

Hybridization of Wide-Angle X-ray and Neutron Diffraction Techniques in the Crystal Structure Analyses of Synthetic Polymers
 and
and
Abstract
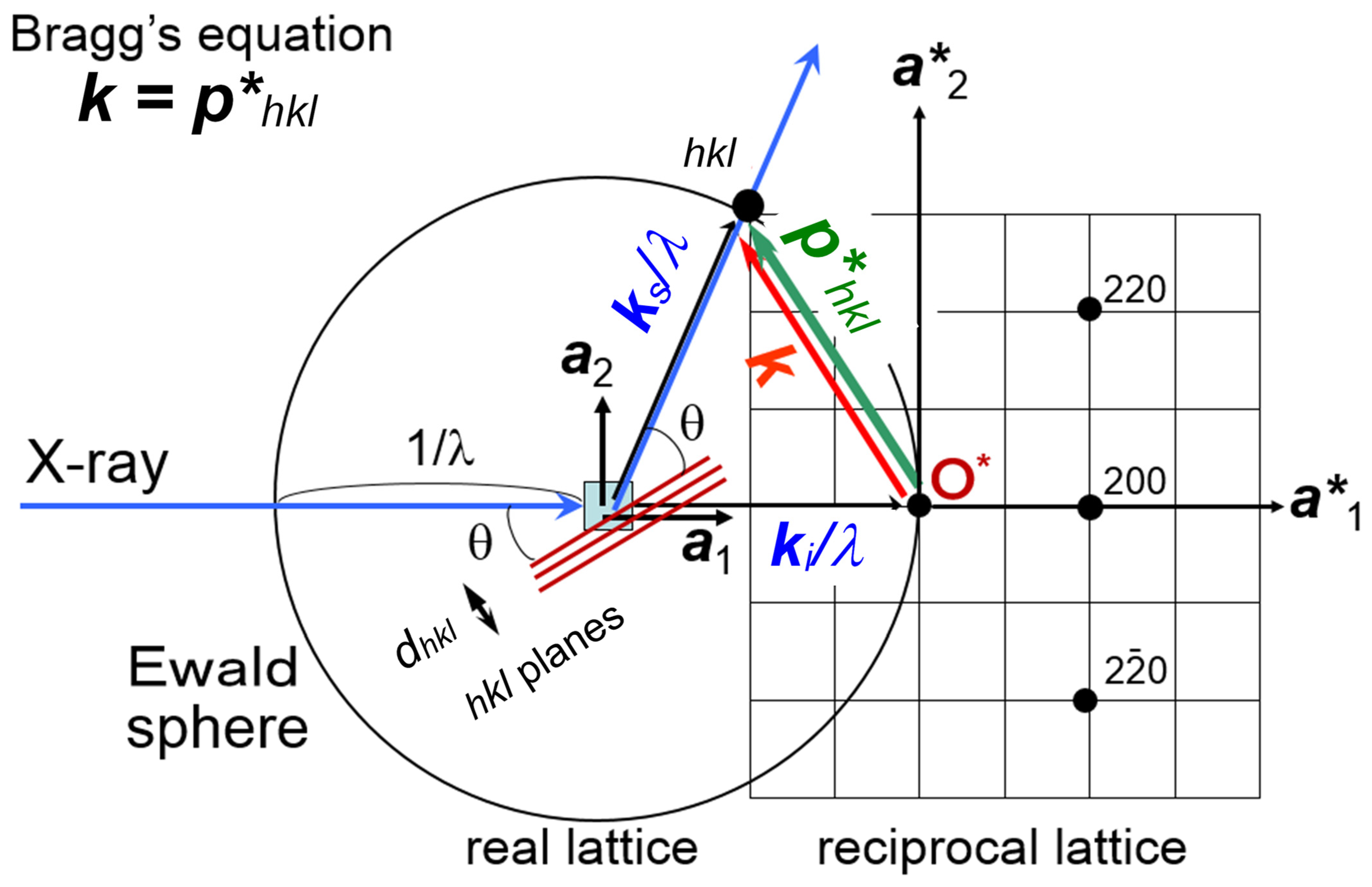
:1. Introduction
2. Experimental Systems
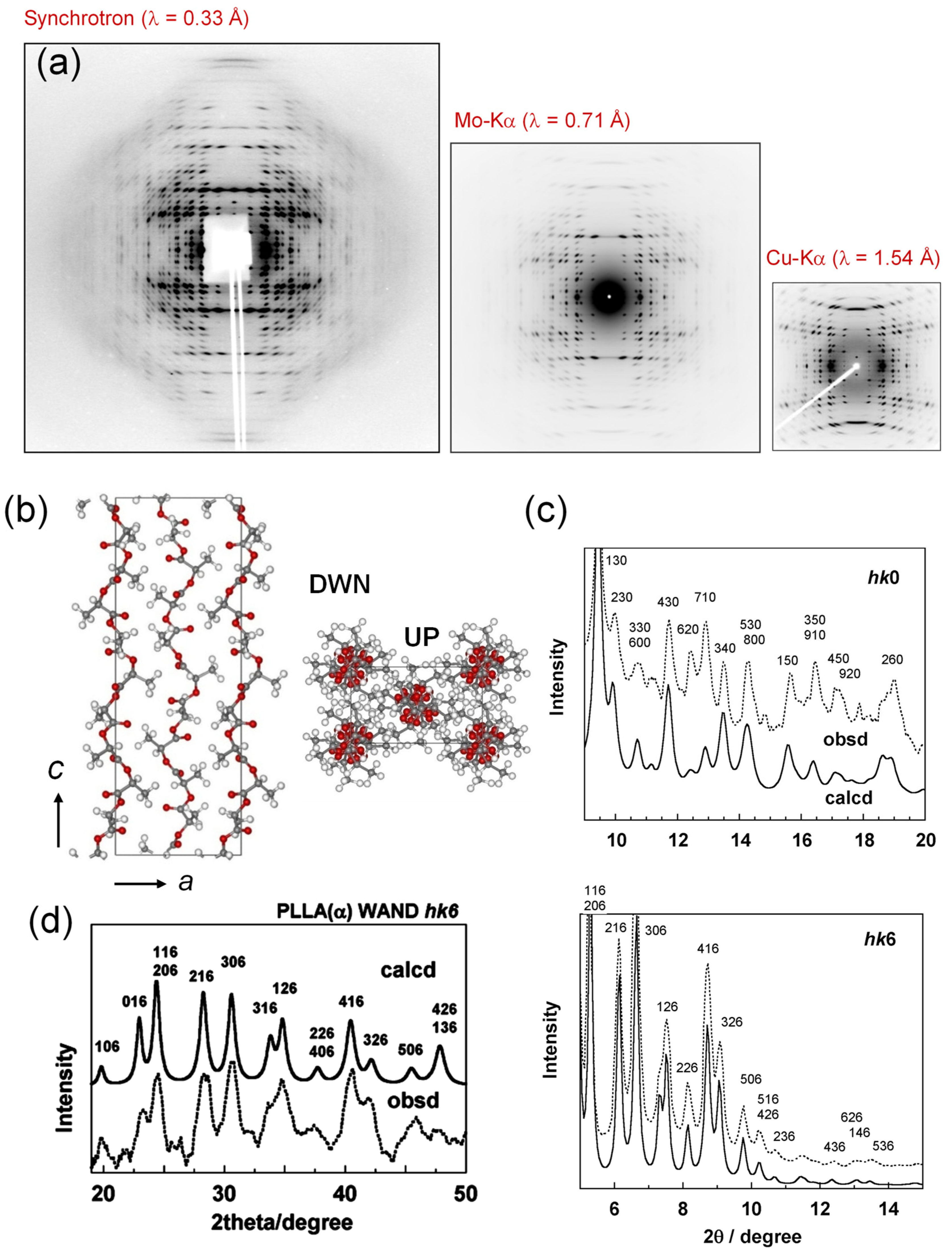
2.1. X-ray Diffraction System
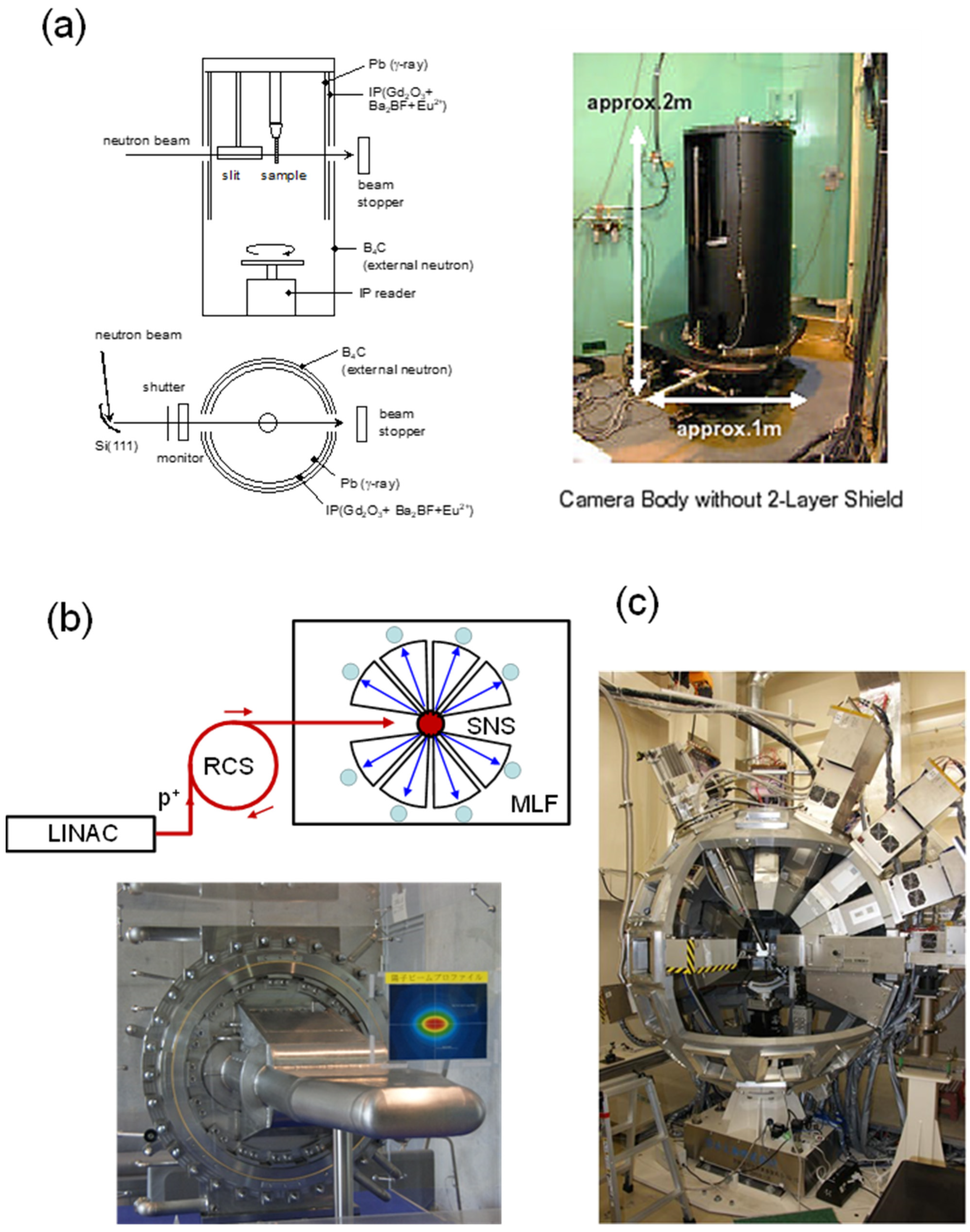
2.2. Neutron Diffraction Systems
2.2.1. Monochromatic Neutron Beam
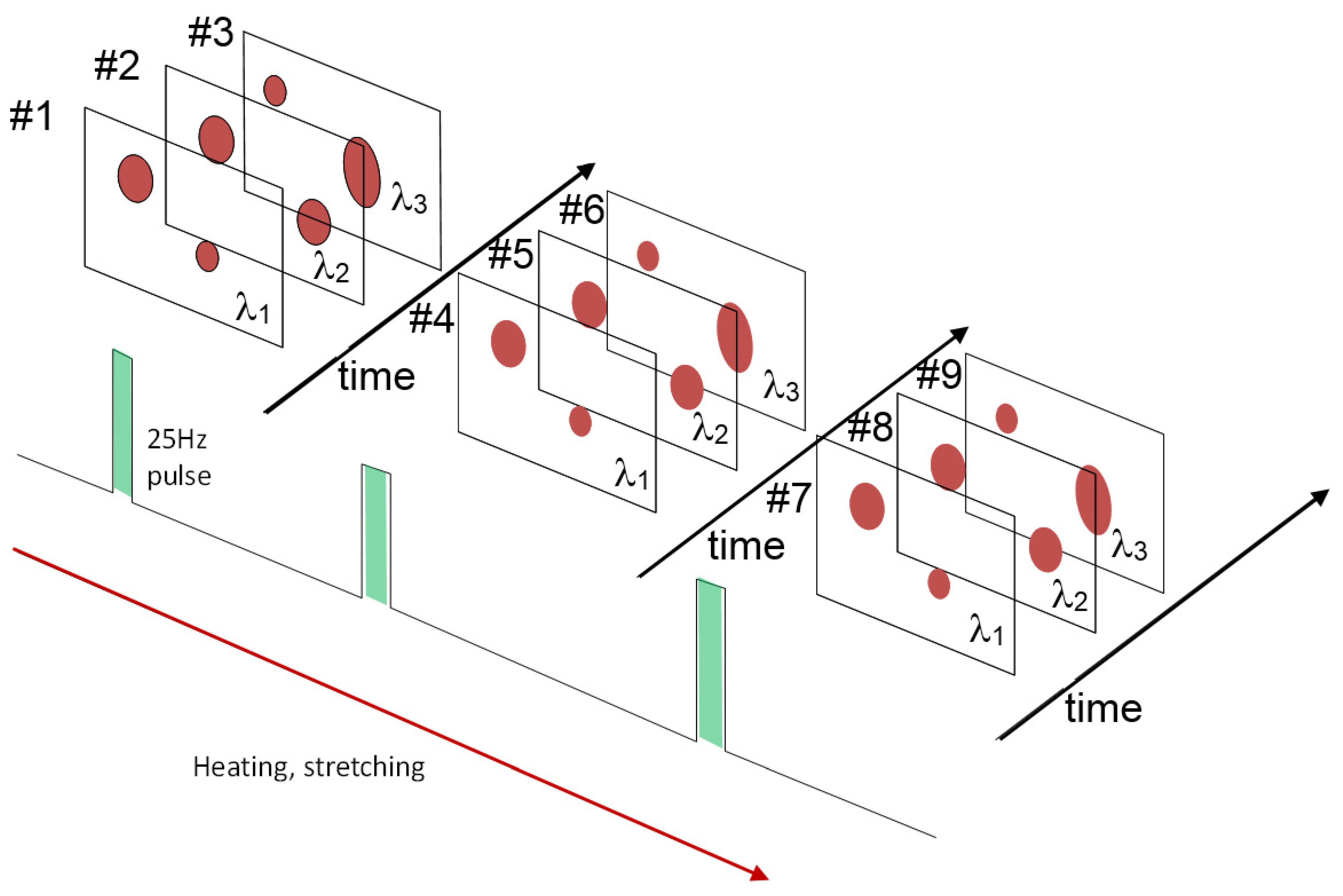
2.2.2. Neutron Pulses
2.2.3. Sample Preparations for WAND Experiments
3. Several Examples of X-ray Crystal Structure Analysis of Polymers
3.1. PLLA α Form
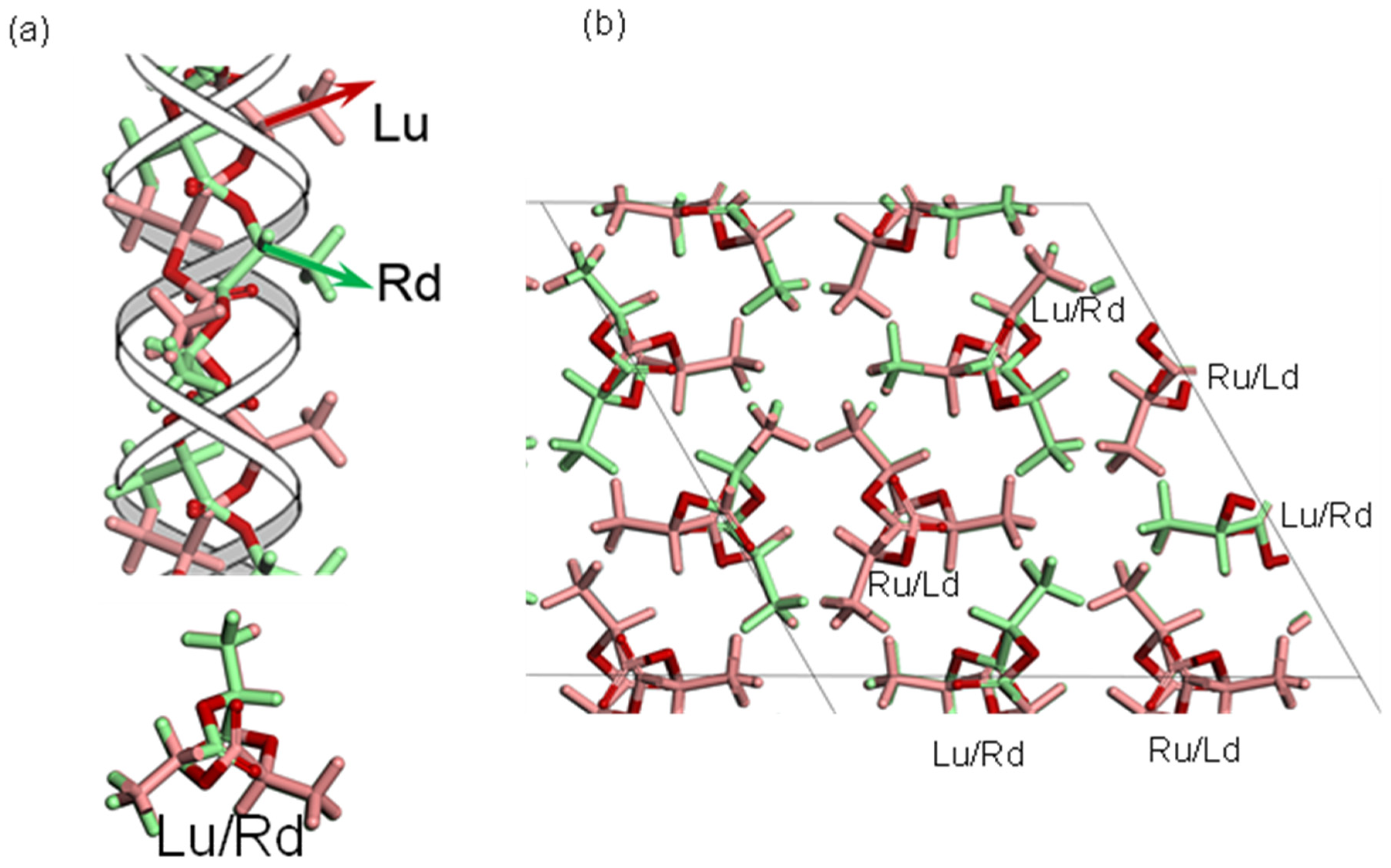
3.2. PLLA/PDLA Stereocomplex
3.3. Poly(3-Hydroxybutyrate)
4. Crystal Structures of Synthetic Polymers by WAND Data Analyses
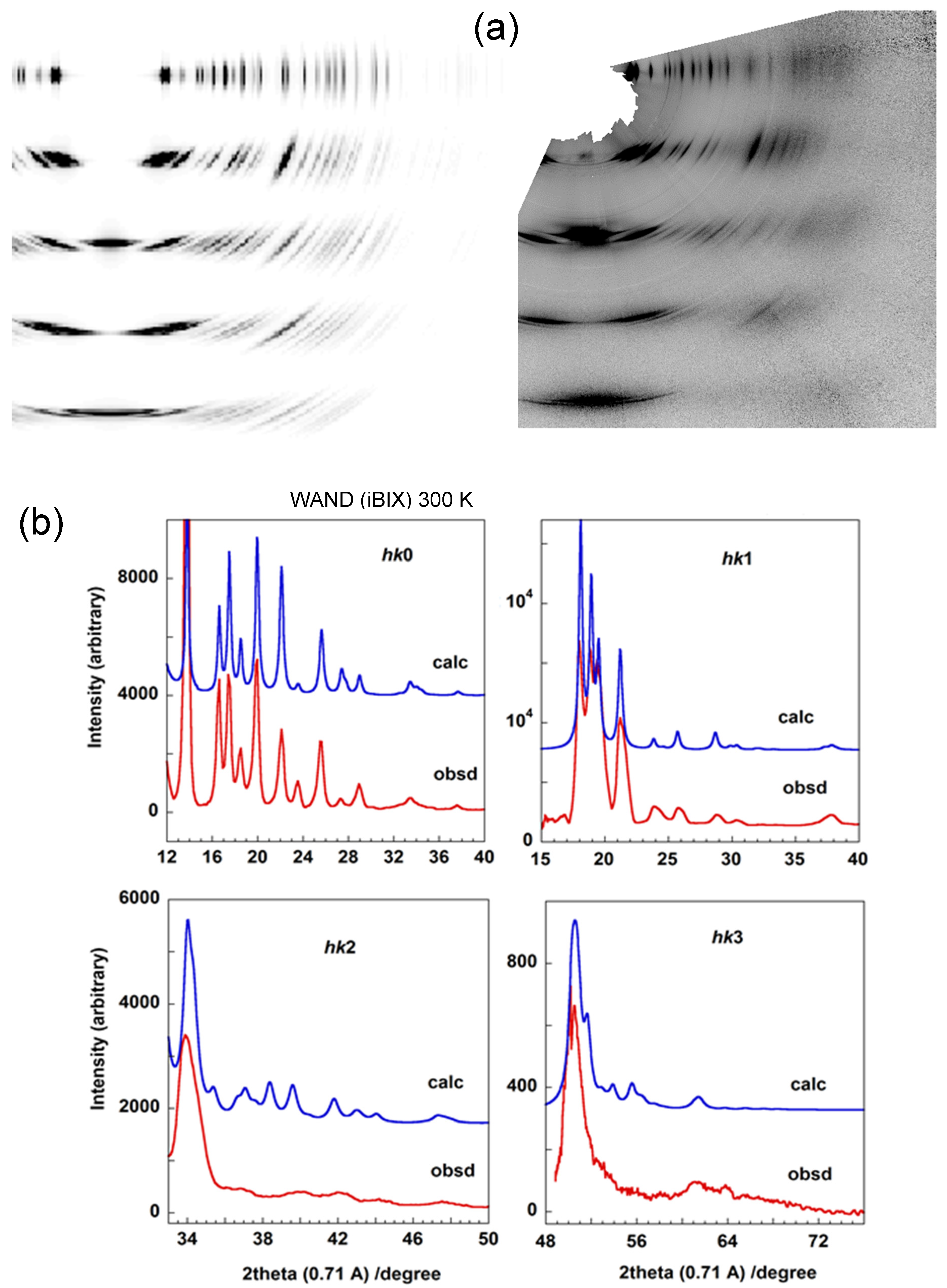
4.1. Orthorhombic Polyethylene
4.2. Trigonal Polyoxymethylene
4.2.1. Crystal Structure of POM
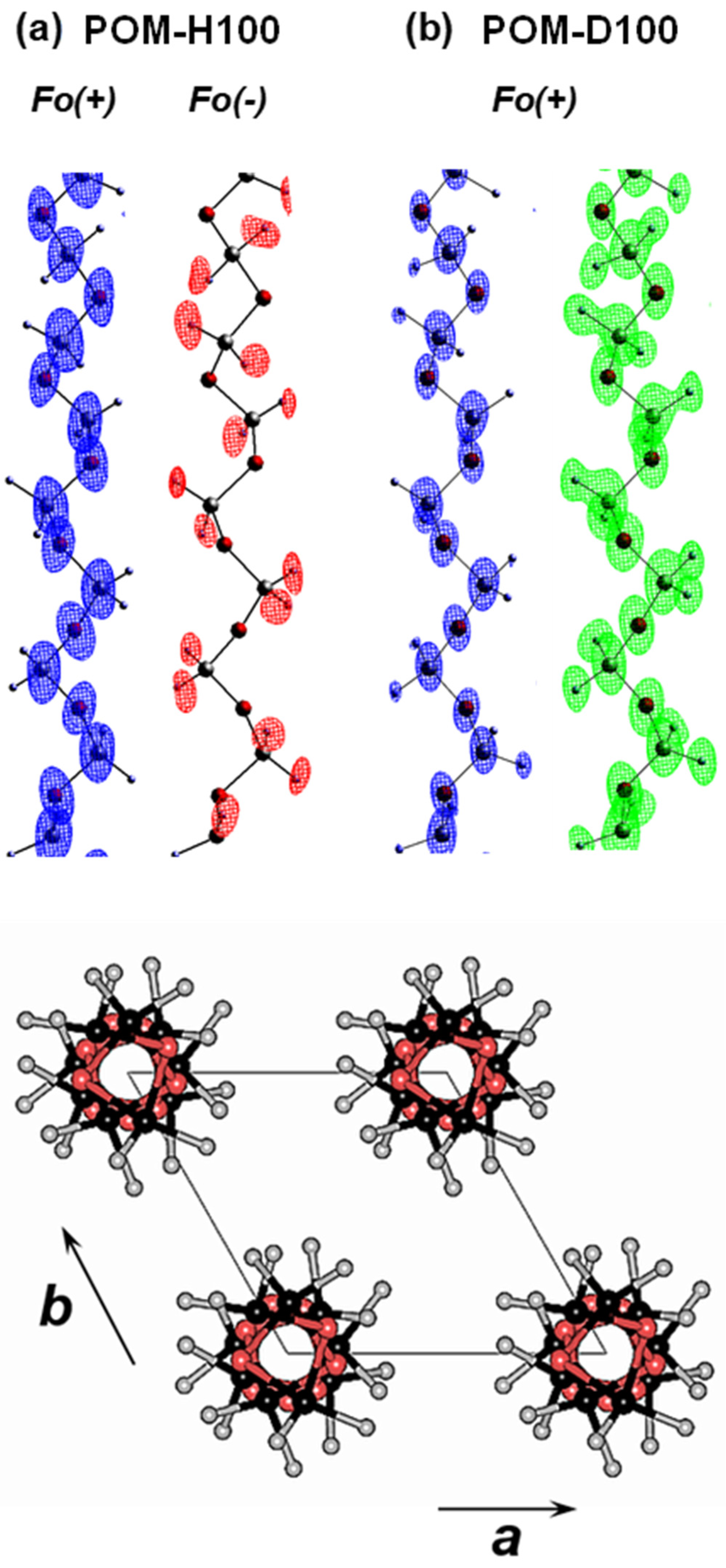
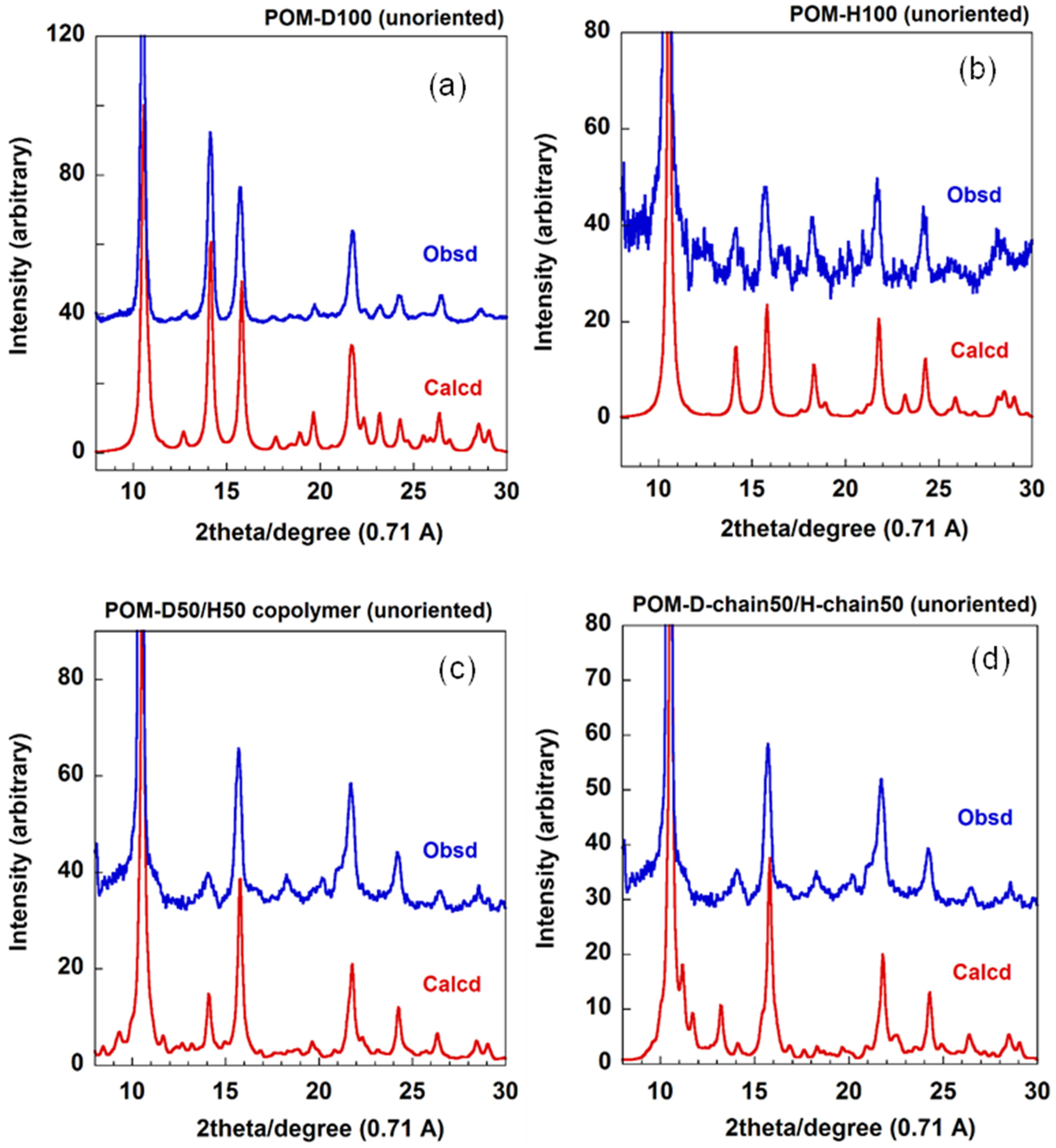
4.2.2. D/H Random Copolymers
5. Dilemma between the WAXD and WAND Analyses
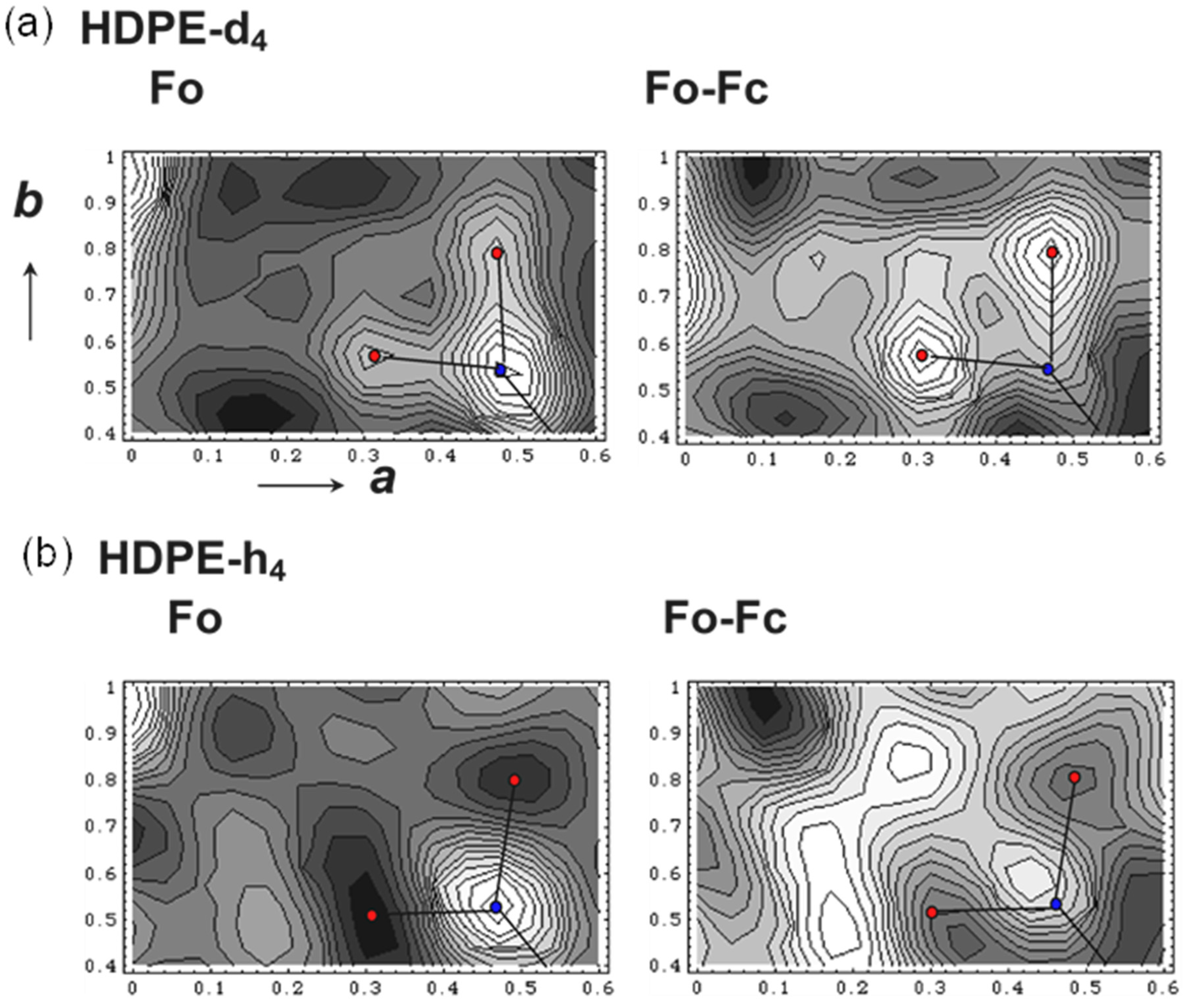
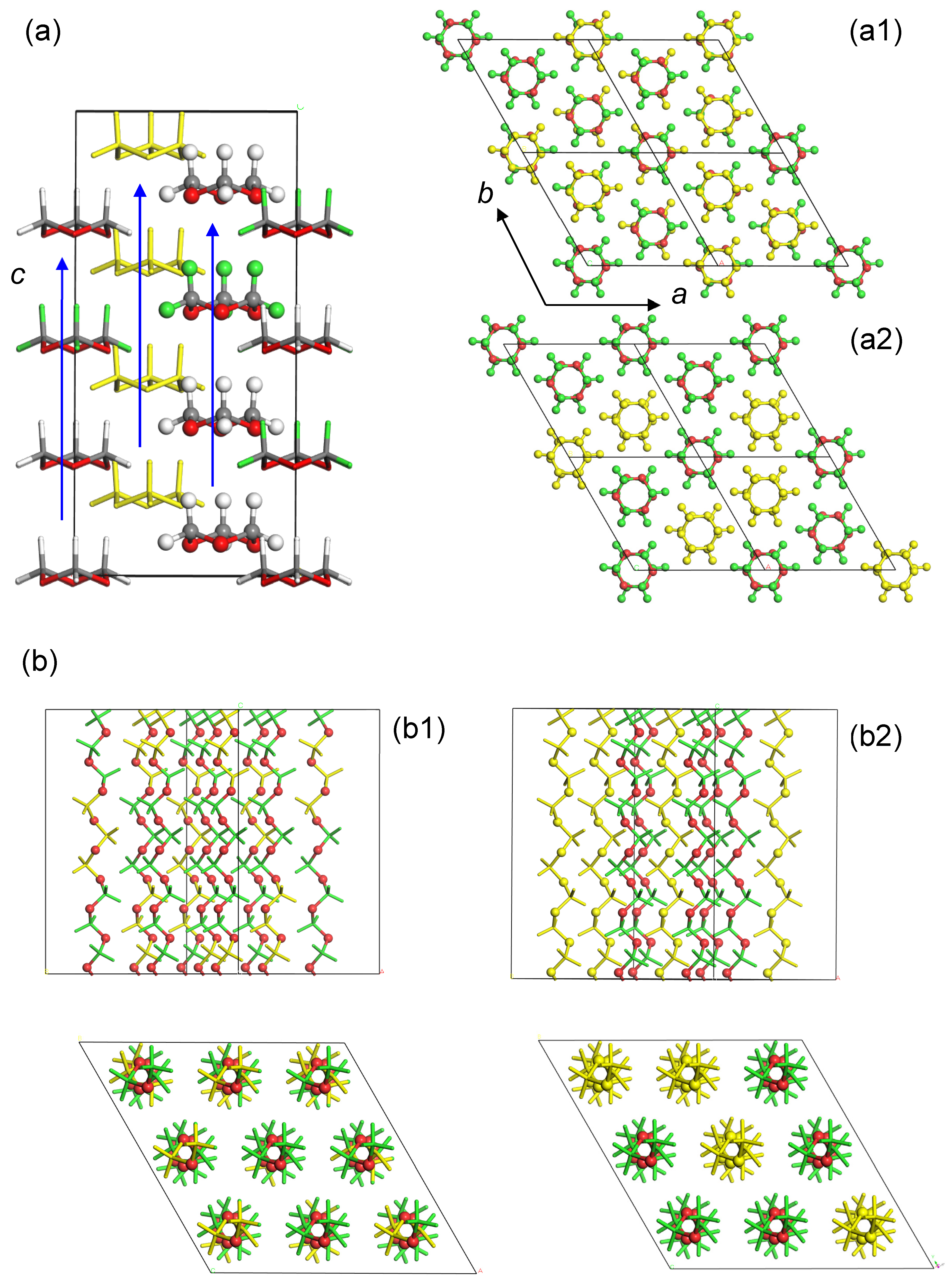
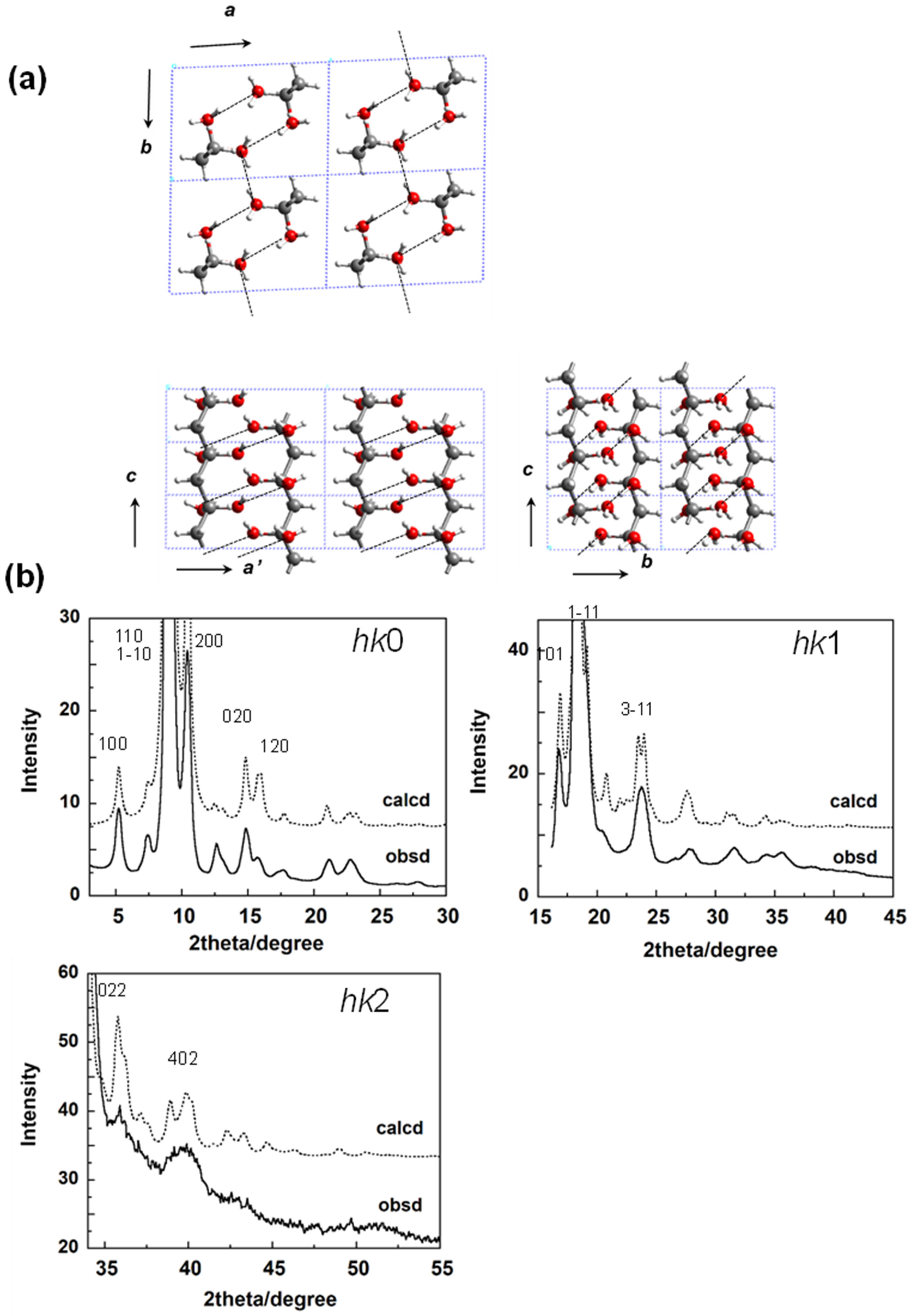
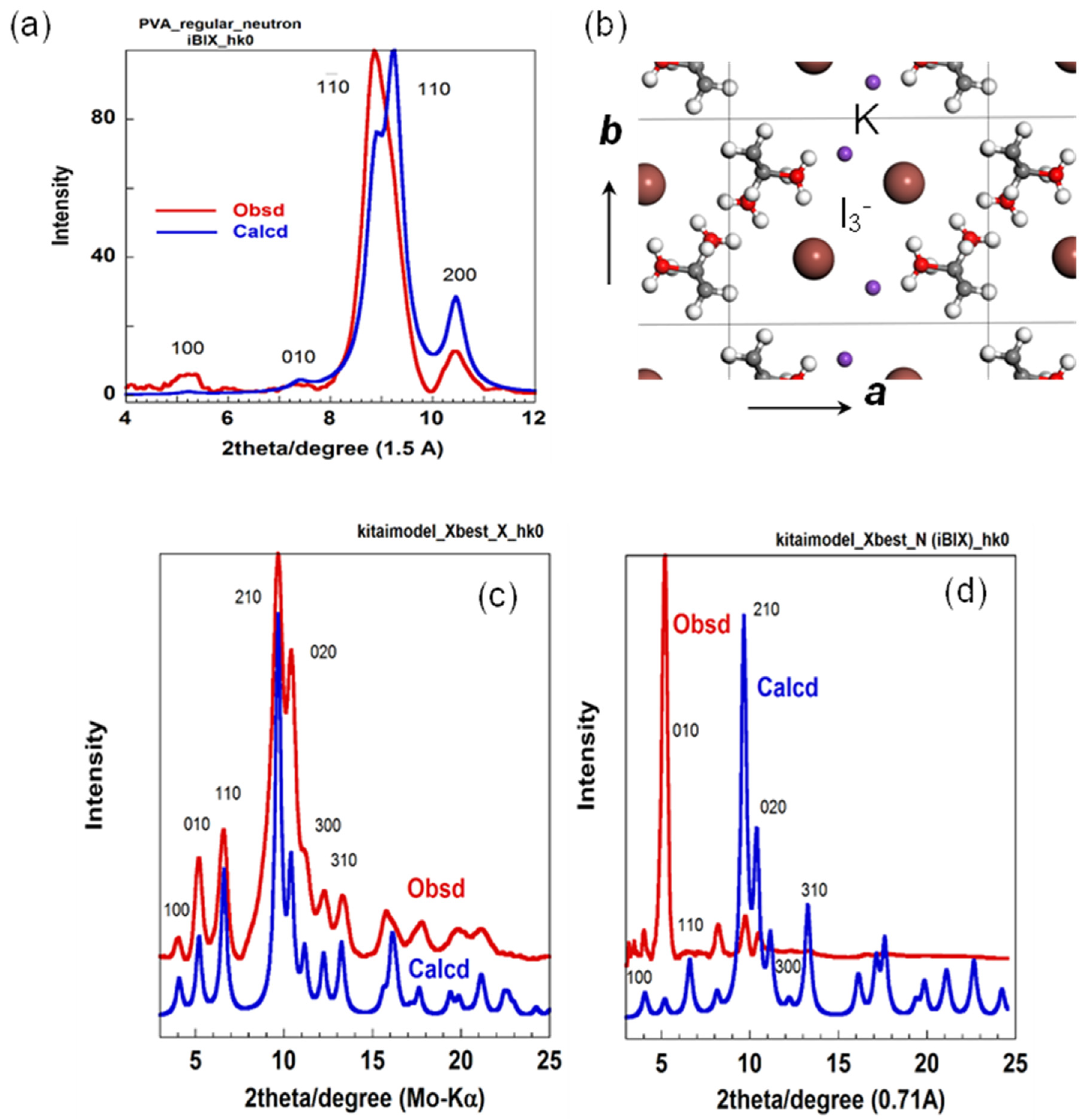
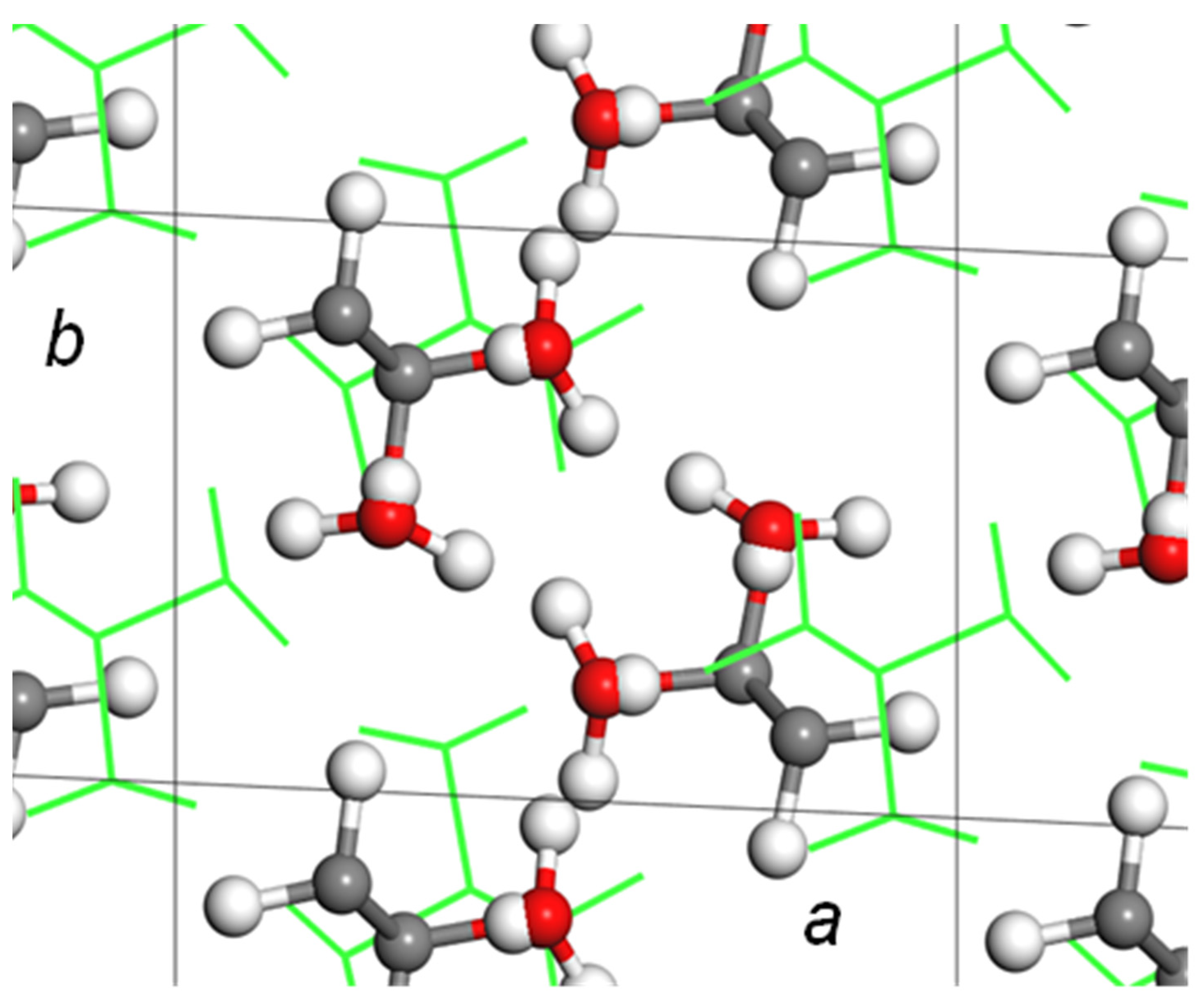
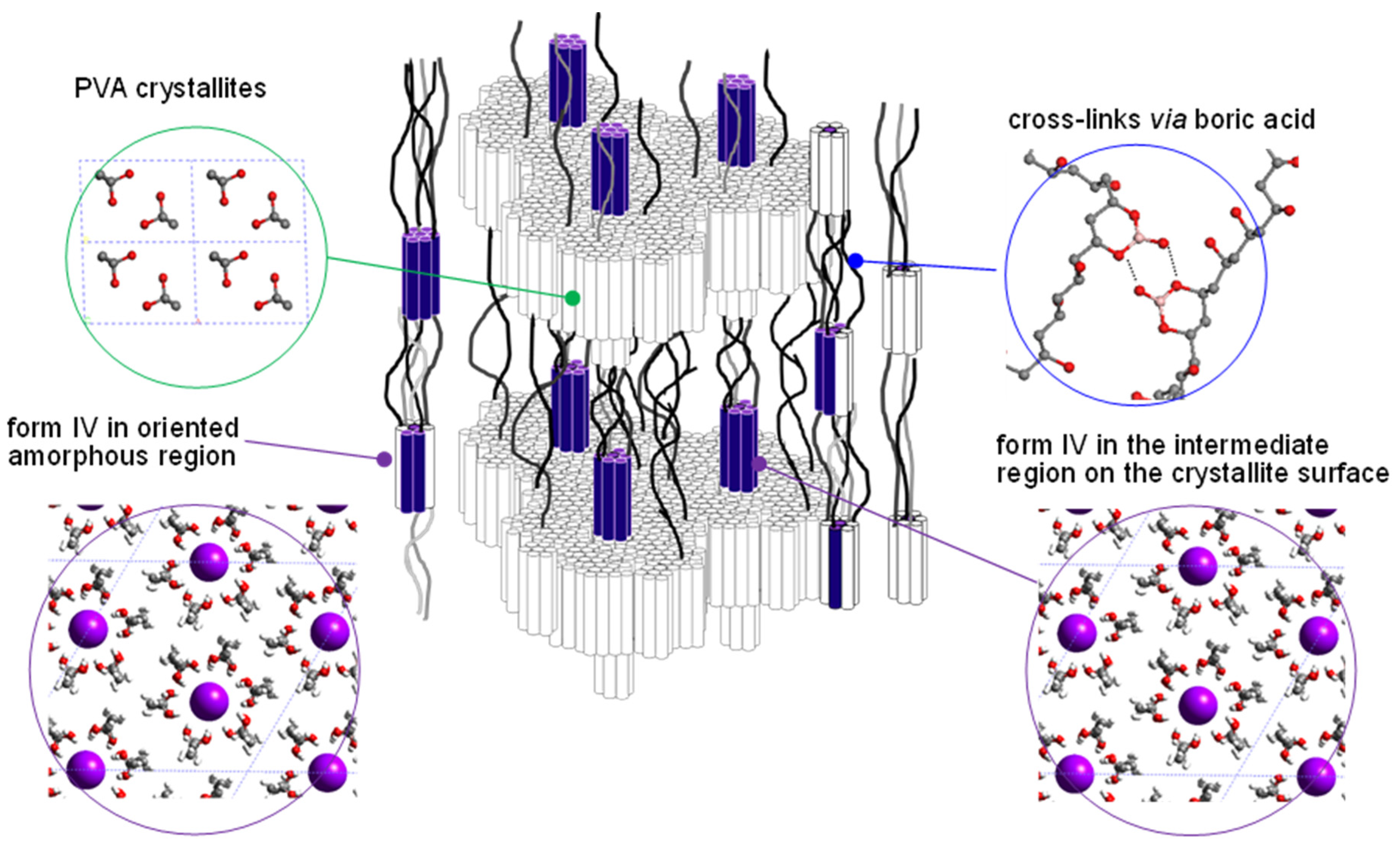
5.1. Crystal Structure of at-PVA
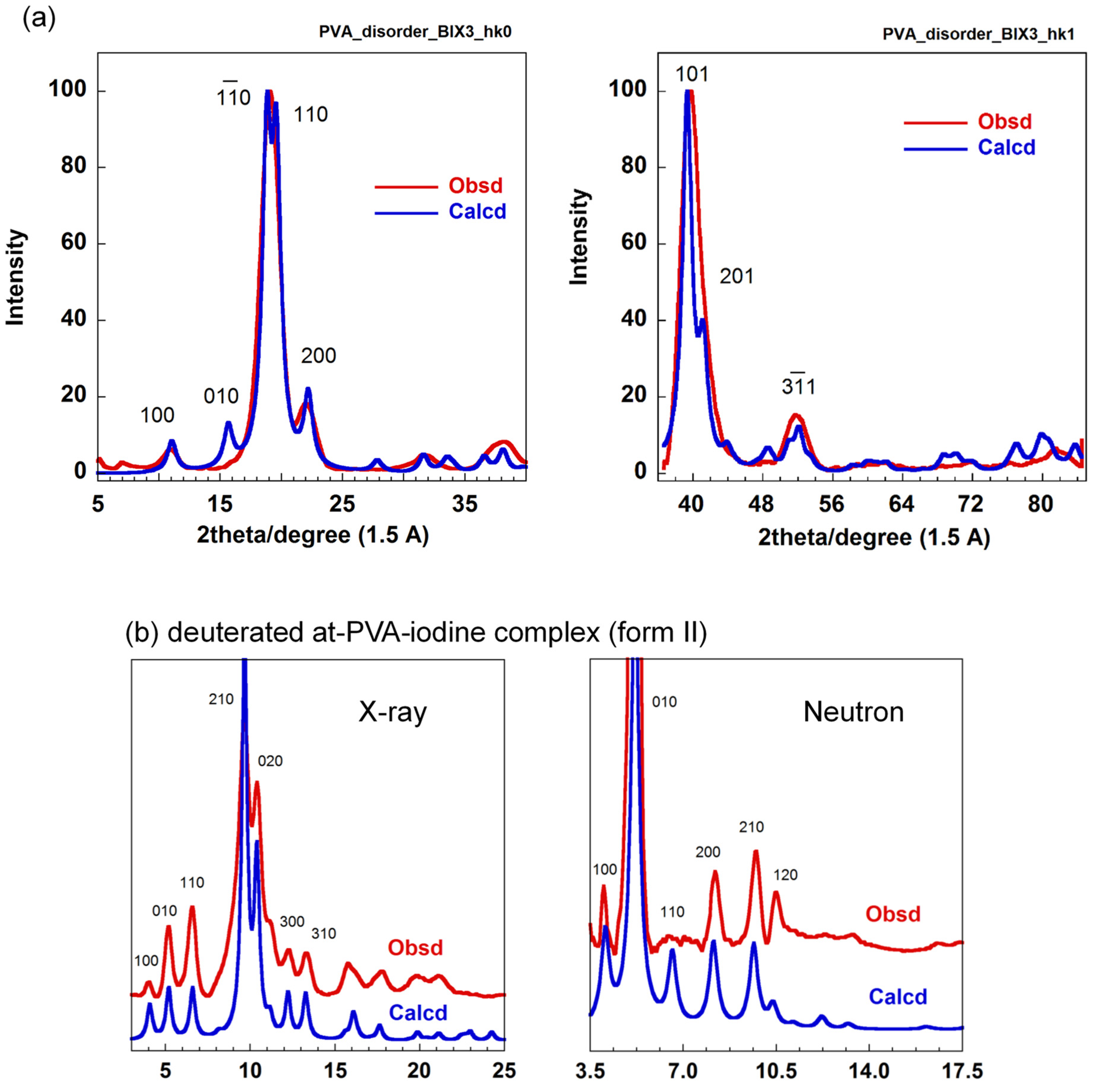
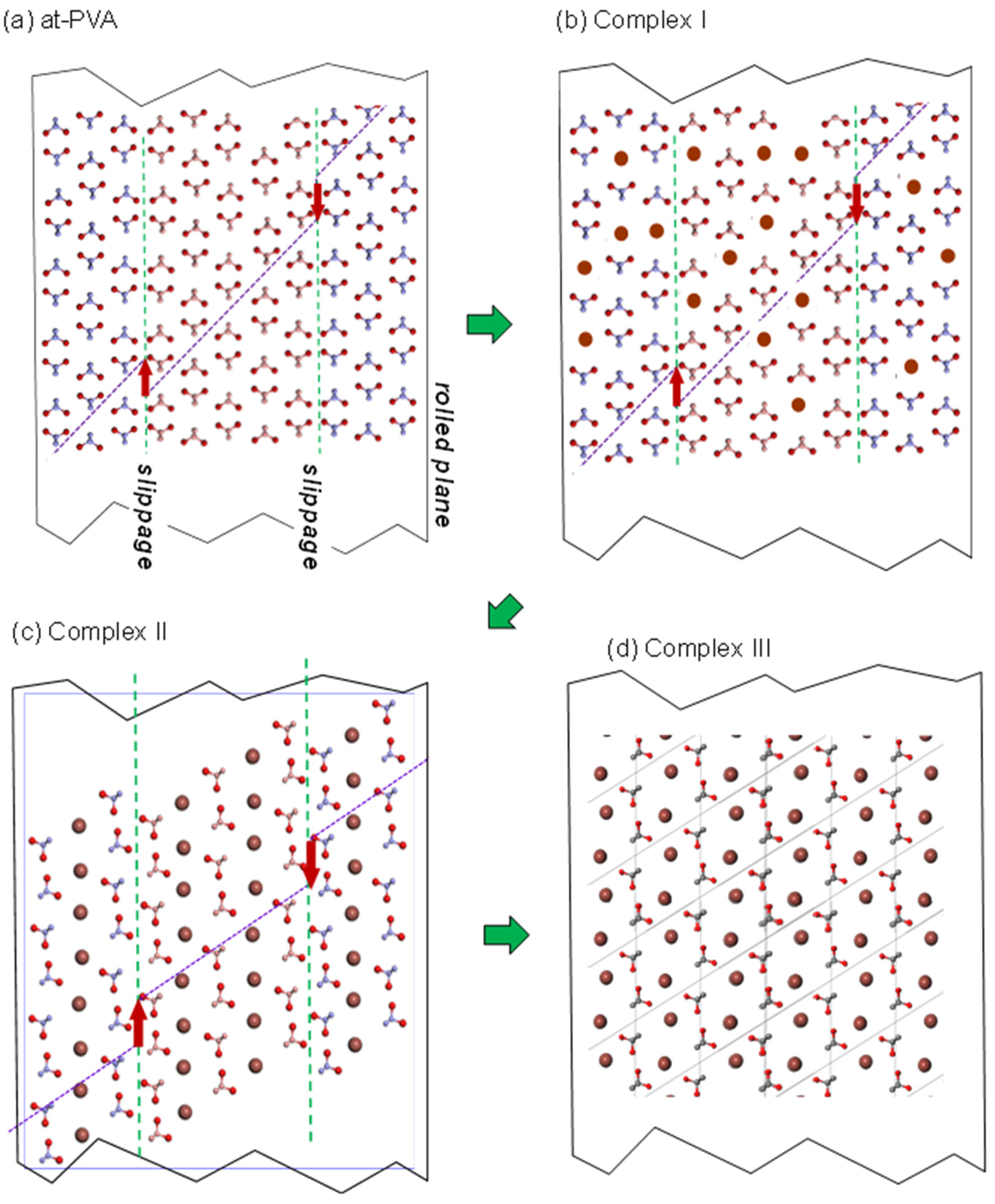
5.2. Crystal Structure of at-PVA-Iodine Complex
5.3. The Combination of WAXD and WAND Data Analysis
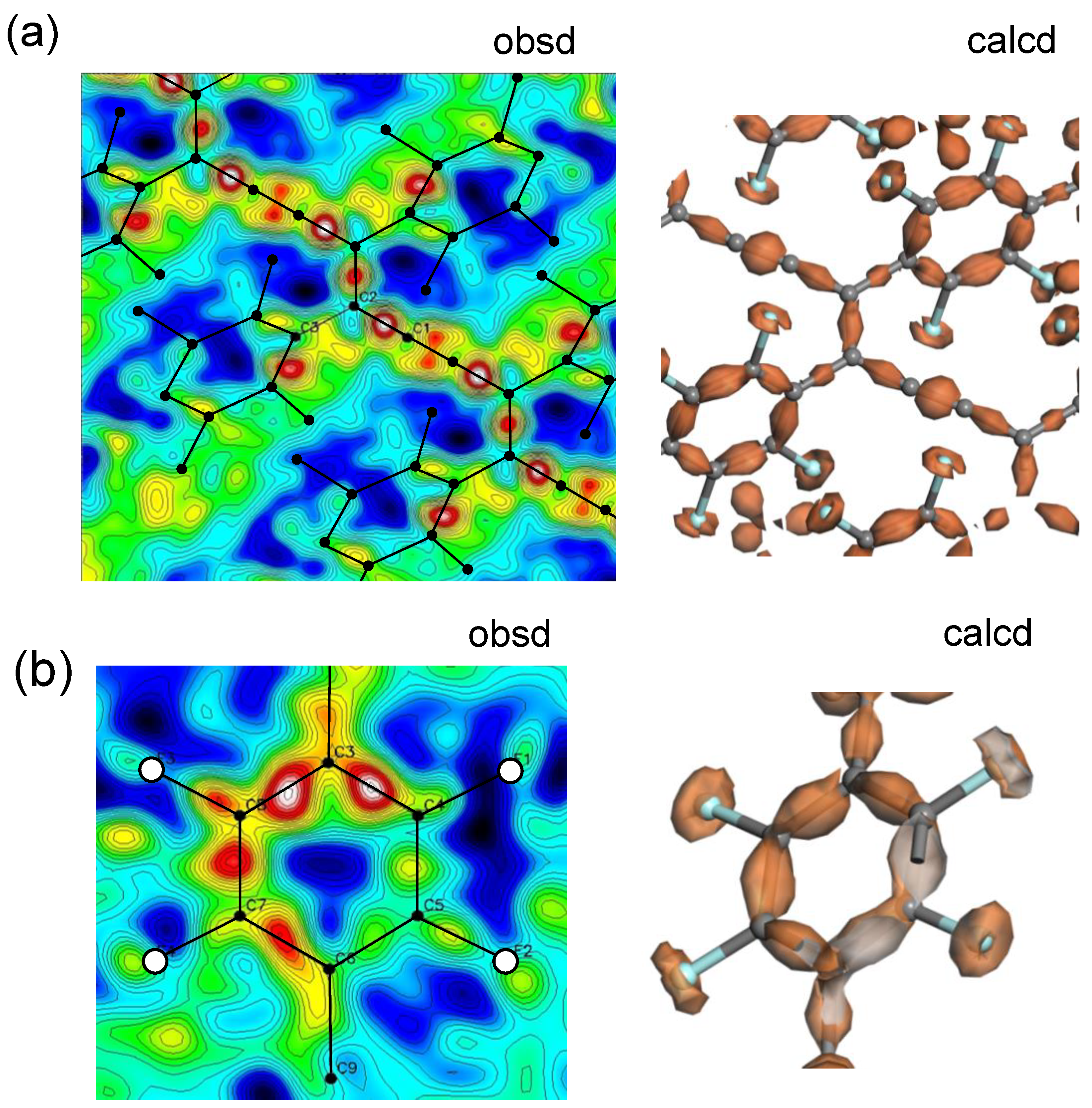
6. Distribution of the Bonded Electron Density along the Polymer Chain (X-N Method)
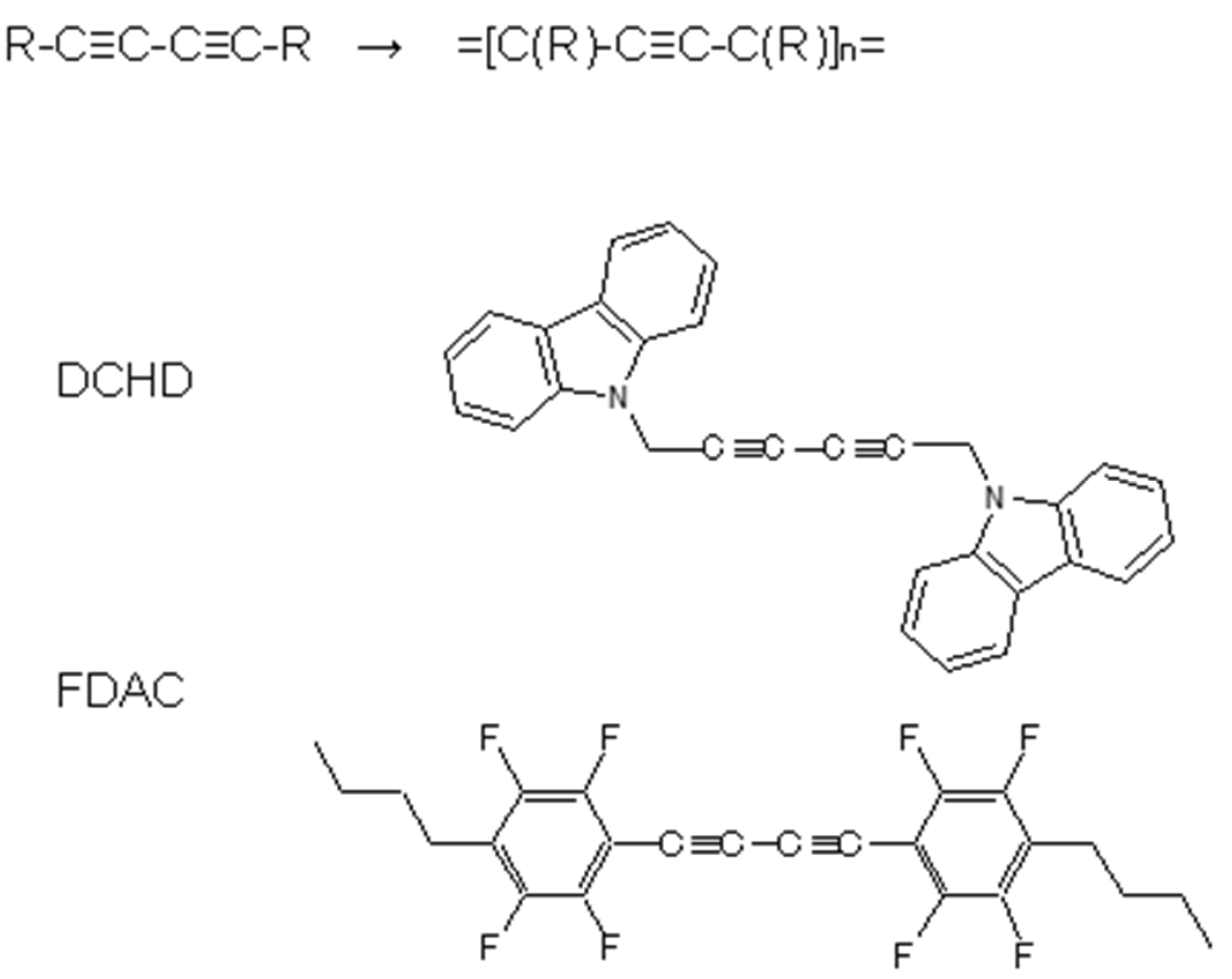
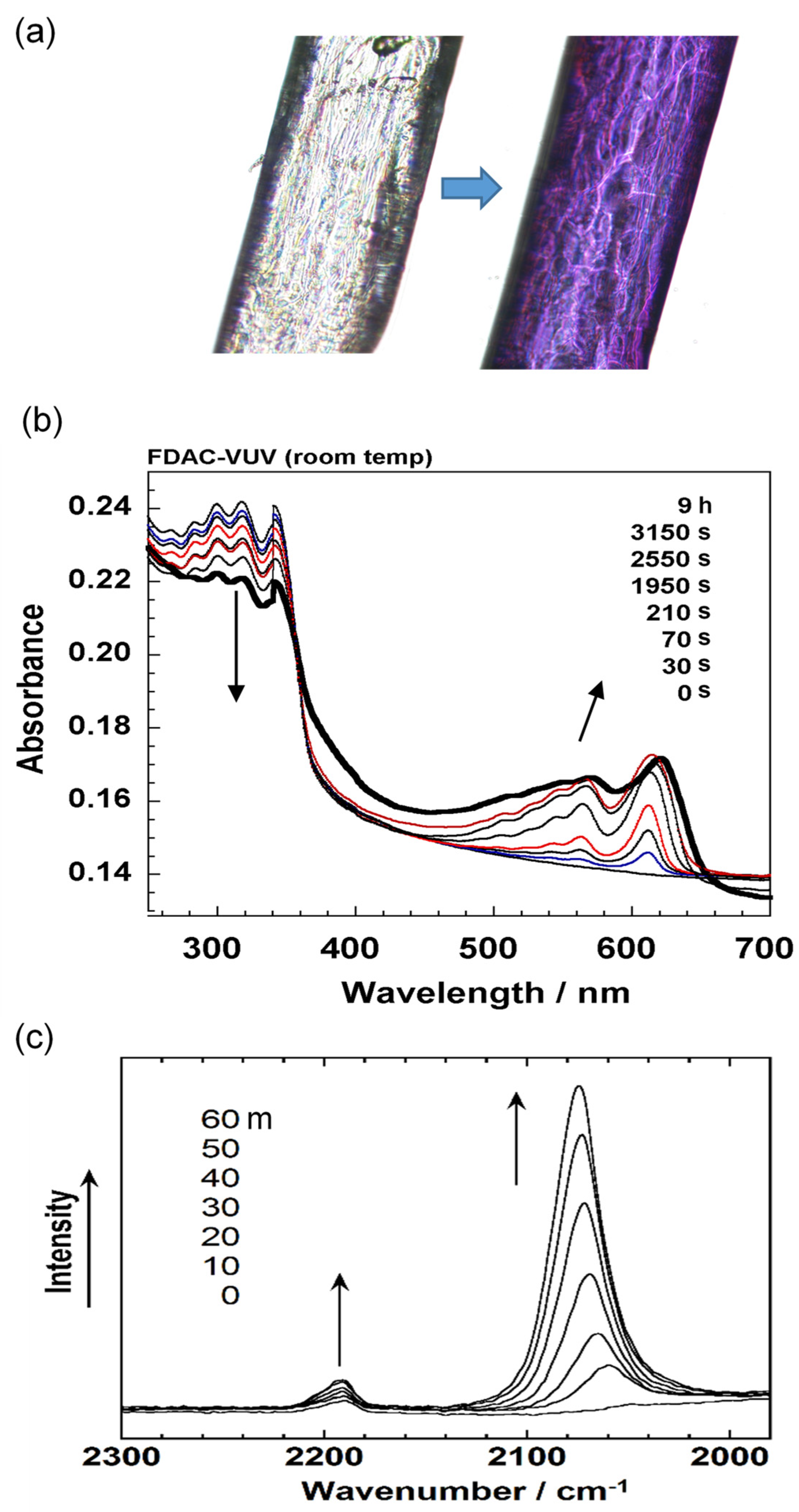
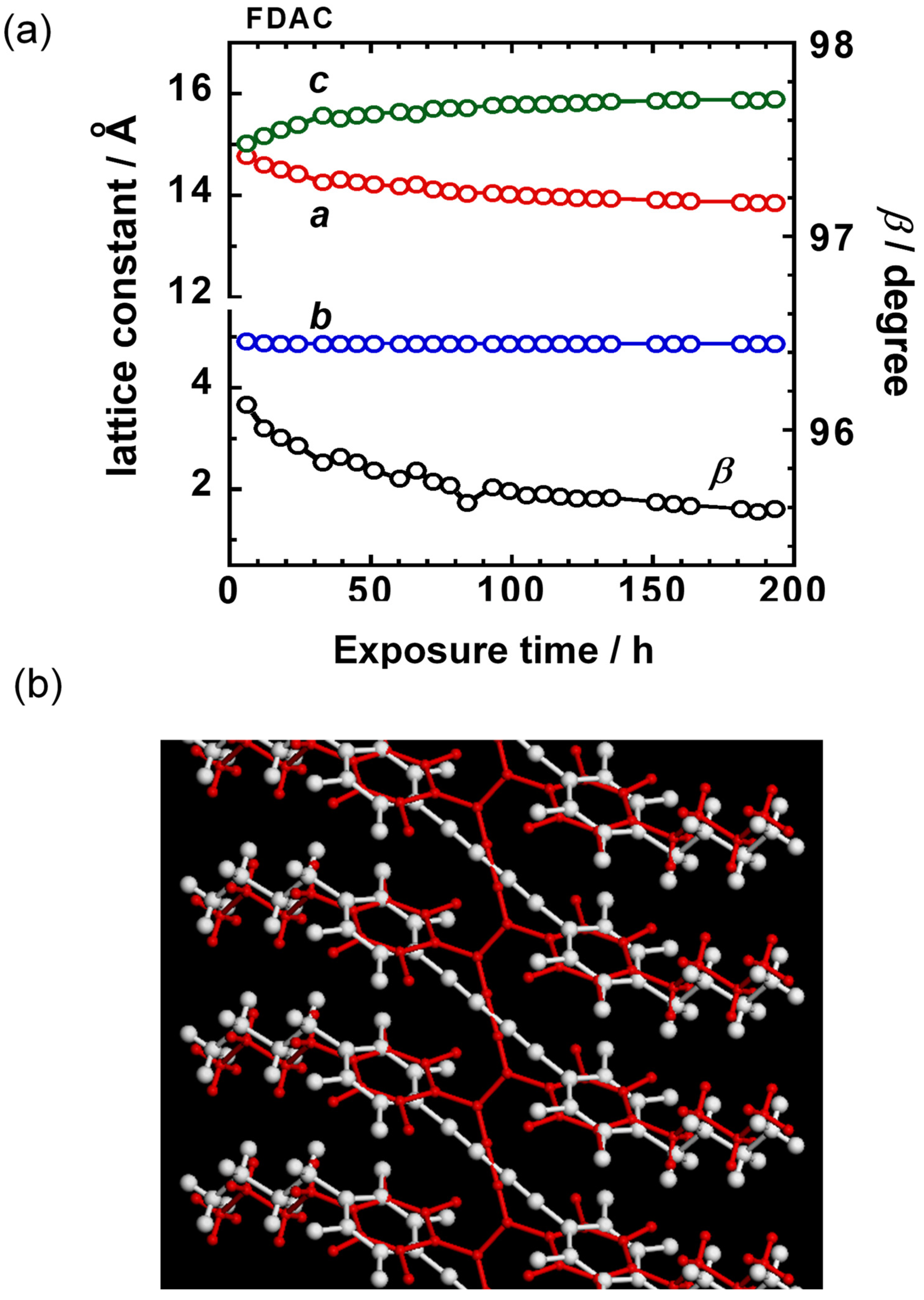
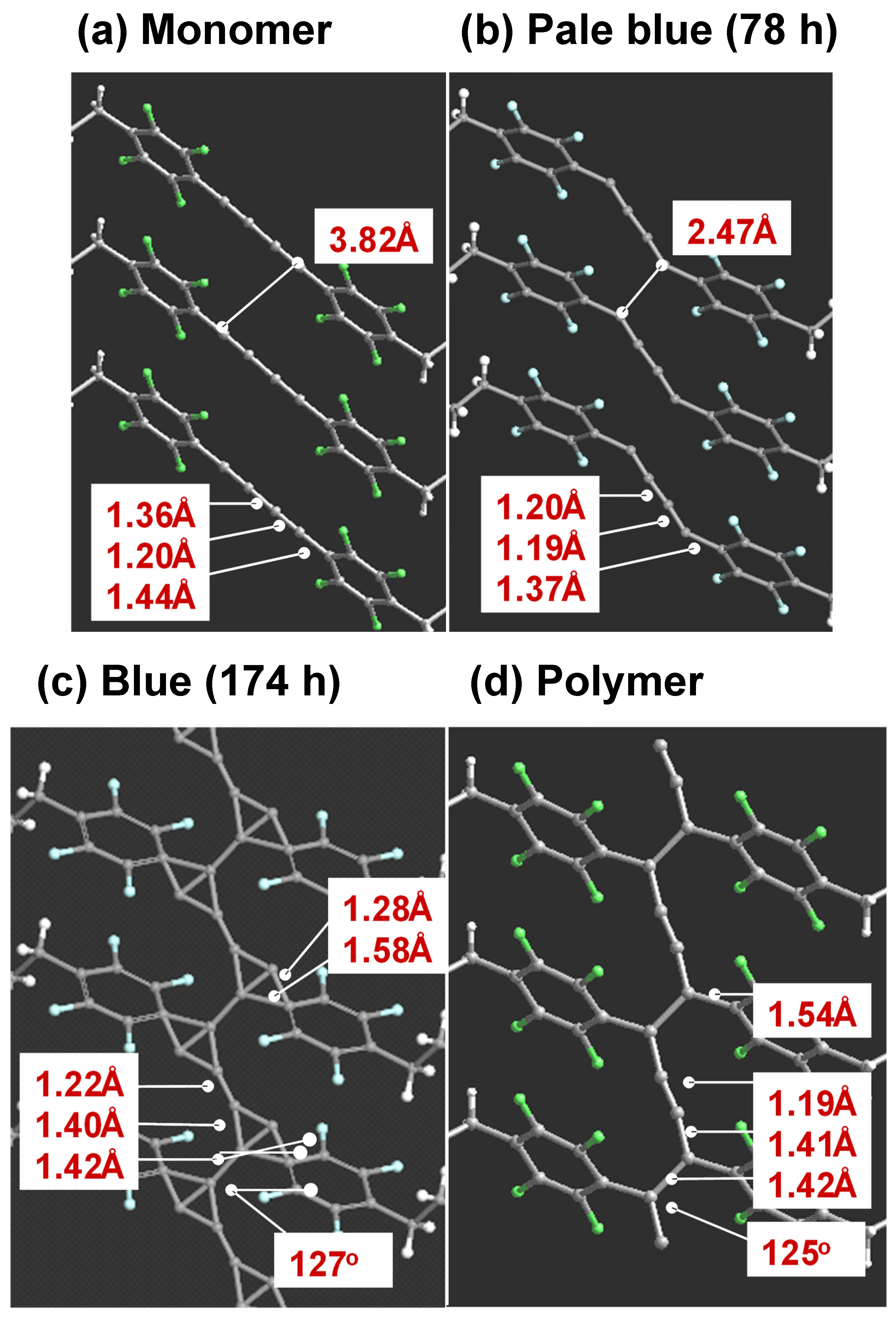
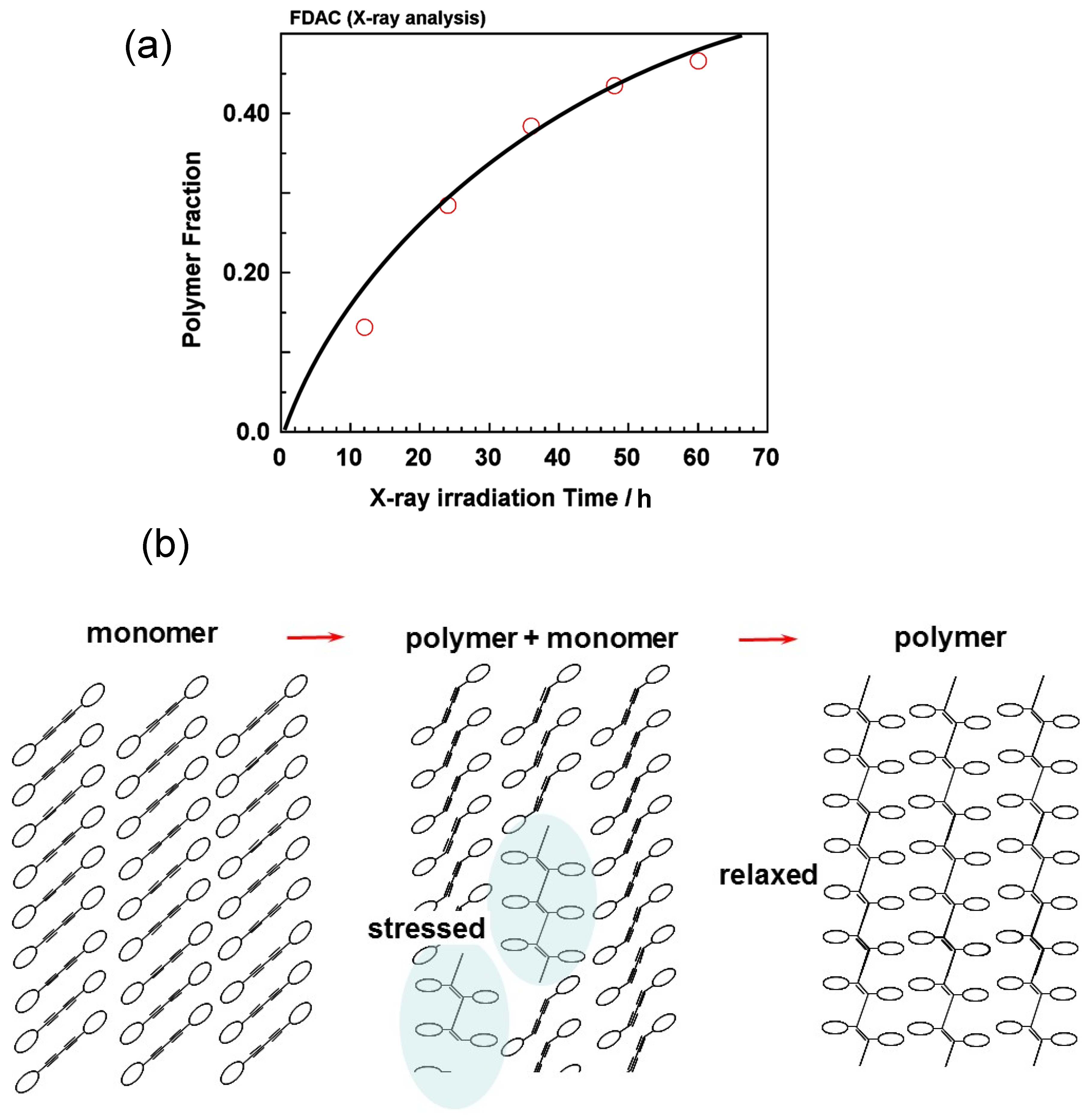
6.1. Photoinduced Solid-State Polymerization of FDAC
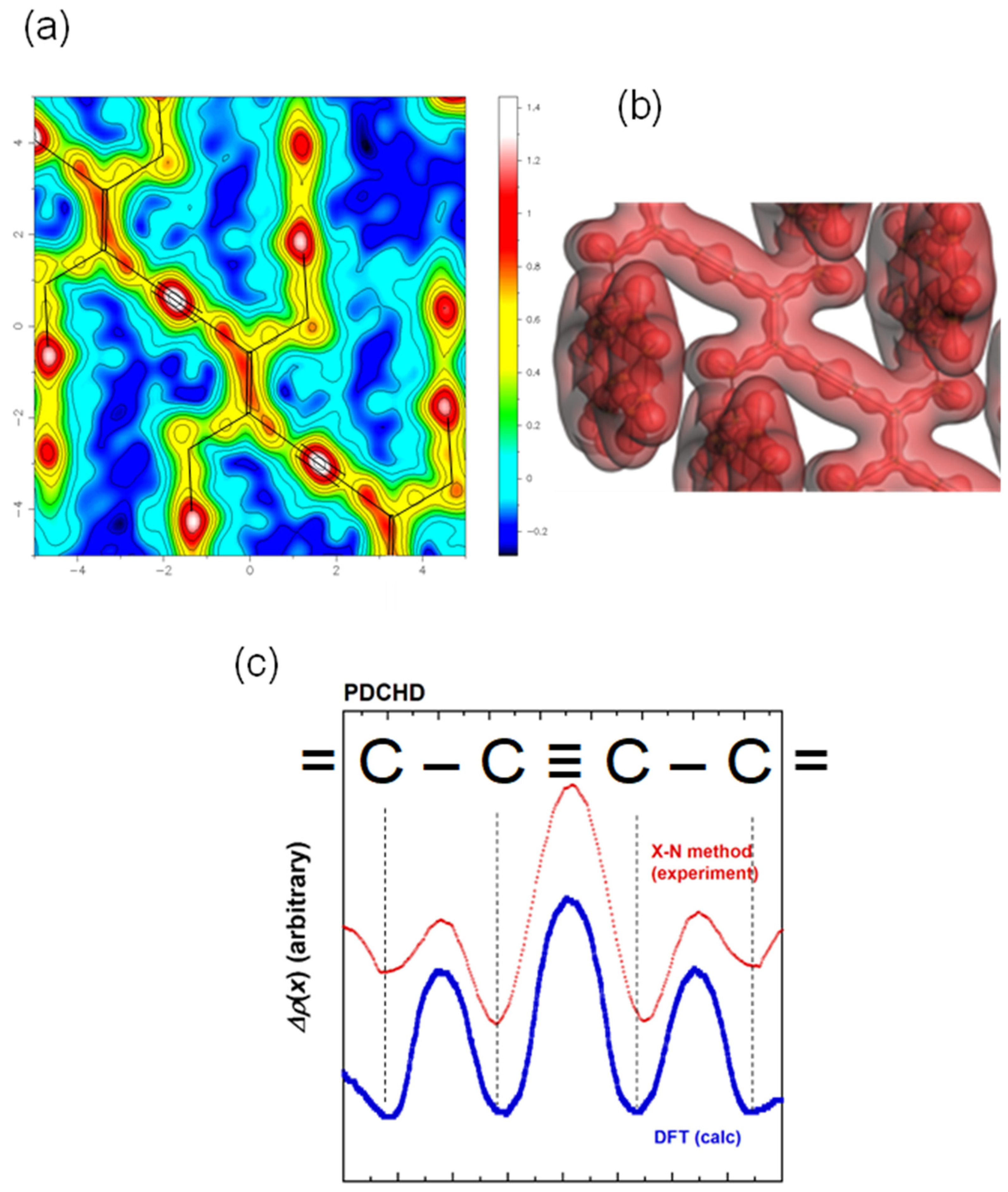
6.2. Bonded Electron Density Distribution along the Polymer Chain
