

Influence of Process Parameters on the Kinetics of the Micelle-to-Vesicle Transition and Ripening of Polystyrene-Block-Polyacrylic Acid


Abstract
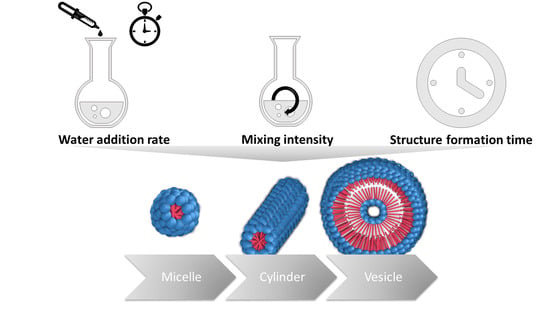
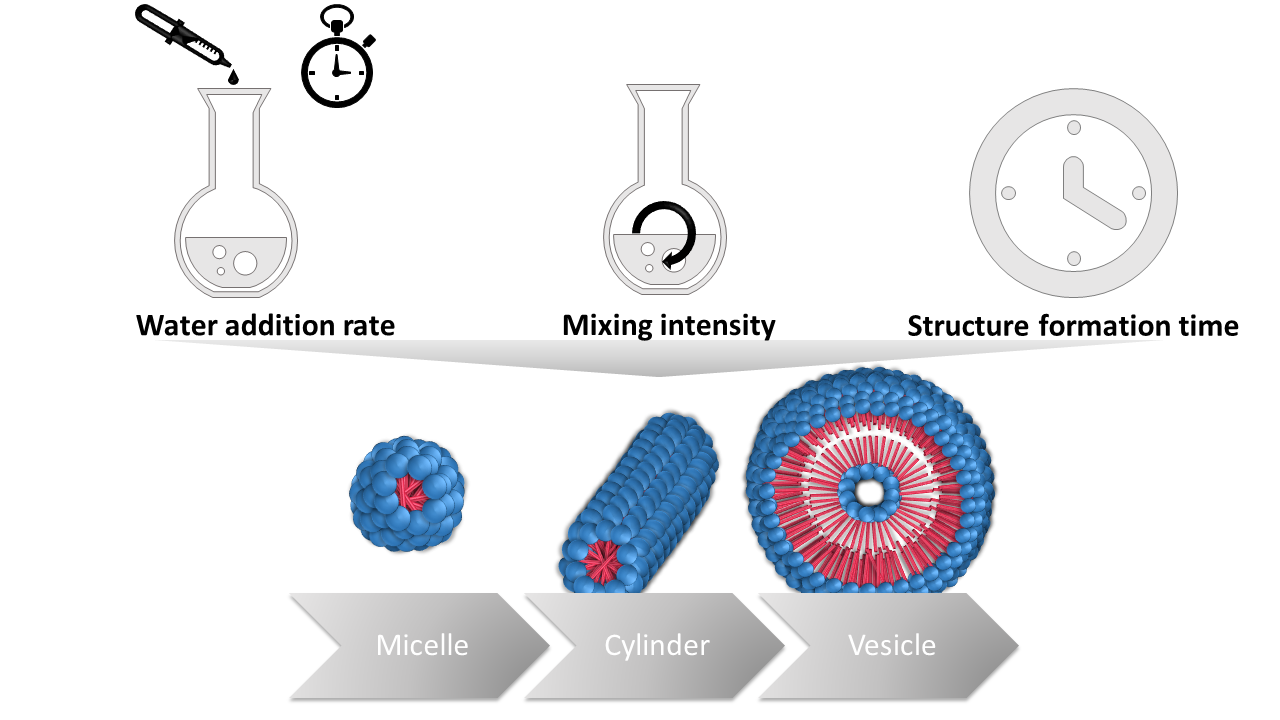
:
1. Introduction
2. Materials and Methods
2.1. Materials
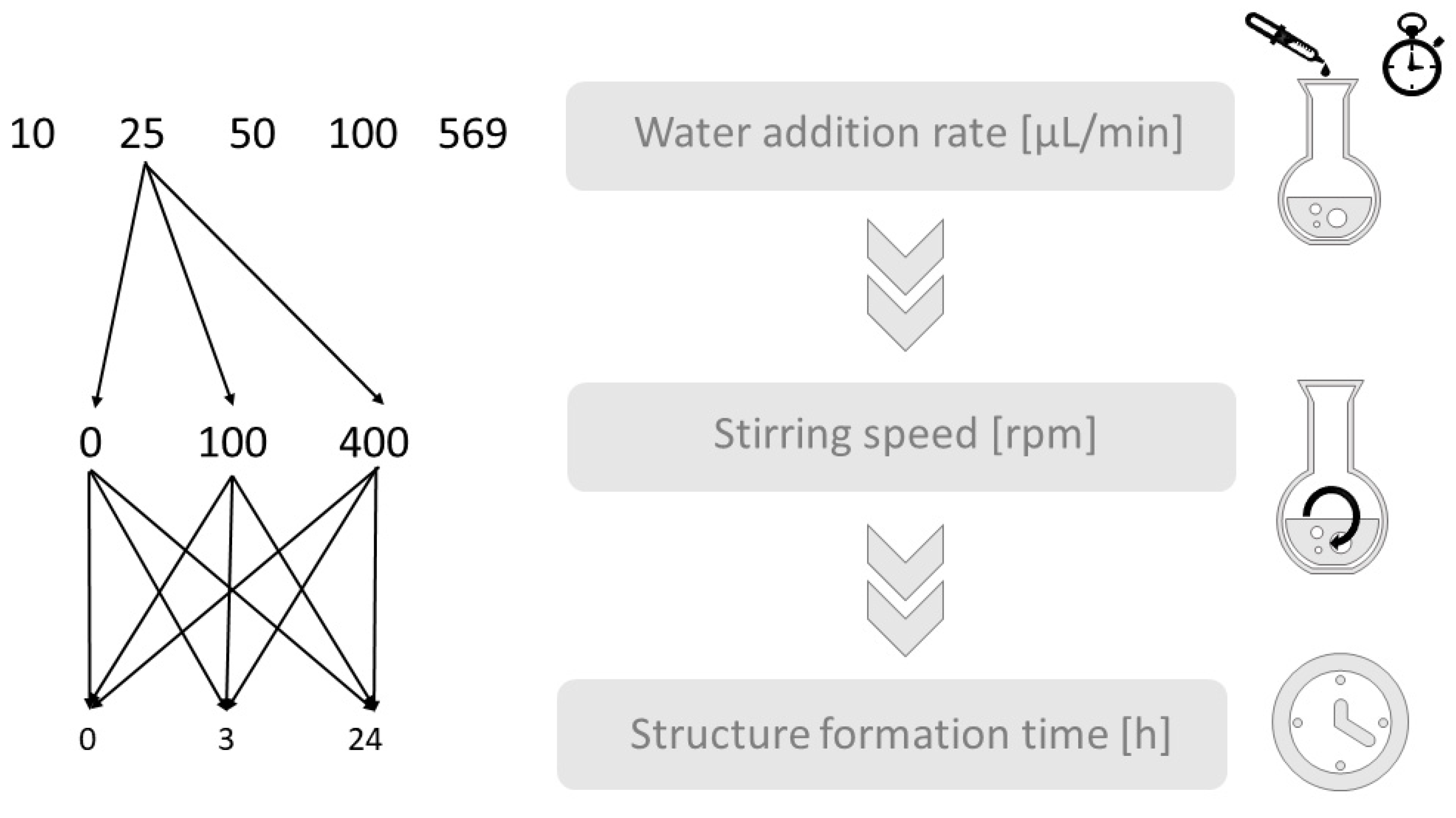
2.2. Preparation of Polymer Structures of PS-b-PAA
2.3. Sample Preparation and Characterization
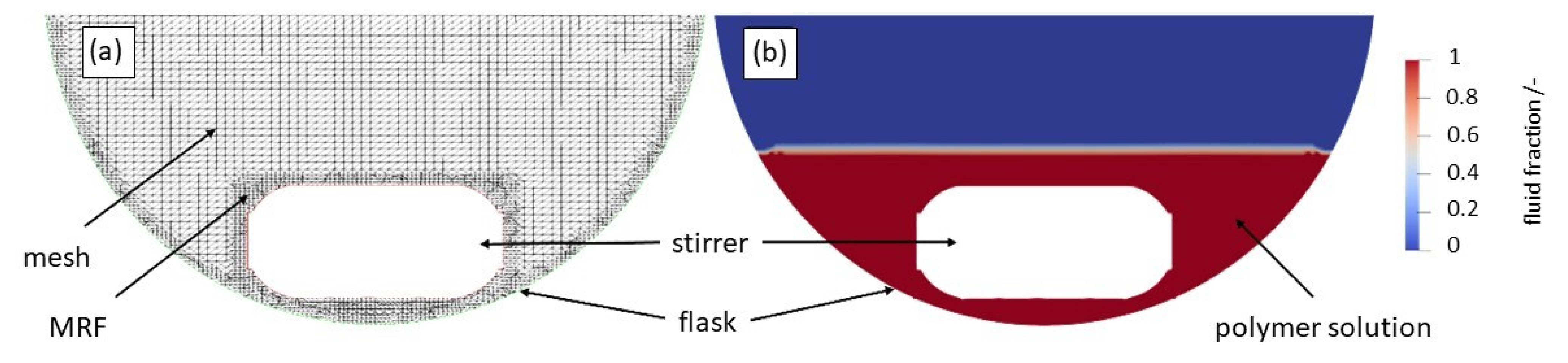
2.4. Computational Fluid Dynamics (CFD)
3. Results and Discussion
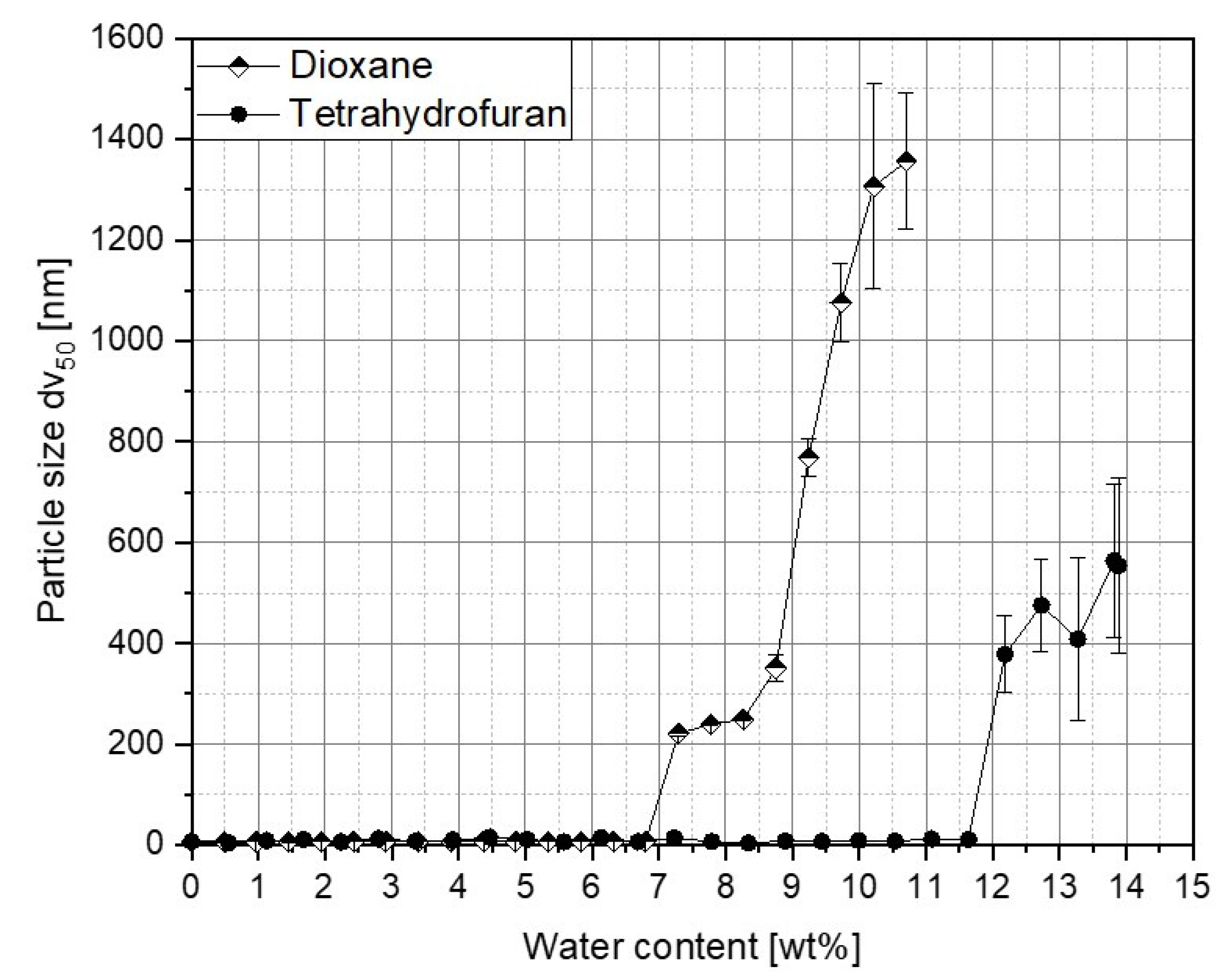
3.1. Determination of Critical Water Content
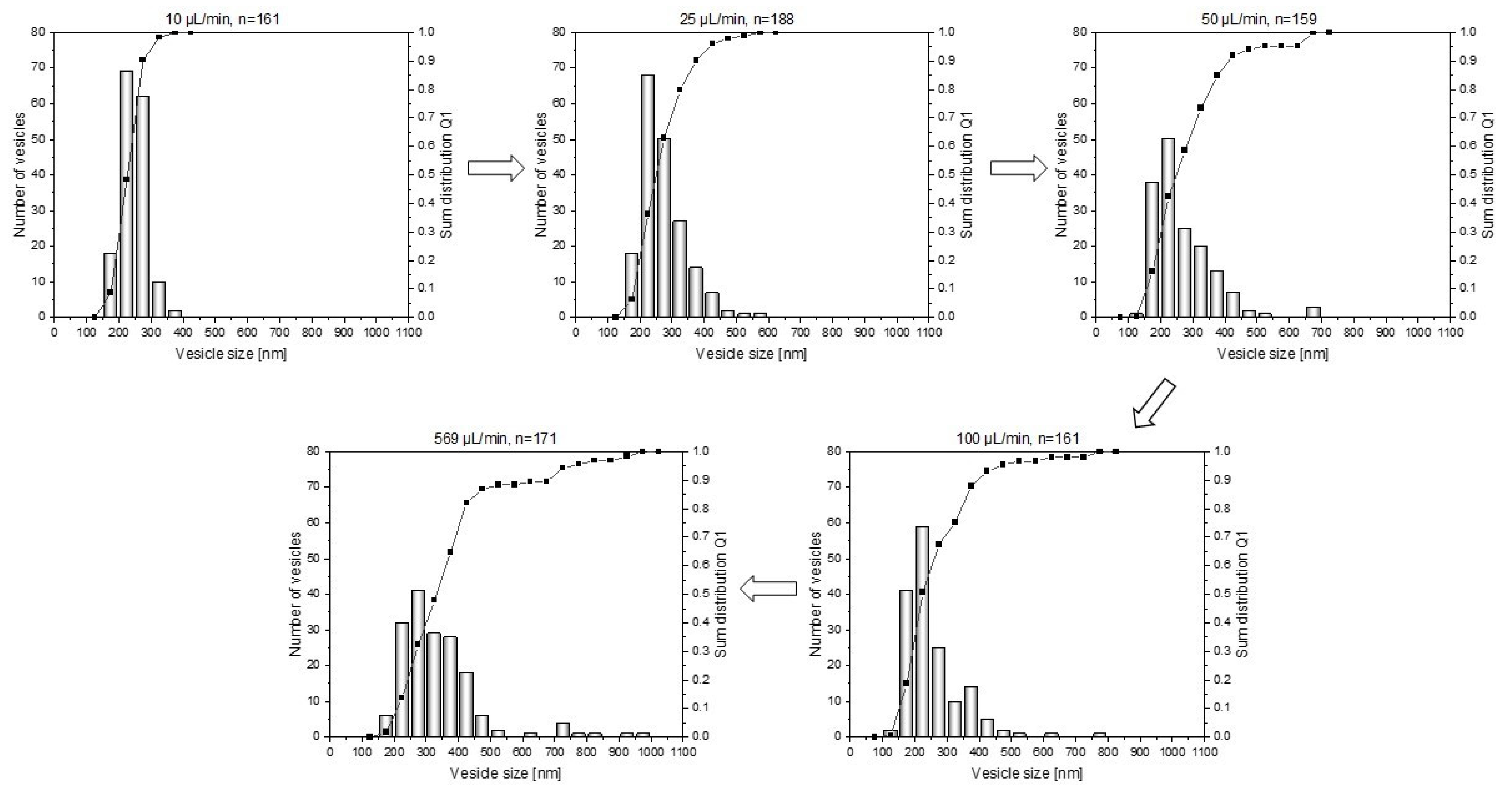
3.2. Influence of Water Addition Rate on the Vesicle Size Distribution
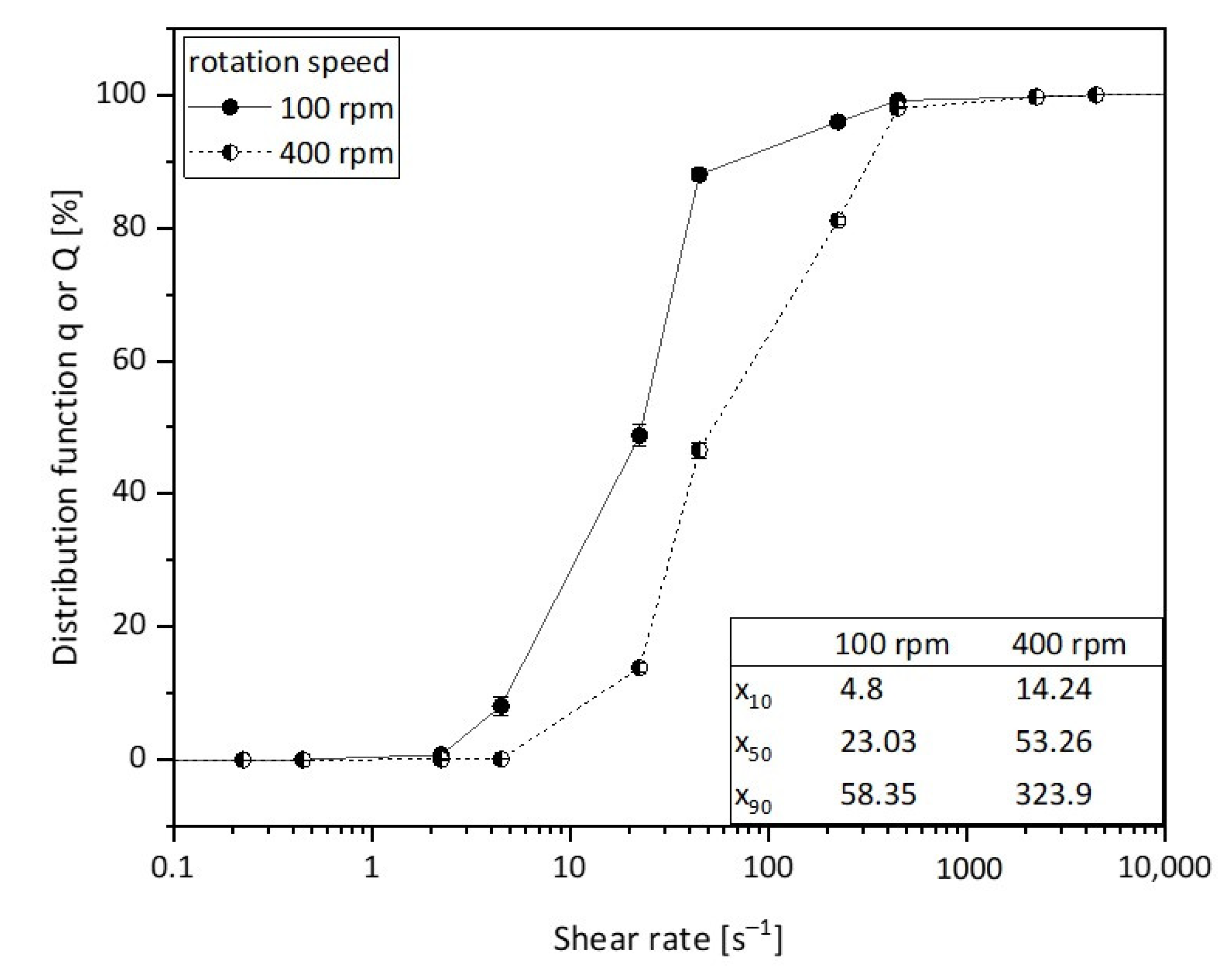
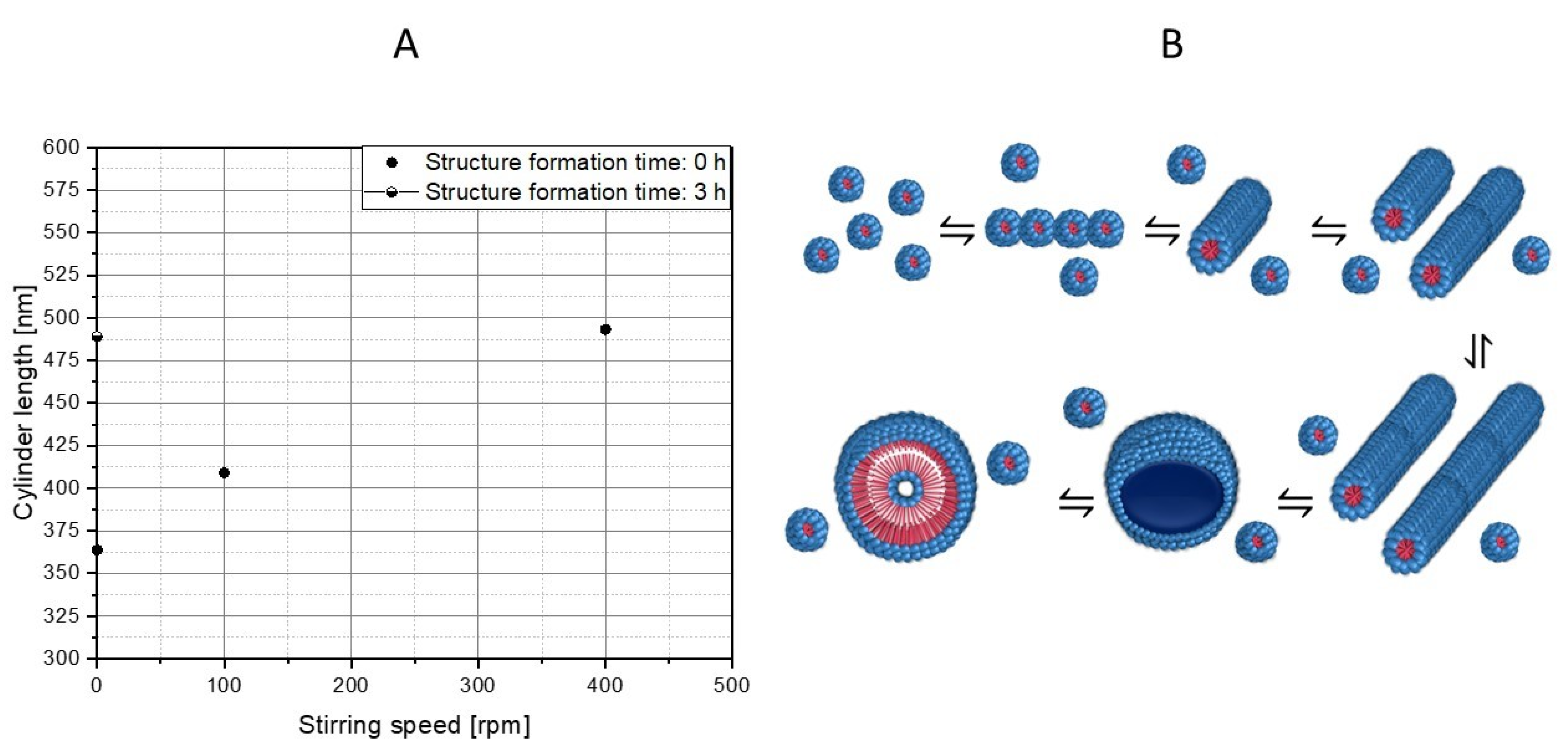
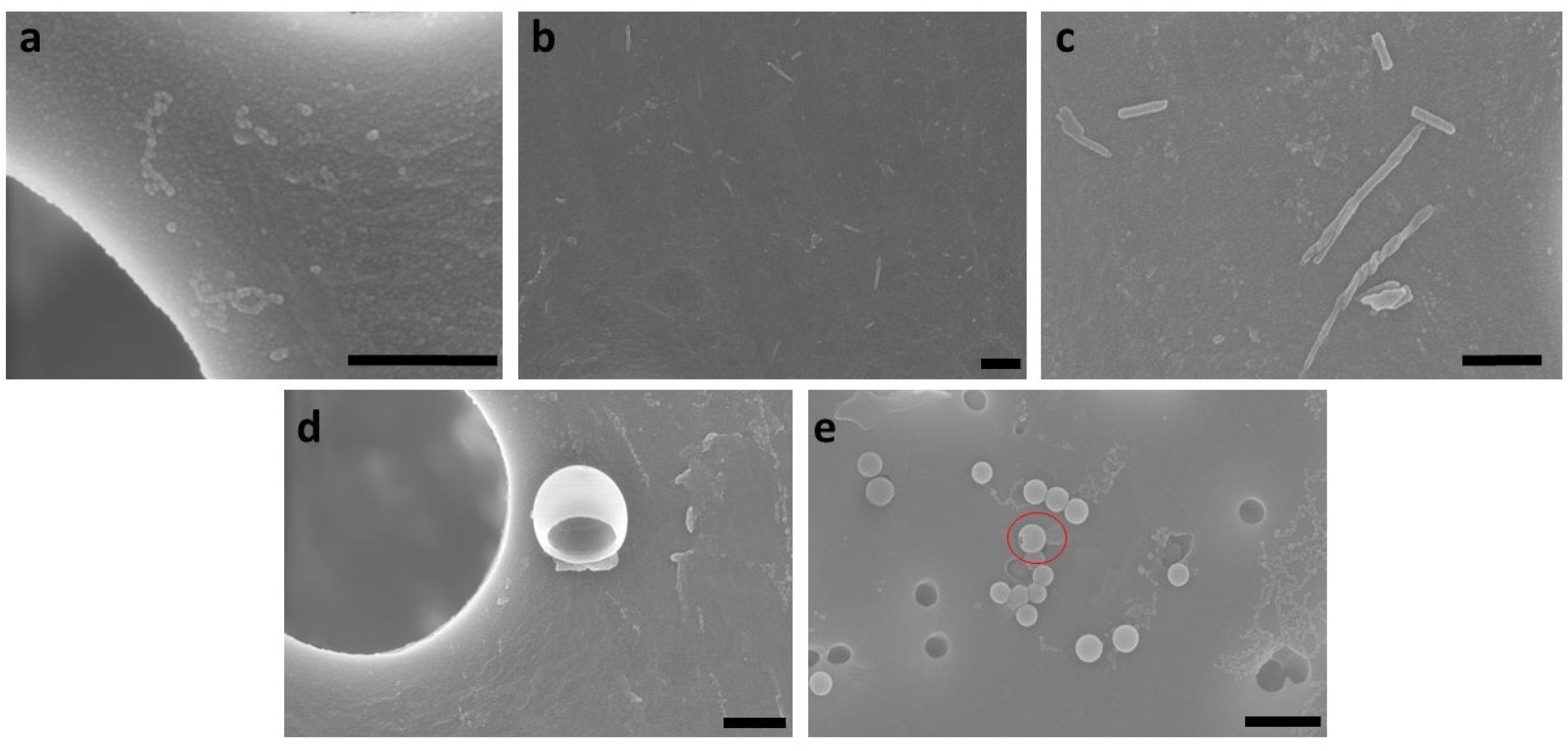
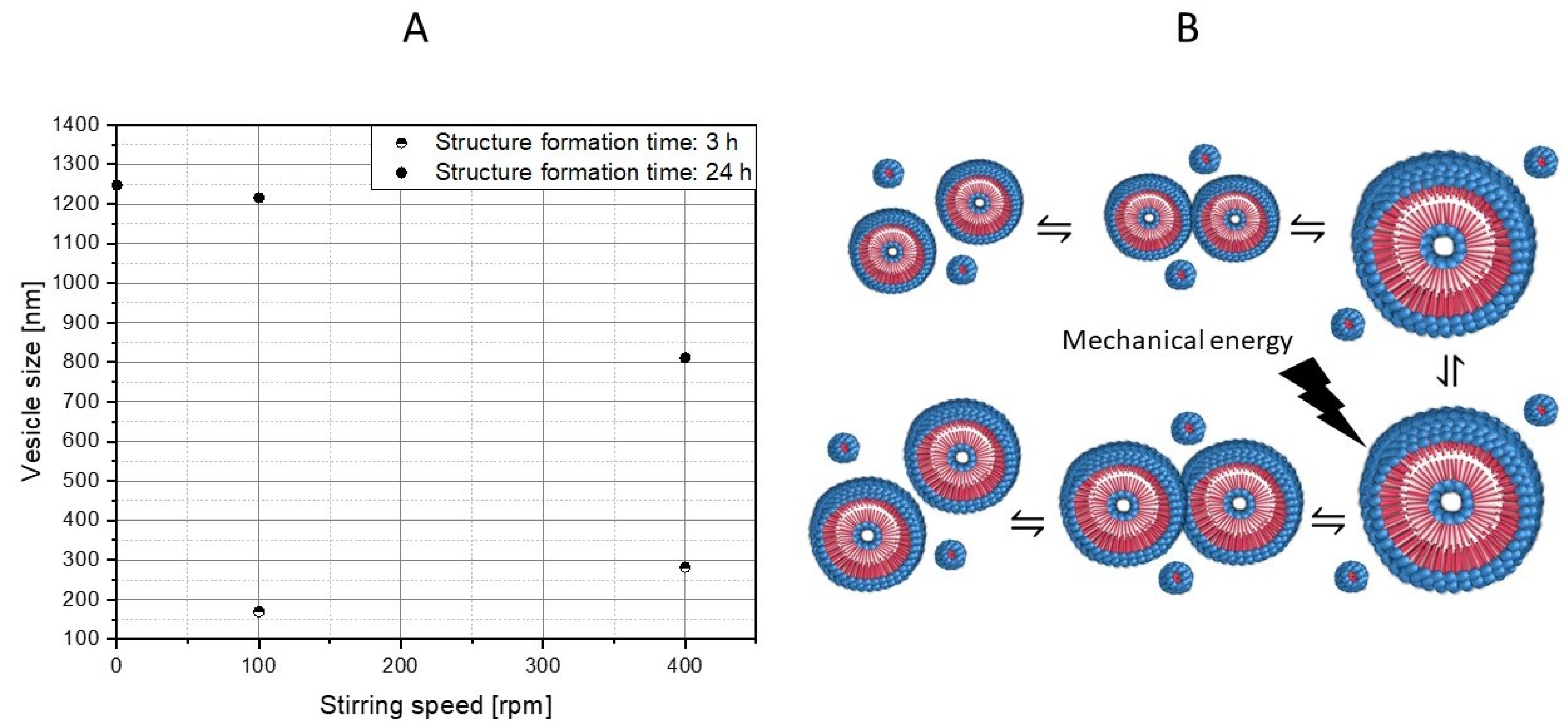
3.3. Influence of Mixing Intensity and Structure Formation Time on the Polymer Structure of PS-b-PAA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef]
- Kickelbick, G. Nanokomposite: Anorganisch + organisch: Homogen und doch so heterogen. Chem. Unserer Zeit 2005, 39, 46–53. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Shen, X.; Zhu, L.; He, J.; Chen, H. Unconventional chain-growth mode in the assembly of colloidal gold nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 8021–8025. