
Development of Polyurethane/Peptide-Based Carriers with Self-Healing Properties


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
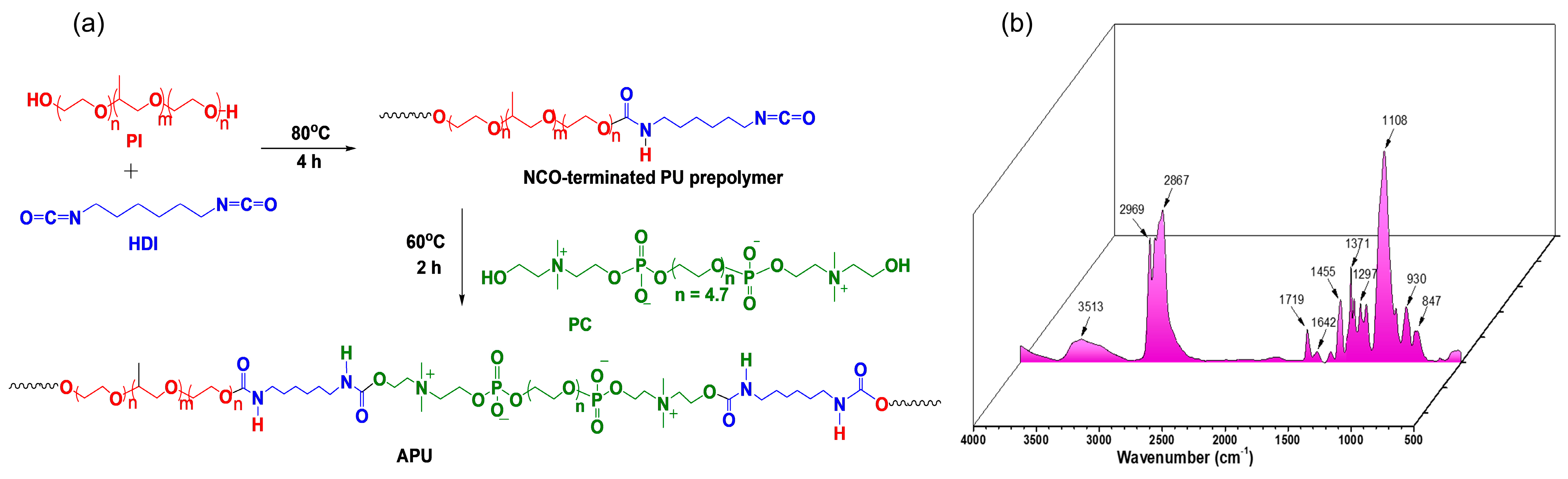
2.2. Synthesis of Amphiphilic Polyurethane
2.3. Preparation of Polyurethane/Peptide-Based Carriers
2.4. Characterization Methods
2.4.1. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Spectroscopy
2.4.2. Dynamic Light Scattering (DLS) Measurements
2.4.3. Rheological Investigations
2.4.4. Gel Permeation Chromatography (GPC)
2.5. Investigation of Gel Stability
2.6. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of Amphiphilic Polyurethane
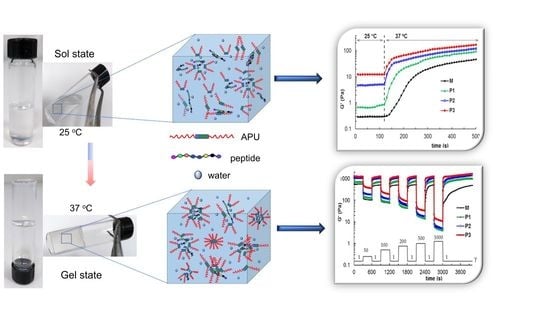
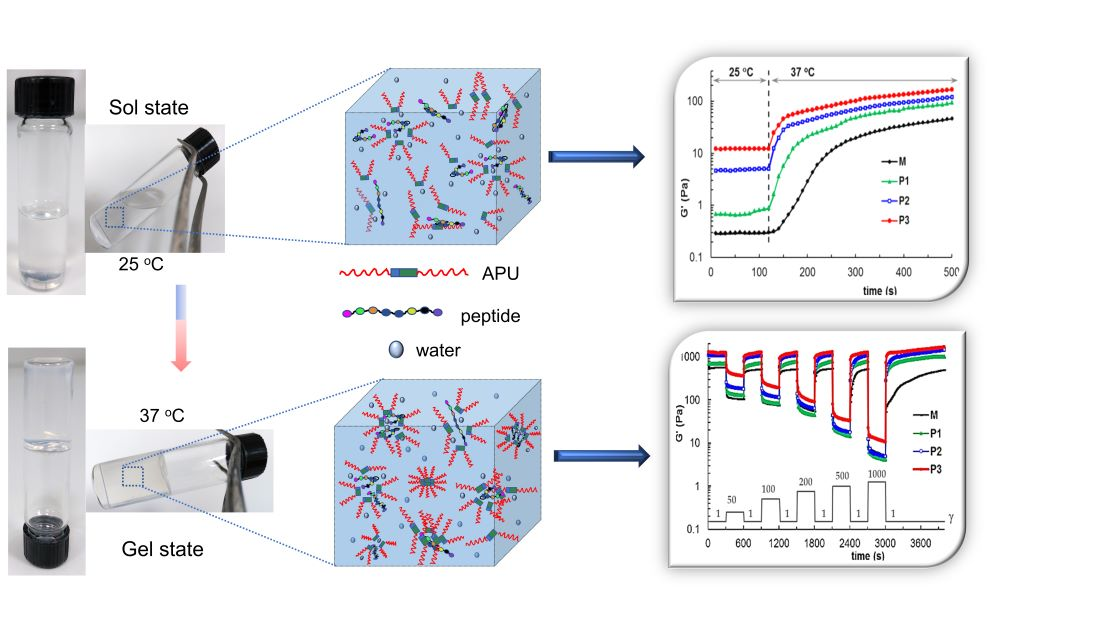
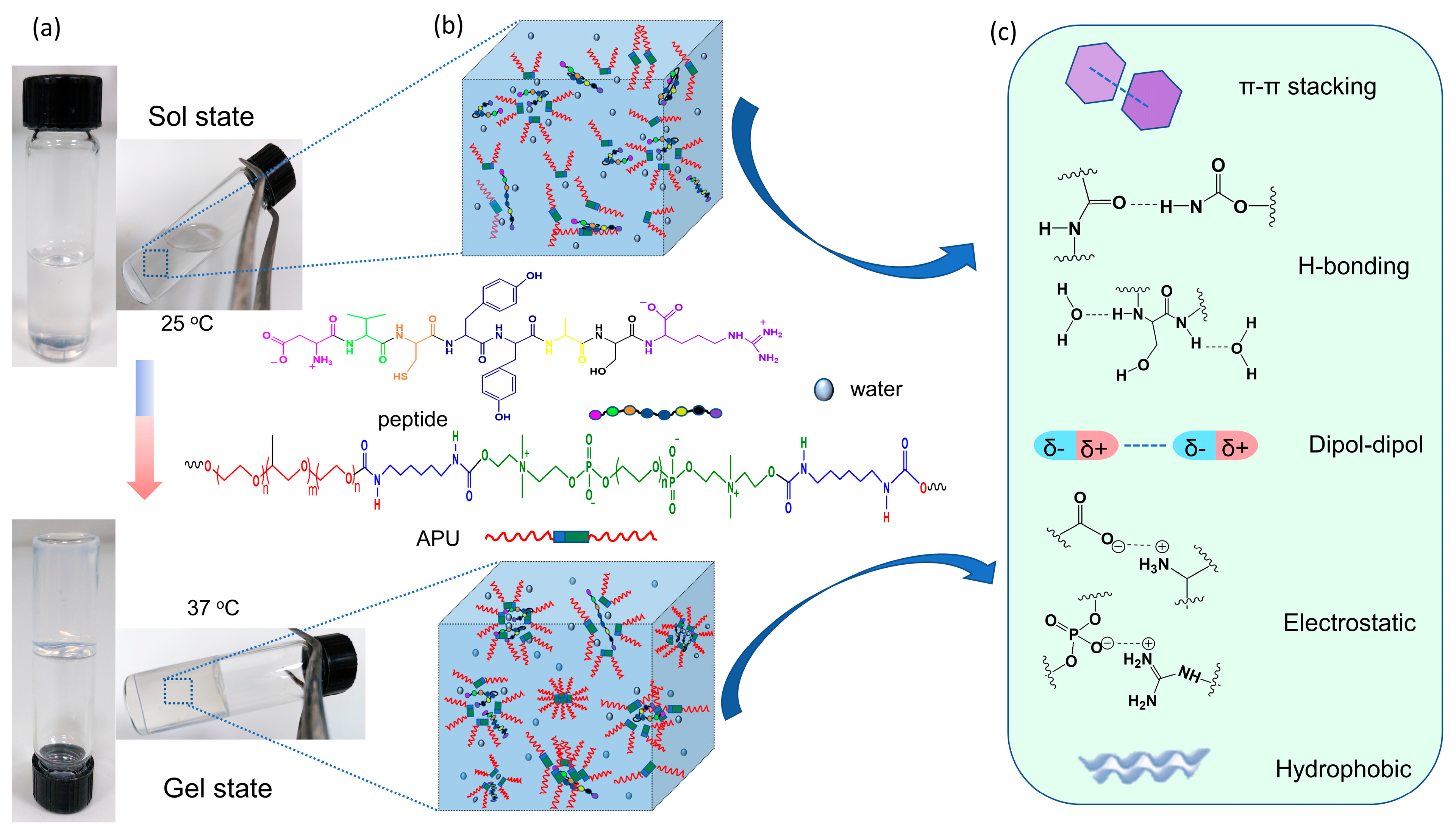
3.2. Development of Gels Based on APU and Peptide
3.3. Study of the Gelation Process
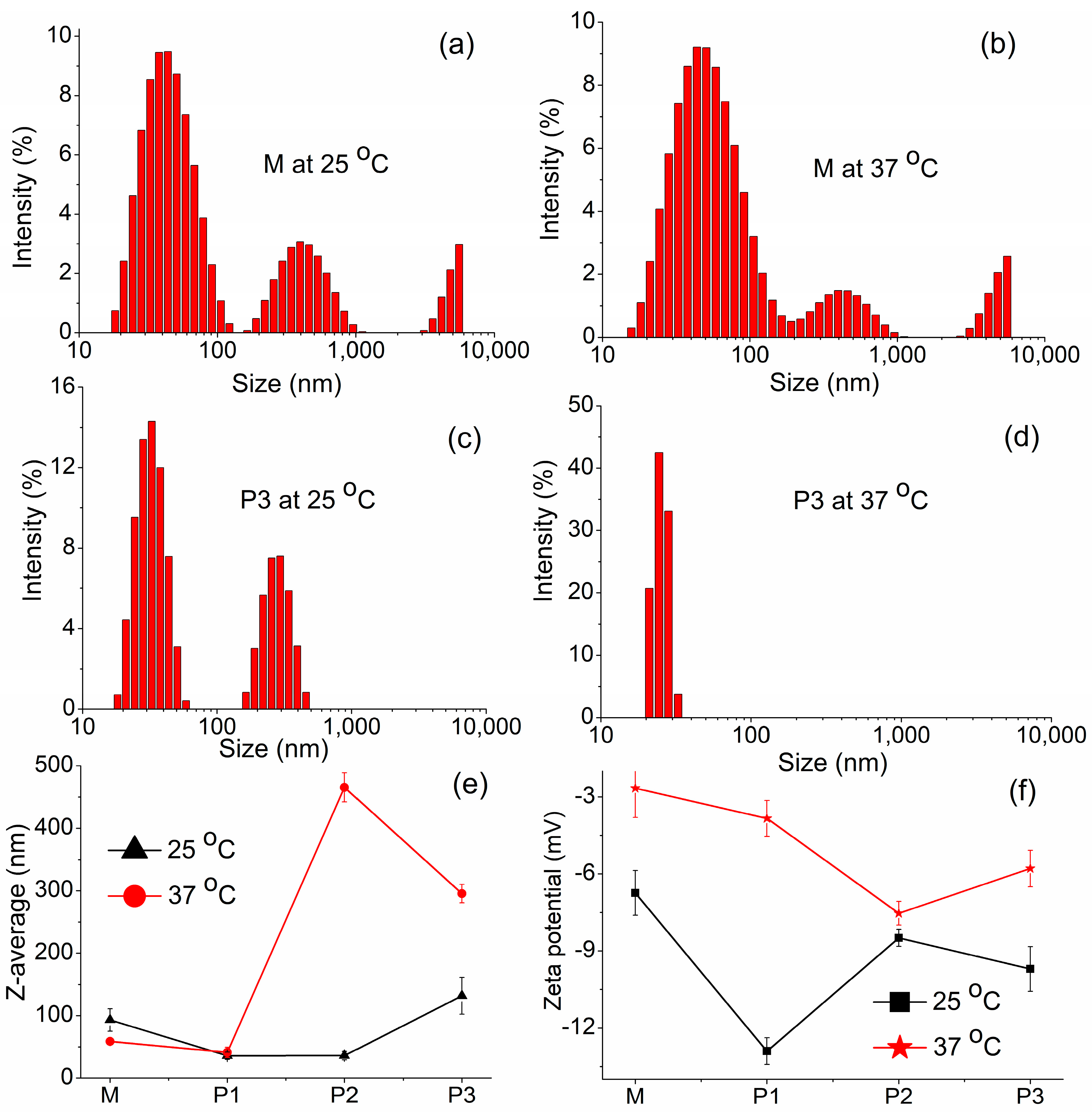
3.3.1. DLS Investigations
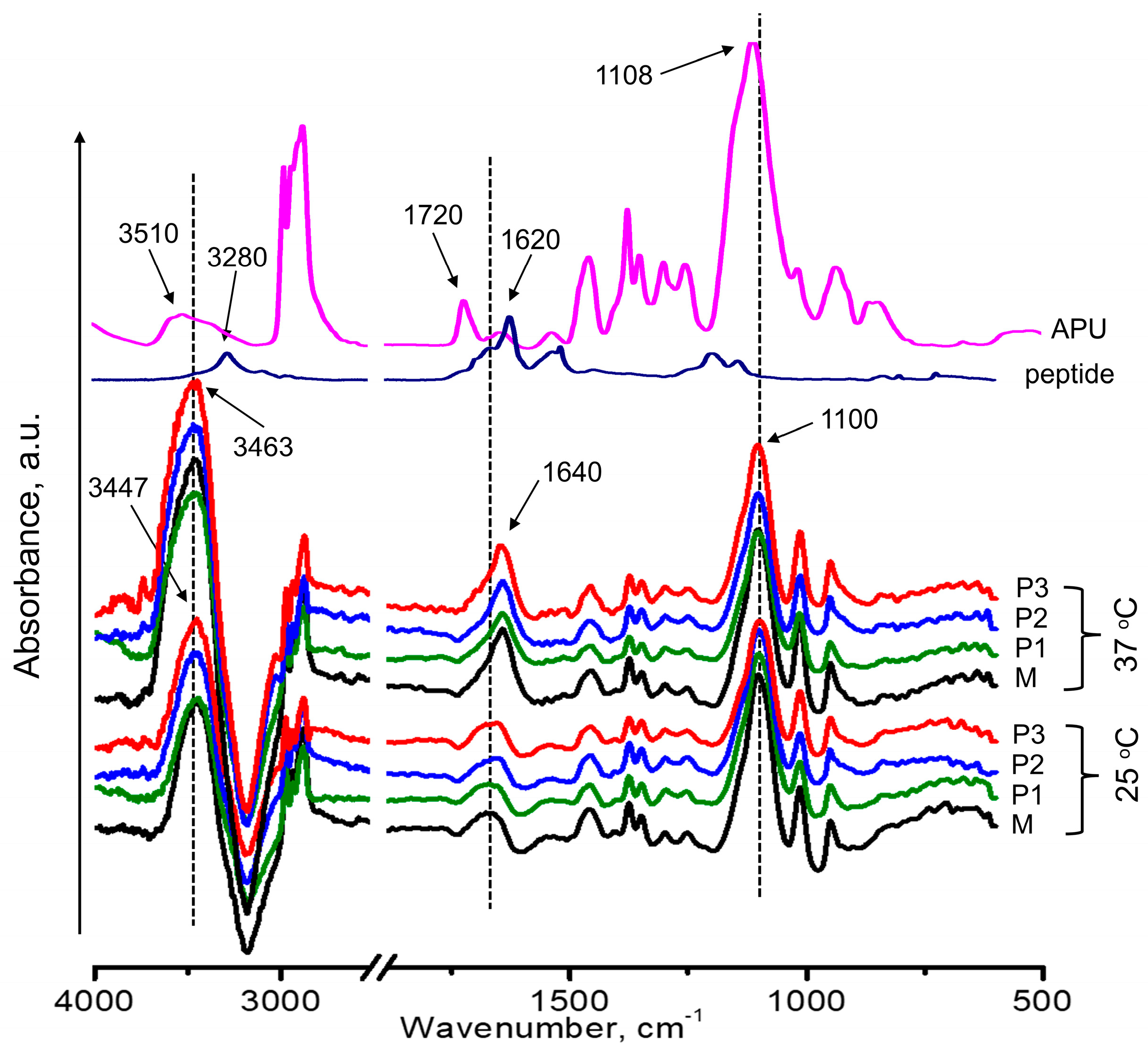
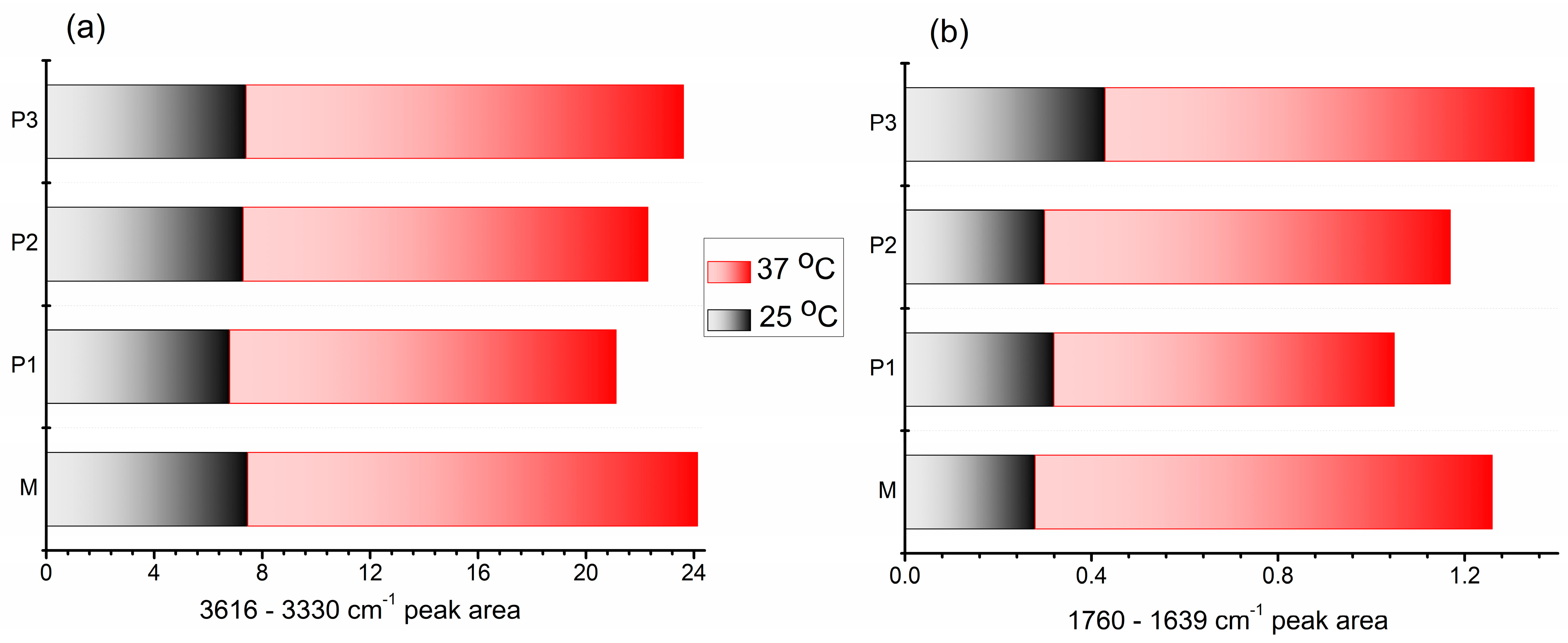
3.3.2. ATR-FTIR Spectroscopy Analysis
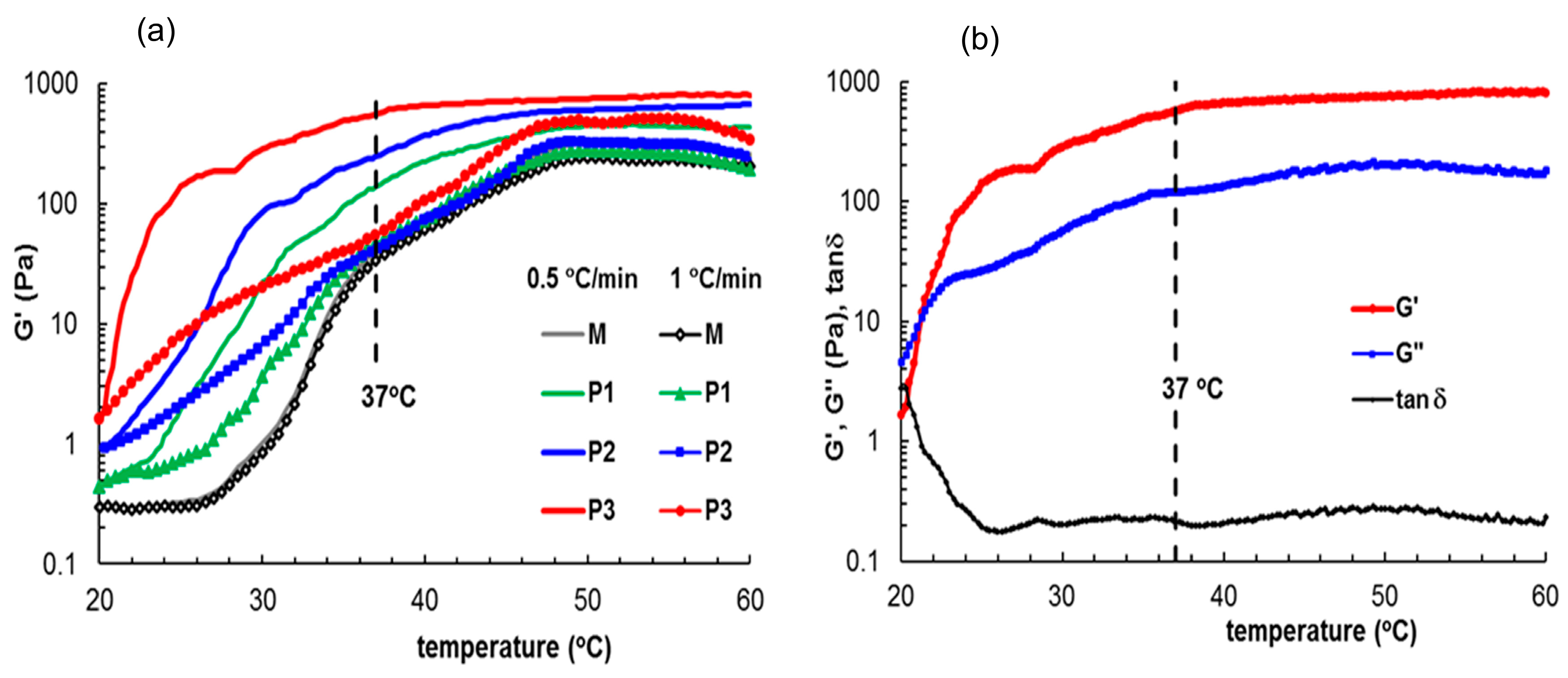
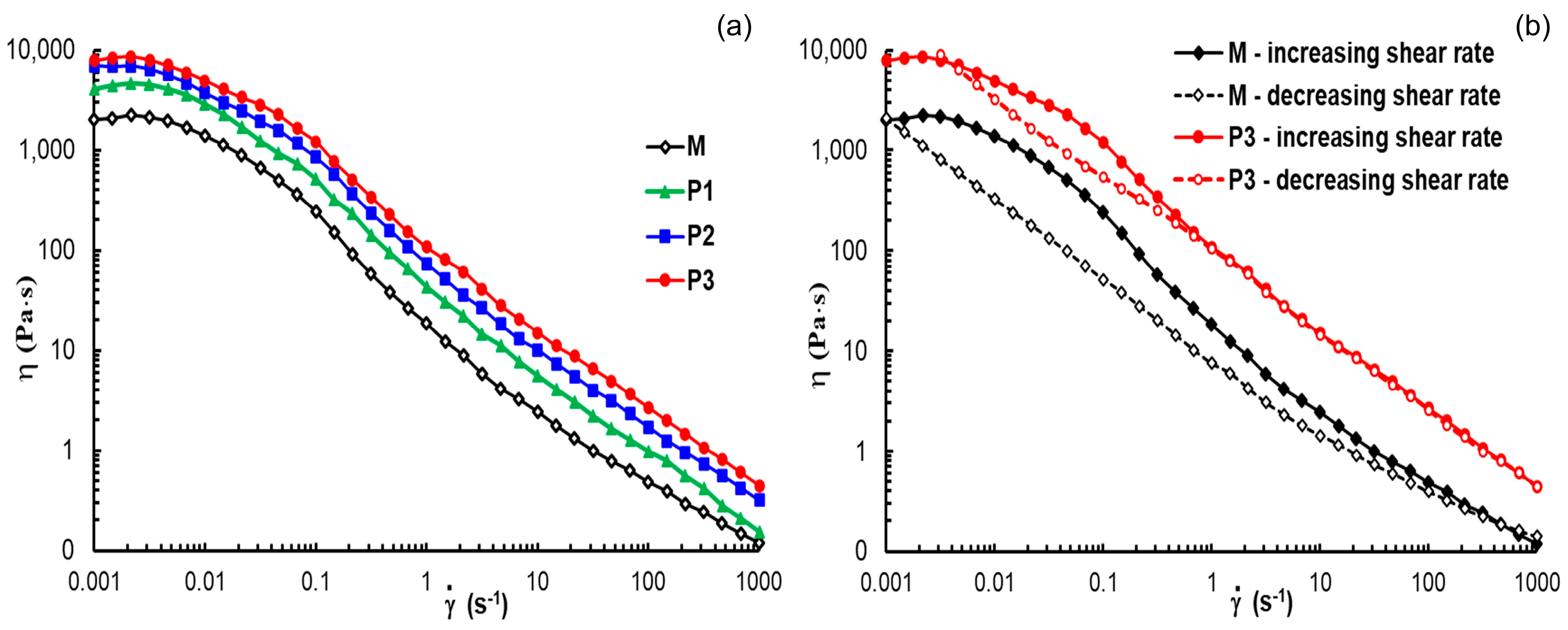
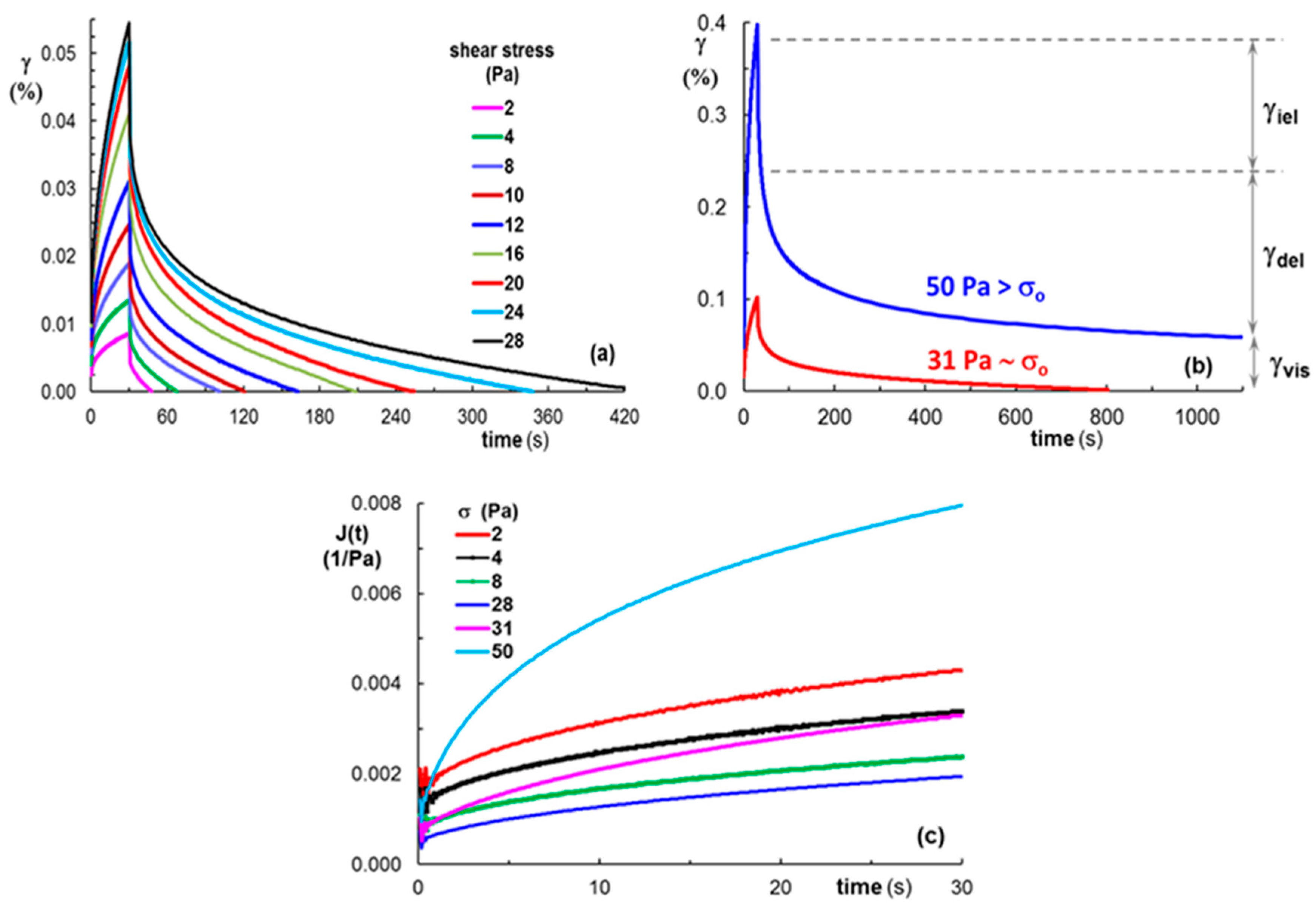
3.3.3. Rheological Behavior
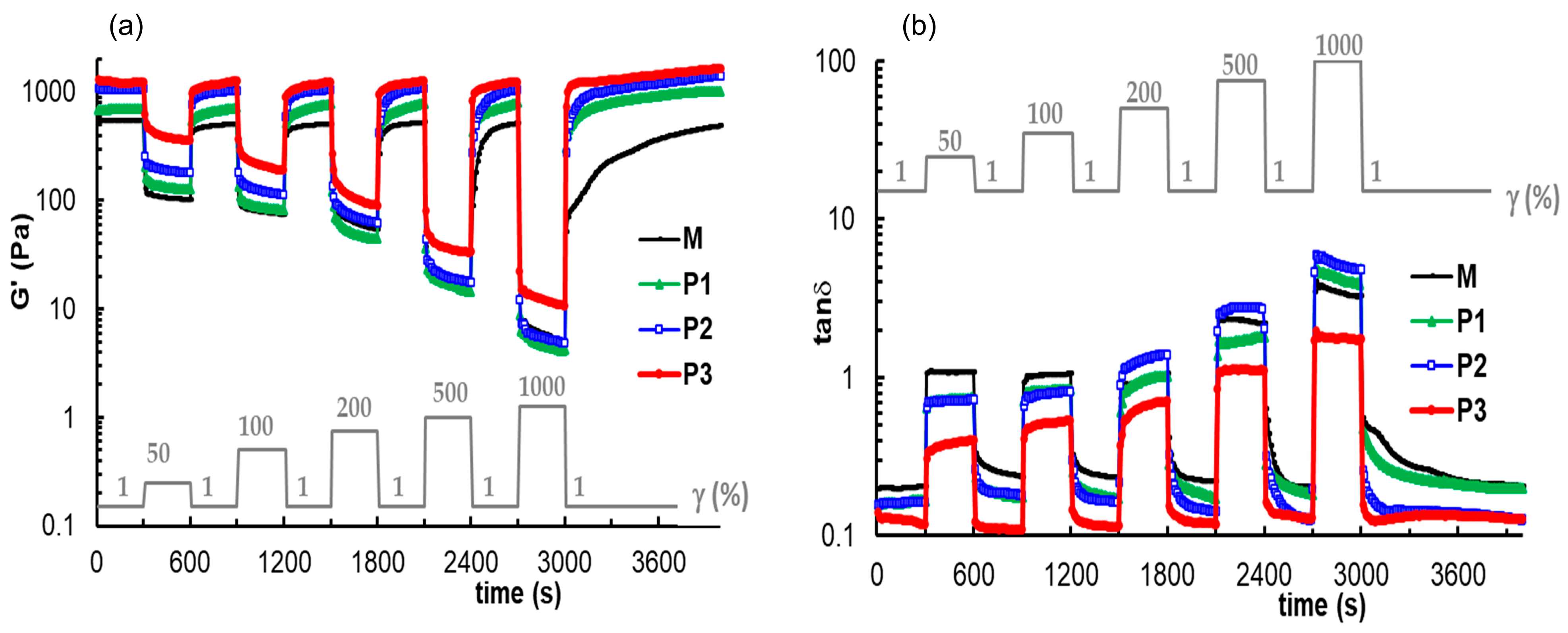
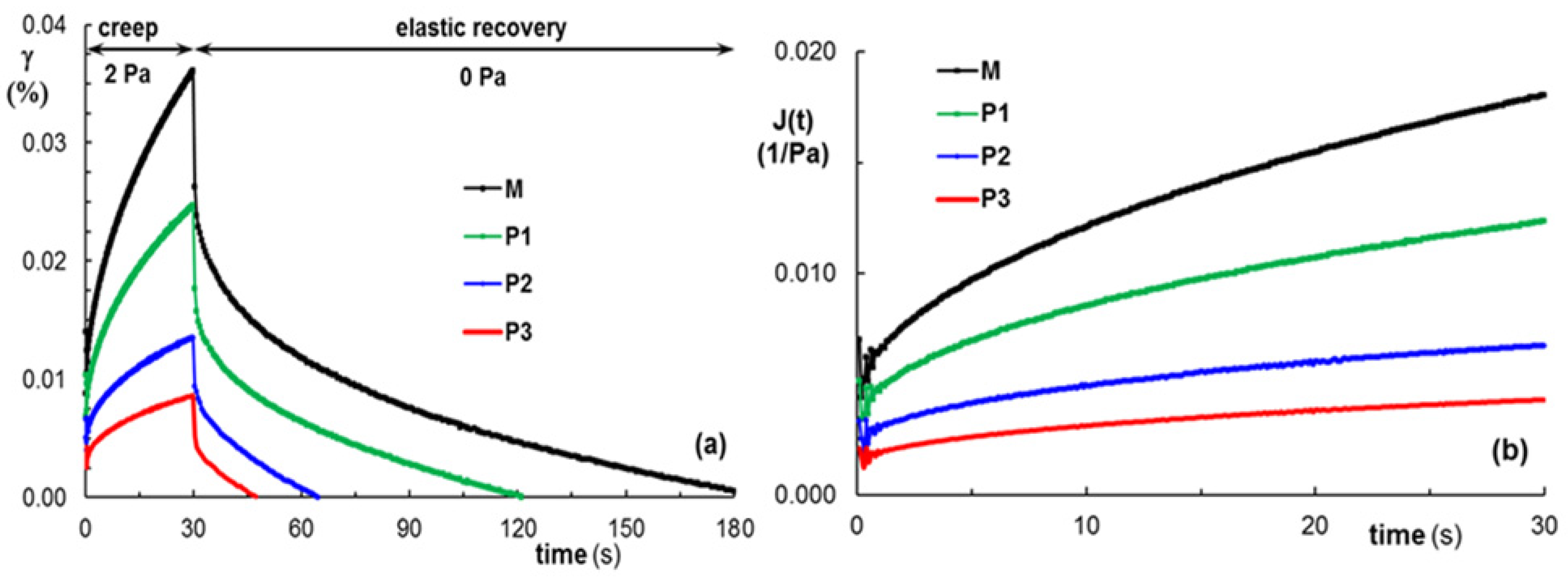
3.4. Self-Healing Behavior of Gels Illustrated in Different Rheological Tests
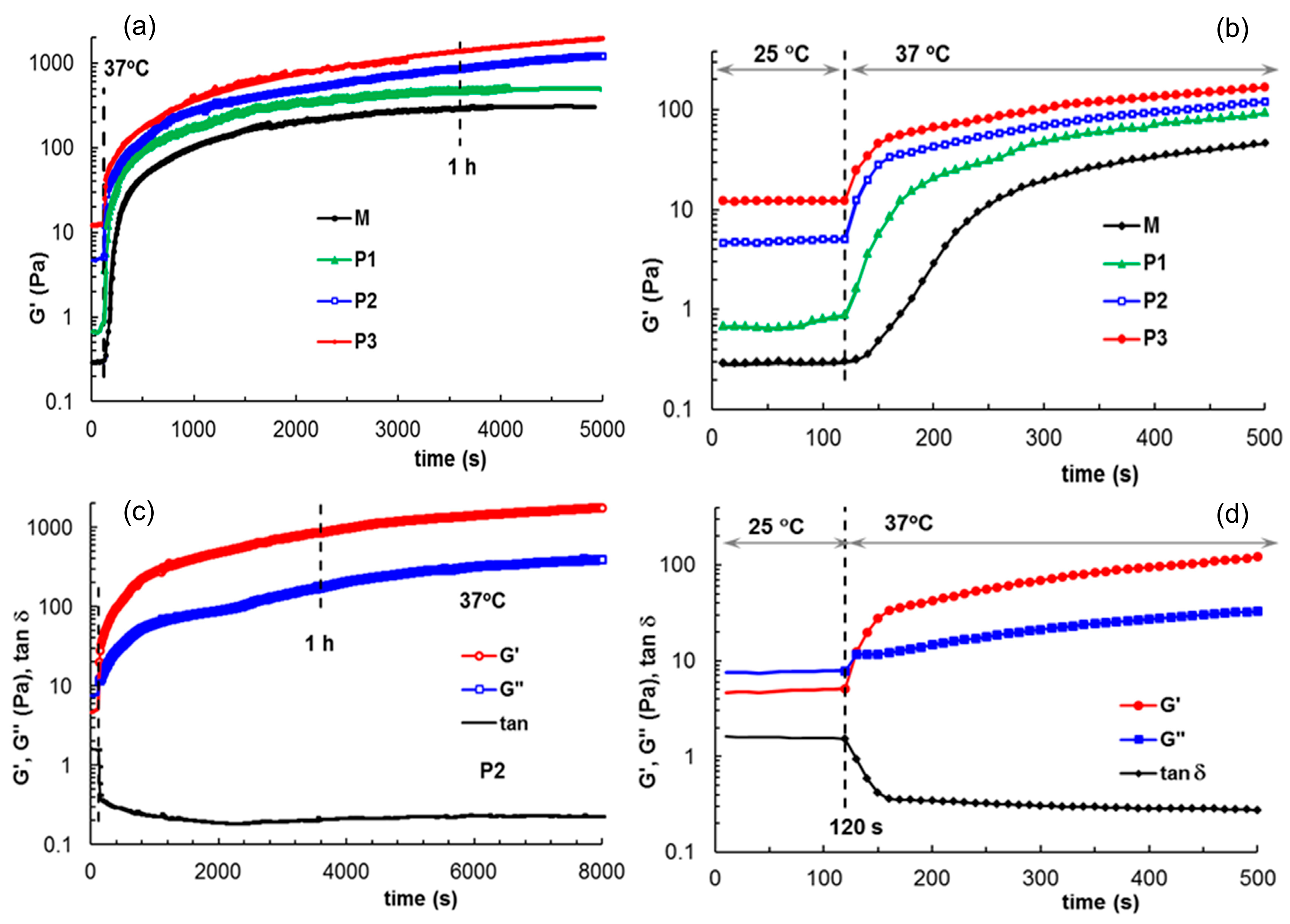
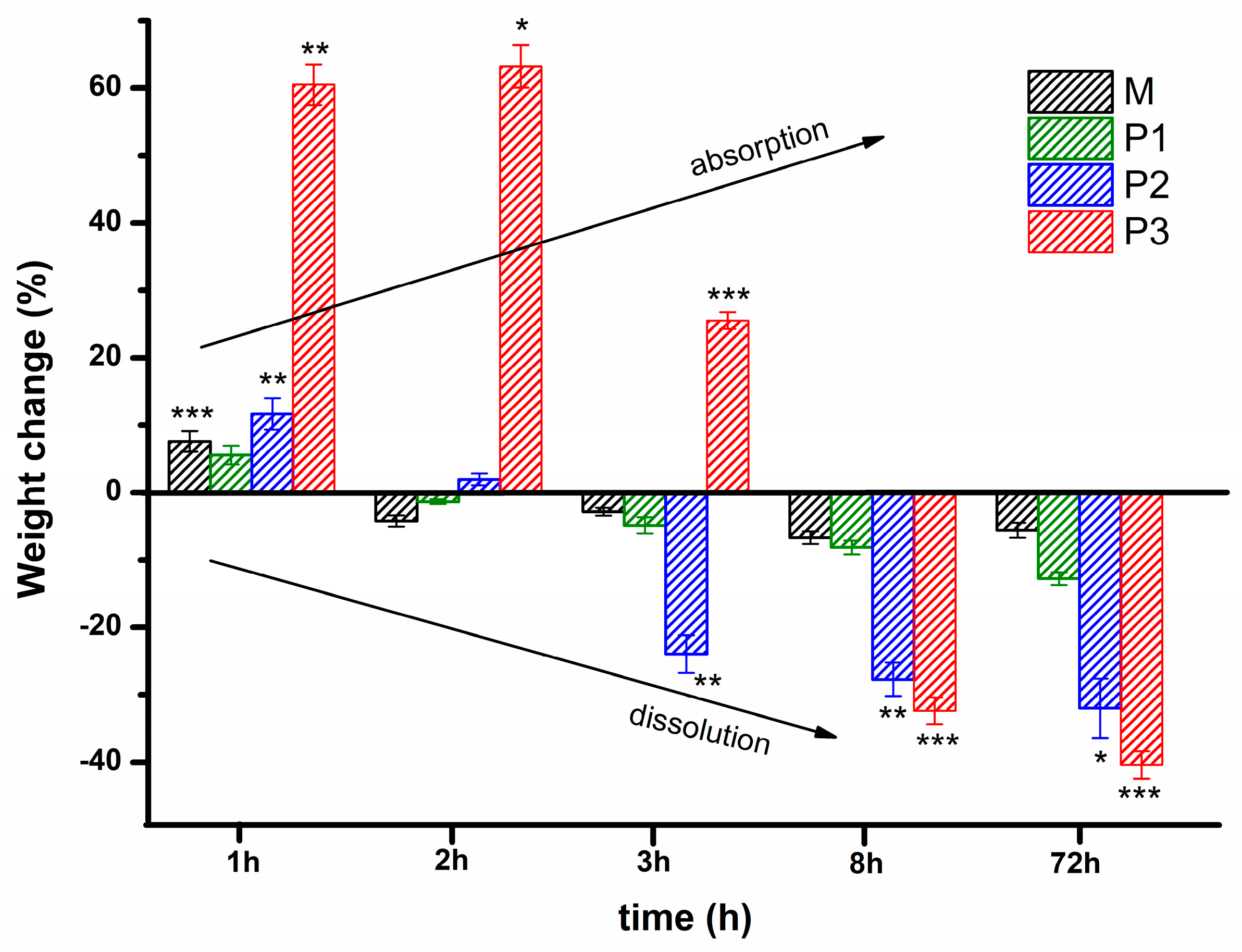
3.5. Investigation of Gel Stability at 37 °C in an Aqueous Environment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, H.-J.; Tsai, Y.-L.; Lin, S.-H.; Hsu, S. Smart polymers for cell therapy and precision medicine. J. Biomed. Sci. 2019, 26, 73. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.d.l.H.; Pennadam, S.; Alexander, C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 2005, 34, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C.G.; Meier, W. Stimuli-Responsive Polymers and Their Applications in Nanomedicine. Biointerphases 2012, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Rumon, M.M.H.; Akib, A.A.; Sultana, F.; Moniruzzaman, M.; Niloy, M.S.; Shakil, M.S.; Roy, C.K. Self-Healing Hydrogels: Development, Biomedical Applications, and Challenges. Polymers 2022, 14, 4539. [Google Scholar] [CrossRef]
- Pathan, N.; Shende, P. Strategic conceptualization and potential of self-healing polymers in biomedical field. Mater. Sci. Eng. C 2021, 125, 112099. [Google Scholar] [CrossRef]
- Bertsch, P.; Diba, M.; Mooney, D.J.; Leeuwenburgh, S.C.G. Self-Healing Injectable Hydrogels for Tissue Regeneration. Chem. Rev. 2023, 123, 834–873. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.; Yuk, H.; Lin, S.; Liu, X.; Parada, G. Soft Materials by Design: Unconventional Polymer Networks Give Extreme Properties. Chem. Rev. 2021, 121, 4309–4372. [Google Scholar] [CrossRef]
