

Constraint Release Rouse Mechanisms in Bidisperse Linear Polymers: Investigation of the Release Time of a Short-Long Entanglement

Abstract
:1. Introduction
2. Modeling
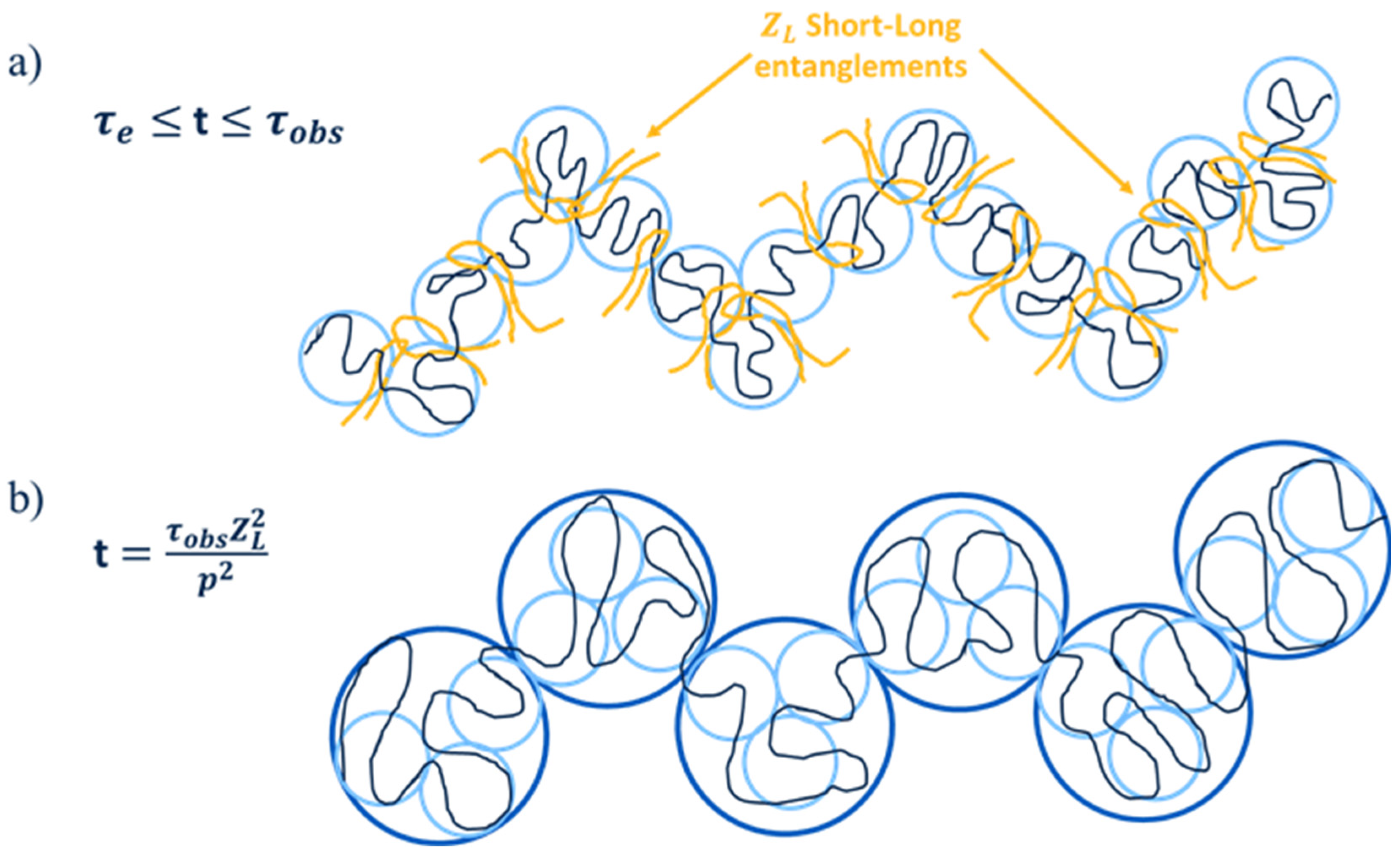
2.1. Description of the CRR Model
2.2. Validity of the CRR Model
3. Materials and Methods
3.1. Bidisperse Blends Composed of Self-Unentangled Long Chains
3.2. Linear Viscoelastic Measurements
4. Results and Discussion
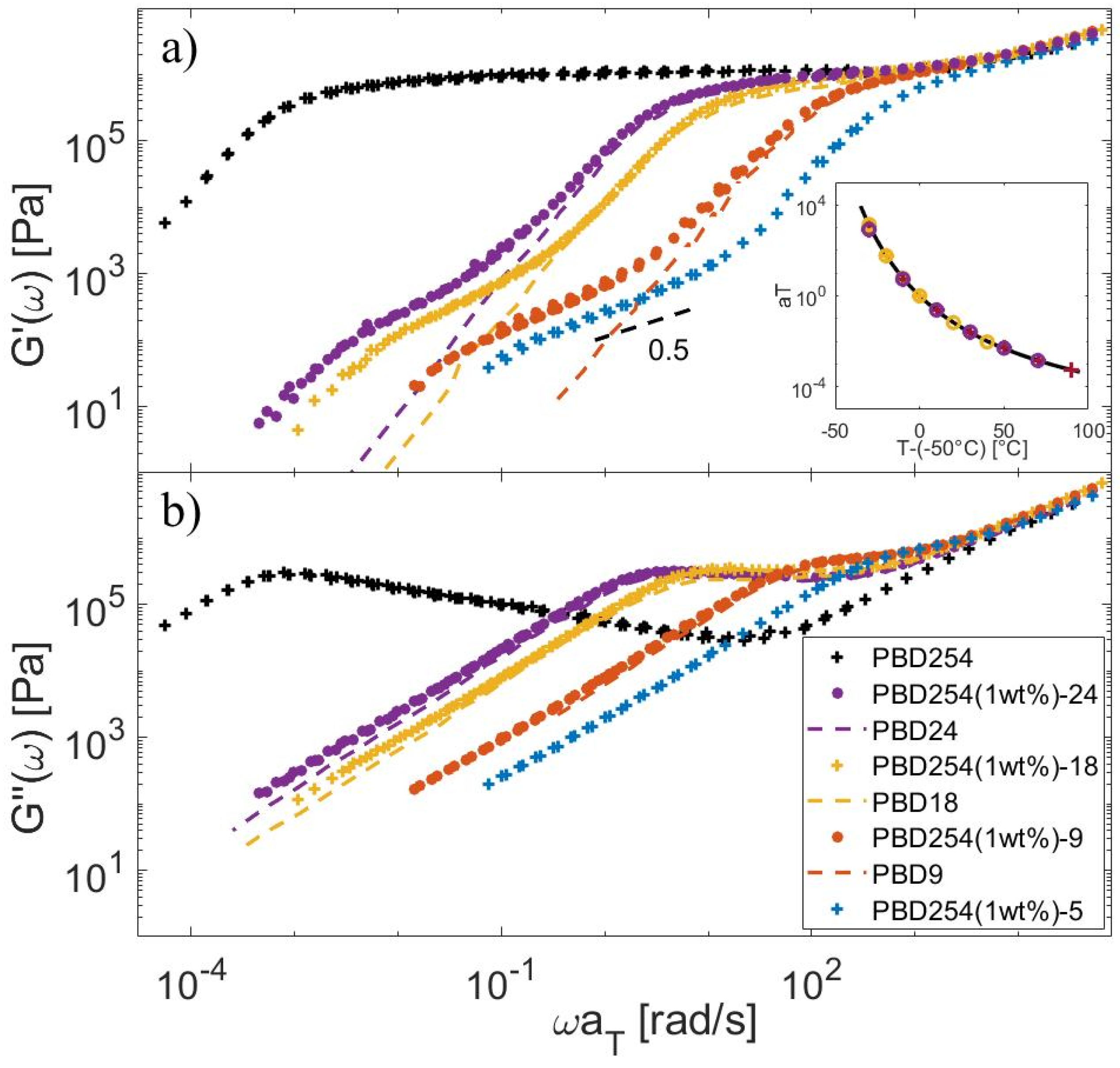
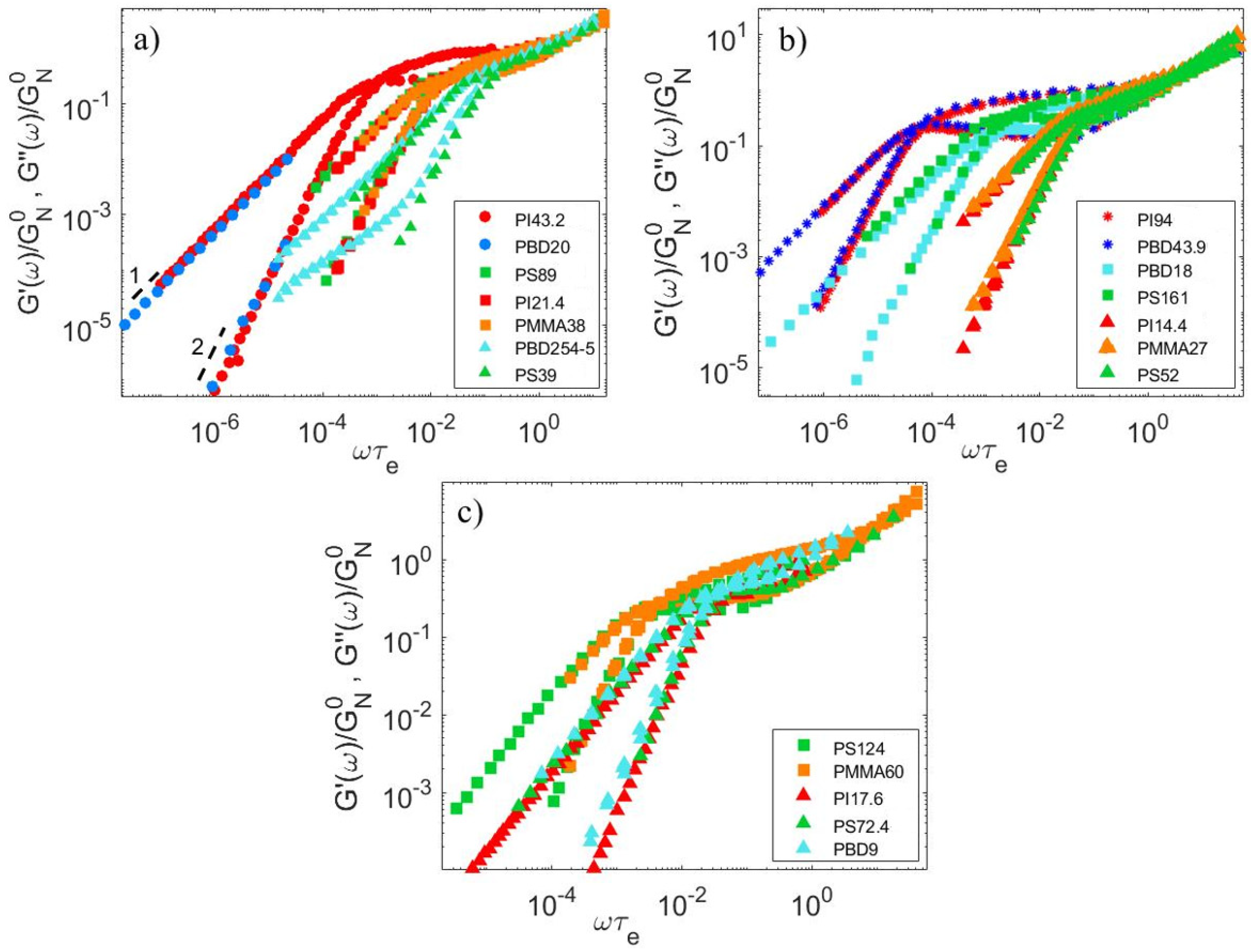
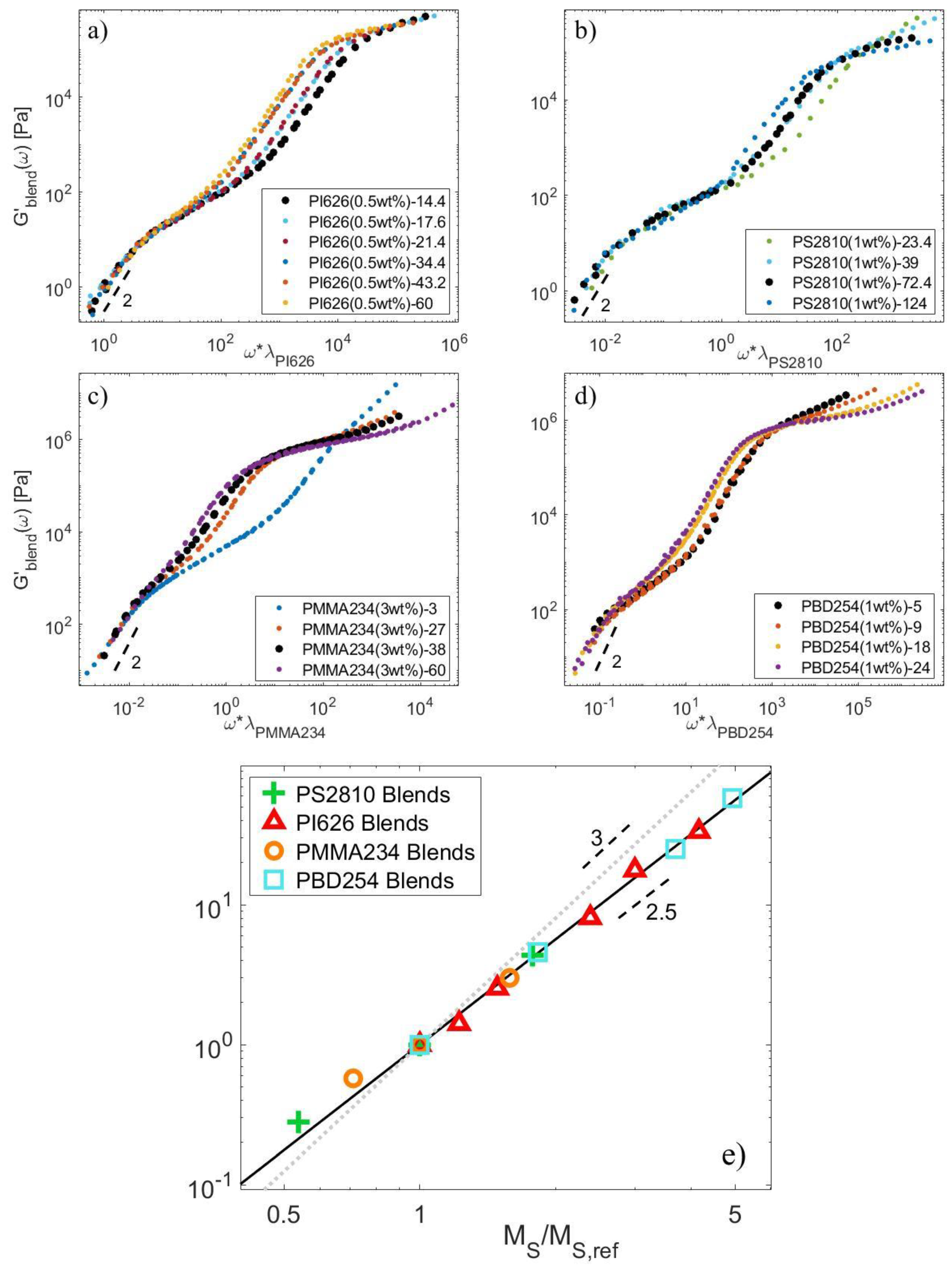
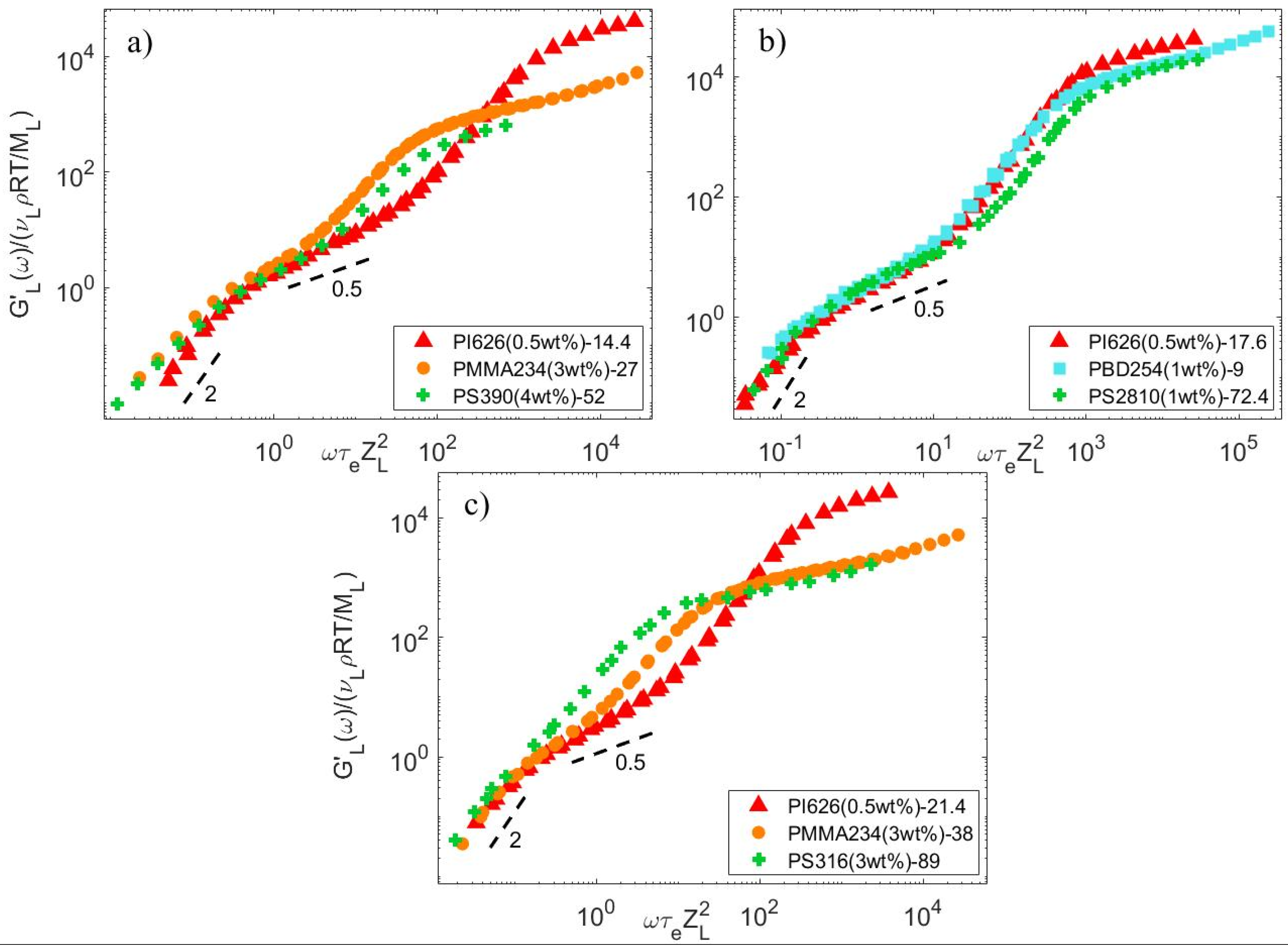
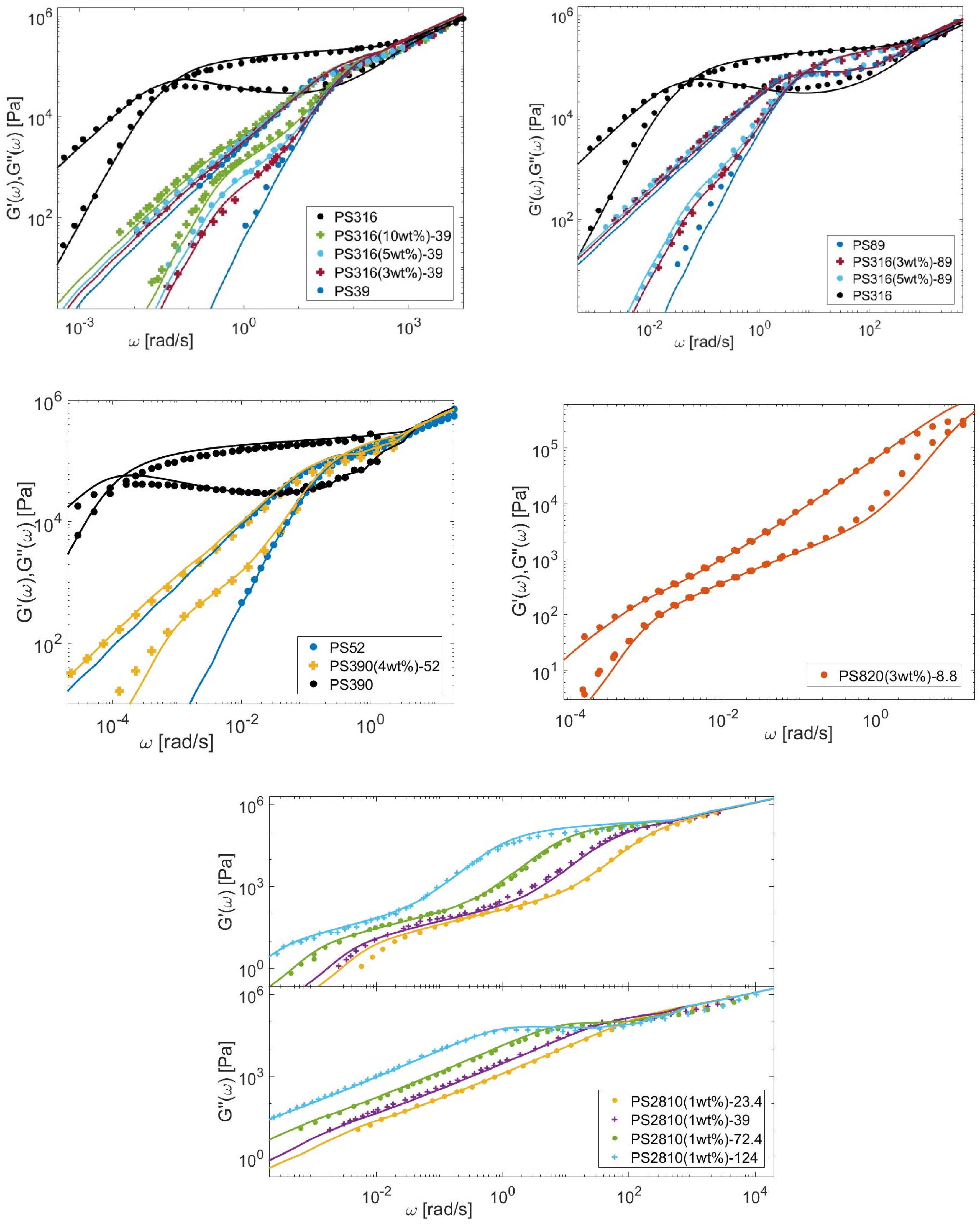
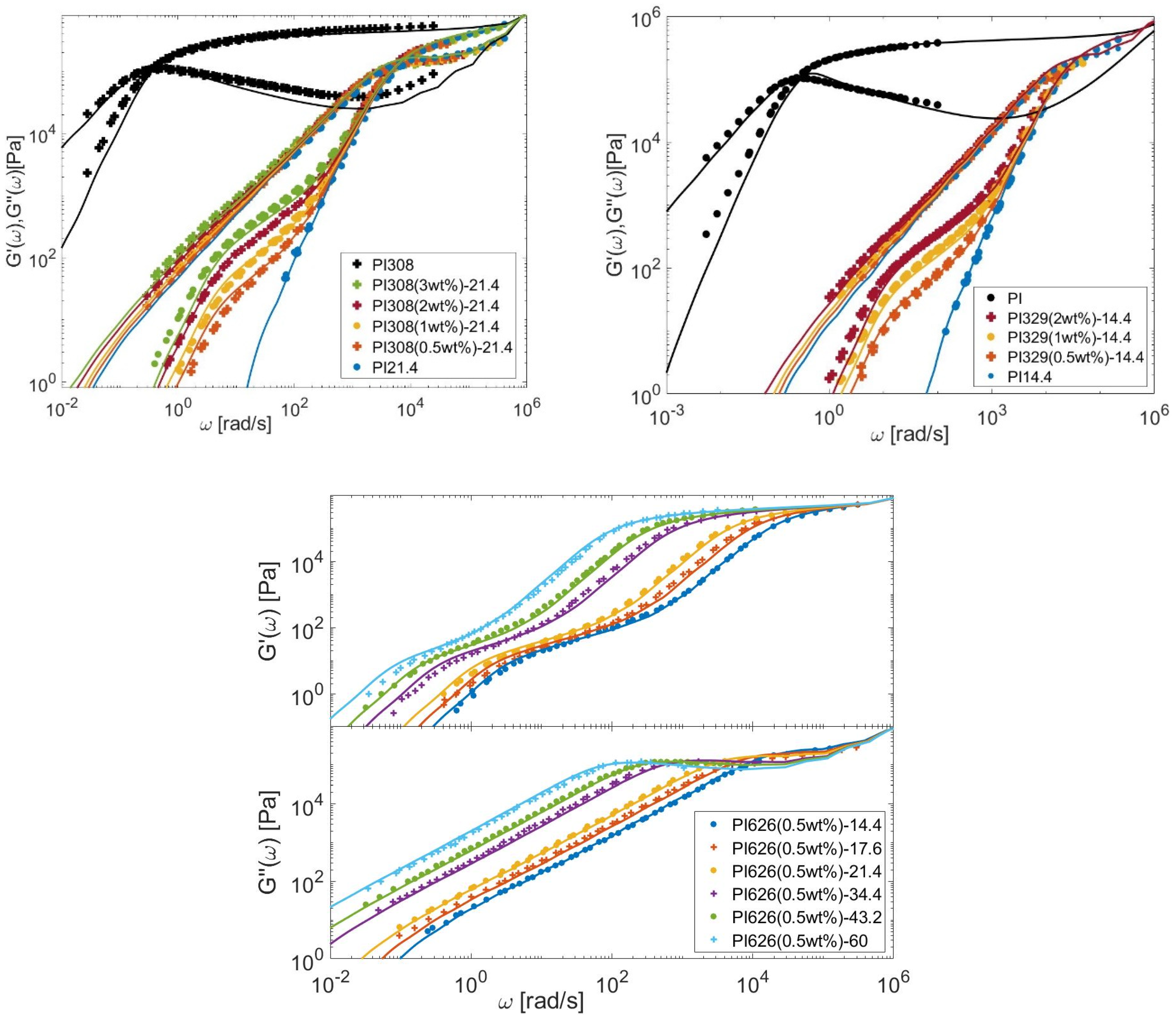
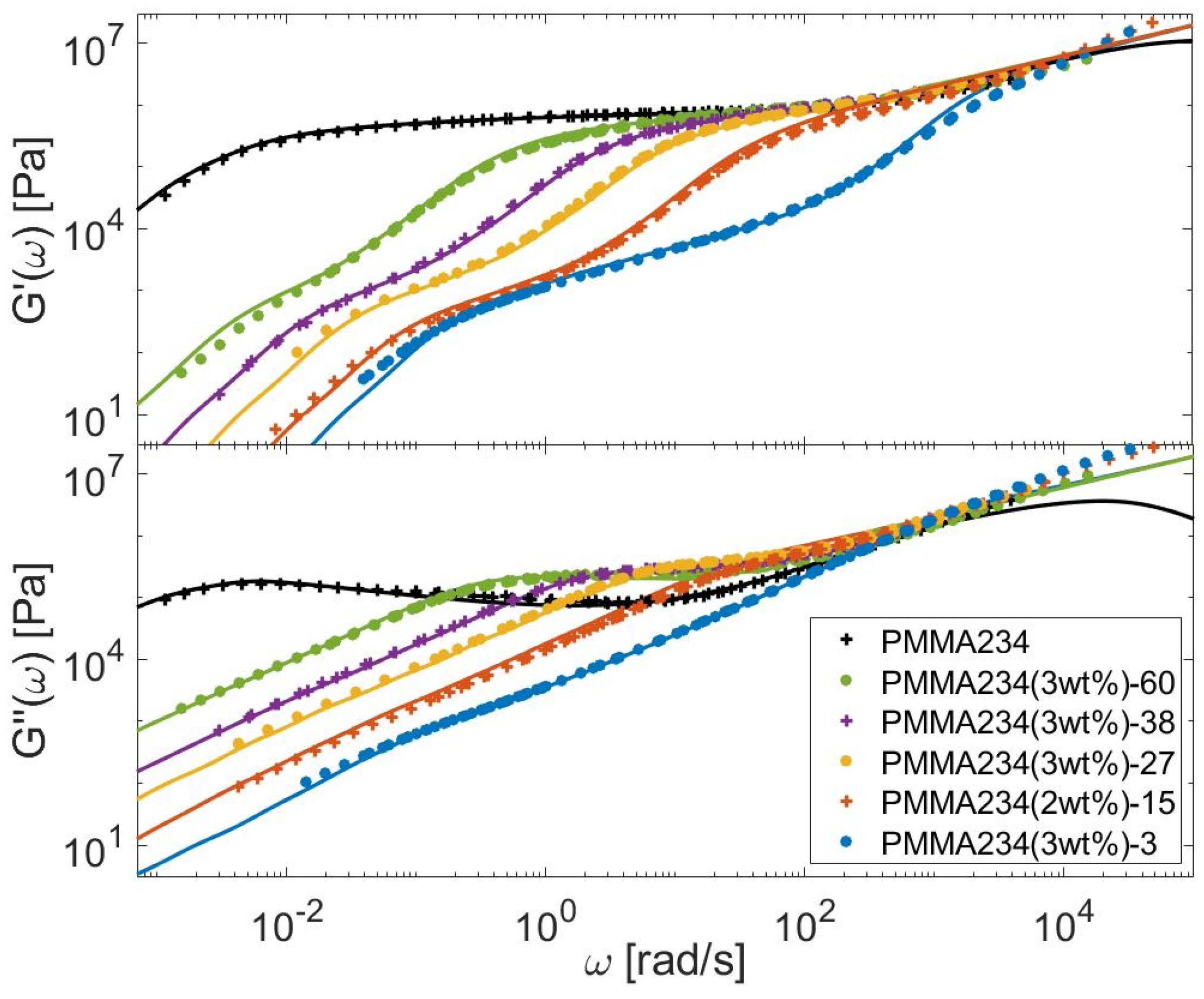
4.1. Linear Viscoelastic Data
4.2. Material Parameters
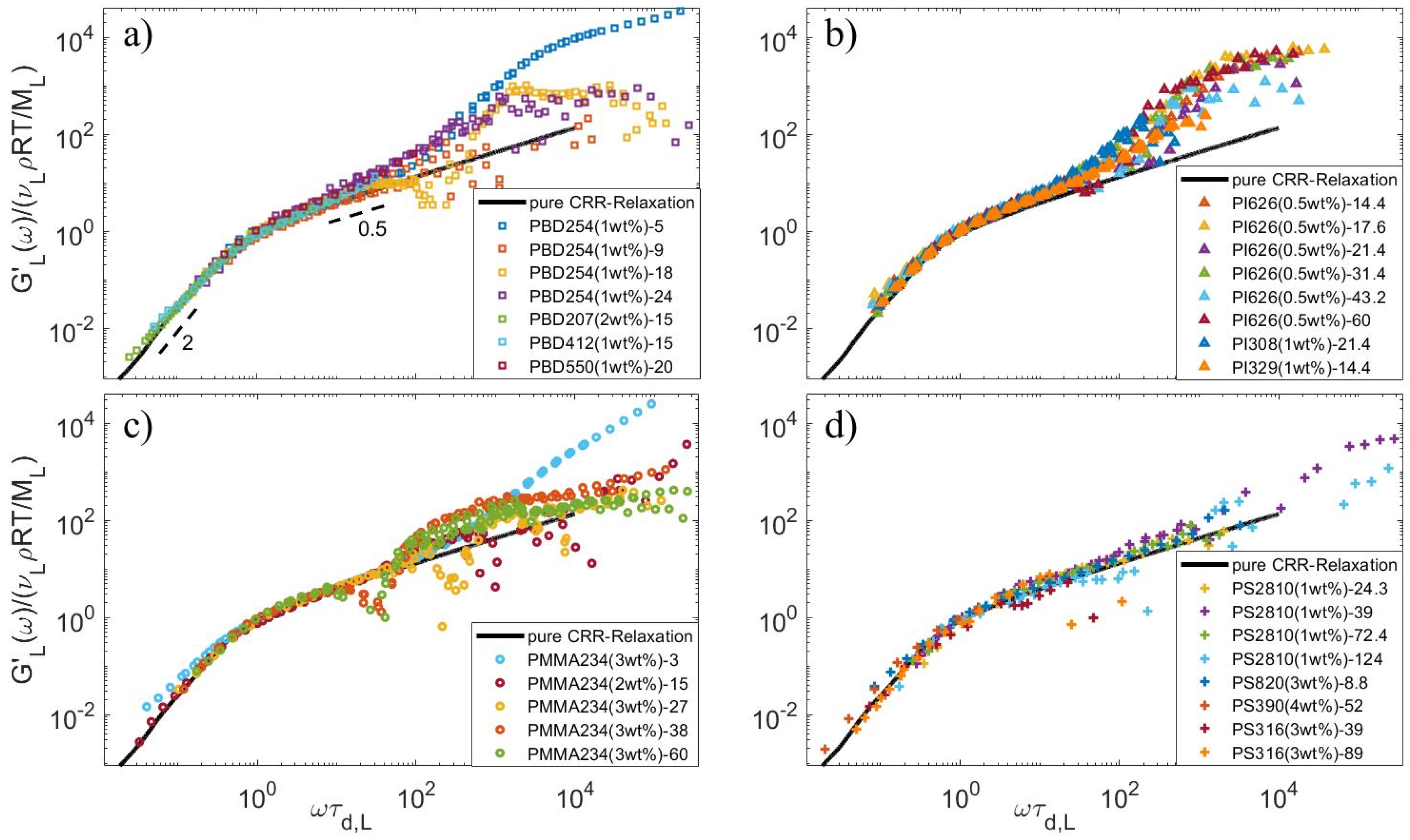
4.3. Determination of the CRR Time of the Long Chains, τCRR,L
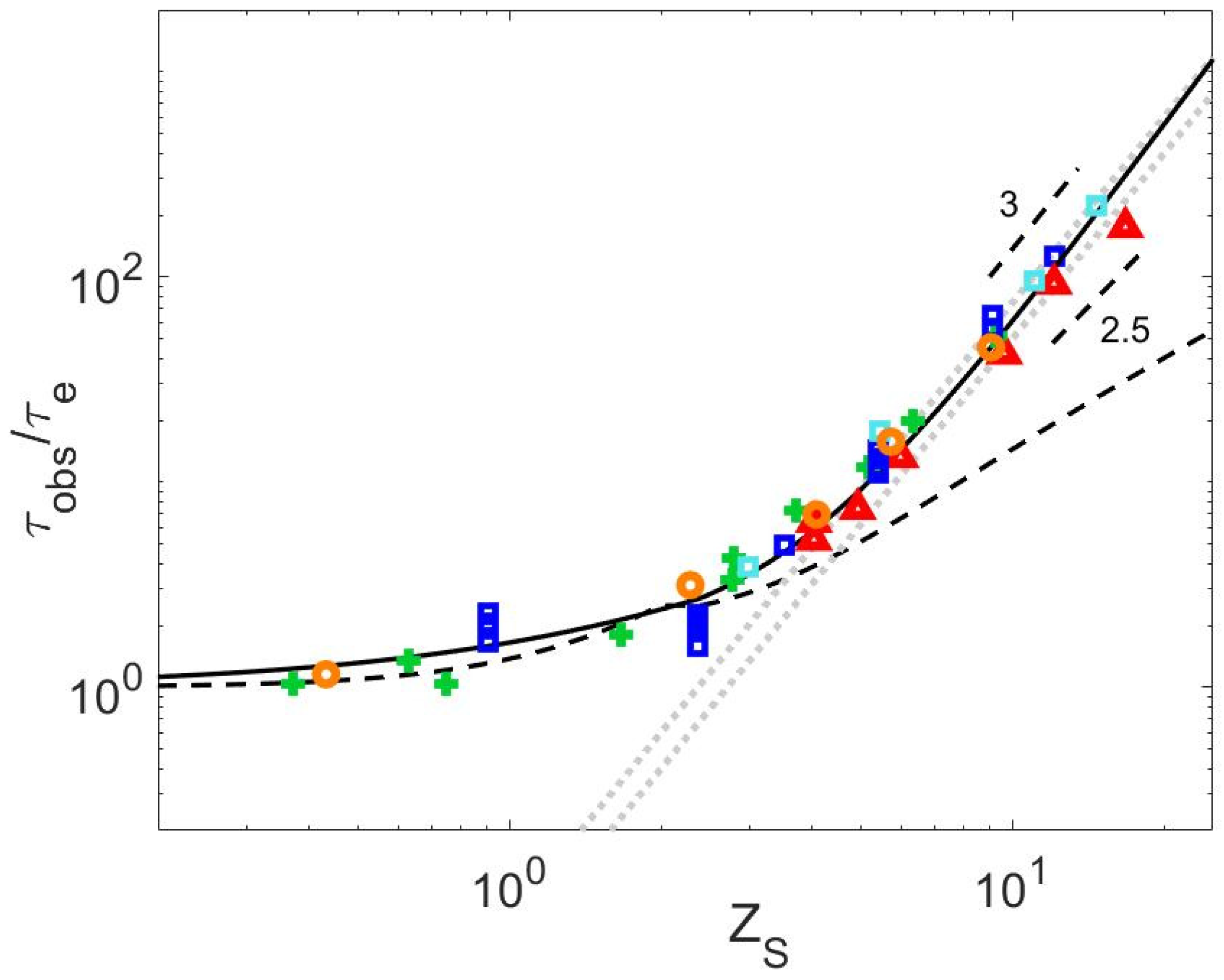
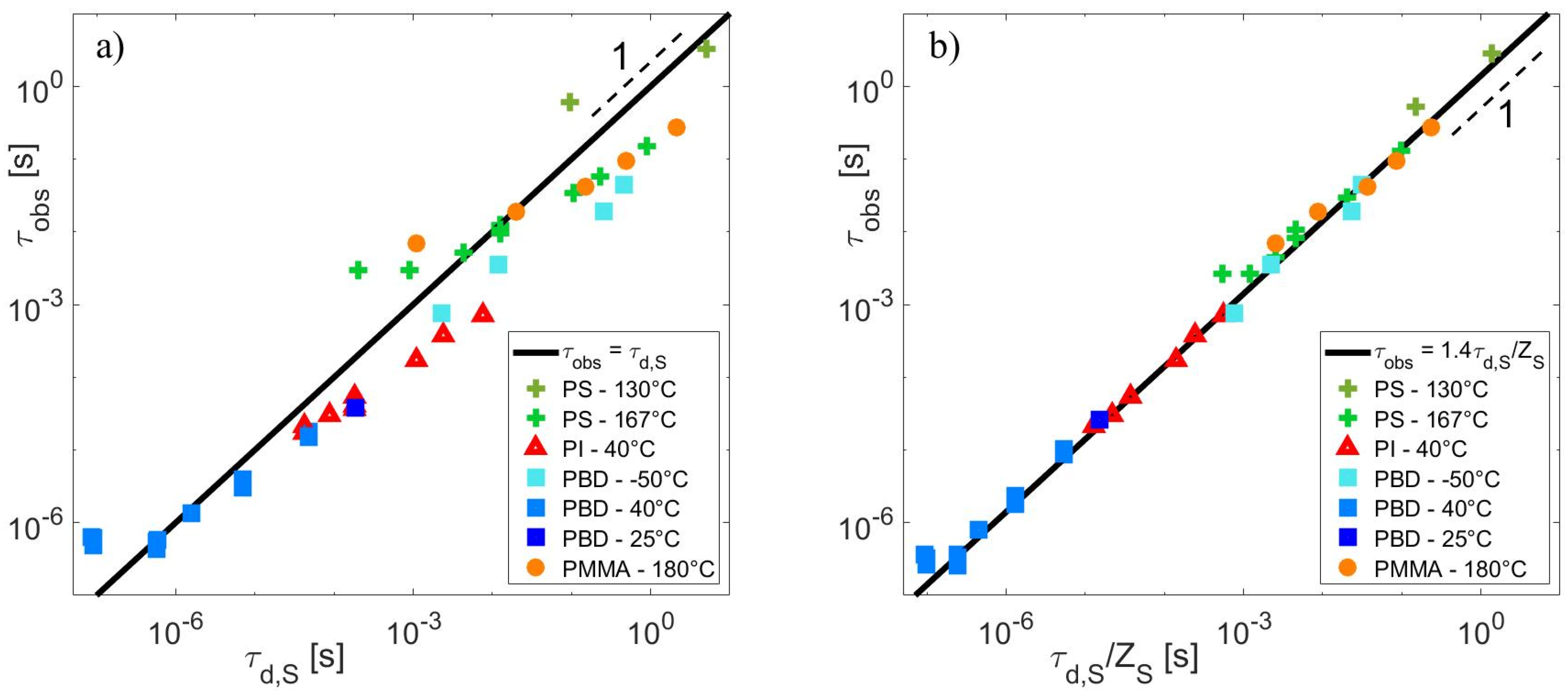
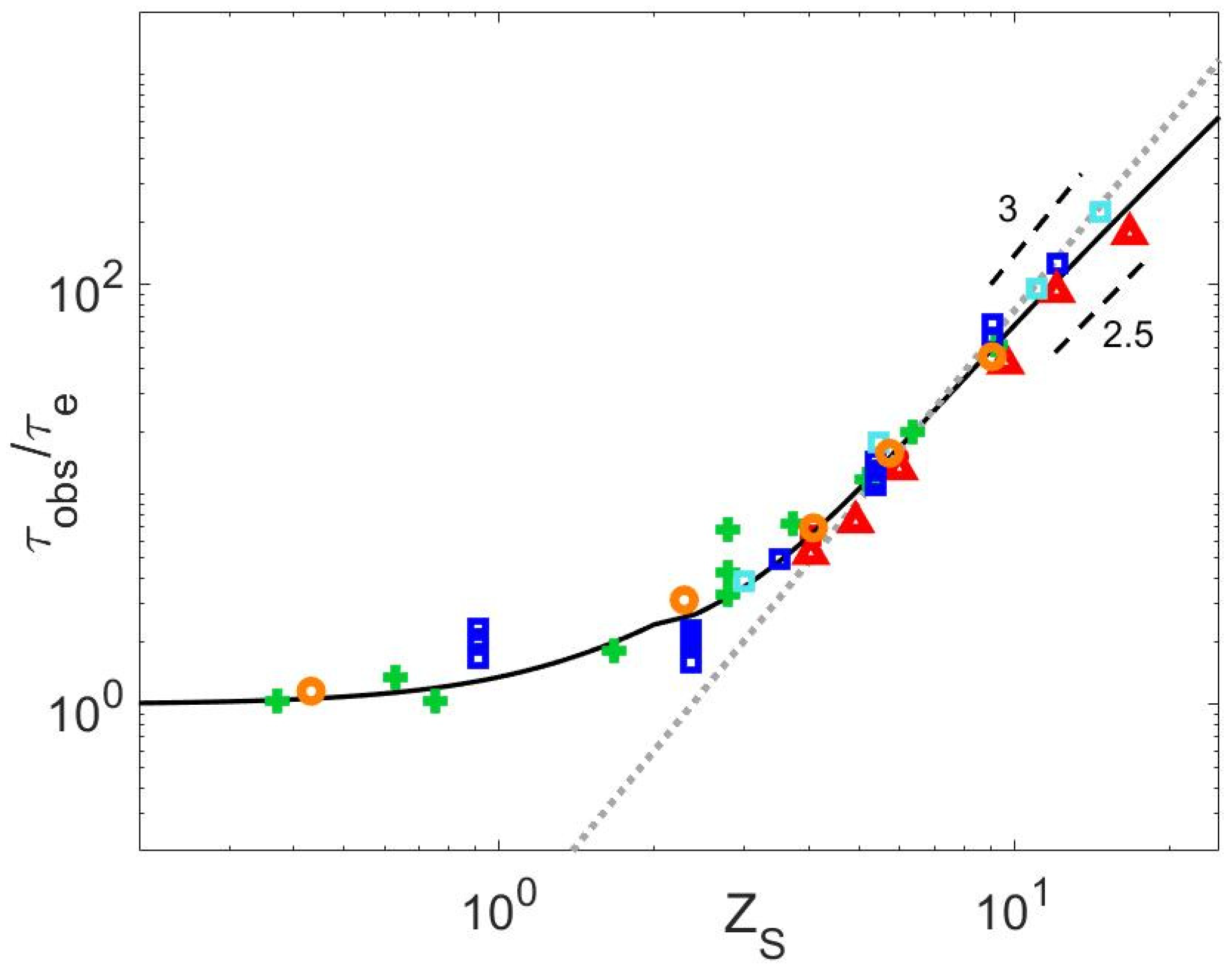
4.4. Relationship between τobs and ZS
4.5. Relationship between τobs and ZS
4.6. Critical Value of the Struglinski–Graessley Criterion for Dilute Binary Blends
5. Modeling the LVE of Self-Unentangled Long Chains Diluted in a Short Chain Matrix
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Gennes, P.G. Reptation of a Polymer Chain in the Presence of Fixed Obstacles. J. Chem. Phys. 1971, 55, 572. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: New York, NY, USA, 1986. [Google Scholar]
- Matsumiya, Y.; Watanabe, H. Entanglement-loosening dynamics resolved through comparison of dielectric and viscoelastic data of type-a Polymers: A review. Rubber Chem. Technol. 2020, 93, 22–60. [Google Scholar] [CrossRef]
