Section 4, the MC results are compared to the predictions of the Flory-Stockmayer theory. Practically, computations of MWD can only be executed for a small system either with the Flory-Stockmayer theory or the MC model. Therefore, a discussion on the effect of finite system size is included. Although the emphasis is on stoichiometric, fully reacted systems, those that are only partially reacted and/or nonstoichiometric are also discussed in
Section 4. Finally, a brief summary is provided in
Section 5.
2. The Flory-Stockmayer Theory of Step-Growth Polymers
Flory and Stockmayer considered a general step-growth polymer that consists of two types of monomers,
A and
B [
8,
9,
10,
11,
12,
13]. All reactions occur between
A and
B. There are
i type-
A monomers, denoted as
,
, and …,
. To simplify the notation,
with
is also used to denote the number of
monomers. Similarly, there are
j type-
B monomers, and the corresponding numbers are
,
, …,
, respectively. The symbol
denotes the functionality of an
monomer, where
, i.e., there are
functional groups on an
monomer, each of which can form a bond with another functional group on a
monomer, where
. The functionality of a
monomer is denoted as
. The Flory-Stockmayer theory can be applied to a polymerized state, where a fraction (
) of all the functional groups on the type-
A monomers have reacted with a fraction (
) of all the functional groups on the type-
B monomers. Therefore,
In this paper, the systems with , and thus, are called stoichiometric systems, while those with and are called nonstoichiometric. The systems with or , or both, equal to 1, are fully reacted.
denotes the number of molecules formed by
monomers of sub-type
and
monomers of sub-type
, with
q running from 1 to
i and
h running from 1 to
j. Here,
is a shorthand of
, which denotes the monomer composition of a given molecule. The Flory-Stockmayer theory predicts that
where
Equation (
2) is called the Stockmayer formula, which gives the number of molecules of any monomer compositions. However, it is practically difficult to compute MWD from the Stockmayer formula, as all the possible combinations in
have to be taken into account. Since
runs from 1 to
for
, and
runs from 1 to
for
, the total number of possible molecules is
. This number is huge when there are many sub-types (i.e., large
i and
j) and/or large numbers (i.e., large
and
) of monomers involved in a polymerization process.
For a molecule with composition
, the total number of monomers is
. Since the Flory-Stockmayer theory does not consider rings, the total number of bonds in this molecule must be
. When
, all the functional groups have reacted, and in a given molecule, the total number of the functional groups on all the type-
A monomers is equal to the total number of the functional groups on all the type-
B monomers. This number must also be equal to the total number of bonds in that molecule. Namely, for
there are two identities,
and
These two identities can help us simplify the Stockmayer formula for stoichiometric, fully reacted systems. Note that in Equation (
2), the terms involving
and
appear as
and
These terms can be dropped out because of Equations (
6) and (
7). As a result, for fully reacted stoichiometric systems with
, the Stockmayer formula is simplified as
with
Computing
is not easy, as it contains many factorials. The calculation can be expedited using the Stirling approximation,
Then, for fully reacted stoichiometric systems, the Stockmayer formula can be approximated logarithmically as
The computation of MWD from
can be further accelerated by noting that not all the combinations in
will yield a molecule. For a fully reacted stoichiometric system where
, Equations (
1), (
6), and (
7) can be used to reduce the total number of
. For the branched PEIs considered in this paper (see
Section 3),
,
,
, and
. The constraints become
and
Equations (
14) and (
15) combined yield
Equations (
15) and (
16) indicate that
and
are totally constrained by
and
in an allowed composition. Furthermore, since
must be nonnegative,
and
cannot be zero at the same time. The time complexity to enumerate all possible molecules is thus
, which is approximately
, with
Z being the system size (i.e., the total number of monomers prior to polymerization). This time complexity is acceptable for small systems. However, if there are multiple sub-types of monomers, then the time complexity will increase exponentially as
, where
w is the number of monomer sub-types. For partially reacted or nonstoichiometric systems where
or
are less than 1, the constraints that help reduce the number of possible
are lost. Then, computing MWD from
has to rely on Equation (
2), and this will become more challenging, even though the Stirling approximation may still be used. In these situations, the MC model described below will serve as a solution as it does not suffer from such limitations and the time complexity of computing MWD with MC simulations is always
, where
k is the number of MC runs needed to obtain the desired statistics. Usually,
k is about
to
.