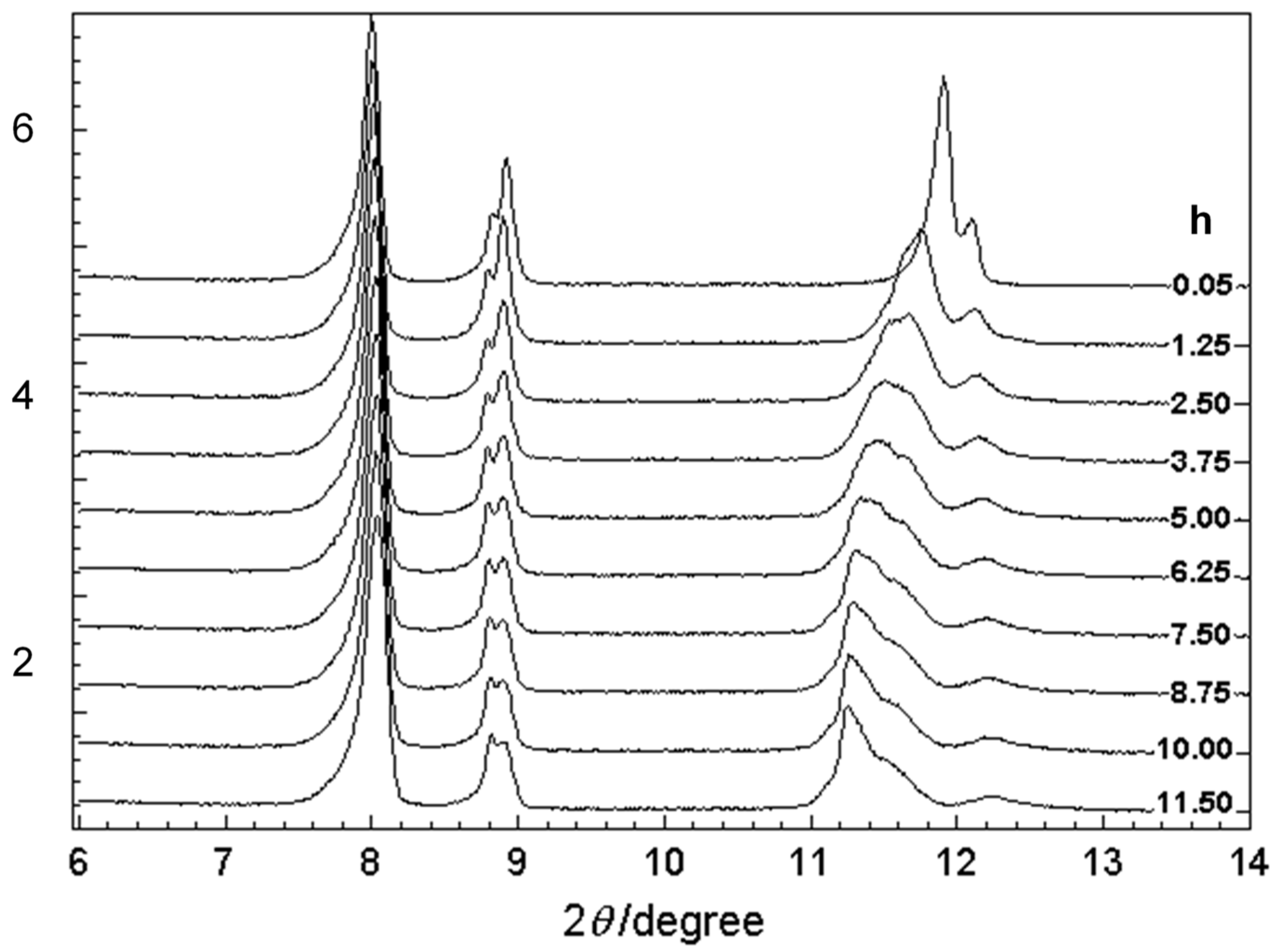
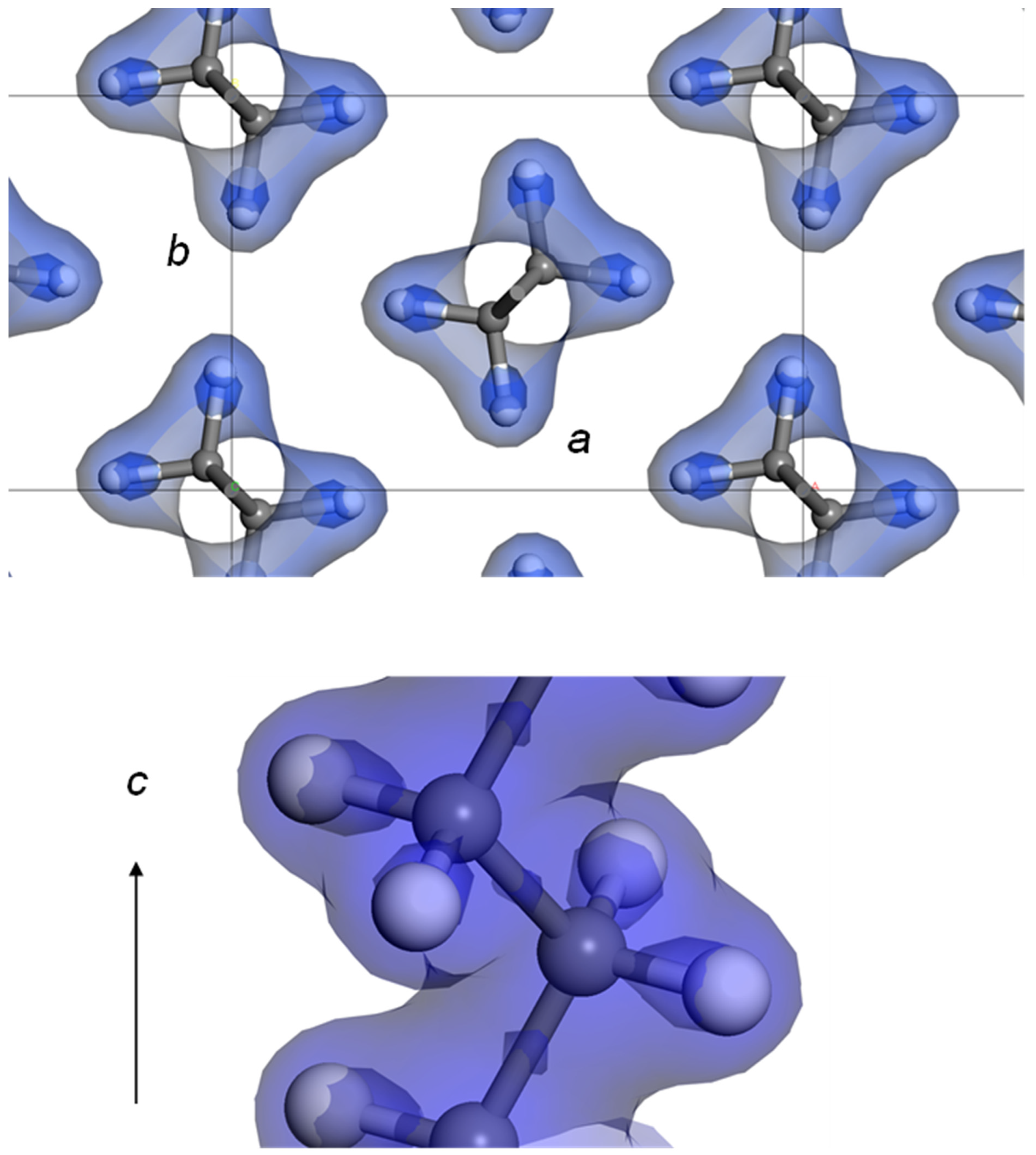
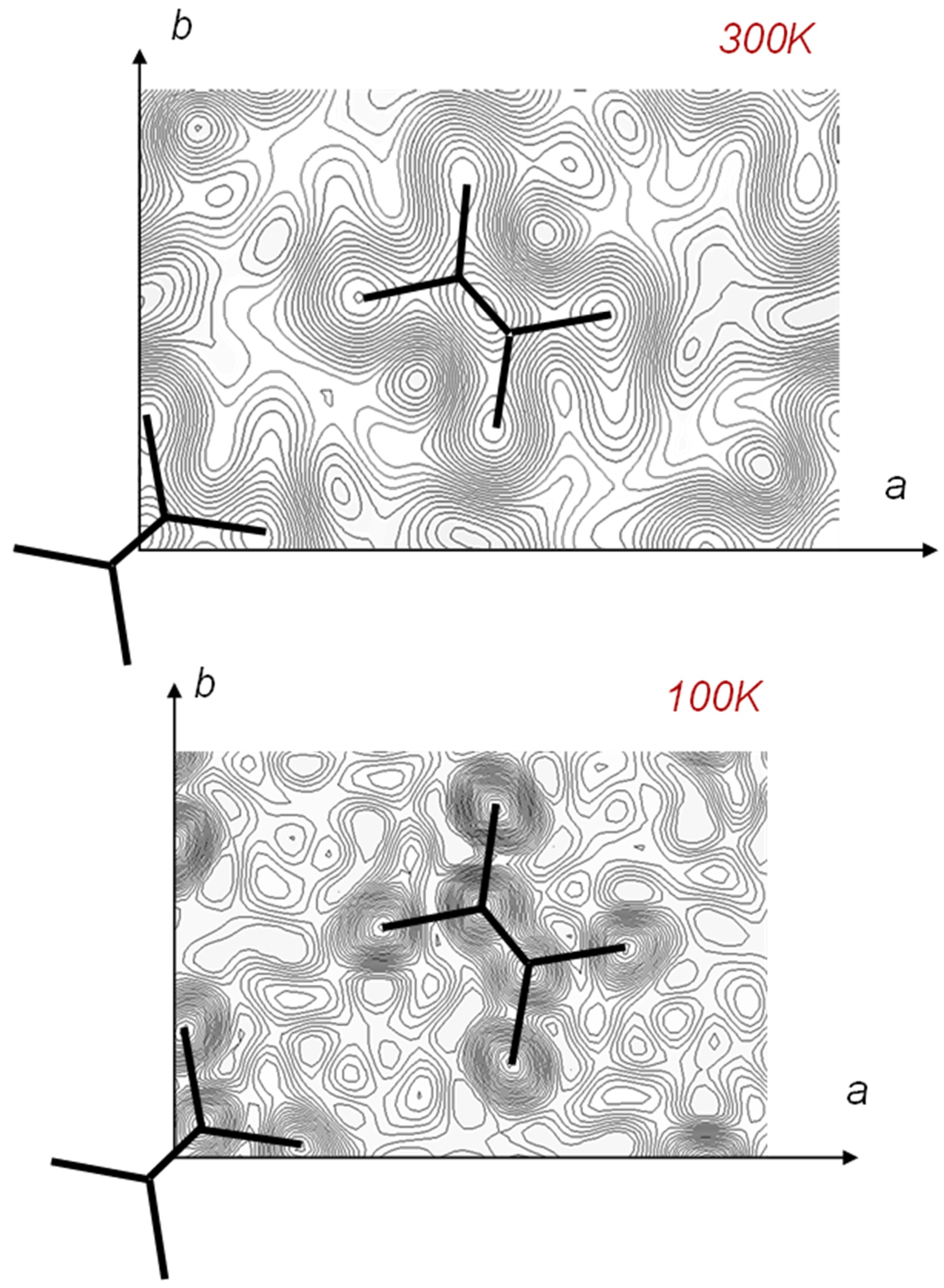
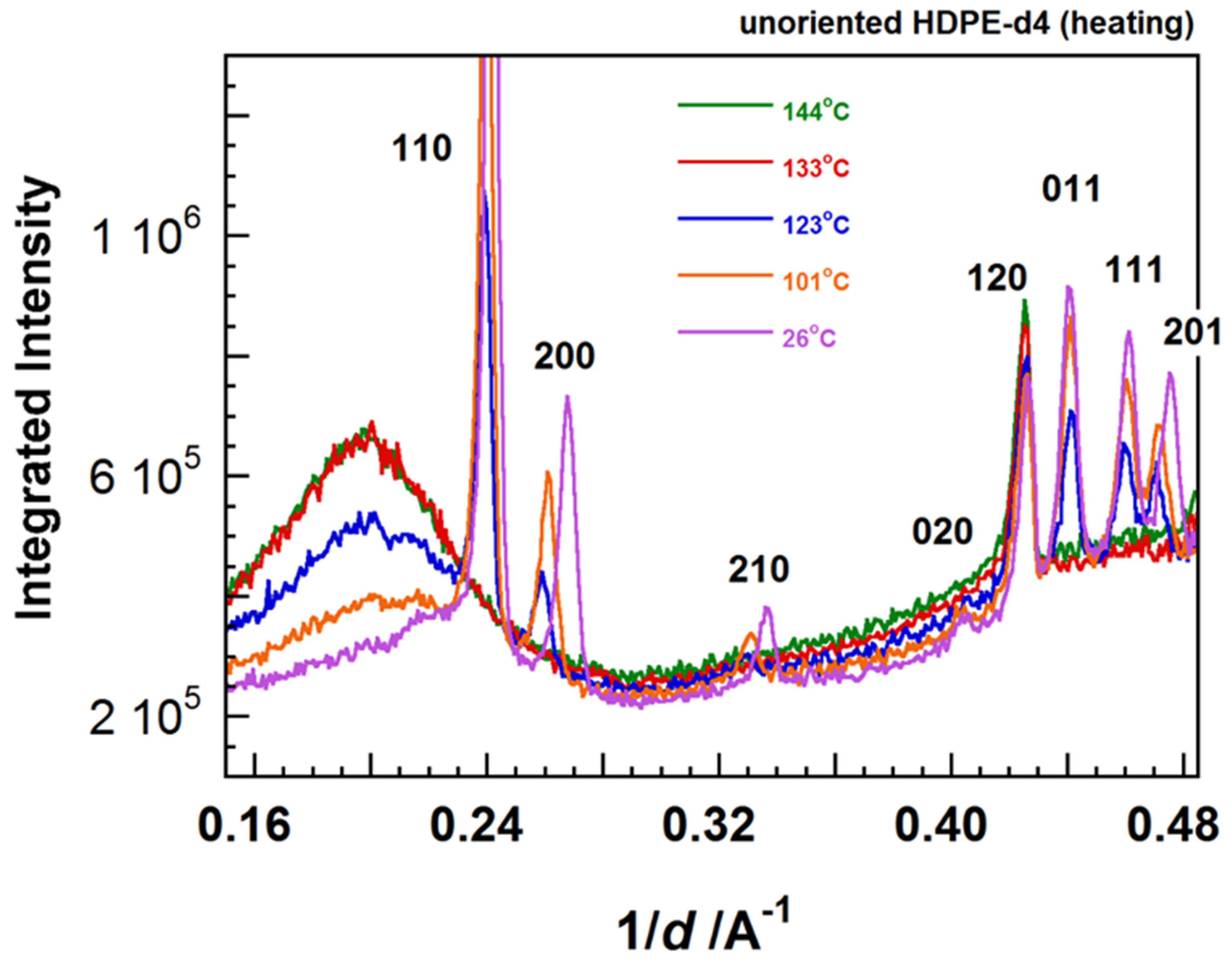
7. Temperature-Dependent WAND Measurements
7.1. Crystal Structure of Orthorhombic Polyethylene at Low Temperature
7.2. Temperature Dependence of WAND Data in the Heating Process
8. WAND Measurements under the Tensile Force
9. Summary and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, L. X-ray Diffraction Methods in Polymer Science; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1969. [Google Scholar] [CrossRef]
- Tadokoro, H. Structure of Crystalline Polymers; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1990. [Google Scholar]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Natta, G.; Corradini, P. Kristallstruktur des isotaktischen polystyrols. Makromol. Chem. 1955, 16, 77–80. [Google Scholar] [CrossRef]
- Tashiro, K. Structural Science of Crystalline Polymers; Springer Nature: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Lengwan, L.; Jakowski, J.; Do, C.; Hong, K. Deuteration and Polymers: Rich History with Great Potential. Macromolecules 2021, 54, 3555–3584. [Google Scholar]
- Werner, K. Forty years in cryoEM of membrane proteins. Microscopy 2022, 71 (Suppl. 1), i30–i50. [Google Scholar]
- Dorset, D.L. Structural Electron Crystallography; Plenum Press: New York, NY, USA, 1995. [Google Scholar] [CrossRef]
- Tashiro, K.; Hanesaka, M.; Ozeki, T. Structure Analysis of Polymer Crystal Based on Wide-Angle X-ray Diffraction Data by Utilizing High-Energy Synchrotron X-ray Beam. Polym. Prepr. Jpn. 2008, 57, 2S18. [Google Scholar]
- Available online: https://www.jaea.go.jp (accessed on 24 November 2022).
- Niimura, N.; Karasawa, Y.; Tanaka, I.; Miyahara, J.; Akahashi, K.; Saito, H.; Koizumi, S.; Hidaka, M. An imaging plate neutron detector. Nucl. Instr. Methods Phys. Res. 1994, A349, 521–525. [Google Scholar] [CrossRef]