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.E.; Eisenberg, A. Kinetic and mechanistic details of the vesicle-to-rod transition in aggregates of PS310-b-PAA52 in dioxane-water mixtures. Polymer 2001, 42, 9111–9120. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1976, 72, 1525. [Google Scholar] [CrossRef]
- Yu, Y.; Eisenberg, A. Control of Morphology through Polymer-Solvent Interactions in Crew-Cut Aggregates of Amphiphilic Block Copolymers. J. Am. Chem. Soc. 1997, 119, 8383–8384. [Google Scholar] [CrossRef]
- Mann, J.; Garnweitner, G.; Schilde, C. Preparation of Self-Assembled Nanoparticle-Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method. Polymers 2023, 15, 444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Eisenberg, A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Eisenberg, A. Structures of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) diblock copolymers. Macromol. Symp. 1997, 113, 221–232. [Google Scholar] [CrossRef]
- Guida, V. Thermodynamics and kinetics of vesicles formation processes. Adv. Colloid Interface Sci. 2010, 161, 77–88. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Thermodynamic vs. Kinetic Aspects in the Formation and Morphological Transitions of Crew-Cut Aggregates Produced by Self-Assembly of Polystyrene-b-poly(acrylic acid) Block Copolymers in Dilute Solution. Macromolecules 1999, 32, 2239–2249. [Google Scholar] [CrossRef]