3. Monte Carlo Model of Polymerization of Branched Polyetherimides (PEIs)
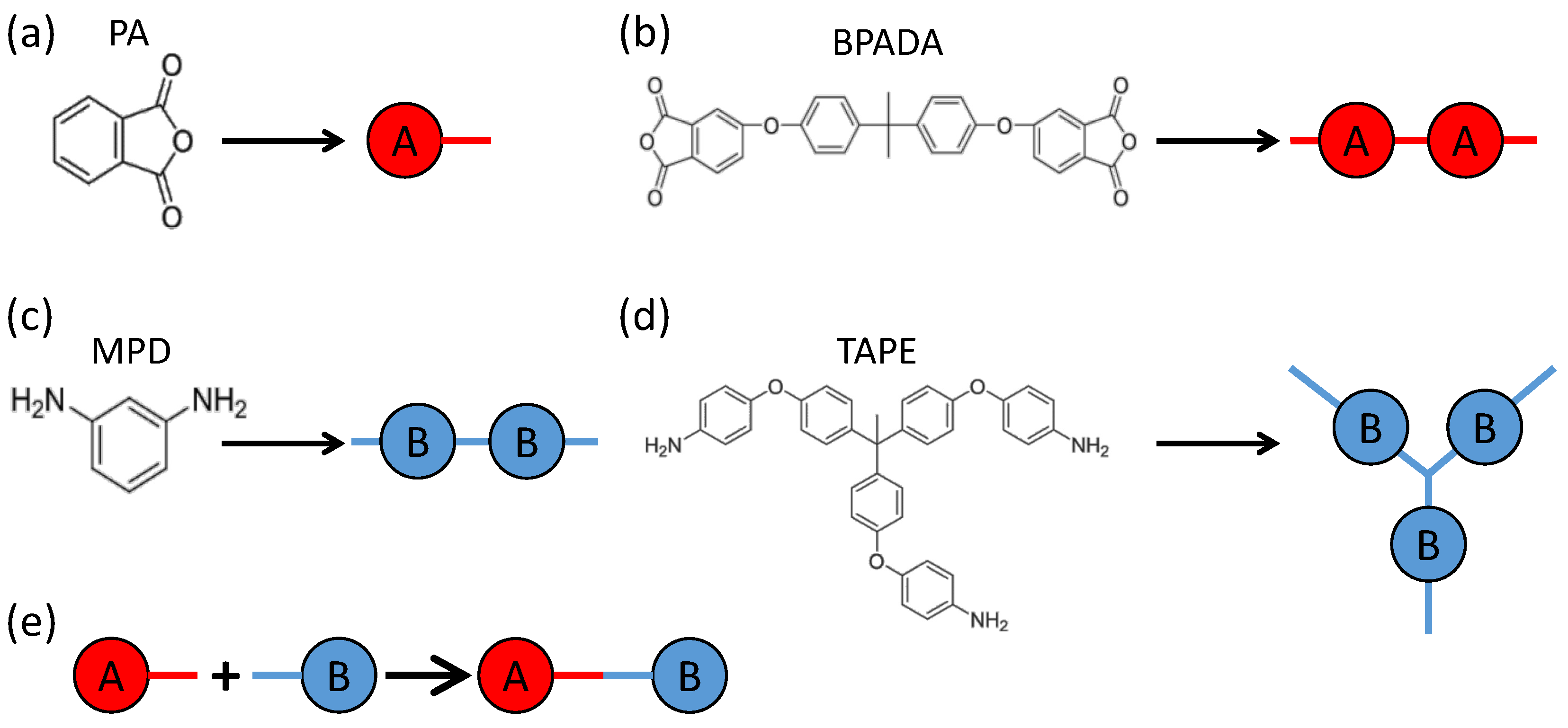
Four types of monomers are involved in the formation of the branched PEIs, including 4,4′-bisphenol A dianhydride (BPADA), m-phenylenediamine (MPD), phthalic anhydride (PA), and tris[4-(4-aminophenoxy)phenyl] ethane (TAPE) [
81]. The chemical structures of these monomers are shown in
Figure 1. The involved reaction is the condensation reaction between an amine group on MPD or TAPE, and a carboxylic anhydride group on BPADA or PA. In the notation of the Flory-Stockmayer theory, PA is monomer
with
, BPADA is monomer
with
, MPD is monomer
with
, and TAPE is monomer
with
. Out of these monomers, PA is an end capper to terminate a chain, and TAPE is a trifunctional branching agent.
Figure 1 shows the representation of these monomers in our MC model. Each functional group containing one carboxylic anhydride is mapped to an
A bead, and that containing one amine is mapped to a
B bead. Each
A bead can react with a
B bead to form a bond (i.e.,
), which describes the condensation reaction between an amine group and a carboxylic anhydride group.
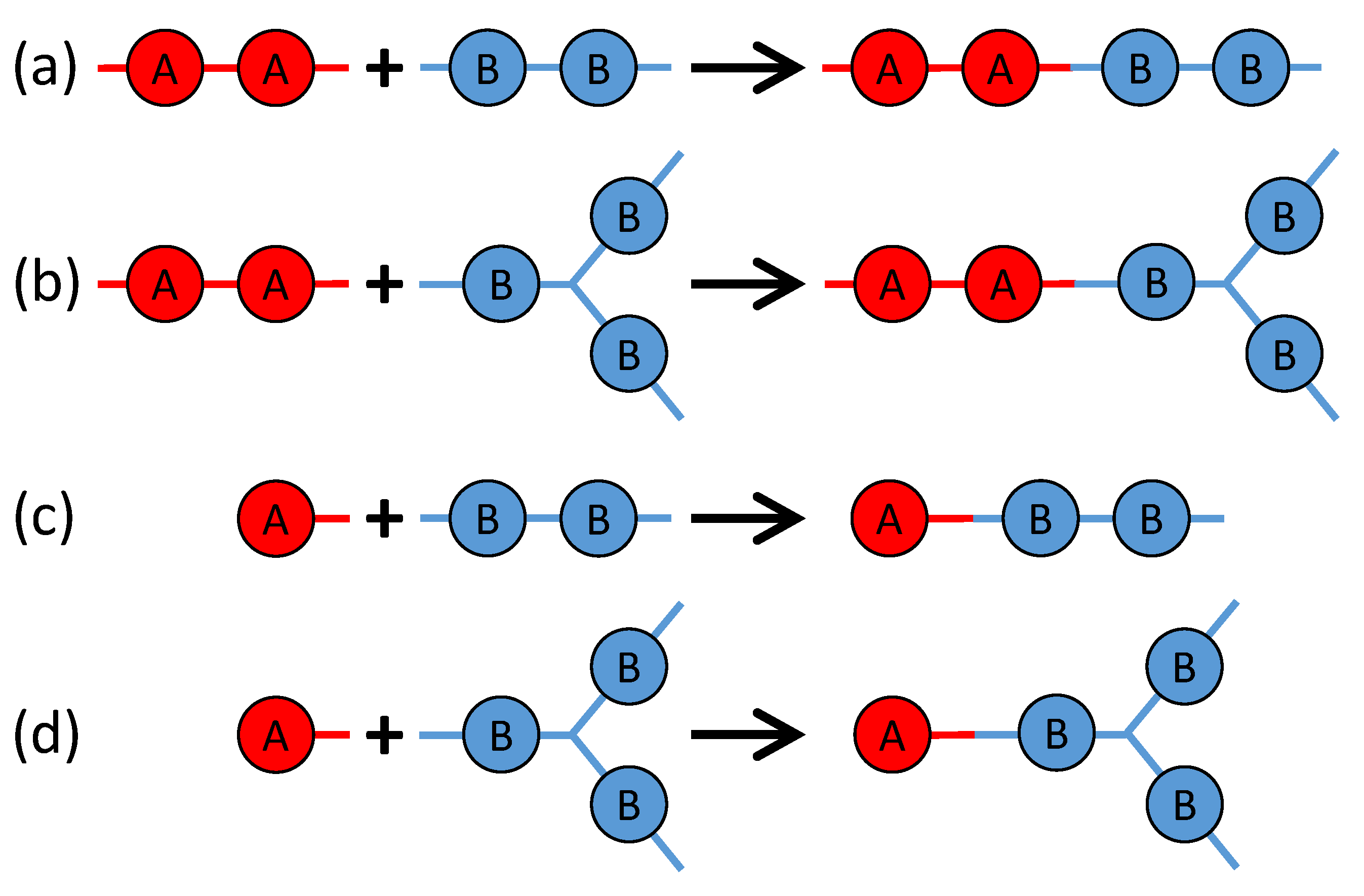
There are four possible reactions among the above four types of monomers, as sketched in
Figure 2. Reaction 1 is between BPADA and MPD, which leads to the formation of a PEI backbone. Reaction 2 is between BPADA and TAPE, which results in branching. Reaction 3 is between PA and MPD, which terminates a PEI chain. Reaction 4 is between PA and TAPE, which consumes one amine group on TAPE and effectively reduces its functionality by 1.