- Ye, E.; Chee, P.L.; Prasad, A.; Fang, X.; Owh, C.; Yeo, V.J.J.; Loh, X.J. Supramolecular soft biomaterials for biomedical applications. Mater. Today 2014, 17, 194–202. [Google Scholar] [CrossRef]
- Kausar, A. Polyurethane Composite Foams in High-Performance Applications: A Review. Polym. Plast. Technol. Eng. 2018, 57, 346–369. [Google Scholar] [CrossRef]
- Mohanty, S.R.; Mohanty, S.; Nayak, S.K.; Samal, S.K. Synthesis and evaluation of novel acrylic and ester-based polyols for transparent polyurethane coating applications. Mater. Today Commun. 2021, 27, 102228. [Google Scholar] [CrossRef]
- Chen, A.T.; Wojcik, R.T. Polyurethane coatings for metal and plastic substrates. Met. Finish. 2010, 108, 108–121. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Raju, K.V.S.N. Structural engineering of polyurethane coatings for high performance applications. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications—A review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, M.; Peng, H.-X. Functionalized MWNT-Doped Thermoplastic Polyurethane Nanocomposites for Aerospace Coating Applications. Macromol. Mater. Eng. 2010, 295, 838–845. [Google Scholar] [CrossRef]
- Engels, H.-W.; Pirkl, H.-G.; Albers, R.; Albach, R.W.; Krause, J.; Hoffmann, A.; Casselmann, H.; Dormish, J. Polyurethanes: Versatile Materials and Sustainable Problem Solvers for Today’s Challenges. Angew. Chem. Int. Ed. 2013, 52, 9422–9441. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mahanwar, P. A brief discussion on advances in polyurethane applications. Adv. Ind. Eng. Polym. Res. 2020, 3, 93–101. [Google Scholar] [CrossRef]
- Davies, P.; Evrard, G. Accelerated ageing of polyurethanes for marine applications. Polym. Degrad. Stab. 2007, 92, 1455–1464. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Patel, R.M.; Wenham, A.; Smith, J.R. Biomedical applications of polyurethane materials and coatings. Trans. IMF 2018, 96, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Wendels, S.; Avérous, L. Biobased polyurethanes for biomedical applications. Bioact. Mater. 2021, 6, 1083–1106. [Google Scholar] [CrossRef]
- Lowinger, M.; Barrett, S.; Zhang, F.; Williams, R. Sustained Release Drug Delivery Applications of Polyurethanes. Pharmaceutics 2018, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Gradinaru, L.M.; Barbalata Mandru, M.; Drobota, M.; Aflori, M.; Butnaru, M.; Spiridon, M.; Doroftei, F.; Aradoaei, M.; Ciobanu, R.C.; Vlad, S. Composite Materials Based on Iron Oxide Nanoparticles and Polyurethane for Improving the Quality of MRI. Polymers 2021, 13, 4316. [Google Scholar] [CrossRef]
