
The Effect of Accelerated Aging on the Molecular Weight and Thermal and Mechanical Properties of Polyester Yarns Containing Ceramic Particles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Post-Spinning Stretching
2.3. Accelerated Aging
2.4. Washing
2.5. Molecular Weight
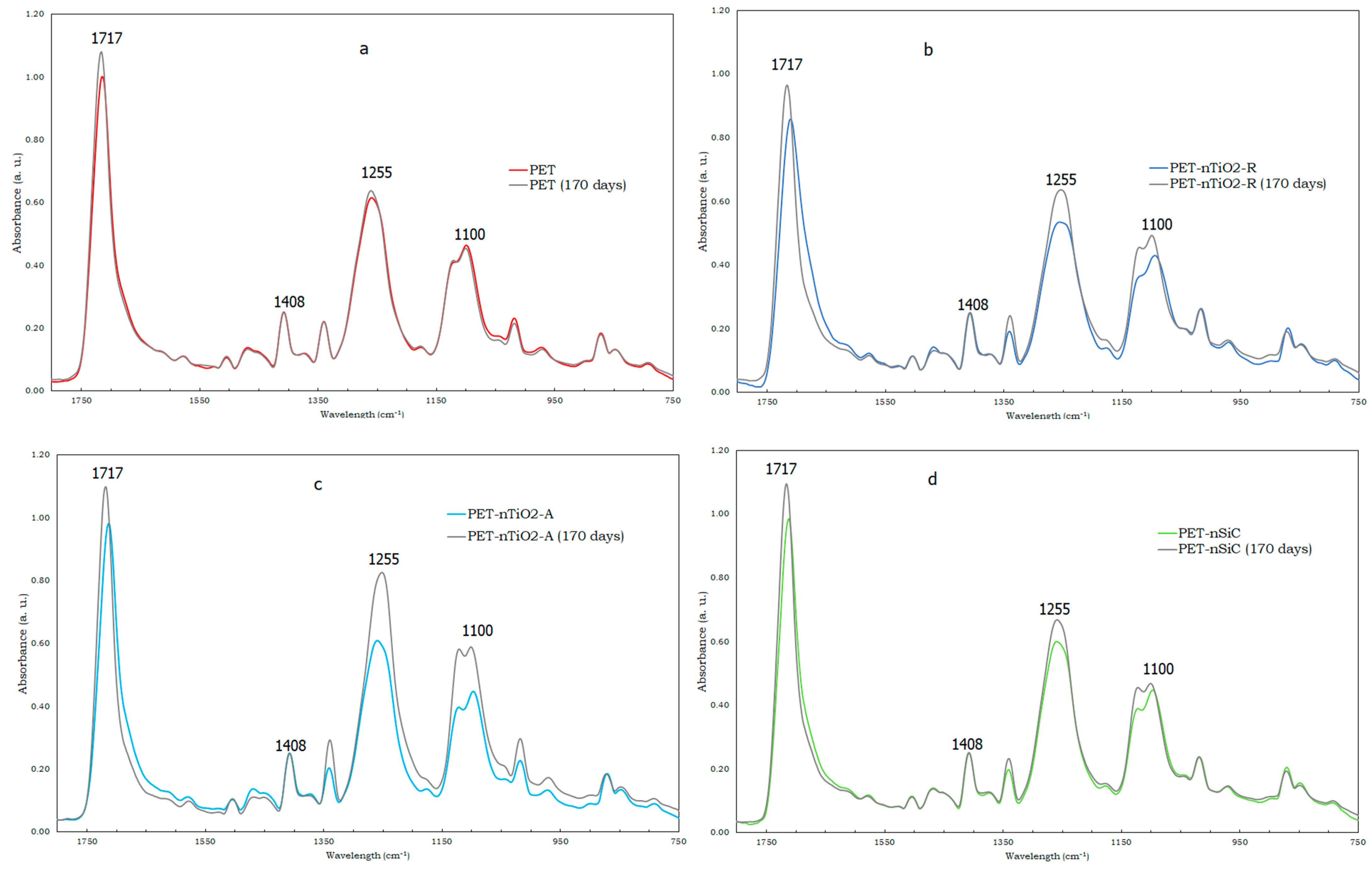
2.6. Fourier Transform Infrared Analysis-Attenuated Total Reflection (FTIR–ATR)
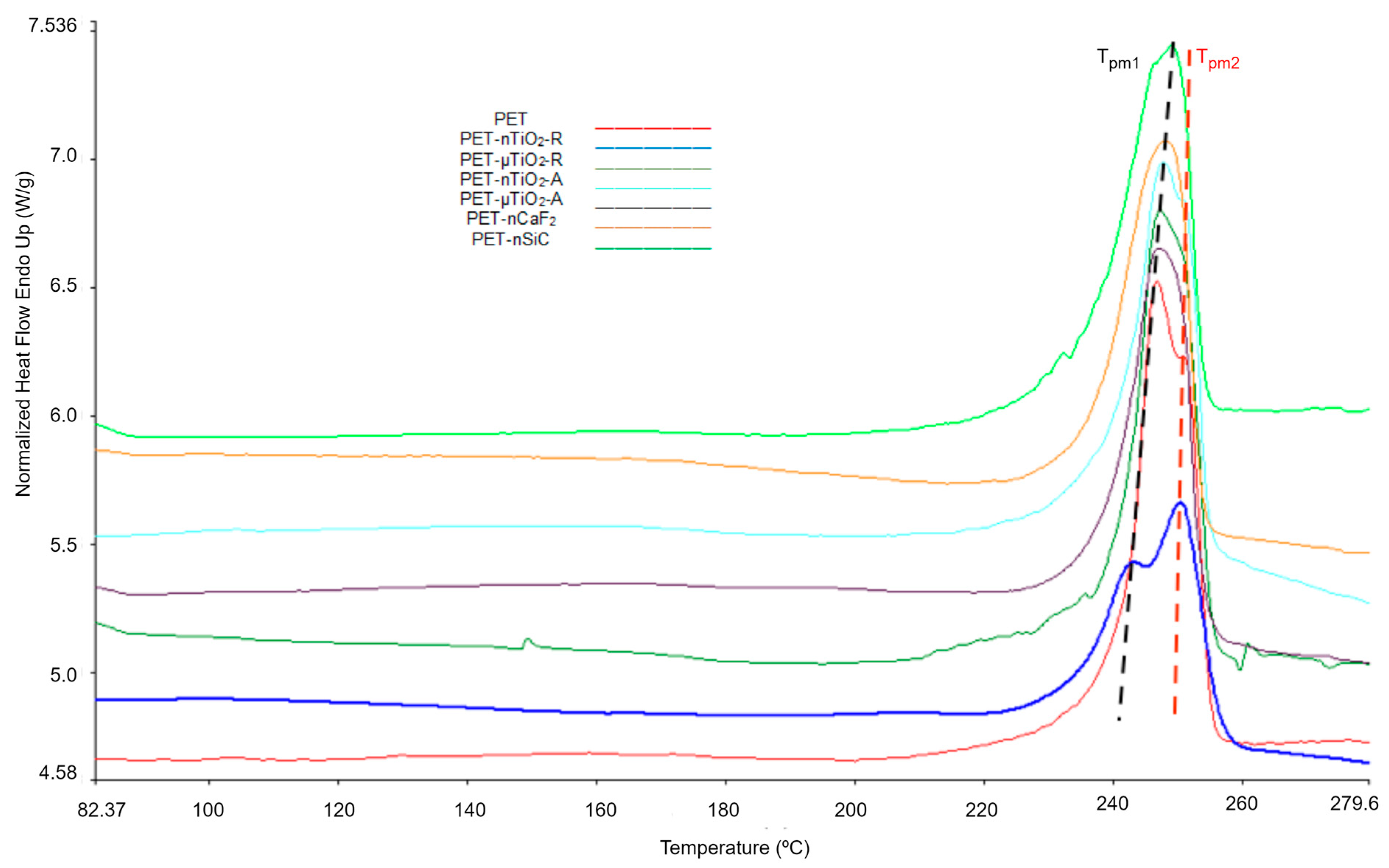
2.7. Thermal Analysis (Differential Scanning Calorimetry)