With the mapping in
Figure 1 and the reaction scheme in
Figure 2, MC simulations are performed to study the branched PEI polymerization. The Gillespie algorithm is adopted to speed up MC simulations. Since only the final chain structures are concerned, the random process in the typical Gillespie algorithm, which determines the time interval after which the next reaction occurs, is neglected. At each MC step, all four reactions will have a probability to occur, and the reaction rate of a particular reaction is determined by a rate constant and the concentration of the unreacted functional groups on the two types of monomers involved in that reaction. Mathematically, the probability of reaction
l is proportional to
where
(
) represents the reactant consisting of
A (
B) beads,
is a rate constant,
(
) is a quantity that depends on the concentration of the reactant
(
), and
indexes the possible reactions sketched in
Figure 2. Specifically,
and
are BPADA,
and
are PA,
and
are MPD, and
and
are TAPE. Since all the four reactions can be reduced to the reaction between an
A bead and a
B bead (i.e., the reaction between a functional group containing one amine and another functional group containing one carboxylic anhydride), as shown in
Figure 1e,
will be set as a constant
k for all the four reactions, and
and
can be expressed as
where
,
,
, and
are the concentrations of active functional groups on each type of monomers. In other words,
(
) is the concentration in terms of the number of unreacted
A (
B) beads on the reactant
(
). The particular reason of writing
and
in this way will be discussed in
Section 4.1.