- Read, D.J.; Shivokhin, M.E.; Likhtman, A.E. Contour length fluctuations and constraint release in entangled polymers: Slip-spring simulations and their implications for binary blend rheology. J. Rheol. 2018, 62, 1017. [Google Scholar] [CrossRef] [Green Version]
- Graessley, W.W. The Constraint Release Concept in Polymer Rheology. Adv. Polym. Sci. 1982, 47, 68–117. [Google Scholar]
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- McLeish, T.C.B. Tube theory of entangled polymer dynamics. Adv. Phys. 2002, 51, 1379–1527. [Google Scholar] [CrossRef]
- Dealy, J.M.; Larson, R.G. Structure and Rheology of Molten Polymers; Hanser Verlag: Munich, German, 2006. [Google Scholar]
- Milner, S.T.; McLeish, T.C.B.; Young, R.N.; Hakiki, A.; Johnson, J.M. Dynamic Dilution, Constraint-Release, and Star-Linear Blends. Macromolecules 1998, 31, 9345–9353. [Google Scholar] [CrossRef]
- Marrucci, G. Relaxation by reptation and tube enlargement: A model for polydisperse polymers. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 159–177. [Google Scholar] [CrossRef]
- des Cloizeaux, J.J. Double Reptation vs. Simple Reptation in Polymer Melts. Europhys. Lett. 1988, 5, 437. [Google Scholar] [CrossRef]
- Tsenoglou, C. Molecular Weight Polydispersity Effects on the Viscoelasticity of Entangled Linear-Polymers. Macromolecules 1991, 24, 1762–1767. [Google Scholar] [CrossRef]
- Struglinski, M.J.; Graessley, W. Effects of Polydispersity on the Linear Viscoelastic Properties of Entangled Polymers. 1. Experimental Observations for Binary Mixtures of Linear Polybutadiene. Macromolecules 1985, 18, 2630–2643. [Google Scholar] [CrossRef]
- Sawada, T.; Qiao, X.; Watanabe, H. Viscoelastic Relaxation of linear polyisoprenes: Examination of constraint release mechanism. Nihon Reoroji Gakkaishi 2006, 35, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Shivokhin, M.E.; Read, D.J.; Kouloumasis, D.; Kocen, R.; Zhuge, F.; Bailly, C.; Hadjichristidis, N.; Likhtman, A.E. Understanding Effect of Constraint Release Environment on End-to-End Vector Relaxation of Linear Polymer Chains. Macromolecules 2017, 50, 4501–4523. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, T.; Taghipour, H.; Grießl, D.; Mehrkhodavandi, P.; Hatzikiriakos, S.; van Ruymbeke, E. Binary Blends of Entangled Star and Linear Poly(hydroxybutyrate): Effect of Constraint Release and Dynamic Tube Dilation. Macromolecules 2017, 50, 2535–2546. [Google Scholar] [CrossRef]
- Yan, Z.-C.; van Ruymbeke, E.; Vlassopoulos, D. Linear Viscoelastic Response of Comb/Linear Polymer Blends: A Three-Step Relaxation Process. Macromolecules 2021, 54, 11047–11060. [Google Scholar] [CrossRef]
- Green, P.; Mills, P.; Palmstroem, C.; Mayer, J.; Kramer, E. Limits of Reptation in Polymer Melts. Phys. Rev. Lett. 1984, 53, 2145–2148. [Google Scholar] [CrossRef]
- Park, S.J.; Larson, R.G. Long-chain dynamics in binary blends of monodisperse linear polymers. J. Rheol. 2006, 50, 21. [Google Scholar] [CrossRef]
- Likhtman, A.E.; McLeish, T.C.B. Quantitative Theory for Linear Dynamics of Linear Entangled Polymers. Macromolecules 2002, 35, 6332–6343. [Google Scholar] [CrossRef]
- Montfort, J.P.; Marin, G.; Monge, P. Effects of Constraint Release on the Dynamics of Entangled Linear Polymer Melts. Macromolecules 1984, 17, 1551–1560. [Google Scholar] [CrossRef]
- Klein, J. Dynamics of Entangled Linear, Branched, and Cyclic Polymers. Macromolecules 1986, 19, 105–118. [Google Scholar] [CrossRef]
- Lentzakis, H.; Costanzo, S.; Vlassopoulos, D.; Colby, R.H.; Read, D.J.; Lee, H.; Chang, T.; van Ruymbeke, E. Constraint Release Mechanisms for H-Polymers Moving in Linear Matrices of Varying Molar Masses. Macromolecules 2019, 52, 3010–3028. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Sakamoto, T.; Kotaka, T. Viscoelastic Properties of Binary Blends of Narrow Molecular Weight Distribution Polystyrenes. 2. Macromolecules 1985, 18, 1008–1015. [Google Scholar] [CrossRef]