2.8. Lineal Density (Count)
2.9. Tenacity and Elongation
2.10. Micrographs
3. Result and Discussion
4. Conclusions
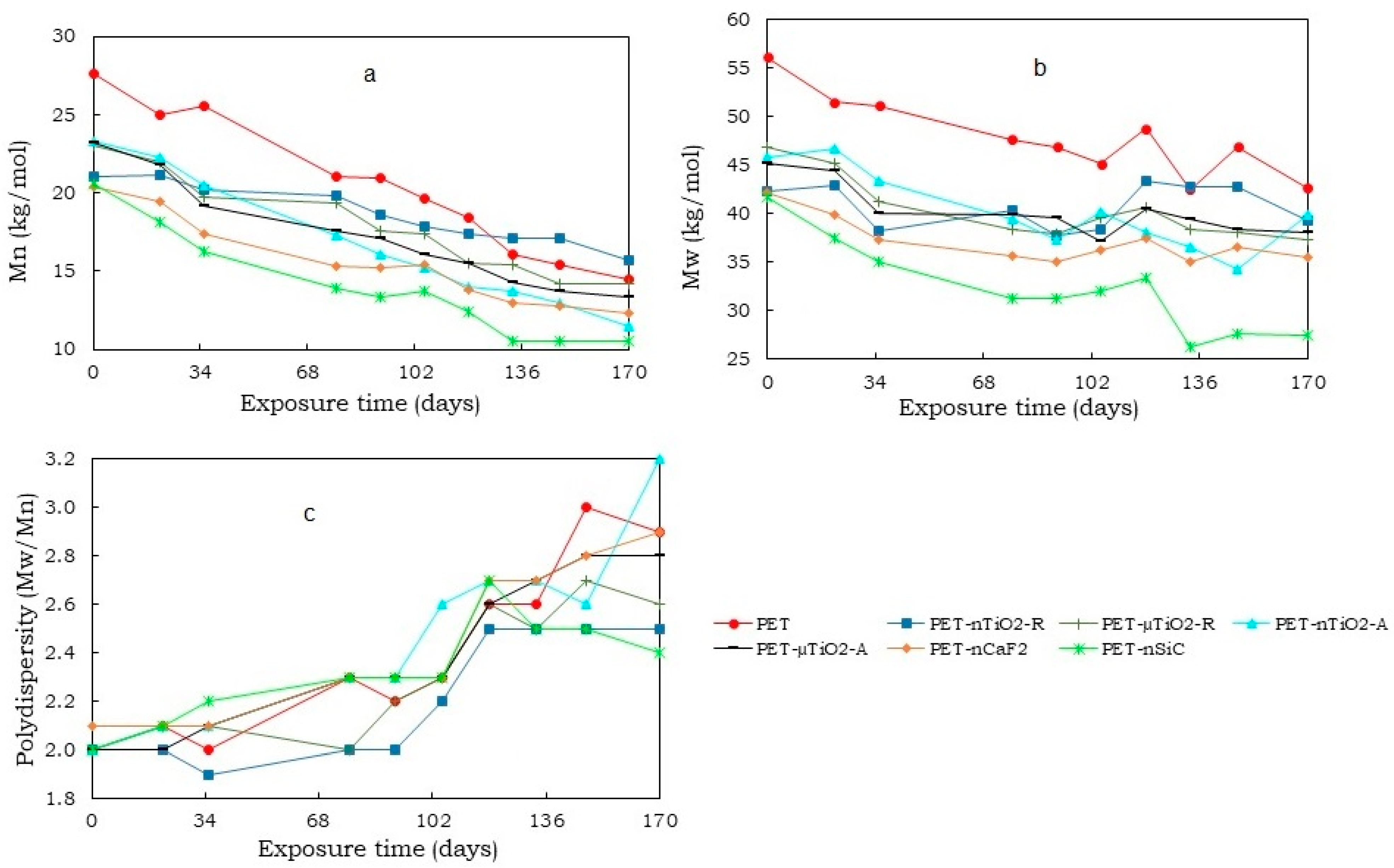
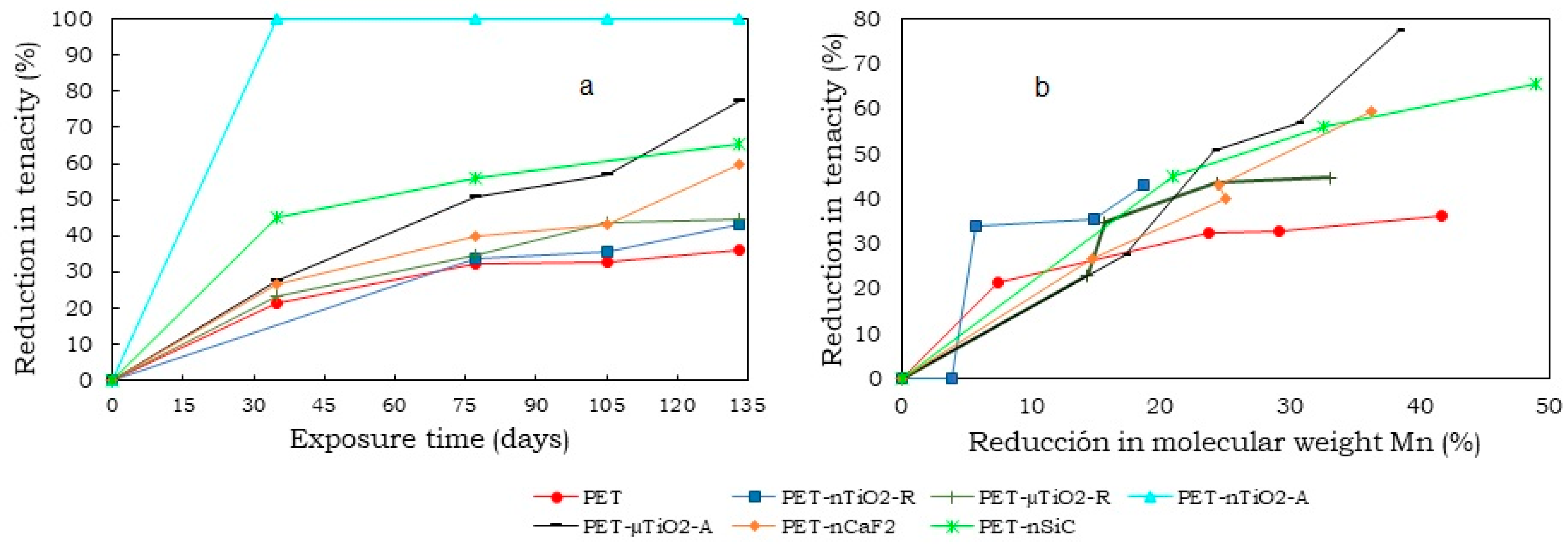
- A decrease in the molecular weight of all yarns that have been subjected to exposure in the climatic chamber was noted. This could be due to the excision of the molecular chains that formed the polymeric matrix. On one hand, the reduction was more evident in yarns with anatase