- Wasanasuk, K.; Tashiro, K.; Hanesaka, M.; Ohhara, T.; Kurihara, K.; Kuroki, R.; Tamada, T.; Ozeki, T.; Kanamoto, T. Crystal Structure Analysis of Poly(l-lactic Acid) α Form On the basis of the 2-Dimensional Wide-Angle Synchrotron X-ray and Neutron Diffraction Measurements. Macromolecules 2011, 44, 6441–6452. [Google Scholar] [CrossRef]
- Wasanasuk, K.; Tashiro, K. Theoretical and Experimental Evaluation of Crystallite Moduli of Various Crystalline Forms of Poly(l-lactic acid). Macromolecules 2012, 45, 7019–7026. [Google Scholar] [CrossRef]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyon, S.H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Tashiro, K.; Kouno, N.; Wang, H.; Tsuji, H. Crystal Structure of Poly(lactic acid) Stereocomplex: Random Packing Model of PDLA and PLLA Chains As Studied by X-ray Diffraction Analysis. Macromolecules 2017, 50, 8048–8065. [Google Scholar] [CrossRef]
- Cartier, L.; Okihara, T.; Lotz, B. Triangular Polymer Single Crystals: Stereocomplexes, Twins, and Frustrated Structures. Macromolecules 1997, 30, 6313–6322. [Google Scholar] [CrossRef]
- Tashiro, K.; Wang, H.; Kouno, N.; Koshobu, J.; Watanabe, K. Confirmation of the X-ray-Analyzed Heterogeneous Distribution of the PDLA and PLLA Chain Stems in the Crystal Lattice of Poly(lactic acid) Stereocomplex on the Basis of the Vibrational Circular Dichroism IR Spectral Measurement. Macromolecules 2017, 50, 8066–8071. [Google Scholar] [CrossRef]
- Iwata, T.; Aoyagi, Y.; Tanaka, T.; Fujita, M.; Takeuchi, A.; Suzuki, Y.; Uesugi, K. Microbeam X-ray Diffraction and Enzymatic Degradation of Poly[(R)-3-hydroxybutyrate] Fibers with Two Kinds of Molecular Conformations. Macromolecules 2006, 39, 5789–5795. [Google Scholar] [CrossRef]
- Yokouchi, M.; Chatani, Y.; Tadokoro, H.; Teranishi, K.; Tani, H. Structural studies of polyesters: 5. Molecular and crystal structures of optically active and racemic poly(β-hydroxybutyrate). Polymer 1973, 14, 267–272. [Google Scholar] [CrossRef]