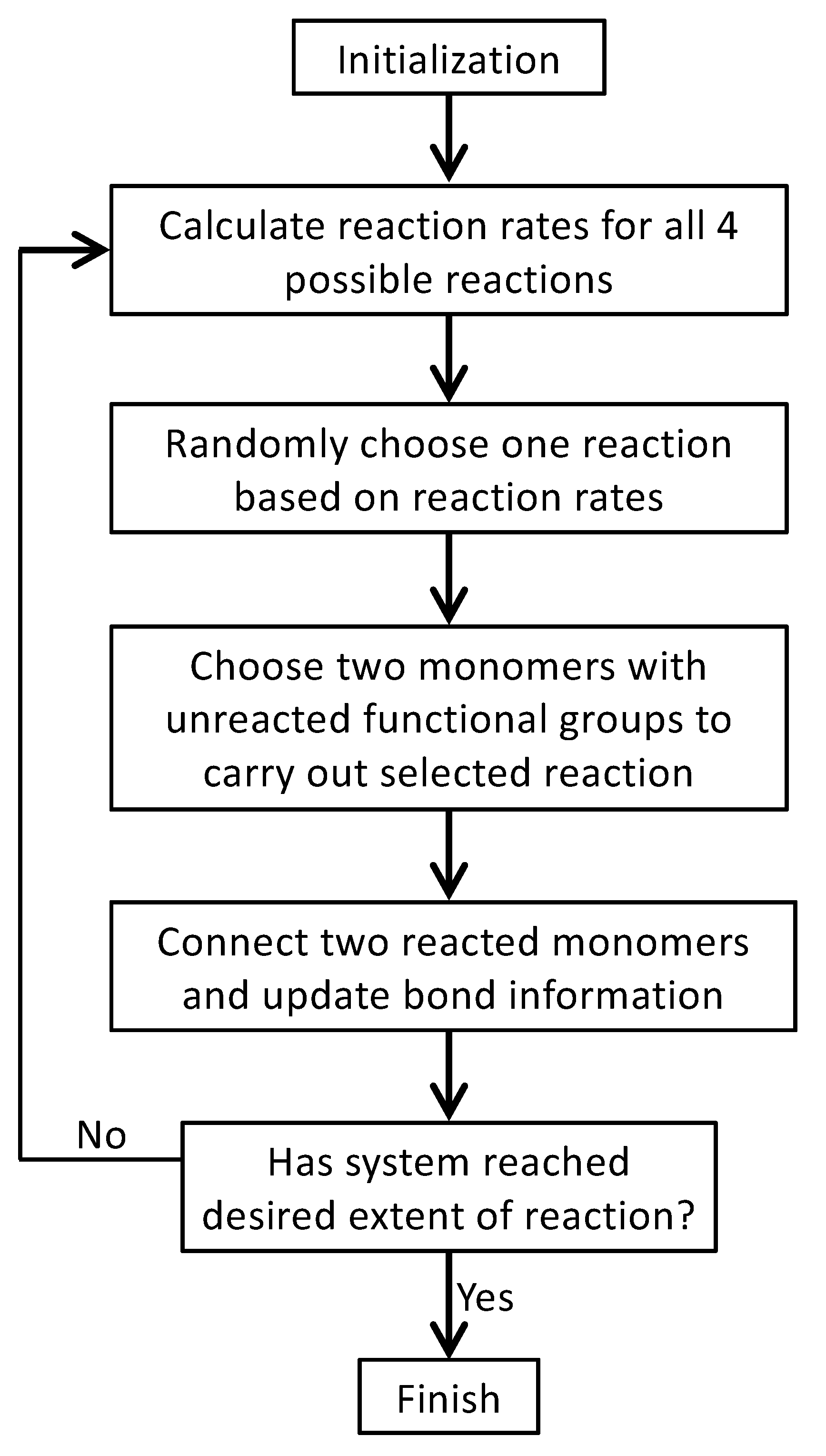
At each MC step, the probability of Reaction
l to be chosen is equal to
. After a reaction is selected, a pair of
and
that have unreacted functional groups (i.e., with unreacted
A and
B beads, respectively) is randomly chosen to react. Then, the system status is updated, including the bond information between the monomers and the identity of monomers with unreacted functional groups. The MC process is repeated for the updated system until no more reactions can occur or when the system has reached a desired extent of reaction. The flow chart of the MC simulation model is shown in
Figure 3. Note that in this model, backward reactions are not allowed, which means that once formed, the bond between an