- Leng, J.; Egelhaaf, S.U.; Cates, M.E. Kinetics of the Micelle-to-Vesicle Transition: Aqueous Lecithin-Bile Salt Mixtures. Biophys. J. 2003, 85, 1624–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voets, I.K.; de Keizer, A.; de Waard, P.; Frederik, P.M.; Bomans, P.H.H.; Schmalz, H.; Walther, A.; King, S.M.; Leermakers, F.A.M.; Stuart, M.A.C. Double-faced micelles from water-soluble polymers. Angew. Chem. Int. Ed. 2006, 45, 6673–6676. [Google Scholar] [CrossRef]
- Pochan, D.J.; Chen, Z.; Cui, H.; Hales, K.; Qi, K.; Wooley, K.L. Toroidal triblock copolymer assemblies. Science 2004, 306, 94–97. [Google Scholar] [CrossRef]
- Yu, H.; Qiu, X.; Nunes, S.P.; Peinemann, K.-V. Biomimetic block copolymer particles with gated nanopores and ultrahigh protein sorption capacity. Nat. Commun. 2014, 5, 4110. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Du, J. Evolution of diverse higher-order membrane structures of block copolymer vesicles. Polym. Chem. 2018, 10, 3020–3029. [Google Scholar] [CrossRef]
- Hashimoto, T.; Mitsumura, N.; Yamaguchi, D.; Takenaka, M.; Morita, H.; Kawakatsu, T.; Doi, M. Nonequilibrium helical-domain morphology in diblock copolymer systems. Polymer 2001, 42, 8477–8481. [Google Scholar] [CrossRef]