TiO2 and SiC nanoparticles (about 50%), possibly due to their higher specific surface area that could have caused the ultraviolet radiation to produce a larger degradation. On the other hand, the rutile TiO2 nanometric had only a reduction of 25.3%.
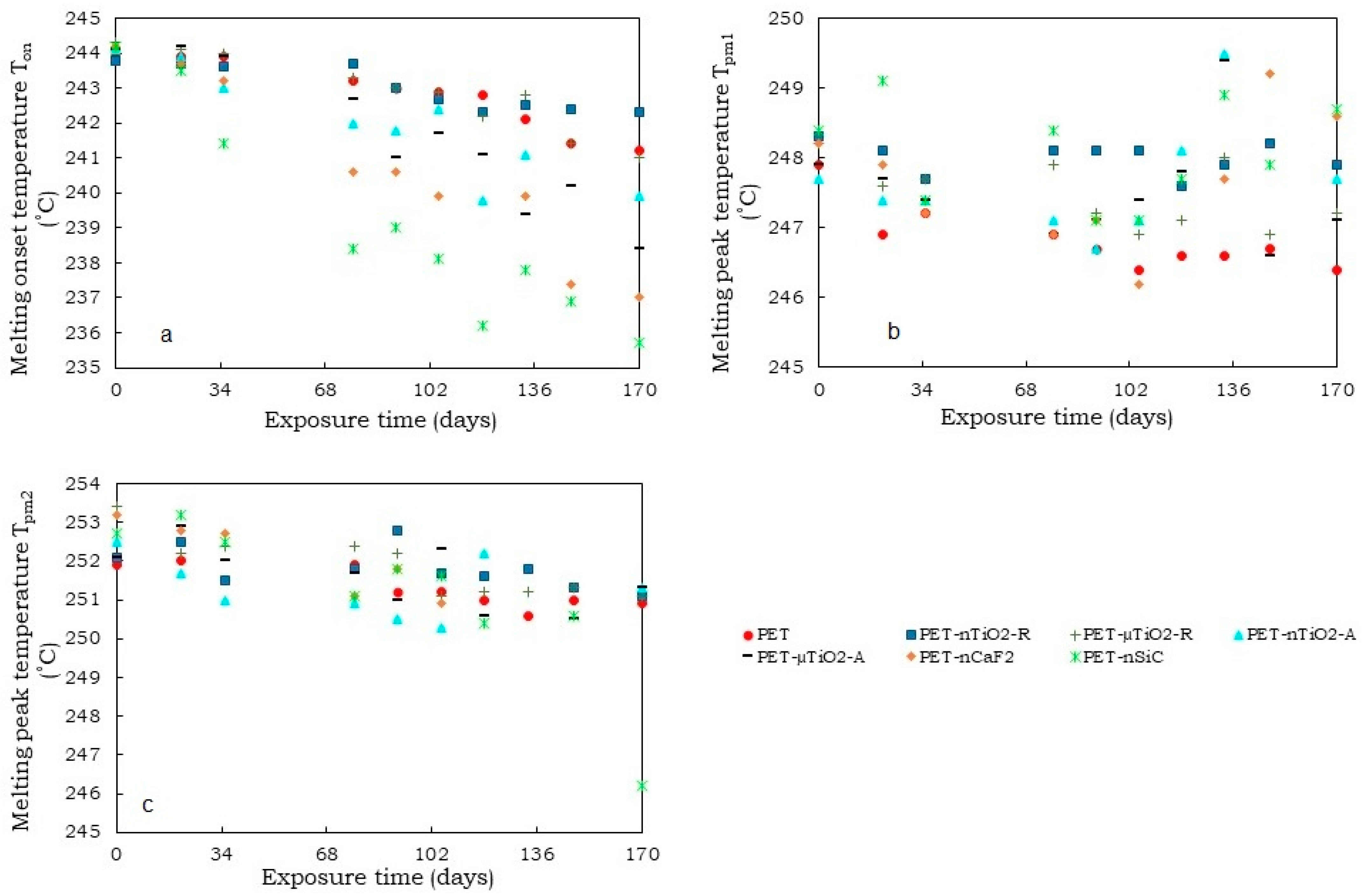
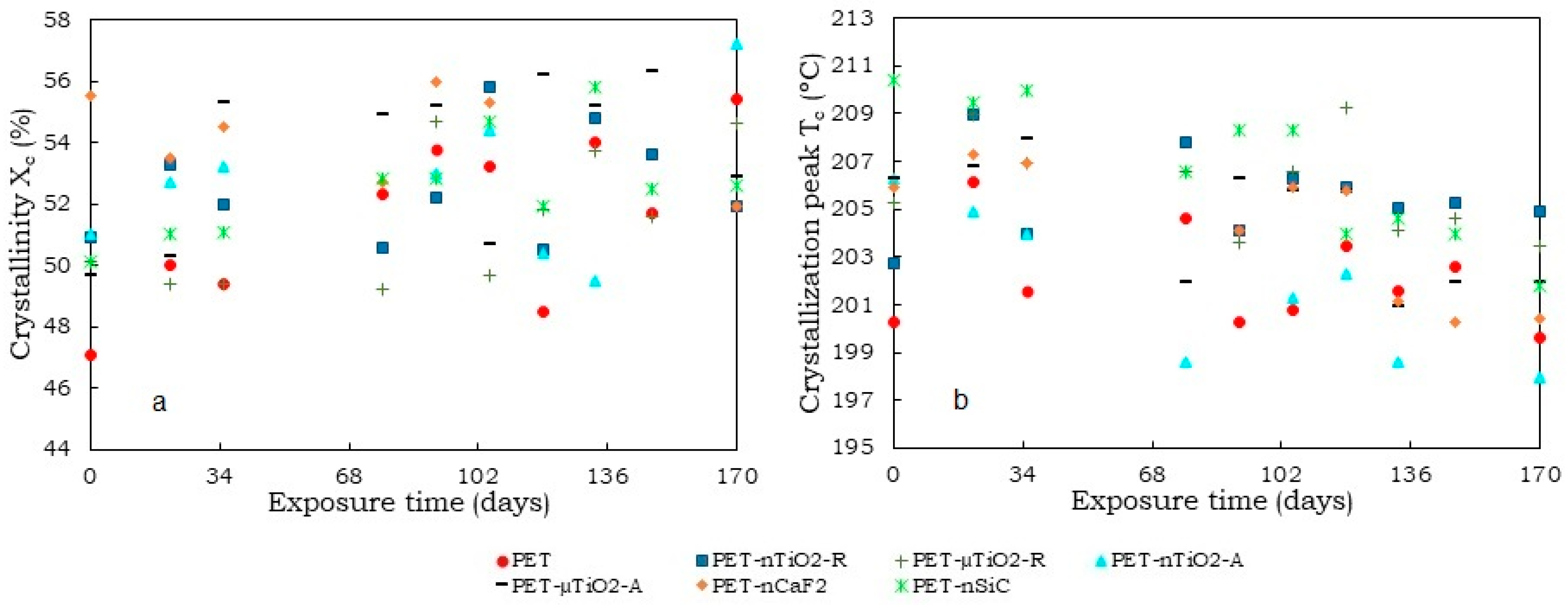
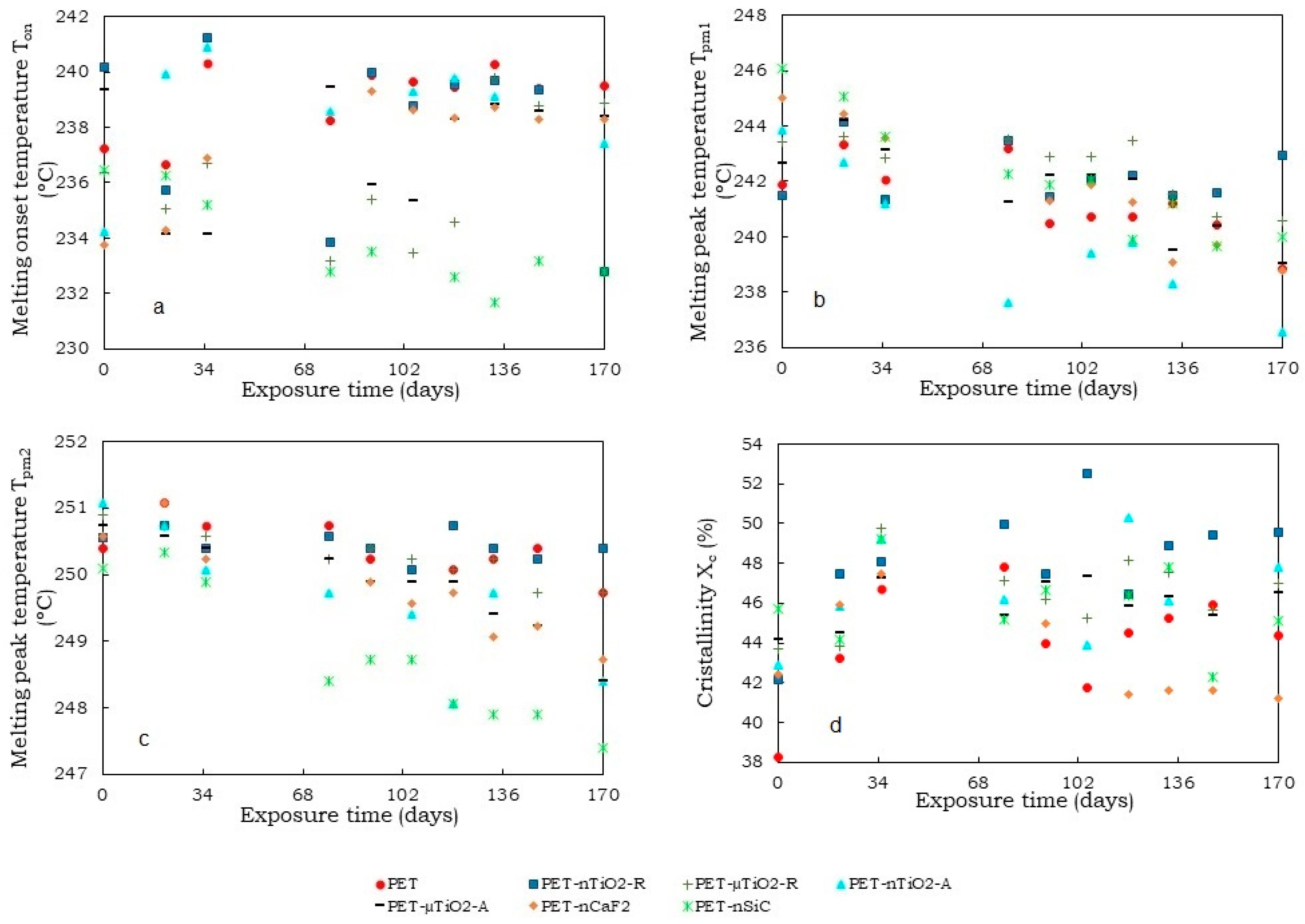
- A trend of increasing crystallinity in the pure PET yarns was observed over time, which could be due to a decrease in the amorphous part of the material. Moreover, the particle size and crystalline form of the ceramic particles did not increase the crystallinity compared to yarns that had not been exposed. Contrarily, in the case of polymer blends containing TiO2, unlike CaF2 and SiC, there was an increase in crystallinity because of the nucleating effect of the ceramic particles used. In this case, the nanoparticles, due to their size, allowed the formation of a greater number of crystals.