- Gradinaru, L.; Ciobanu, C.; Vlad, S.; Bercea, M.; Popa, M. Synthesis and rheology of thermoreversible polyurethane hydrogels. Open Chem. 2012, 10, 1859–1866. [Google Scholar] [CrossRef]
- Vlad, S.; Tanase, C.; Macocinschi, D.; Ciobanu, C.; Balaes, T.; Filip, D.; Gostin, I.N.; Gradinaru, L.M. Antifungal behaviour of polyurethane membranes with zinc oxide nanoparticles. Dig. J. Nanomater. Biostruct. 2012, 7, 51–58. [Google Scholar]
- Cook, M.T.; Haddow, P.; Kirton, S.B.; McAuley, W.J. Polymers Exhibiting Lower Critical Solution Temperatures as a Route to Thermoreversible Gelators for Healthcare. Adv. Funct. Mater. 2021, 31, 2008123. [Google Scholar] [CrossRef]
- Yu, J.; Qiu, H.; Yin, S.; Wang, H.; Li, Y. Polymeric Drug Delivery System Based on Pluronics for Cancer Treatment. Molecules 2021, 26, 3610. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K. Recent progress in biomedical applications of Pluronic (PF127): Pharmaceutical perspectives. J. Control. Release 2015, 209, 120–138. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Villa, C. Poloxamer Hydrogels for Biomedical Applications. Pharmaceutics 2019, 11, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitto-Barry, A.; Barry, N.P.E. Pluronic® block-copolymers in medicine: From chemical and biological versatility to rationalisation and clinical advances. Polym. Chem. 2014, 5, 3291–3297. [Google Scholar] [CrossRef] [Green Version]
- Zarrintaj, P.; Ramsey, J.D.; Samadi, A.; Atoufi, Z.; Yazdi, M.K.; Ganjali, M.R.; Amirabad, L.M.; Zangene, E.; Farokhi, M.; Formela, K.; et al. Poloxamer: A versatile tri-block copolymer for biomedical applications. Acta Biomater. 2020, 110, 37–67. [Google Scholar] [CrossRef]
- Boffito, M.; Grivet Brancot, A.; Lima, O.; Bronco, S.; Sartori, S.; Ciardelli, G. Injectable thermosensitive gels for the localized and controlled delivery of biomolecules in tissue engineering/regenerative medicine. Biomed. Sci. Eng. 2019, 3, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Boffito, M.; Gioffredi, E.; Chiono, V.; Calzone, S.; Ranzato, E.; Martinotti, S.; Ciardelli, G. Novel polyurethane-based thermosensitive hydrogels as drug release and tissue engineering platforms: Design and in vitro characterization. Polym. Int. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Colucci, F.; Mancini, V.; Mattu, C.; Boffito, M. Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study. Pharmaceutics 2021, 13, 464. [Google Scholar] [CrossRef] [PubMed]