- Smart, T.; Lomas, H.; Massignani, M.; Flores-Merino, M.; Perez, L.; Battaglia, G. Block copolymer nanostructures. Nano Today 2008, 3, 38–46. Available online: https://www.observatorioplastico.com/ficheros/noticias/30133944nano1.pdf (accessed on 11 February 2022). [CrossRef]
- Hillmyer, M.A.; Lipic, P.M.; Hajduk, D.A.; Almdal, K.; Bates, F.S. Self-Assembly and Polymerization of Epoxy Resin-Amphiphilic Block Copolymer Nanocomposites. J. Am. Chem. Soc. 1997, 119, 2749–2750. [Google Scholar] [CrossRef]
- Riess, G. Micellization of block copolymers. Prog. Polym. Sci. 2003, 28, 1107–1170. [Google Scholar] [CrossRef] [Green Version]
- Onaca, O.; Enea, R.; Hughes, D.W.; Meier, W. Stimuli-Responsive Polymersomes as Nanocarriers for Drug and Gene Delivery. Macromol. Biosci. 2009, 9, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-poly(ethylene oxide) block copolymer micelles as a novel drug delivery vehicle for neurotrophic agents FK506 and L-685,818. Bioconjug. Chem. 1998, 9, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhong, Z.; Feijen, J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar] [CrossRef]
- Litvinchuk, S.; Lu, Z.; Rigler, P.; Hirt, T.D.; Meier, W. Calcein release from polymeric vesicles in blood plasma and PVA hydrogel. Pharm. Res. 2009, 26, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.; Schmidt, M.; Ebenhoch, J.; Baumann, F.; Deubzer, B.; Weis, J. Design and Synthesis of Molecular Reactors for the Preparation of Topologically Trapped Gold Cluster. Adv. Mater. 1999, 11, 761–766. [Google Scholar] [CrossRef]