- Wang, H.; Tashiro, K. Reinvestigation of Crystal Structure and Intermolecular Interactions of Biodegradable Poly(3-Hydroxybutyrate) α-Form and the Prediction of Its Mechanical Property. Macromolecules 2016, 49, 581–594. [Google Scholar] [CrossRef]
- Sato, H.; Murakami, R.; Padermshoke, A.; Hirose, F.; Senda, K.; Noda, I.; Ozaki, Y. Infrared Spectroscopy Studies of CHO Hydrogen Bondings and Thermal Behavior of Biodegradable Poly(hydroxyalkanoate). Macromolecules 2004, 37, 7203–7213. [Google Scholar] [CrossRef]
- Tashiro, K.; Kobayashi, M.; Tadokoro, H. Vibrational Spectra and Theoretical Three-Dimensional Elastic Constants of Isotactic Polypropylene Crystal: An Important Role of Anharmonic Vibrations. Polym. J. 1992, 24, 899–916. [Google Scholar] [CrossRef]
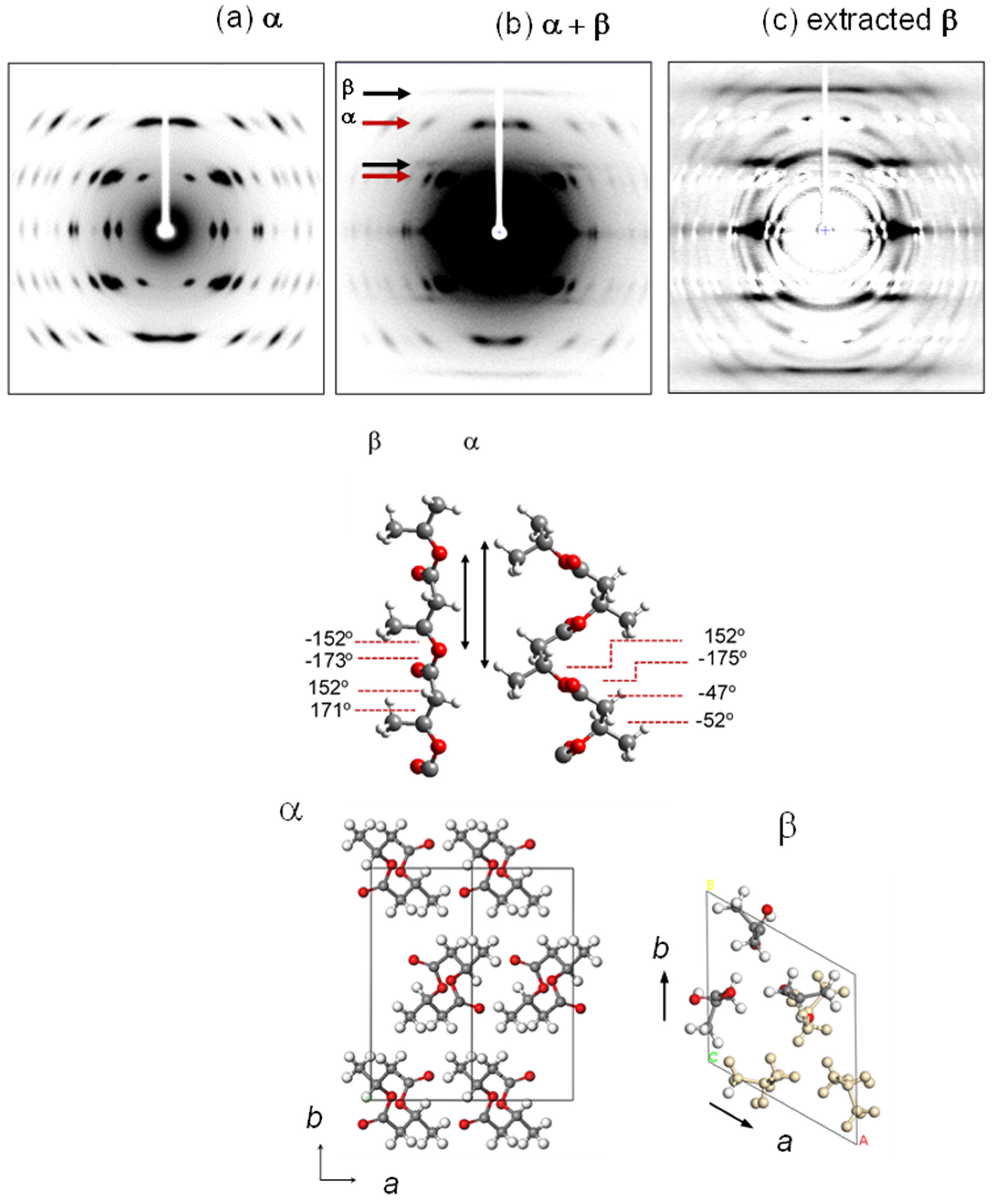
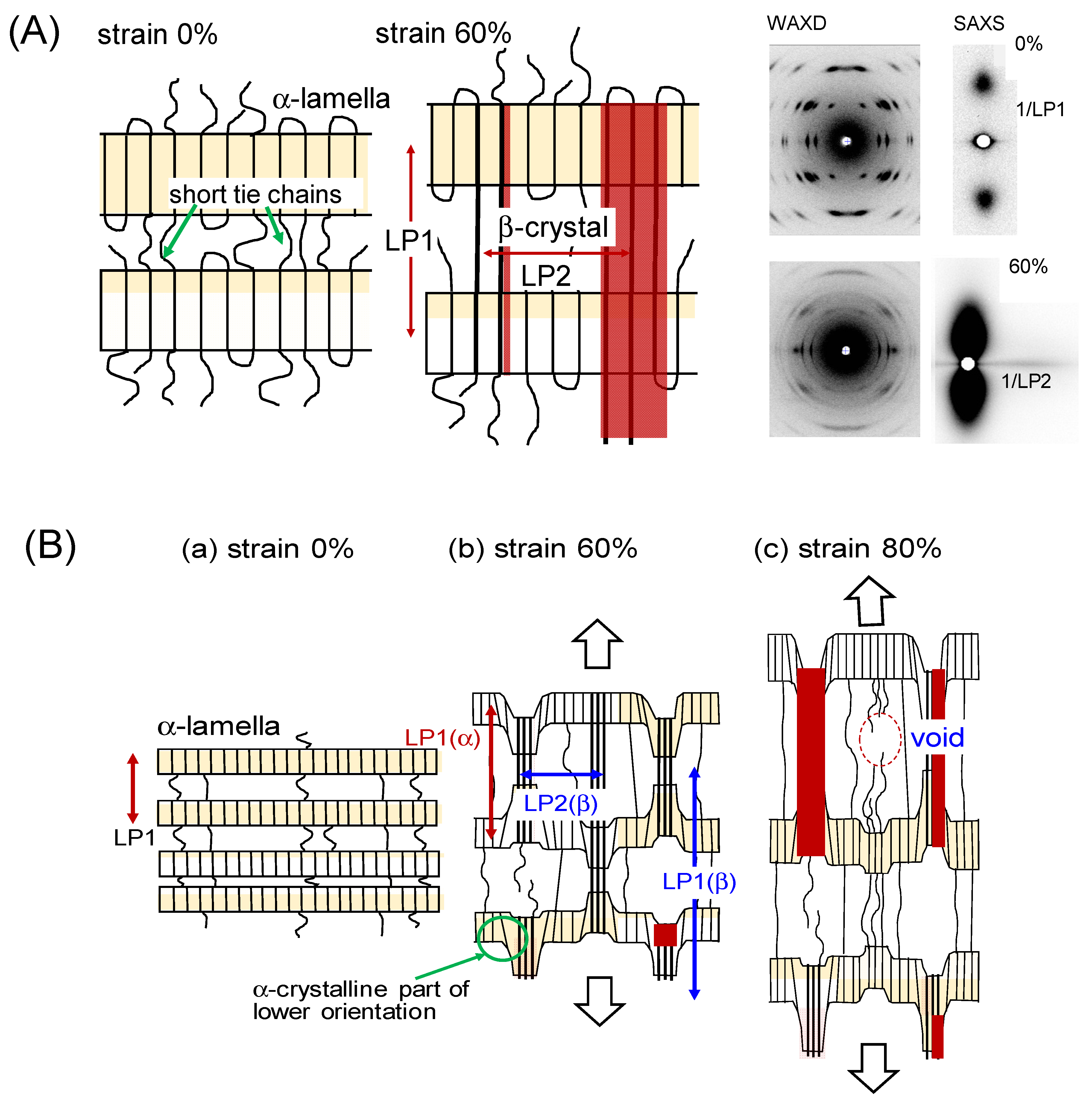
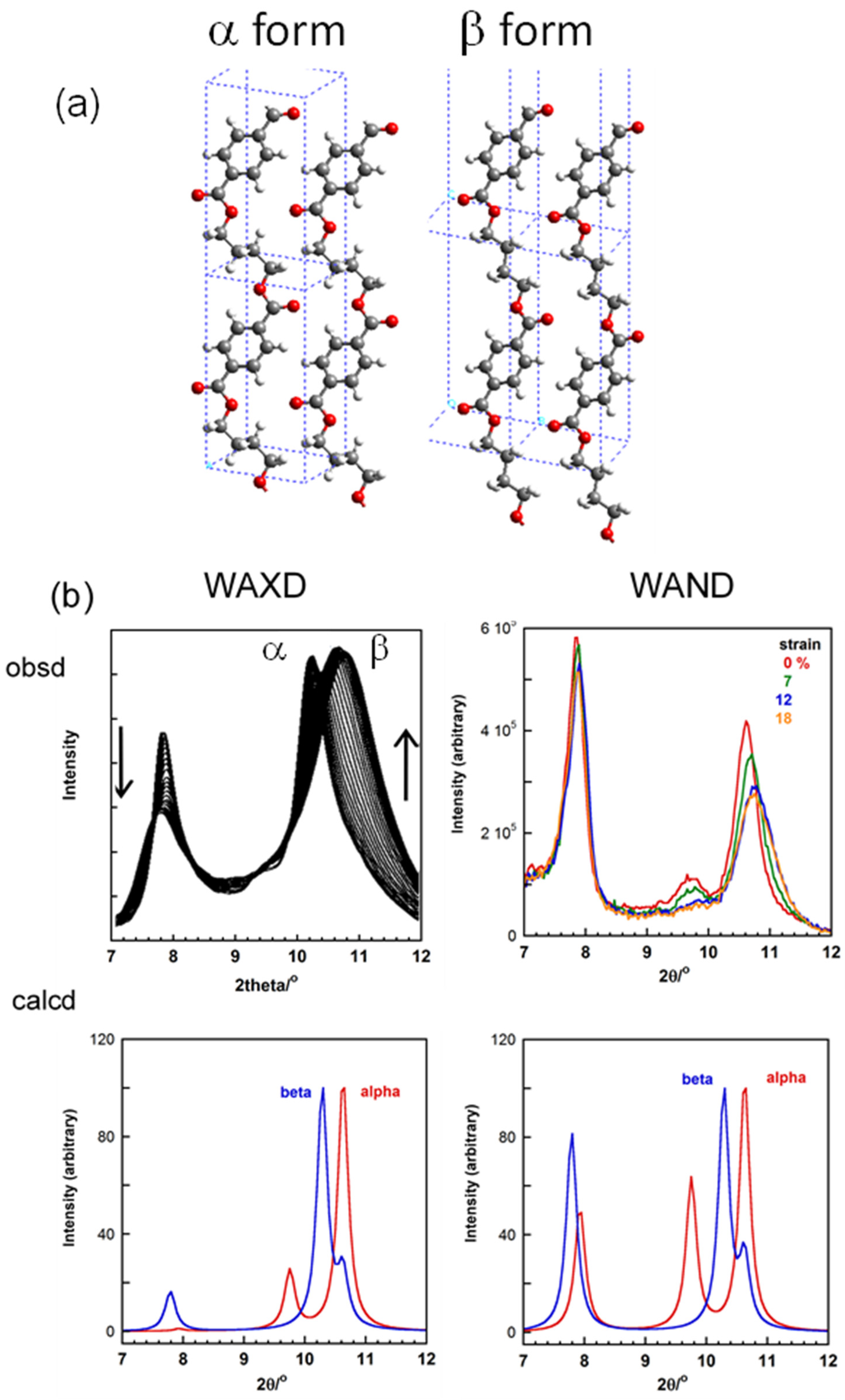
- Phongtamrug, S.; Tashiro, K. X-ray Crystal Structure Analysis of Poly(3-hydroxybutyrate) β-Form and the Proposition of a Mechanism of the Stress-Induced α-to-β Phase Transition. Macromolecules 2019, 52, 2995–3009. [Google Scholar] [CrossRef]
- Tashiro, K.; Tanaka, I.; Ohhara, T.; Niimura, N.; Fujiwara, S.; Kamae, T. Extraction of Hydrogen Atom Positions in Poly-ethylene Crystal Lattice from the Wide-Angle Neutron Diffraction Data Collected by 2-Dimensional Imaging Plate System: A Comparison with the X-ray and Electron Diffraction Results. Macromolecules 2004, 37, 4109–4117. [Google Scholar] [CrossRef]
- Tashiro, K.; Hanesaka, M.; Yamamoto, H.; Wasanasuk, K.; Jayaratri, P.; Yoshizawa, Y.; Tanaka, I.; Niimura, N.; Kusaka, K.; Hosoya, T.; et al. Accurate Structure Analyses of Polymer Crystals on the Basis of Wide-Angle X-ray and Neutron Diffractions. Kobunshi Ronbunshu 2014, 71, 508–526. [Google Scholar] [CrossRef]
- Uchida, T.; Tadokoro, H. Structural studies of polyethers. IV. Structure analysis of the polyoxymethylene molecule by three-dimensional fourier syntheses. J. Polym. Sci. Part A-2 Polym. Phys. 1967, 5, 63–81. [Google Scholar] [CrossRef]