- Laurano, R.; Abrami, M.; Grassi, M.; Ciardelli, G.; Boffito, M.; Chiono, V. Using Poloxamer® 407 as Building Block of Amphiphilic Poly(ether urethane)s: Effect of its Molecular Weight Distribution on Thermo-Sensitive Hydrogel Performances in the Perspective of Their Biomedical Application. Front. Mater. 2020, 7, 594515. [Google Scholar] [CrossRef]
- Bonilla-Hernández, M.; Zapata-Catzin, G.A.; Castillo-Cruz, O.d.J.; Vargas-Coronado, R.F.; Cervantes-Uc, J.M.; Xool-Tamayo, J.F.; Borges-Argaez, R.; Hernández-Baltazar, E.; Cauich-Rodríguez, J.V. Synthesis and characterization of metformin-pluronic based polyurethanes for controlled drug delivery. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 656–667. [Google Scholar] [CrossRef]
- Loh, X.J.; Gan, H.X.; Wang, H.; Tan, S.J.E.; Neoh, K.Y.; Jean Tan, S.S.; Diong, H.F.; Kim, J.J.; Sharon Lee, W.L.; Fang, X.; et al. New thermogelling poly(ether carbonate urethane)s based on pluronics F127 and poly(polytetrahydrofuran carbonate). J. Appl. Polym. Sci. 2014, 131, 39924. [Google Scholar] [CrossRef]
- Almasian, A.; Najafi, F.; Eftekhari, M.; Shams Ardekani, M.R.; Sharifzadeh, M.; Khanavi, M. Preparation of Polyurethane/Pluronic F127 Nanofibers Containing Peppermint Extract Loaded Gelatin Nanoparticles for Diabetic Wounds Healing: Characterization, In Vitro, and In Vivo Studies. Evid.-Based Complement. Altern. Med. 2021, 2021, 6646702. [Google Scholar] [CrossRef] [PubMed]
- Volkmer, E.; Leicht, U.; Moritz, M.; Schwarz, C.; Wiese, H.; Milz, S.; Matthias, P.; Schloegl, W.; Friess, W.; Goettlinger, M.; et al. Poloxamer-based hydrogels hardening at body core temperature as carriers for cell based therapies: In vitro and in vivo analysis. J. Mater. Sci. Mater. Med. 2013, 24, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, L.M.; Ciobanu, C.; Vlad, S.; Bercea, M.; Popa, M. Thermoreversible Poly(isopropyl lactate diol)-Based Polyurethane Hydrogels: Effect of Isocyanate on Some Physical Properties. Ind. Eng. Chem. Res. 2012, 51, 12344–12354. [Google Scholar] [CrossRef]
- Ciobanu, C.; Gradinaru, L.M.; Drobota, M.; Quaini, F.; Falco, A.; Frati, C.; Graiani, G.; Madeddu, D.; Lagrasta, C.; Vlad, S.; et al. Injectable Thermoreversible Hydrogels Based on Amphiphilic Polyurethanes: Structure-Property Correlations. J. Hydrogels 2015, 1, 12–25. [Google Scholar] [CrossRef]
- Ciobanu, C.; Gradinaru, L.M.; Drobota, M.; Vlad, S.; Bercea, M.; Popa, M. Influence of Diisocyanate Structure on Properties of Some Thermoreversible Polyurethane Hydrogels. J. Hydrogels 2015, 1, 41–49. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Ciobanu, C.; Vlad, S.; Bercea, M. Rheological investigation of thermoreversible polyurethane hydrogels. Rev. Roum. Chim. 2016, 61, 411–417. [Google Scholar]