- Schäfer, S.; Kickelbick, G. Double Reversible Networks: Improvement of Self-Healing in Hybrid Materials via Combination of Diels-Alder Cross-Linking and Hydrogen Bonds. Macromolecules 2018, 51, 6099–6110. [Google Scholar] [CrossRef]
- Shen, H.; Eisenberg, A. Morphological Phase Diagram for a Ternary System of Block Copolymer PS310-b-PAA52 /Dioxane/H2O. J. Phys. Chem. B 1999, 103, 9473–9487. [Google Scholar] [CrossRef]
- Schillen, K.; Brown, W.; Johnsen, R.M. Micellar Sphere-to-Rod Transition in an Aqueous Triblock Copolymer System. A Dynamic Light Scattering Study of Translational and Rotational Diffusion. Macromolecules 1994, 27, 4825–4832. [Google Scholar] [CrossRef]
- Burke, S.E.; Eisenberg, A. Kinetics and Mechanisms of the Sphere-to-Rod and Rod-to-Sphere Transitions in the Ternary System PS310-b-PAA52/Dioxane/Water. Langmuir 2001, 17, 6705–6714. [Google Scholar] [CrossRef]
- Yang, C.; Gao, L.; Lin, J.; Wang, L.; Cai, C.; Wei, Y.; Li, Z. Toroid Formation through a Supramolecular “Cyclization Reaction” of Rodlike Micelles. Angew. Chem. Int. Ed. 2017, 56, 5546–5550. [Google Scholar] [CrossRef]