- Tashiro, K.; Hanesaka, M.; Ohhara, T.; Ozeki, T.; Kitano, T.; Nishu, T.; Kurihara, K.; Tamada, T.; Kuroki, R.; Fujiwara, S.; et al. Structural Refinement and Extraction of Hydrogen Atomic Positions in Polyoxymethylene Crystal Based on the First Successful Measurements of 2-Dimensional High-Energy Synchrotron X-ray Diffraction and Wide-Angle Neutron Diffraction Patterns of Hydrogenated and Deuterated Species. Polym. J. 2007, 39, 1253–1273. [Google Scholar] [CrossRef] [Green Version]
- Busetti, V.; Mammi, M.; Carazzolo, G. A three-dimensional refinement of crystalline structure of trioxane. Z. Für Krist. 1963, 119, 310–318. [Google Scholar] [CrossRef]
- Kobayashi, M. Raman study of polar phonons in piezoelectric trioxane crystal. J. Chem. Phys. 1982, 76, 1187–1193. [Google Scholar] [CrossRef]
- Kummara, S.; Tashiro, K.; Monma, T.; Horita, K. Isotope Effect on the Melt−Isothermal Crystallization of Polyoxymethylene D/H Random Copolymers and D/H Blend Samples. Macromolecules 2015, 48, 8070–8081. [Google Scholar] [CrossRef]
- Tashiro, K.; Kusaka, K.; Yamamoto, H.; Hanesaka, M. Introduction of Disorder in the Crystal Structures ofAtacticPoly(vinyl Alcohol) and Its Iodine Complex To Solve a Dilemma between X-ray and Neutron Diffraction Data Analyses. Macromolecules 2020, 53, 6656–6671. [Google Scholar] [CrossRef]
- Bunn, C.W. Crystal Structure of Polyvinyl Alcohol. Nature 1948, 161, 929–930. [Google Scholar] [CrossRef]
- Nitta, I.; Taguchi, I.; Chatani, Y. Crystal structure of P. V. A. Ann. Rep. Inst. Fiber Sci. 1957, 10, 1. [Google Scholar]
- Tashiro, K.; Kobayashi, M.; Tadokoro, H. Polarized Raman spectra of doubly oriented poly(vinyl alcohol). Polym. Bull. 1978, 1, 61–66. [Google Scholar] [CrossRef]
- Sakurada, I.; Fuchino, K.; Okada, N. Crystal Structure of Polyvinyl Alcohol. Bull. Inst. Chem. Res. Kyoto Univ. 1950, 23, 78–79. [Google Scholar]
- Miyasaka, K. PVA-Iodine complexes: Formation, structure, and properties. Adv. Polym. Sci. 1993, 108, 91–129. [Google Scholar] [CrossRef]