- Watanabe, H.; Sakamoto, T.; Kotaka, T. Entanglements in Linear Polystyrenes. Macromolecules 1985, 18, 1436–1442. [Google Scholar] [CrossRef]
- Shahid, T.; Huang, Q.; Oosterlinck, F.; Clasen, C.; van Ruymbeke, E. Dynamic dilution exponent in monodisperse entangled polymer solutions. Soft Matter 2017, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.K.; Rasmussen, H.K.; Hassager, O.; McKinley, G.H. Elongational viscosity of monodisperse and bidisperse polystyrene melts. J. Rheol. 2006, 50, 453. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Ishida, S.; Matsumiya, Y.; Inoue, T. Viscoelastic and Dielectric Behavior of Entangled Blends of Linear Polyisoprenes Having Widely Separated Molecular Weights: Test of Tube Dilation Picture. Macromolecules 2004, 37, 1937–1951. [Google Scholar] [CrossRef]
- Watanabe, H.; Ishida, S.; Matsumiya, Y.; Inoue, T. Test of Full and Partial Tube Dilation Pictures in Entangled Blends of Linear Polyisoprenes. Macromolecules 2004, 37, 6619–6631. [Google Scholar] [CrossRef]
- Matsumiya, Y.; Kumazawa, K.; Nagao, M.; Urakawa, O.; Watanabe, H. Dielectric Relaxation of Monodisperse Linear Polyisoprene: Contribution of Constraint Release. Macromolecules 2013, 46, 6067–6080. [Google Scholar] [CrossRef]
- Viovy, J.L.; Rubinstein, M.; Colby, R.H. Constraint release in polymer melts. Tube reorganization versus tube dilation. Macromolecules 1991, 24, 3587–3596. [Google Scholar] [CrossRef]
- Hawke, L.G.D. Viscoelastic properties of linear associating poly(n-butyl acrylate) chains. J. Rheol. 2016, 60, 297. [Google Scholar] [CrossRef]