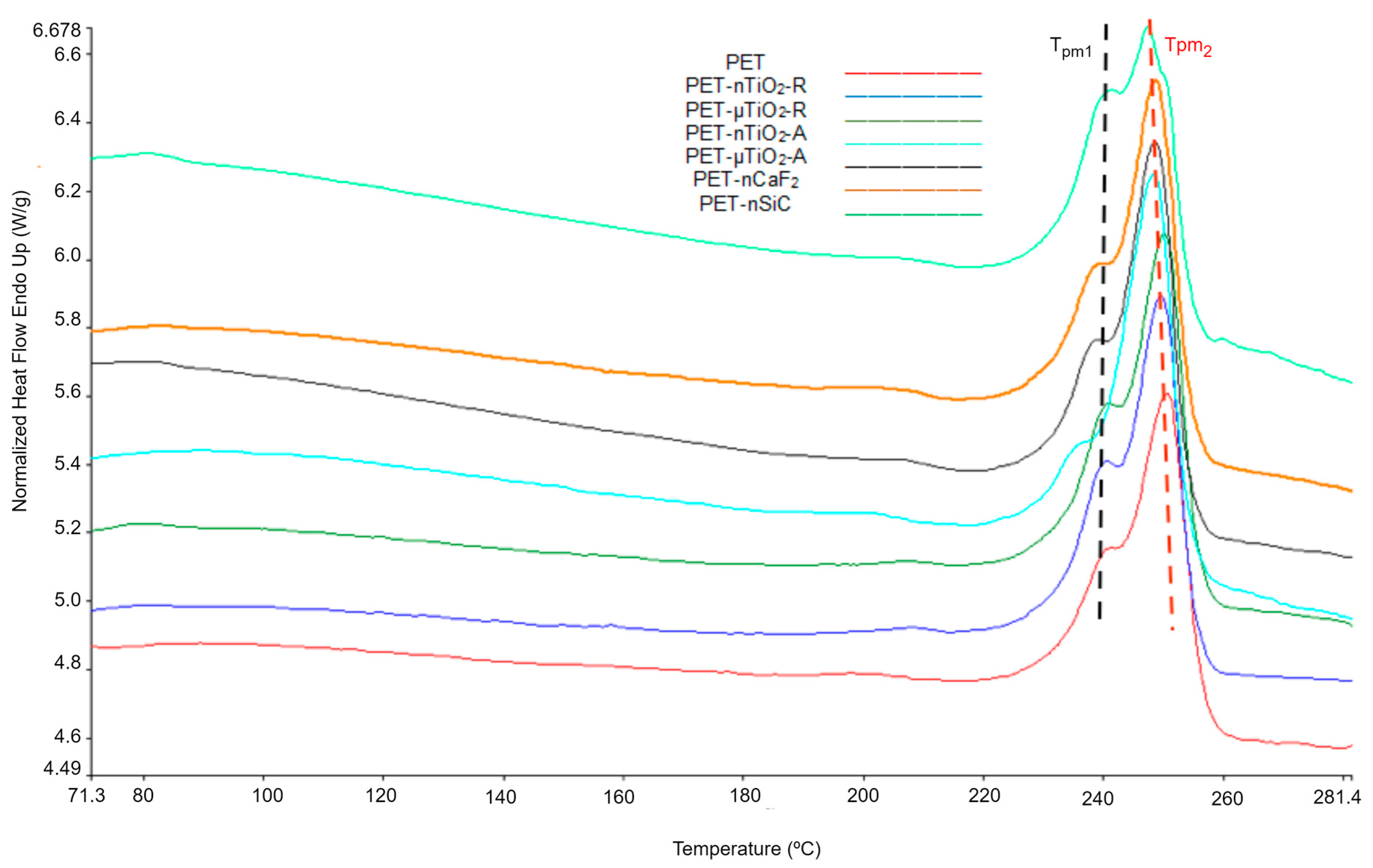
- Although an increase in nonisothermal crystallization temperature was observed in yarns with ceramic particles compared to pure PET, a tendency was found for it to decrease at higher exposure times because of a possible decrease in the concentration of the nucleating agents in the polymer melt because of the exposure in the climatic chamber.