- Tashiro, K.; Kitai, H.; Saharin, S.M.; Shimazu, A.; Itou, T. Quantitative Crystal Structure Analysis of Poly(vinyl Alcohol)-Iodine Complexes on the Basis of 2D X-ray Diffraction, Raman Spectra, and Computer Simulation Techniques. Macromolecules 2015, 48, 2138–2148. [Google Scholar] [CrossRef]
- Tashiro, K.; Takahama, T.; Wang, M.-F. X-ray Study of Poly(vinyl Alcohol)-Iodine Complex Prepared from the Dilute Iodine Solution as a Hint to Know the Inner Structure of Polarizer. Polymer 2021, 233, 124180. [Google Scholar] [CrossRef]
- Takahama, T.; Saharin, S.M.; Tashiro, K. Details of the intermolecular interactions in poly(vinyl alcohol)-iodine complexes as studied by quantum chemical calculations. Polymer 2016, 99, 566–579. [Google Scholar] [CrossRef]
- Wegner, G. Solid-State Polymerization Mechanisms. Pure Appl. Chem. 1977, 49, 443–453. [Google Scholar] [CrossRef]
- Tashiro, K.; Nishimura, H.; Kobayashi, M. First Success in Direct Analysis of Microscopic Deformation Mechanism of Polydiacetylene Single Crystal by the X-ray Imaging-Plate System. Macromolecules 1996, 29, 8188–8196. [Google Scholar] [CrossRef]
- Tashiro, K.; Kusaka, K.; Hosoya, T.; Ohhara, T.; Hanesaka, M.; Yoshizawa, Y.; Yamamoto, H.; Niimura, N.; Tanaka, I.; Kurihara, K.; et al. Structure Analysis and Derivation of Deformed Electron Density Distribution of Polydiacetylene Giant Single Crystal by the Combination of X-ray and Neutron Diffraction Data. Macromolecules 2018, 51, 3911–3922. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tashiro, K.; Okada, S. In-situ Observation of Molecular and Crystal Structural Changes in Photo-induced Solid-state Polymerization Reaction of a Wholly-conjugated Diacetylene Compound. Polym. Prepr. Jpn. 2011, 60, 720. [Google Scholar]
- Enkelman, V. Structural aspects of the topochemical polymerization of diacetylenes. Adv. Polym. Sci. 1984, 63, 91–136. [Google Scholar]
- Nakamoto, S.; Tashiro, K.; Matsumoto, A. Quantitative evaluation of stress distribution in bulk polymer samples through the comparison of mechanical behaviors between giant single-crystal and semicrystalline samples of poly(trans-1,4-diethyl muconate). J. Polym. Sci. Part B Polym. Phys. 2003, 41, 444–453. [Google Scholar] [CrossRef]