- Dietzen, D.J. Amino Acids, Peptides, and Proteins. In Principles and Applications of Molecular Diagnostics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 345–380. ISBN 9780128160619. [Google Scholar]
- Golinska, M.D.; Włodarczyk-Biegun, M.K.; Werten, M.W.T.; Stuart, M.A.C.; de Wolf, F.A.; de Vries, R. Dilute Self-Healing Hydrogels of Silk-Collagen-Like Block Copolypeptides at Neutral pH. Biomacromolecules 2014, 15, 699–706. [Google Scholar] [CrossRef]
- Mocanu, C.S.; Petre, B.A.; Darie-Ion, L.; Drochioiu, G.; Niculaua, M.; Stoica, I.; Homocianu, M.; Nita, L.E.; Gradinaru, V.R. Structural Characterization of a New Collagen Biomimetic Octapeptide with Nanoscale Self-Assembly Potential: Experimental and Theoretical Approaches. Chempluschem 2022, 87, e202100462. [Google Scholar] [CrossRef]
- Munteanu, I.G.; Grădinaru, V.R.; Apetrei, C. Sensitive Detection of Rosmarinic Acid Using Peptide-Modified Graphene Oxide Screen-Printed Carbon Electrode. Nanomaterials 2022, 12, 3292. [Google Scholar] [CrossRef]
- Li, Y.; Qin, M.; Cao, Y.; Wang, W. Designing the mechanical properties of peptide-based supramolecular hydrogels for biomedical applications. Sci. China Phys. Mech. Astron. 2014, 57, 849–858. [Google Scholar] [CrossRef]
- Jain, R.; Roy, S. Designing a bioactive scaffold from coassembled collagen–laminin short peptide hydrogels for controlling cell behaviour. RSC Adv. 2019, 9, 38745–38759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Singla, A.; Lee, Y. Biomedical applications of collagen. Int. J. Pharm. 2001, 221, 1–22. [Google Scholar] [CrossRef]
- Rezvani Ghomi, E.; Nourbakhsh, N.; Akbari Kenari, M.; Zare, M.; Ramakrishna, S. Collagen-based biomaterials for biomedical applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2021, 109, 1986–1999. [Google Scholar] [CrossRef]
- Benhardt, H.; Sears, N.; Touchet, T.; Cosgriff-Hernandez, E. Synthesis of Collagenase-Sensitive Polyureas for Ligament Tissue Engineering. Macromol. Biosci. 2011, 11, 1020–1030. [Google Scholar] [CrossRef]
- Aluri, R.; Jayakannan, M. Development of Tyrosine-Based Enzyme-Responsive Amphiphilic Poly(ester-urethane) Nanocarriers for Multiple Drug Delivery to Cancer Cells. Biomacromolecules 2017, 18, 189–200. [Google Scholar] [CrossRef]
- Ding, X.; Chin, W.; Lee, C.N.; Hedrick, J.L.; Yang, Y.Y. Peptide-Functionalized Polyurethane Coatings Prepared via Grafting-To Strategy to Selectively Promote Endothelialization. Adv. Healthc. Mater. 2018, 7, 1700944. [Google Scholar] [CrossRef]
- Mandru, M.; Bercea, M.; Gradinaru, L.M.; Ciobanu, C.; Drobota, M.; Vlad, S.; Albulescu, R. Polyurethane/poly(vinyl alcohol) hydrogels: Preparation, characterization and drug delivery. Eur. Polym. J. 2019, 118, 137–145. [Google Scholar] [CrossRef]