- Finally, a reduction in tensile strength greater than that of the original PET was observed for all of the yarns that were subjected to the aging treatment. This could be a consequence of the high number of ceramic particles present in the composite materials and the breakage of the molecular chains of the substrate. The yarn with the anatase TiO2 micrometric showed the highest brittleness, which also experienced the greatest decrease in its molecular weight.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yokoyama, T.; Masuda, H.; Suzuki, M.; Ehara, K.; Nogi, K.; Fuji, M.; Fukui, T.; Suzuki, H.; Tatami, J.; Hayashi, K.; et al. Basic Properties and Measuring Methods of Nanoparticles. In Nanoparticle Technology Handbook; Nogi, K., Hosokawa, M., Naito, M., Yokoyama, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 3–48. ISBN 9780444563361. [Google Scholar]
- Mishra, R.; Militky, J.; Baheti, V.; Huang, J.; Kale, B.; Venkataraman, M.; Bele, V.; Arumugam, V.; Zhu, G.; Wang, Y. The Production, Characterization and Applications of Nanoparticles in the Textile Industry. Text. Prog. 2014, 46, 133–226. [Google Scholar] [CrossRef]
- Sankauskaite, A.; Rubežiene, V.; Kubiliene, D.; Abraitiene, A.; Baltušnikaite-Guzaitiene, J.; Dubinskaite, K. Investigation of Thermal Behavior of 3D PET Knits with Different Bioceramic Additives. Polymers 2020, 12, 1319. [Google Scholar] [CrossRef]
- Montazer, M.; Pakdel, E. Functionality of Nano Titanium Dioxide on Textiles with Future Aspects: Focus on Wool. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 293–303. [Google Scholar] [CrossRef]
- Almaieli, L.M.A.; Khalaf, M.M.; Gouda, M.; Alhayyani, S.; Abou Taleb, M.F.; Abd El-Lateef, H.M. Titanium Dioxide/Chromium Oxide/Graphene Oxide Doped into Cellulose Acetate for Medical Applications. Polymers 2023, 15, 485. [Google Scholar] [CrossRef]
- Taga, Y. Titanium Oxide Based Visible Light Photocatalysts: Materials Design and Applications. Thin Solid Films 2009, 517, 3167–3172. [Google Scholar] [CrossRef]
- Pakdel, E.; Daoud, W.A.; Wang, X. Assimilating the Photo-Induced Functions of TiO2-Based Compounds in Textiles: Emphasis on the Sol-Gel Process. Text. Res. J. 2015, 85, 1404–1428. [Google Scholar] [CrossRef]
- Akhsin Muflikhun, M.; Kuncoro Adi, R.; Nonato, C.; Santos, G. Nanocomposite Material Synthesized Via Horizontal Vapor Phase Growth Technique: Evaluation and Application Perspective. In 21st Century Nanostructured Materials—Physics, Chemistry, Classification, and Emerging Applications in Industry, Biomedicine, and Agriculture; Phuong, P., Ed.; IntechOpen: London, UK, 2022. [Google Scholar]
- Doualan, J.L.; Camy, P.; Moncorgé, R.; Daran, E.; Couchaud, M.; Ferrand, B. Latest Developments of Bulk Crystals and Thin Films of Rare-Earth Doped CaF2 for Laser Applications. J. Fluor. Chem. 2007, 128, 459–464. [Google Scholar] [CrossRef]
- Pandurangappa, C.; Lakshminarasappa, B.N.; Nagabhushana, B.M. Synthesis and Characterization of CaF2 Nanocrystals. J. Alloys Compd. 2010, 489, 592–595. [Google Scholar] [CrossRef]
- Park, S.W.; Choi, H.M.; Shin, D.S.; Oh, T.H. Effect of SiC and ZnO Nanoparticles on UV Absorbance and Heat Transfer of PET Composite Film. Fibers Polym. 2018, 19, 188–194. [Google Scholar] [CrossRef]
- He, R.; Zhou, N.; Zhang, K.; Zhang, X.; Zhang, L.; Wang, W.; Fang, D. Progress and Challenges towards Additive Manufacturing of SiC Ceramic. J. Adv. Ceram. 2021, 10, 637–674. [Google Scholar] [CrossRef]
- Chen, X.; Cheng, G.; Zhang, J.; Guo, F.; Zhou, H.; Liao, C.; Wang, H.; Zhang, X.; Dong, S. Residual Stress Variation in SiCf/SiC Composite during Heat Treatment and Its Effects on Mechanical Behavior. J. Adv. Ceram. 2020, 9, 567–575. [Google Scholar] [CrossRef]
- Riba-Moliner, M.; Mijas, G.; Sánchez-Loredo, M.G.; Cayuela, D. Obtaining of a PET-CaF2 Hybrid Multifilament: Non-Isothermal Crystallization Studies. Polym. Test. 2020, 86, 106449. [Google Scholar] [CrossRef]
- Riba-Moliner, M.; Mijas, G.; Roig, D.; Cayuela, D. Obtaining of a PET/SiC Hybrid Multifilament: Non-Isothermal Crystallization Studies. J. Text. Inst. 2021, 112, 1779–1787. [Google Scholar] [CrossRef]
- Cayuela, D.; Cot, M.; Riva, M.; Sanchez, R.J.; Sánchez-Loredo, M.G.; Algaba, I.; Manich, A.M. Effect of Surface Treatment of Titanium Dioxide Nanoparticles on Non-Isothermal Crystallization Behavior, Viscoelastic Transitions and Cold Crystallization of Poly(Ethylene Terephthalate) Nanocomposites. J. Macromol. Sci. Part A 2014, 51, 831–841. [Google Scholar] [CrossRef]
- Manich, A.M.; Cot, M.A.; Algaba, I.; Cayuela, D. Effect of the Presence of an Ester of Montanic Acids with Multifunctional Alcohols in the Composites of Titanium Dioxide Nanoparticles with Poly(Ethylene Terephthalate) in Their Non-Isothermal Crystallization. J. Macromol. Sci. Part A Pure Appl. Chem. 2015, 52, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Manich, A.M.; Cot, M.A.; Algaba, I.; Cayuela, D. Effect of the Titania Polymorph and Concentration of Dispersant on Thermal Transitions of PET/Titania Composites, Determined by Thermomechanical Analysis (TMA) and Differential Scanning Calorimetry (DSC). In Proceedings of the XXXVI Biennial Meeting of the Royal Spanish Society of Chemistry, Sitges, Spain, 25–29 June 2017. [Google Scholar]