- Tashiro, K.; Minami, S.; Wu, G.; Kobayashi, M. Quasiharmonic treatment of infrared and raman vibrational frequency shifts induced by tensile deformation of polymer chains. II. Application to the polyoxymethylene and isotactic polypropylene single chains and the three-dimensional orthorhombic polyethylene crystal. J. Polym. Sci. Part B Polym. Phys. 1992, 30, 1143–1155. [Google Scholar] [CrossRef]
- Coppens, P.; Hermansson, K. X-ray Charge Densities and Chemical Bonding; Oxford University Press Inc.: Oxford, UK, 1997. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, K.; Yamamoto, H.; Ohhara, T.; Kurihara, K.; Kuroki, R.; Tamada, T. Temperature Dependent Measurement of 2D-Wide-Angle Neutron Diffraction Diagrams of Uniaxially-oriented Deuterated Polyethylene Sample and Quantitative Analysis of Thermal Motion of Hydrogen Atoms in the Crystal Lattice. Polym. Prepr. Jpn. 2011, 60, 717. [Google Scholar]
- Tashiro, K.; Ishino, K.; Ohta, T. Temperature dependence of crystal structure of uniaxially-oriented polyethylene analysed by an X-ray imaging plate system. Polymer 1999, 40, 3469–3478. [Google Scholar] [CrossRef]
- Jakeways, R.; Smith, T.; Ward, I.M.; Wilding, M.A. Reversible crystal deformation and conformational changes in poly(tetramethylene terephthalate). J. Polym. Sci. Polym. Lett. Ed. 1976, 14, 41–46. [Google Scholar] [CrossRef]
- Tashiro, K.; Nakai, Y.; Kobayashi, M.; Tadokoro, H. Solid-State Transition of Poly(butylene terephthalate) Induced by Mechanical Deformation. Macromolecules 1980, 13, 137–145. [Google Scholar] [CrossRef]
- Tashiro, K.; Yamamoto, H.; Yoshioka, T.; Ninh, T.H.; Tasaki, M.; Shimada, S.; Nakatani, T.; Iwamoto, H.; Ohta, N.; Masunaga, H. Hierarchical Structural Change in the Stress-Induced Phase Transition of Poly(tetramethylene terephthalate) As Studied by the Simultaneous Measurement of FTIR Spectra and 2D Synchrotron Undulator WAXD/SAXS Data. Macromolecules 2014, 47, 2052–2061. [Google Scholar] [CrossRef]