- Hannecart, C.; Shahid, T.; Vlassopoulos, D.; Oosterlinck, F.; Clasen, C.; van Ruymbeke, E. Decoding the steady elongational viscosity of monodisperse linear polymers using tube-based modeling. J. Rheol. 2022, 66, 197. [Google Scholar] [CrossRef]
- van Ruymbeke, E.; Bailly, C.; Keunings, R.; Vlassopoulos, D. A General Methodology to Predict the Linear Rheology of Branched Polymers. Macromolecules 2006, 39, 6248–6259. [Google Scholar] [CrossRef]
- van Ruymbeke, E.; Vlassopoulos, D.; Kapnistos, M.; Liu, C.-Y.; Bailly, C. Proposal to Solve the Time-Stress Discrepancy of Tube Models. Macromolecules 2010, 43, 525–531. [Google Scholar] [CrossRef]
- Schwarzl, F.; Staverman, A.J. Higher Approximation Methods For The Relaxation Spectrum From Static And Dynamic Measurements Of Visco-Elastic Materials. Appl. Sci. Res. Sect. A 1953, 4, 127–141. [Google Scholar] [CrossRef]
- Watanabe, H.; Urakawa, O.; Kotaka, T. Slow Dielectric Relaxation of Entangled Linear cis-Polyisoprenes with Asymmetrically Inverted Dipoles. 2. Behavior in a Short Matrix. Macromolecules 1994, 27, 3525–3536. [Google Scholar] [CrossRef]
- Watanabe, H. Slow Dynamics in Homopolymer Liquids. Polym. J. 2009, 41, 929–950. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Urakawa, O.; Kotaka, T. Slow Dielectric Relaxation of Entangled Linear cis-Polyisoprenes with Asymmetrically Inverted Dipoles. 1. Bulk systems. Macromolecules 1993, 26, 5073–5083. [Google Scholar] [CrossRef]
- Park, S.J.; Larson, R.G. Tube Dilation and Reptation in Binary Blends of Monodisperse Linear Polymers. Macromolecules 2004, 37, 597–604. [Google Scholar] [CrossRef]
- Wang, S.; Elkasabi, Y.; Wang, S.-Q. Rheological Study of Chain Dynamics in Dilute Binary Polymer Mixtures. Macromolecules 2005, 38, 125–133. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-forming Liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Liu, C.-Y.; He, J. New Linearized Relation for the Universal Viscosity-Temperature Behavior of Polymer Melts. Macromolecules 2006, 39, 8867–8869. [Google Scholar] [CrossRef]