- Cayuela, D.; Cot, M.; Algaba, I.; Manich, A.M. Effect of Different Dispersing Agents in the Non-Isothermal Kinetics and Thermomechanical Behavior of PET/TiO2 Composites. J. Macromol. Sci. Part A 2016, 53, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Leija, R.J.; Riba-Moliner, M.; Cayuela-Marín, D.; Domínguez-Espinós, O.; Sánchez-Loredo, M.G. Surface Effect of Two Different Calcium Fluoride Fillers on the Non-Isothermal Crystallization Behavior of Poly(Ethylene Terephthalate). J. Macromol. Sci. Part B 2014, 53, 173–190. [Google Scholar] [CrossRef]
- Searle, N. Weathering. In Encyclopedia of Polymer Science and Technology; Mark, H., Kroschwitz, J., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2002; pp. 629–659. [Google Scholar]
- Jones, M.S. Effects of UV Radiation on Building Materials. In Proceedings of the UV Workshop, Christchurch (Published in Current Proceedings); Building Research Association of New Zealand (BRANZ): Judgeford, New Zealand, 2002; pp. 26–28. [Google Scholar]
- Fotopoulou, K.N.; Karapanagioti, H.K. Degradation of Various Plastics in the Environment. In Handbook of Environmental Chemistry; Springer: Berlin/Heidelberg, Germany, 2019; Volume 78, pp. 71–92. [Google Scholar]
- Shokrieh, M.M.; Bayat, A. Effects of Ultraviolet Radiation on Mechanical Properties of Glass/Polyester Composites. J. Compos. Mater. 2007, 41, 2443–2455. [Google Scholar] [CrossRef]
- Jacques, L.F.E. Accelerated and Outdoor/Natural Exposure Testing of Coatings. Prog. Polym. Sci. 2000, 25, 1337–1362. [Google Scholar] [CrossRef]
- Bae, H.; Lee, M.; Kim, W.; Rhee, C. Dispersion Properties of TiO2 Nano-Powder Synthesized by Homogeneous Precipitation Process at Low Temperatures. Colloids Surfaces A Physicochem. Eng. Asp. 2003, 220, 169–177. [Google Scholar] [CrossRef]
- Nguyen, T.-C.; Nguyen, T.-D.; Vu, D.-T.; Dinh, D.-P.; Nguyen, A.-H.; Ly, T.-N.-L.; Dao, P.-H.; Nguyen, T.-L.; Bach, L.-G.; Thai, H. Modification of Titanium Dioxide Nanoparticles with 3-(Trimethoxysilyl)Propyl Methacrylate Silane Coupling Agent. J. Chem. 2020, 2020, 1381407. [Google Scholar] [CrossRef]
- Prastomo, N.; Ayad, M.; Kawamura, G.; Matsuda, A. Synthesis and Characterization of Polyaniline Nanofiber/TiO2 Nanoparticles Hybrids. J. Ceram. Soc. Japan 2011, 119, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Gatto, S. Photocatalytic Activity Assessment of Micro-Sized TiO2 Used as Powders and as Starting Material for Porcelain Gres Tiles Production. Ph.D. Thesis, Università Degli Studi Di Milano, Milan, Italy, 2014. [Google Scholar]
- Nanostructured & Amorphous Materials Silicon Carbide Nanopowder. Available online: https://www.nanoamor.com/inc/sdetail/621 (accessed on 18 February 2023).
- Gacén Esbec, I. Modificación de La Estructura Fina de La Fibras de PET En El Termofijado y En Su Tintura Posterior. Tintura Competitiva de Sustratos Termofijados a Temperaturas Vecinas. Ph.D. Thesis, Universidad Politécnica de Cataluña, Barcelona, Spain, 2004. [Google Scholar]
- UNE-EN 13392:2001; Textiles. Monofilamentos. Determinación de La Densidad Lineal. AENOR: Madrid, Spain, 2001.
- ISO 2062:2009; Determinación de La Fuerza o Carga de Rotura y Del Alargamiento En La Rotura de Hilos Individuales Con Un Equipo de Velocidad Constante de Alargamiento (CRE). AENOR: Madrid, Spain, 2010.
- Aflori, M.; Drobota, M. Modification of Polyethylene Terephthalate. In Poly(Ethylene Terephthalate) Based Blends, Composites and Nanocomposites; Visakh, P.M., Liang, M., Eds.; Elsevier: Oxford, UK, 2015; pp. 15–39. ISBN 9780323266987. [Google Scholar]
- Skandan, G.; Chen, Y.J.; Glumac, N.; Kear, B.H. Synthesis of Oxide Nanoparticles in Low Pressure Flames. Nanostructured Mater. 1999, 11, 149–158. [Google Scholar] [CrossRef]
- Luttrell, T.; Halpegamage, S.; Tao, J.; Kramer, A.; Sutter, E.; Batzill, M. Why Is Anatase a Better Photocatalyst than Rutile?—Model Studies on Epitaxial TiO2 Films. Sci. Rep. 2014, 4, 4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Song, H.; Meng, X.; Shen, T.; Sun, J.; Han, W.; Wang, X. Effects of Particle Size on the Structure and Photocatalytic Performance by Alkali-Treated TiO2. Nanomaterials 2020, 10, 546. [Google Scholar] [CrossRef] [Green Version]
- Hagihara, H.; Oishi, A.; Funabashi, M.; Kunioka, M.; Suda, H. Free-Volume Hole Size Evaluated by Positron Annihilation Lifetime Spectroscopy in the Amorphous Part of Poly(Ethylene Terephthalate) Degraded by a Weathering Test. Polym. Degrad. Stab. 2014, 110, 389–394. [Google Scholar] [CrossRef]
- Seo, K.S.; Cloyd, J.D. Kinetics of Hydrolysis and Thermal Degradation of Polyester Melts. J. Appl. Polym. Sci. 1991, 42, 845–850. [Google Scholar] [CrossRef]
- Awad, S.A.; Khalaf, E.M. Improvement of the Chemical, Thermal, Mechanical and Morphological Properties of Polyethylene Terephthalate–Graphene Particle Composites. Bull. Mater. Sci. 2018, 41, 67. [Google Scholar] [CrossRef] [Green Version]