- Liu, C.; Yao, L.; Wang, H.; Phua, Z.R.; Song, X.; Chen, H. Bridging the Gap in the Micellar Transformation from Cylinders to Vesicles. Small 2014, 10, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Yi, X.; Zhu, W.; Gao, H. Packing of flexible nanofibers in vesicles. Extrem. Mech. Lett. 2018, 19, 20–26. [Google Scholar] [CrossRef]
- Du, J.; O’Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544. [Google Scholar] [CrossRef]
- Rank, A.; Hauschild, S.; Förster, S.; Schubert, R. Preparation of monodisperse block copolymer vesicles via a thermotropic cylinder-vesicle transition. Langmuir 2009, 25, 1337–1344. [Google Scholar] [CrossRef]
- Geng, Z.; Xiong, B.; Wang, L.; Wang, K.; Ren, M.; Zhang, L.; Zhu, J.; Yang, Z. Moebius strips of chiral block copolymers. Nat. Commun. 2019, 10, 4090. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Jiang, T.; Lin, J.; Cai, C. Toroid formation through self-assembly of graft copolymer and homopolymer mixtures: Experimental studies and dissipative particle dynamics simulations. Langmuir 2013, 29, 8417–8426. [Google Scholar] [CrossRef]
- Wei, K.; Su, L.; Chen, G.; Jiang, M. Does PNIPAM block really retard the micelle-to-vesicle transition of its copolymer? Polymer 2011, 52, 3647–3654. [Google Scholar] [CrossRef]
- Sundararaman, A.; Stephan, T.; Grubbs, R.B. Reversible restructuring of aqueous block copolymer assemblies through stimulus-induced changes in amphiphilicity. J. Am. Chem. Soc. 2008, 130, 12264–12265. [Google Scholar] [CrossRef] [PubMed]