- Fox, T.J.; Flory, P. The Glass Temperature and Related Properties of Polystyrene. Influence of Molecular Weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Wang, S.; Wang, S.-Q.; Halasa, A.; Hsu, W.-L. Relaxation Dynamics in Mixtures of Long and Short Chains: Tube Dilation and Impeded Curvilinear Diffusion. Macromolecules 2003, 36, 5355–5371. [Google Scholar] [CrossRef]
- Shchetnikava, V.; Slot, J.; van Ruymbeke, E. Comparative Analysis of Different Tube Models for Linear Rheology of Monodisperse Linear Entangled Polymers. Polymers 2019, 11, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Ruymbeke, E.; Shchetnikava, V.; Matsumiya, Y.; Watanabe, H. Dynamic Dilution Effect in Binary Blends of Linear Polymers with Well-Separated Molecular Weights. Macromolecules 2014, 47, 7653–7665. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
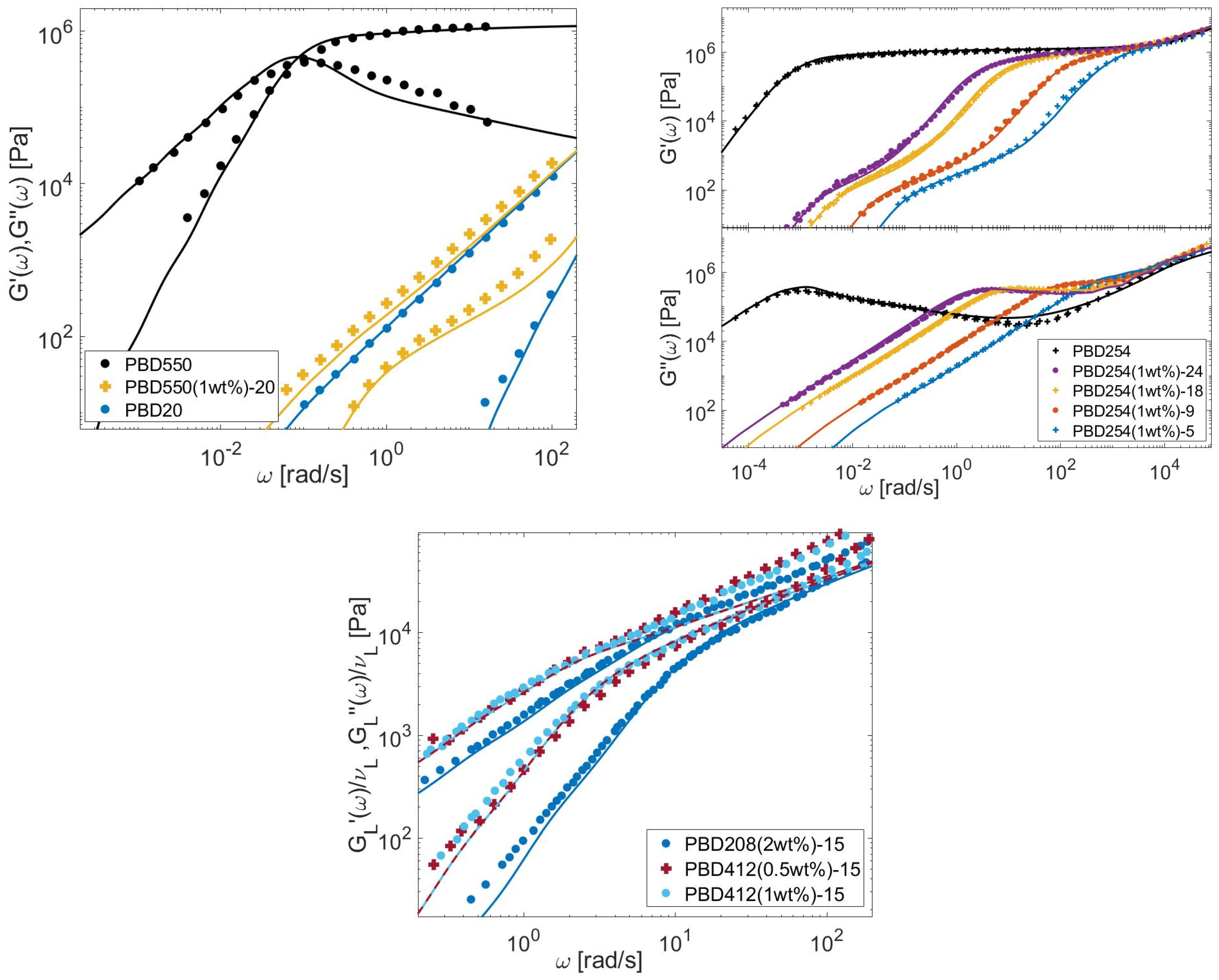{kind=link}
| Sample | ML [kg/mol] (PDI) | MS [kg/mol] (PDI) | υL [%] | ZLυL | Tg(MS) [°C] | Tg(blend) [°C] | Tref [°C] |
|---|---|---|---|---|---|---|---|
| PMMA234-60 | 234 (1.11) | 59.8 (1.08) | 3 | 1.1 | 120 | 120 | 180 |
| PMMA234-38 | 37.8 (1.11) | 3 | 1.1 | 120 | 120 | 180 | |
| PMMA234-27 | 26.9 (1.09) | 3 | 1.1 | 120 | 120 | 180 | |
| PMMA234-15 | 15.1 (1.08) | 2 | 0.71 | 113 | 113 | 173 | |
| PMMA234-3 | 2.85 (1.09) | 3 | 1.1 | 90 | 90 | 150 |
| Sample | ML [kg/mol] (PDI) 1,2:1,4 ratio | MS [kg/mol] (PDI) 1,2:1,4 ratio | υL [%] | ZLυL | Tref [°C] |
|---|---|---|---|---|---|
| PBD254-24 | 254 (1.02) 0.06:0.94 | 24.3 (1.01) 0.07:0.93 | 1 | 1.5 | −50 |
| PBD254-18 | 18.2 (1.01) 0.07:0.93 | 1 | 1.5 | −50 | |
| PBD254-9 | 9.00 (1.02) 0.09:0.91 | 1 | 1.5 | −50 | |
| PBD254-5 | 4.93 (1.03) 0.1:0.9 | 1 | 1.5 | −50 |