- Pires, H.M.; Mendes, L.C.; Cestari, S.P.; Pita, V.J.R.R. Effect of Weathering and Accelerated Photoaging on PET/PC (80/20 Wt/Wt%) Melt Extruded Blend. Mater. Res. 2015, 18, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Tri, P.; Prud’homme, R.E. Nanoscale Analysis of the Photodegradation of Polyester Fibers by AFM-IR. J. Photochem. Photobiol. A Chem. 2019, 371, 196–204. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectroscopy of Polymers, VIII: Polyesters and the Rule of Three. Spectroscopy 2022, 37, 25–28. [Google Scholar] [CrossRef]
- Ehrenstein, G.; Riedel, G.; Trawiel, P. Thermal Analysis of Plastics; Hanser: Munich, Germany, 2004; ISBN 1-56990-362-X. [Google Scholar]
- De Clerck, K.; Rahier, H.; Van Mele, B.; Kiekens, P. Thermal Properties Relevant to the Processing of PET Fibers. J. Appl. Polym. Sci. 2003, 89, 3840–3849. [Google Scholar] [CrossRef]
- Wadey, B.L. Plasticizers. In Encyclopedia of Physical Science and Technology; Meyers, R.A., Ed.; Academic Press: Millbrae, CA, USA, 2003; pp. 441–456. ISBN 978-0-12-227410-7. [Google Scholar]
- Todorov, L.V.; Martins, C.I.; Viana, J.C. Characterization of PET Nanocomposites with Different Nanofillers. Solid State Phenom. 2009, 151, 113–117. [Google Scholar] [CrossRef]
- Gacen Guillen, J. Fibras de Poliéster, 2nd ed.; Universidad Politécnica de Catalunya: Terrassa, Spain, 1991; ISBN 84-7653-080-3. [Google Scholar]
- Peacock, A.J.; Calhoun, A.R. Polymer Chemistry: Properties and Applications; Hanser Gardner Publications: Cincinnati, OH, USA, 2006; ISBN 9781569903971. [Google Scholar]
- Shayestehfar, S.; Yazdanshenas, M.E.; Khajavi, R.; Rashidi, A.S. Physical and Mechanical Properties of Nylon 6/Titanium Dioxide Micro and Nano-Composite Multifilament Yarns. J. Eng. Fiber. Fabr. 2014, 9, 158–167. [Google Scholar]
- Selvin, T.P.; Kuruvilla, J.; Sabu, T. Mechanical Properties of Titanium Dioxide-Filled Polystyrene Microcomposites. Mater. Lett. 2004, 58, 281–289. [Google Scholar] [CrossRef]
- Rusu, M.; Sofian, N.; Rusu, D. Mechanical and Thermal Properties of Zinc Powder Filled High Density Polyethylene Composites. Polym. Test. 2001, 20, 409–417. [Google Scholar] [CrossRef]
- Burke, M.; Young, R.J.; Stanford, J.L. The Relationship between Structure and Properties in Titanium Dioxide Filled Polypropylene. Polym. Bull. 1993, 30, 361–368. [Google Scholar] [CrossRef]
- Haupert, F.; Wetzel, B. Reinforcement of Thermosetting Polymers by the Incorporation of Micro- and Nanoparticles. In Polymer Composites: From Nano- to Macro-Scale; Springer: Boston, MA, USA, 2005; pp. 45–62. ISBN 978-0-387-26213-0. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Ceramic Particle | Ceramic Particle (%) | Dispersant (%) | Name of the Sample |
|---|---|---|---|---|
| PET | - | 0 | 0 | PET |
| TiO2 rutile nm | 1.8 | 1.8 | PET-nTiO2-R | |
| TiO2 rutile µm | 2.0 | 2.0 | PET-µTiO2-R | |
| TiO2 anatase nm | 2.0 | 2.0 | PET-nTiO2-A | |
| TiO2 anatase µm | 2.0 | 2.0 | PET-µTiO2-A | |
| CaF2 nm | 2.0 | 2.0 | PET-nCaF2 | |
| SiC nm | 2.0 | 2.0 | PET-nSiC |
| Particle type | Rutile Nanometric | Rutile Micrometric | Anatase Nanometric | Anatase Micrometric | |||
| Commercial reference | TiO2 MT-500HD Tayca Corporation | TiO2 KRONOS 2360 | TiO2 AMT-600 Tayca Corporation | TiO2 KRONOS 1071 | |||
| Particle size (nm) | 30 | 190 | 30 | 220 | |||
| Specific surface area (m2/g) | 48 | 13–17 | 52 | 9–11 | |||
| Composition (%) | TiO2 | 85 | TiO2 | ≥ 92 | TiO2 80–98 | TiO2 | ≥96 |
| Al2O3 | 1–15 | Al2O3 | 3–3.8 | Al2O3 | 1–1.2 | ||
| ZrO2 | 1–10 | SiO2 | 2.4–3 | SiO2 | 0.5–0.7 | ||
| C | 0.18–0.2 | P2O5 | 0.3–0.4 | ||||
| C | 0.15–0.25 | ||||||
| Melting temperature (°C) | 1843 | ||||||
| Sample | Kn (mol/kg·day) (10−4) | C (mol/kg) (10−2) | R2 |
|---|---|---|---|
| PET | 2.0 ± 0.2 | 3.4 ± 0.2 | 0.946 |
| PET-nTiO2-R | 0.9 ± 0.1 | 4.6 ± 0.1 | 0.941 |
| PET-µTiO2-R | 1.7 ± 0.1 | 4.2 ± 0.1 | 0.949 |
| PET-nTiO2-A | 2.6 ± 0.1 | 4.0 ± 0.1 | 0.984 |
| PET-µTiO2-A | 1.9 ± 0.1 | 4.3 ± 0.1 | 0.982 |
| PET-nCaF2 | 1.9 ± 0.1 | 4.9 ± 0.1 | 0.970 |
| PET-nSiC | 2.9 ± 0.1 | 4.9 ± 0.3 | 0.947 |
| Sample | Count (tex) | Tenacity | Elongation | ||
|---|---|---|---|---|---|
| Value (cN/tex) | CV (%) | Value (%) | CV (%) | ||
| PET | 16.0 ± 0.2 | 35.9 | 4.4 | 23.1 | 9.6 |
| PET-nTiO2-R | 17.2 ± 0.2 | 17.9 | 12.2 | 24.5 | 20.6 |
| PET-µTiO2-R | 13.3 ± 0.1 | 33.1 | 5.5 | 20.4 | 12.7 |
| PET-nTiO2-A | 18.5 ± 0.6 | 21.4 | 8.0 | 27.1 | 14.4 |
| PET-µTiO2-A | 16.9 ± 0.4 | 31.0 | 4.5 | 21.5 | 9.7 |
| PET-nCaF2 | 15.3 ± 0.1 | 28.5 | 7.5 | 21.9 | 13.8 |
| PET-nSiC | 16.4 ± 0.9 | 24.3 | 4.7 | 23.0 | 7.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mijas, G.; Riba-Moliner, M.; Cayuela, D. The Effect of Accelerated Aging on the Molecular Weight and Thermal and Mechanical Properties of Polyester Yarns Containing Ceramic Particles. Polymers 2023, 15, 1348. https://doi.org/10.3390/polym15061348
Mijas G, Riba-Moliner M, Cayuela D. The Effect of Accelerated Aging on the Molecular Weight and Thermal and Mechanical Properties of Polyester Yarns Containing Ceramic Particles. Polymers. 2023; 15(6):1348. https://doi.org/10.3390/polym15061348
Chicago/Turabian StyleMijas, Gabriela, Marta Riba-Moliner, and Diana Cayuela. 2023. "The Effect of Accelerated Aging on the Molecular Weight and Thermal and Mechanical Properties of Polyester Yarns Containing Ceramic Particles" Polymers 15, no. 6: 1348. https://doi.org/10.3390/polym15061348