- Moughton, A.O.; Patterson, J.P.; O’Reilly, R.K. Reversible morphological switching of nanostructures in solution. Chem. Commun. 2011, 47, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Aubrecht, K.B.; Grubbs, R.B. Thermally induced changes in amphiphilicity drive reversible restructuring of assemblies of ABC triblock copolymers with statistical polyether blocks. J. Am. Chem. Soc. 2011, 133, 1058–1065. [Google Scholar] [CrossRef]
- Heyn, T.R.; Mayer, J.; Neumann, H.R.; Selhuber-Unkel, C.; Kwade, A.; Schwarz, K.; Keppler, J.K. The threshold of amyloid aggregation of beta-lactoglobulin: Relevant factor combinations. J. Food Eng. 2020, 283, 110005. [Google Scholar] [CrossRef]
- Christopher, J.; Greenshields. OpenFOAM User Guide. Available online: http://foam.sourceforge.net/docs/Guides-a4/OpenFOAMUserGuide-A4.pdf (accessed on 15 May 2022).
- Nayak, J.N.; Aralaguppi, M.I.; Naidu, B.V.K.; Aminabhavi, T.M. Thermodynamic Properties of Water + Tetrahydrofuran and Water + 1,4-Dioxane Mixtures at (303.15, 313.15, and 323.15) K. J. Chem. Eng. Data 2004, 49, 468–474. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; Taylor & Francis: Boca Raton, FL, USA, 2007; ISBN 978-0-8493-7248-3. [Google Scholar]
- Yu, K.; Zhang, L.; Eisenberg, A. Novel Morphologies of “Crew-Cut” Aggregates of Amphiphilic Diblock Copolymers in Dilute Solution. Langmuir 1996, 12, 5980–5984. [Google Scholar] [CrossRef]