| Sample | ML [kg/mol] (PDI) | MS [kg/mol] (PDI) | υL [%] | ZLυL | Tref [°C] | Tg(blend) [°C] | Ref |
|---|---|---|---|---|---|---|---|
| PS316-39 | 316 (1.07) | 38.9 (1.07) | 3; 5; 10 | 0.68; 1.1; 2.3 | 167 | 103.9 *; 103.9 *; 104.1 * | [24] |
| PS316-89 | 316 (1.07) | 88.6 (1.07) | 3; 5 | 0.68; 1.1 | 167 | 106.6; 105.3 * | [24] |
| PS2810-23.4 | 2810 (1.09) | 23.4 (1.07) | 1 | 2.0 | 167 | 107; 100.9 * | [25] |
| PS2810-39 | 2810 (1.09) | 38.9 (1.07) | 1 | 2.0 | 167 | 107; 103.8 * | [25] |
| PS2810-72.4 | 2810 (1.09) | 72.4 (1.06) | 1 | 2.0 | 167 | 106.6; 105.1 * | [25] |
| PS2810-124 | 2810 (1.09) | 124 (1.05) | 1 | 2.0 | 167 | 106.6; 105.7 * | [25] |
| PS820-9 | 820 (1.02) | 8.8 (1.1) | 3 | 1.8 | 130 | 98.1 | [26] |
| PS390-52 | 390 (1.06) | 51.7 (1.03) | 4 | 1.1 | 130 | 106.6; 104.5 * | [27] |
| Sample | ML [kg/mol] (PDI) | MS [kg/mol] (PDI) | υL [%] | ZLυL | Tref [°C] | Ref |
|---|---|---|---|---|---|---|
| PI308-21.4 | 308 (1.08) | 21.4 (1.04) | 0.5; 1; 2; 3 | 0.43; 0.86; 1.7; 2.6 | 40 | [28] |
| PI329-14.4 | 329 (1.06) | 14.4 (1.03) | 0.5; 1; 2 | 0.46; 0.92; 1.8 | 40 | [30] |
| PI626-14.4 | 626 (1.06) | 14.4 (1.03) | 0.5 | 0.88 | 40 | [14] |
| PI626-17.6 | 626 (1.06) | 17.6 (1.04) | 0.5 | 0.88 | 40 | [14] |
| PI626-21.4 | 626 (1.06) | 21.4 (1.04) | 0.5 | 0.88 | 40 | [14] |
| PI626-34.4 | 626 (1.06) | 34.4 (1.04) | 0.5 | 0.88 | 40 | [14] |
| PI626-43.2 | 626 (1.06) | 43.2 (1.03) | 0.5 | 0.88 | 40 | [14] |
| PI626-60 | 626 (1.06) | 59.9 (1.05) | 0.5 | 0.88 | 40 | [14] |
| Sample | ML [kg/mol] (PDI) 1,2:1,4 ratio | MS [kg/mol] (PDI) 1,2:1,4 ratio | υL [%] | ZLυL | Tref [°C] | Ref |
|---|---|---|---|---|---|---|
| PBD550-20 | 550 | 20 | 1 | 3.3 | 25 | [40] |
| PBD208-15 | 208 (1.01) 0.08:0.92 | 15.5 (1.10) | 2 | 2.5 | 40 | [41] |
| PBD412-15 | 412 (1.01) 0.08:0.92 | 15.5 (1.10) | 0.5; 1 | 1.2; 2.5 | 40 | [41] |