- Hebling, J.; Bianchi, L.; Basso, F.G.; Scheffel, D.L.; Soares, D.G.; Carrilho, M.R.O.; Pashley, D.H.; Tjäderhane, L.; de Souza Costa, C.A. Cytotoxicity of dimethyl sulfoxide (DMSO) in direct contact with odontoblast-like cells. Dent. Mater. 2015, 31, 399–405. [Google Scholar] [CrossRef] [Green Version]
- de Abreu Costa, L.; Henrique Fernandes Ottoni, M.; dos Santos, M.; Meireles, A.; Gomes de Almeida, V.; de Fátima Pereira, W.; Alves de Avelar-Freitas, B.; Eustáquio Alvim Brito-Melo, G. Dimethyl Sulfoxide (DMSO) Decreases Cell Proliferation and TNF-α, IFN-γ, and IL-2 Cytokines Production in Cultures of Peripheral Blood Lymphocytes. Molecules 2017, 22, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockbank, K.G. Removal of Potentially Cytotoxic DMSO from Cell Therapy Cryopreservation Formulations. MOJ Cell Sci. Rep. 2016, 3, 119–120. [Google Scholar] [CrossRef]
- Nita, L.E.; Chiriac, A.; Bercea, M.; Wolf, B.A. Synergistic behavior of poly(aspartic acid) and Pluronic F127 in aqueous solution as studied by viscometry and dynamic light scattering. Colloids Surf. B Biointerfaces 2013, 103, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Touchet, T.J.; Cosgriff-Hernandez, E.M. Hierarchal structure–property relationships of segmented polyurethanes. In Advances in Polyurethane Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 3–22. ISBN 9780081006221. [Google Scholar]
- Pandya, H.; Mahanwar, P. Fundamental insight into anionic aqueous polyurethane dispersions. Adv. Ind. Eng. Polym. Res. 2020, 3, 102–110. [Google Scholar] [CrossRef]
- Aikawa, T.; Yokota, K.; Kondo, T.; Yuasa, M. Intermolecular Interaction between Phosphatidylcholine and Sulfobetaine Lipid: A Combination of Lipids with Antiparallel Arranged Headgroup Charge. Langmuir 2016, 32, 10483–10490. [Google Scholar] [CrossRef]
- Zhang, F.; Hu, C.; Kong, Q.; Luo, R.; Wang, Y. Peptide-/Drug-Directed Self-Assembly of Hybrid Polyurethane Hydrogels for Wound Healing. ACS Appl. Mater. Interfaces 2019, 11, 37147–37155. [Google Scholar] [CrossRef]
- Zheng, J.; Shen, T.; Ma, J.; Liang, L.; Lu, M. Physicochemical studies on micelle formation of amphiphilic polyurethane in aqueous solution. Chem. Phys. Lett. 2011, 502, 211–216. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, Z.; Yin, Z.; Qian, H.; Ma, D. Amide II and Amide III Bands in Polyurethane Model Soft and Hard Segments. Polym. Bull. 2008, 60, 97–101. [Google Scholar] [CrossRef]