A bead and a
B bead cannot break. However, the model permits the formation of rings, loops, and networks.
5. Conclusions
MC simulations are used to study the formation of branched polymers via a step-growth polymerization mechanism, and the method is applied to the polymerization of the branched PEIs from BPADA (backbone monomer), MPD (backbone monomer), PA (chain terminator), and TAPE (branching agent). All the reactions for this system can be reduced to a condensation reaction between an amine group and a carboxylic anhydride group, and thus, they can be characterized by one reaction rate. The results show that as assumed in the Flory-Stockmayer theory, the reaction rate is determined by the concentrations of the active functional groups on the monomers involved in a specific reaction, and not by the concentrations of the monomers themselves [
8,
9,
10,
11,
12,
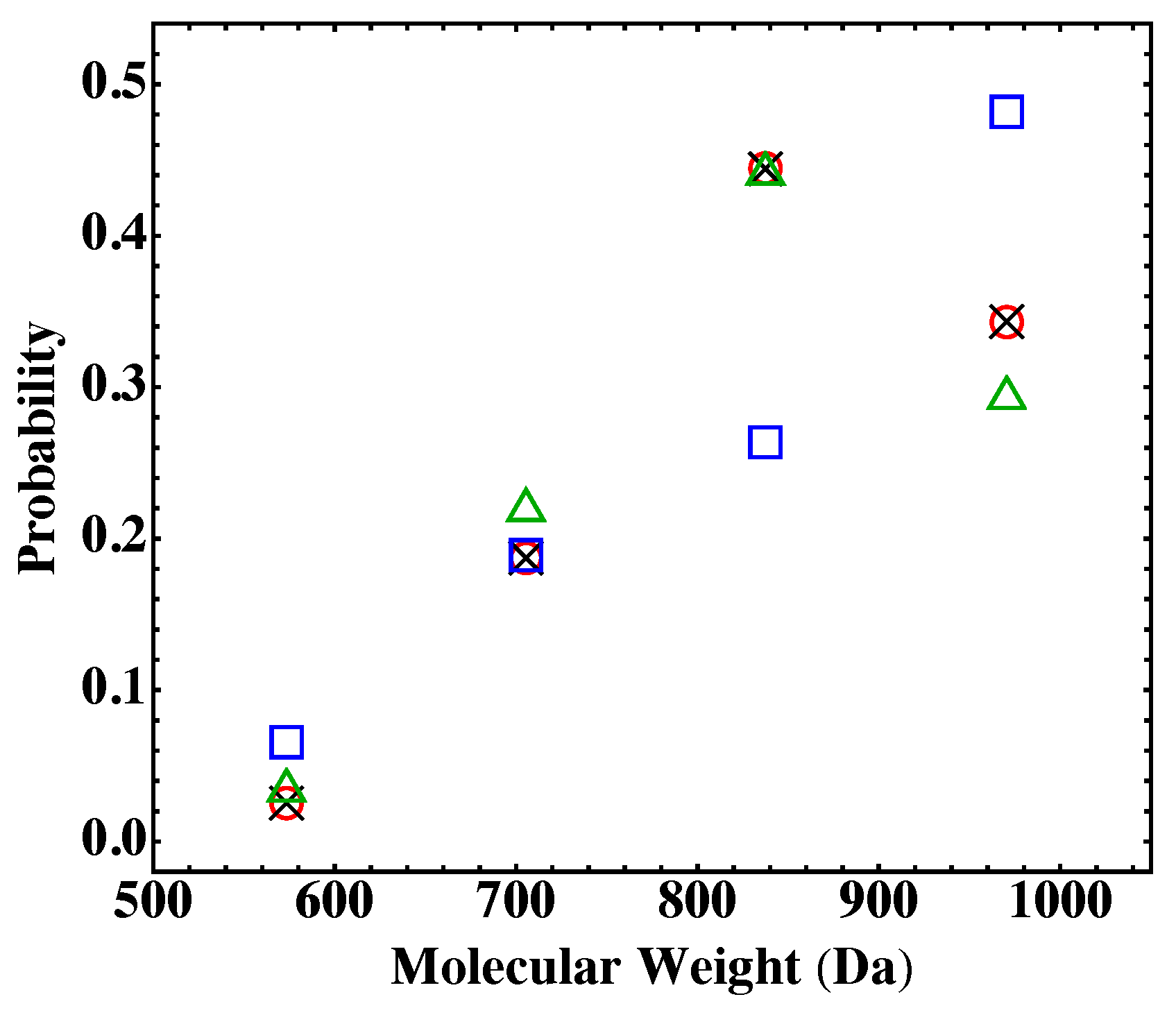
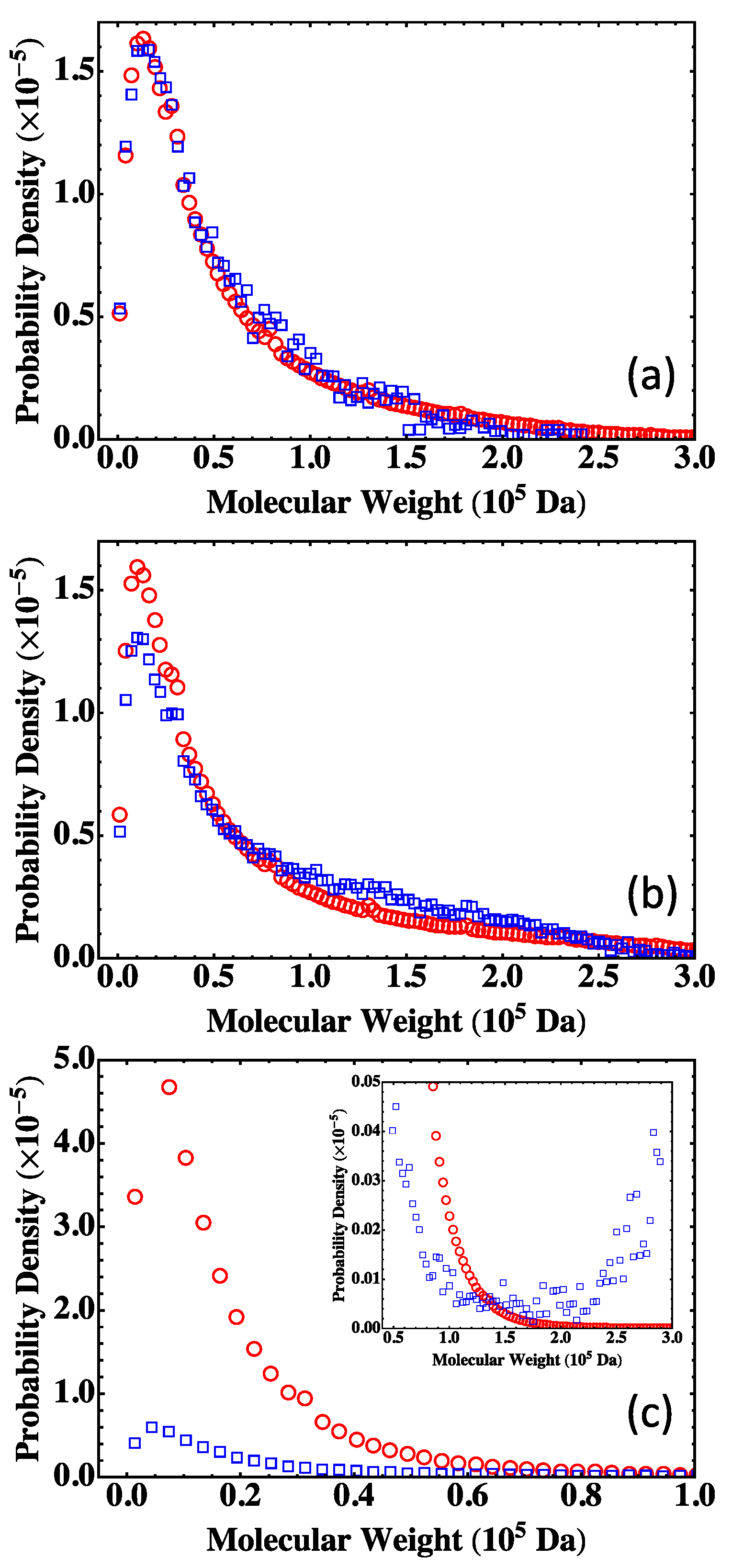
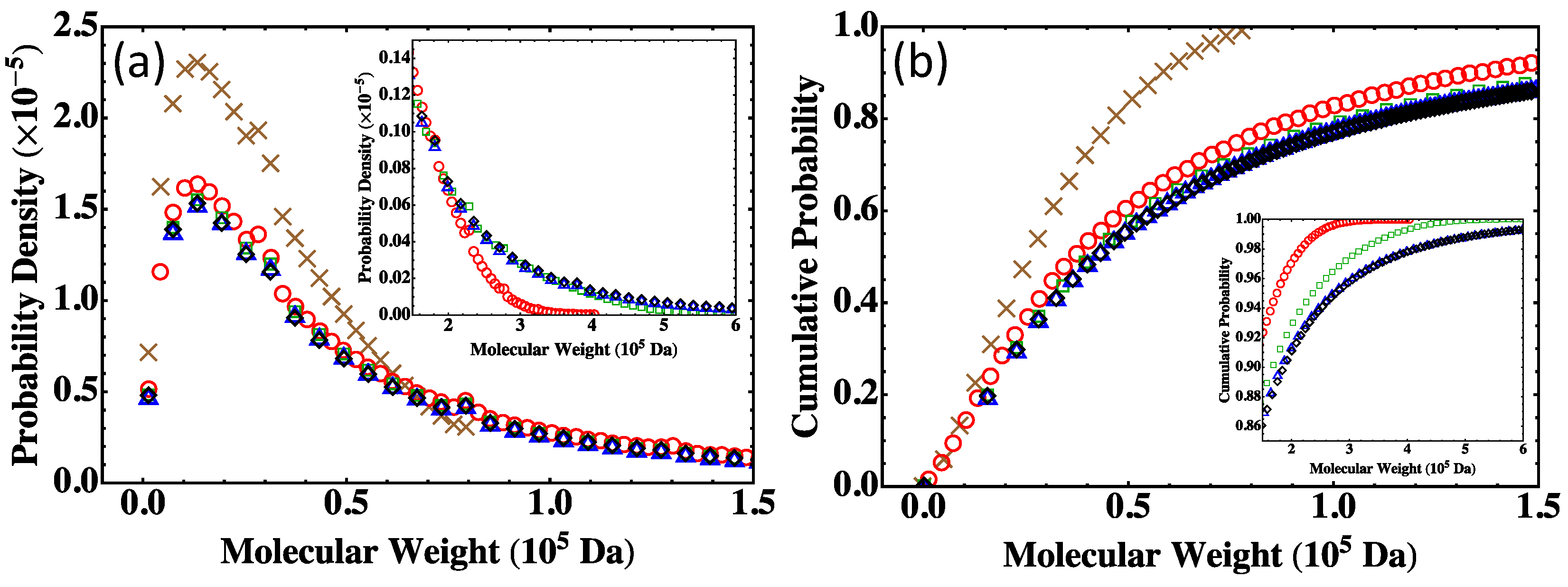
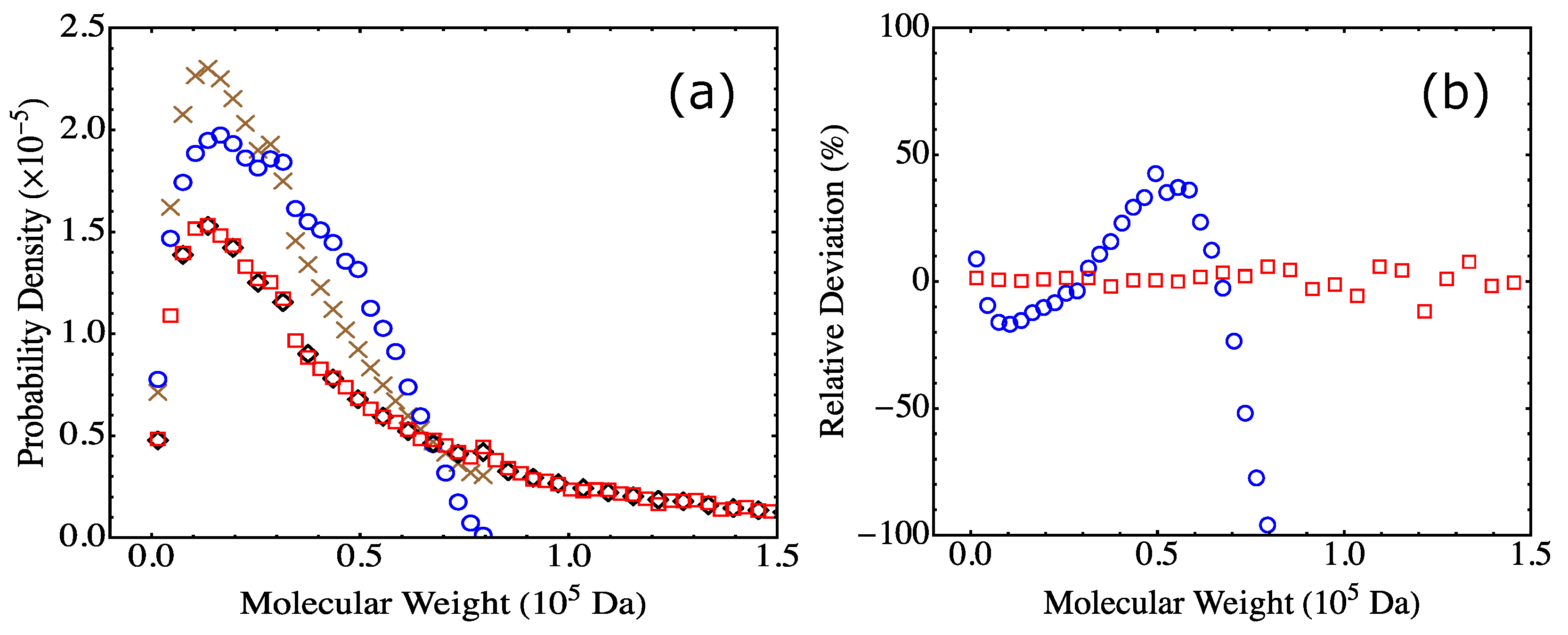
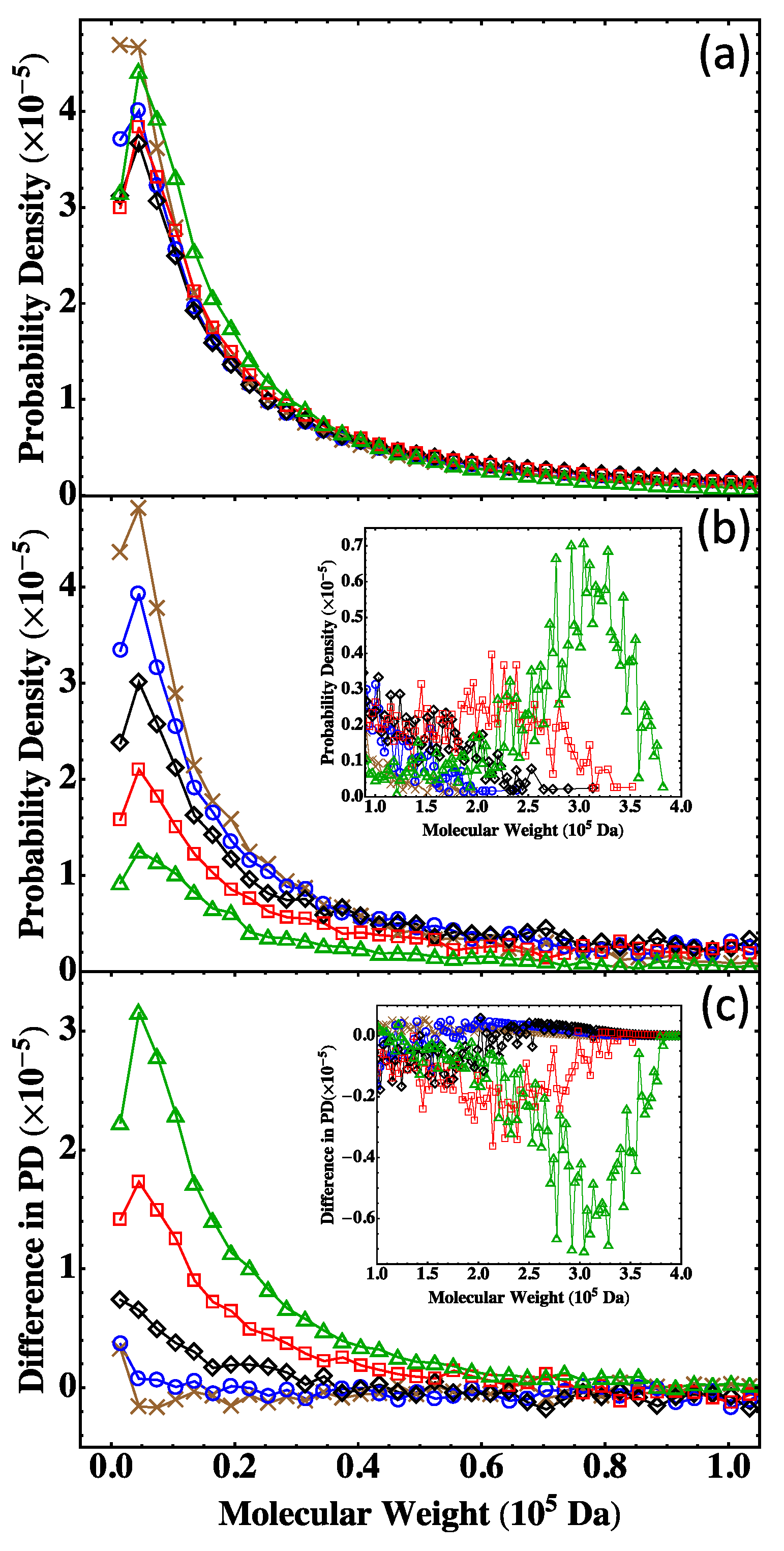
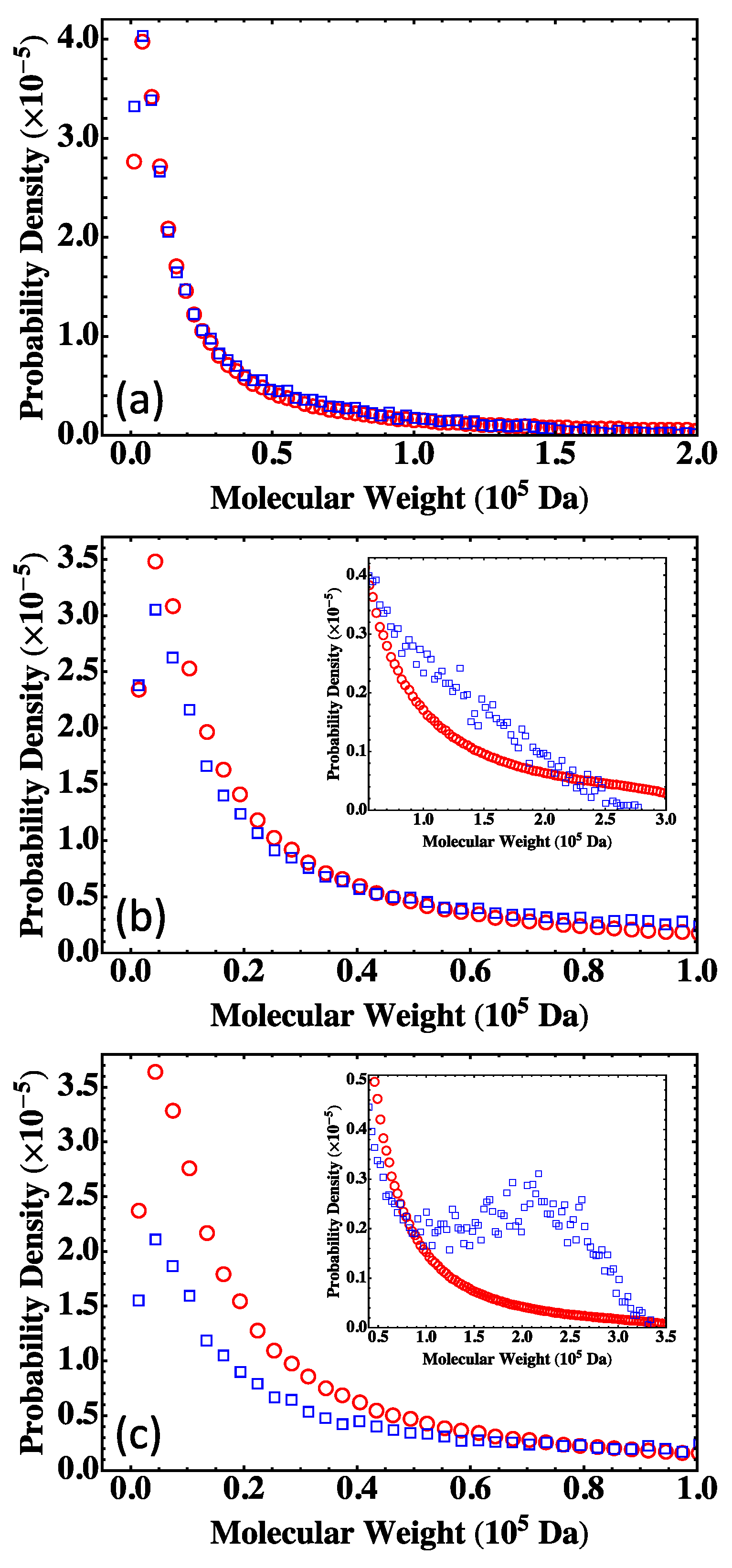
13]. A practical approach of using the Flory-Stockmayer theory to compute MWD has been suggested. In particular, the Stockmayer formula on MWD is simplified to a much more tractable form for fully reacted stoichiometric systems. The MC results are then compared to the predictions of the Flory-Stockmayer theory. Both theory and simulations accurately predict MWD for systems well below the gel point that is set by the functionality of the branching agent, though ring formation is not considered by the Flory-Stockmayer theory but is allowed in MC simulations. The agreement between the theory and simulations thus indicates that ring formation is negligible for systems that are well below the gel point. However, for systems close to or above the gel point, the Flory-Stockmayer theory is not applicable, as many cyclic polymers can be produced, and rings and loops can form in highly branched networks. The theory significantly underestimates the fraction of polymers with high molecular weights. For these systems, the MC simulations can still be used to quickly compute MWD that can be used to describe experimental measurements, including the average molecular weights.
The results further indicate that in the MC simulations, a system with only a few hundred to a few thousand monomers, but at the same molar ratios of participating monomers, is large enough to yield converging results on MWD for the region of molecular weight relevant to typical experiments (from 0 to about Da in the case of the branched PEIs). These conclusions have been thoroughly confirmed with simulations for fully reacted, partially reacted, stoichiometric, and nonstoichiometric systems. The MC model presented here is expected to be applicable to a wide range of step-growth polymers.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}