| ML [kg/mol] (PDI) | 43.9 (1.01) −100°C | 99.1 (1.01) −100 °C | 208 (1.01) −100 °C | 412 (1.01) −100 °C | |
|---|---|---|---|---|---|
| MS [kg/mol] (PDI) | |||||
| 1.5 (/), Tg: −89 °C | 3%; 5% | 1%; 2%; 3% | 0.5%; 0.8%; 1% | 0.3%; 0.5%; 0.8% | |
| 3.9 (1.10), Tg: −102 °C | 2%; 3% | 0.75%; 1%; 2% | 0.5%; 0.75%; 1% | 0.5%; 0.75% | |
| 5.8 (1.06), Tg: −102 °C | 0.8%; 1%; 2%; 3% | / | / | / | |
| 8.9 (1.04), Tg: −102 °C | 2%; 3%; 5% | 0.5%; 1%; 2% | 0.5%; 1% | 0.2%; 0.5% | |
| 15.5 (1.10), (/) | / | 0.75%; 1%; 2% | 0.5%; 0.75%; 1% | 0.1%; 0.2%; 0.5%; 0.75% | |
| Sample Set | Tref [°C] | Me [kg/mol] | τe [s] | [kPa] | Ref of the Data |
|---|---|---|---|---|---|
| PS–130 | 130 | 14.0 | 0.39 | 230 | [26,27] |
| PS–167 | 167 | 14.0 | 0.0025 | 210 | [24,25] |
| PI–40 | 40 | 3.575 | 2.7 × 10−6 | 440 | [14,28,29,30] |
| PMMA–180 | 180 | 6.6 | 0.006 | 720 | / |
| PBD–25 | 25 | 1.65 | 2.1 × 10−7 | 1250 | [40] |
| PBD–40 | 40 | 1.65 | 1.6 × 10−7 | 1250 | [41] |
| PBD–−50 | −50 | 1.65 | 2.0 × 10−4 | 1250 | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannecart, C.; Clasen, C.; van Ruymbeke, E. Constraint Release Rouse Mechanisms in Bidisperse Linear Polymers: Investigation of the Release Time of a Short-Long Entanglement. Polymers 2023, 15, 1569. https://doi.org/10.3390/polym15061569
Hannecart C, Clasen C, van Ruymbeke E. Constraint Release Rouse Mechanisms in Bidisperse Linear Polymers: Investigation of the Release Time of a Short-Long Entanglement. Polymers. 2023; 15(6):1569. https://doi.org/10.3390/polym15061569
Chicago/Turabian StyleHannecart, Céline, Christian Clasen, and Evelyne van Ruymbeke. 2023. "Constraint Release Rouse Mechanisms in Bidisperse Linear Polymers: Investigation of the Release Time of a Short-Long Entanglement" Polymers 15, no. 6: 1569. https://doi.org/10.3390/polym15061569