- Li, Y.-J.; Nakamura, N.; Wang, Y.-F.; Kodama, M.; Nakaya, T. Synthesis and Hemocompatibilities of New Segmented Polyurethanes and Poly(urethane urea)s with Poly(butadiene) and Phosphatidylcholine Analogues in the Main Chains and Long-Chain Alkyl Groups in the Side Chains. Chem. Mater. 1997, 9, 1570–1577. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, D.; Li, J.; Tan, H.; Fu, Q. Synthesis and hemocompatibity evaluation of segmented polyurethane end-capped with both a fluorine tail and phosphatidylcholine polar headgroups. Biofouling 2011, 27, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Li, Y. Recent progress of phospholipid polymers. Des. Monomers Polym. 2003, 6, 309–351. [Google Scholar] [CrossRef] [Green Version]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiriac, A.P.; Ghilan, A.; Serban, A.; Macsim, A.; Bargan, A.; Doroftei, F.; Chiriac, V.M.; Nita, L.E.; Rusu, A.G.; Sandu, A. Preparation of an Antioxidant Assembly Based on a Copolymacrolactone Structure and Erythritol following an Eco-Friendly Strategy. Antioxidants 2022, 11, 2471. [Google Scholar] [CrossRef]
- Luo, H.; Jiang, K.; Liang, X.; Liu, H.; Li, Y. Small molecule-mediated self-assembly behaviors of Pluronic block copolymers in aqueous solution: Impact of hydrogen bonding on the morphological transition of Pluronic micelles. Soft Matter 2020, 16, 142–151. [Google Scholar] [CrossRef]
- Nambam, J.S.; Philip, J. Effects of Interaction of Ionic and Nonionic Surfactants on Self-Assembly of PEO–PPO–PEO Triblock Copolymer in Aqueous Solution. J. Phys. Chem. B 2012, 116, 1499–1507. [Google Scholar] [CrossRef]
- Hirashima, Y.; Sato, H.; Suzuki, A. ATR-FTIR Spectroscopic Study on Hydrogen Bonding of Poly(N-isopropylacrylamide-co-sodium acrylate) Gel. Macromolecules 2005, 38, 9280–9286. [Google Scholar] [CrossRef]
- Dinkgreve, M.; Paredes, J.; Denn, M.M.; Bonn, D. On different ways of measuring “the” yield stress. J. Nonnewton. Fluid Mech. 2016, 238, 233–241. [Google Scholar] [CrossRef]
- Lee, W.-J.; Oh, H.-G.; Cha, S.-H. A Brief Review of Self-Healing Polyurethane Based on Dynamic Chemistry. Macromol. Res. 2021, 29, 649–664. [Google Scholar] [CrossRef]
- Strandman, S.; Zhu, X.X. Self-Healing Supramolecular Hydrogels Based on Reversible Physical Interactions. Gels 2016, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, M.A.; Khoda, B. Rheological analysis of bio-ink for 3D bio-printing processes. J. Manuf. Process. 2022, 76, 708–718. [Google Scholar] [CrossRef]
- Hsiao, S.-H.; Hsu, S. Synthesis and Characterization of Dual Stimuli-Sensitive Biodegradable Polyurethane Soft Hydrogels for 3D Cell-Laden Bioprinting. ACS Appl. Mater. Interfaces 2018, 10, 29273–29287. [Google Scholar] [CrossRef]
- Malekpour, A.; Chen, X. Printability and Cell Viability in Extrusion-Based Bioprinting from Experimental, Computational, and Machine Learning Views. J. Funct. Biomater. 2022, 13, 40. [Google Scholar] [CrossRef]