- Cui, H.; Chen, Z.; Wooley, K.L.; Pochan, D.J. Origins of toroidal micelle formation through charged triblock copolymer self-assembly. Soft Matter 2009, 5, 1269–1278. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PS * | PAA * | PS-b-PAA | |
|---|---|---|---|
| Mn [g/mol] | 27,617 | 1761 | 29,378 |
| PDI | ≤1.1 |
| Water Addition Rate [µL/min] | Average Vesicle Size [nm] | Polydispersity |
|---|---|---|
| 10 | 248 | 0.5 |
| 25 | 272 | 0.9 |
| 50 | 265 | 1.3 |
| 100 | 255 | 1.3 |
| 569 | 388 | 1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mann, J.; Mayer, J.K.; Garnweitner, G.; Schilde, C. Influence of Process Parameters on the Kinetics of the Micelle-to-Vesicle Transition and Ripening of Polystyrene-Block-Polyacrylic Acid. Polymers 2023, 15, 1695. https://doi.org/10.3390/polym15071695
Mann J, Mayer JK, Garnweitner G, Schilde C. Influence of Process Parameters on the Kinetics of the Micelle-to-Vesicle Transition and Ripening of Polystyrene-Block-Polyacrylic Acid. Polymers. 2023; 15(7):1695. https://doi.org/10.3390/polym15071695
Chicago/Turabian StyleMann, Jil, Julian K. Mayer, Georg Garnweitner, and Carsten Schilde. 2023. "Influence of Process Parameters on the Kinetics of the Micelle-to-Vesicle Transition and Ripening of Polystyrene-Block-Polyacrylic Acid" Polymers 15, no. 7: 1695. https://doi.org/10.3390/polym15071695