- Shi, J.; Wu, B.; Li, S.; Song, J.; Song, B.; Lu, W.F. Shear stress analysis and its effects on cell viability and cell proliferation in drop-on-demand bioprinting. Biomed. Phys. Eng. Express 2018, 4, 045028. [Google Scholar] [CrossRef]
- Ishida-Ishihara, S.; Takada, R.; Furusawa, K.; Ishihara, S.; Haga, H. Improvement of the cell viability of hepatocytes cultured in three-dimensional collagen gels using pump-free perfusion driven by water level difference. Sci. Rep. 2022, 12, 20269. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Temperature (°C) | DH (nm) | DH (nm) | Z-Average (nm) | PDI | Zeta Potential (mV) | Conductivity (mS/cm) |
|---|---|---|---|---|---|---|---|
| Peak 1 | Peak 2 | ||||||
| M | 25 | 46.91 ± 19.53 | 439.2 ± 168.1 | 93.04 ± 17.91 | 0.206 | −6.75 ± 0.87 | 0.0112 |
| 37 | 56.42 ± 30.11 | 440.6 ± 173.7 | 58.72 ± 4.93 | 0.799 | −2.66 ± 1.14 | 0.0125 | |
| P1 | 25 | 30.76 ± 10.84 | 295.2 ± 81.94 | 35.87 ± 5.76 | 0.318 | −12.9 ± 0.52 | 0.0184 |
| 37 | 33.99 ± 9.52 | 221.3 ± 46.63 | 41.25 ± 8.06 | 0.255 | −3.84 ± 0.7 | 0.00943 | |
| P2 | 25 | 35.32 ± 14.8 | 321.6 ± 76.26 | 36.03 ± 6.8 | 0.359 | −8.49 ± 0.33 | 0.00842 |
| 37 | 21.23 ± 2.34 | - | 465.5 ± 23.27 | 0.406 | −7.53 ± 0.46 | 0.0203 | |
| P3 | 25 | 32.86 ± 8.03 | 282.9 ± 66.74 | 131.7 ± 29.51 | 0.256 | −9.7 ± 0.87 | 0.00502 |
| 37 | 25.26 ± 2.99 | - | 295.3 ± 14.76 | 0.641 | −5.79 ± 0.71 | 0.00568 |
| Sample Code | Tgelation a (°C) | Tgelation a (°C) | G′ b (Pa) | G″ b (Pa) | γL c (%) | σo c (Pa) | ηo (Pa·s) | n | Thixotropic Area (Pa·s) |
|---|---|---|---|---|---|---|---|---|---|
| (0.5 °C/min) | (1 °C/min) | ||||||||
| M | 29.4 | 34 | 547 | 110 | 2.16 | 20.6 | 2220 | −0.829 | 128.67 |
| P1 | 27.5 | 31 | 696 | 119 | 4.59 | 21.2 | 4690 | −0.815 | 72.56 |
| P2 | 25.8 | 27.6 | 1030 | 139 | 9.89 | 28.3 | 6980 | −0.817 | 51.16 |
| P3 | 21.2 | 23.5 | 1180 | 153 | 10.23 | 55.5 | 8600 | −0.816 | 49.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gradinaru, L.M.; Bercea, M.; Lupu, A.; Gradinaru, V.R. Development of Polyurethane/Peptide-Based Carriers with Self-Healing Properties. Polymers 2023, 15, 1697. https://doi.org/10.3390/polym15071697
Gradinaru LM, Bercea M, Lupu A, Gradinaru VR. Development of Polyurethane/Peptide-Based Carriers with Self-Healing Properties. Polymers. 2023; 15(7):1697. https://doi.org/10.3390/polym15071697
Chicago/Turabian StyleGradinaru, Luiza Madalina, Maria Bercea, Alexandra Lupu, and Vasile Robert Gradinaru. 2023. "Development of Polyurethane/Peptide-Based Carriers with Self-Healing Properties" Polymers 15, no. 7: 1697. https://doi.org/10.3390/polym15071697