- Yokouchi, M.; Sakakibara, Y.; Chatani, Y.; Tadokoro, H.; Tanaka, T.; Yoda, K. Structures of Two Crystalline Forms of Poly(butylene terephthalate) and Reversible Transition between Them by Mechanical Deformation. Macromolecules 1976, 9, 266–273. [Google Scholar] [CrossRef]
- Hall, I.; Pass, M. Chain conformation of poly(tetramethylene terephthalate) and its change with strain. Polymer 1976, 17, 807–816. [Google Scholar] [CrossRef]
- Desborough, I.; Hall, I. A comparison of published crystalline structures of poly(tetramethylene terephthalate). Polymer 1977, 18, 825–830. [Google Scholar] [CrossRef]
- Tashiro, K.; Yamamoto, H.; Kummara, S.; Takahama, T.; Aoyama, K.; Sekiguchi, H.; Iwamoto, H. High-Electric-Field-Induced Hierarchical Structure Change of Poly(vinylidene fluoride) as Studied by the Simultaneous Time-Resolved WAXD/SAXS/FTIR Measurements and Computer Simulations. Macromolecules 2021, 54, 2334–2352. [Google Scholar] [CrossRef]
- Tashiro, K.; Yamamoto, H.; Kummara, S.; Tahara, D.; Aoyama, K.; Sekiguchi, H. Electric-Field-Induced Phase Transition and Crystal Structural Change of the Oriented Poly(vinylidene Fluoride) β Form as Clarified by the In Situ Synchrotron Wide-Angle X-ray Diffraction Measurement. Macromolecules 2022, 55, 6644–6660. [Google Scholar] [CrossRef]
- Kusaka, K.; Hosoya, T.; Yamada, T.; Tomoyori, K.; Ohhara, T.; Katagiri, M.; Kurihara, K.; Tanaka, I.; Niimura, N. Evaluation of performance for IBARAKI biological crystal diffractometer iBIX with new detectors. J. Synchrotron Radiat. 2013, 20, 994–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, T.; Nakamura, T.; Katagiri, M.; Birumachi, A.; Ebine, M.; Soyama, K. Development of a new detector and DAQ systems for iBIX. Nucl. Instrum. Methods Phys. Res. A 2009, 600, 217–219. [Google Scholar] [CrossRef]
- Niimura, N. Analysis of Transient Phenomena in Crystalline and Liquid structures Using Neutron Time-of-Flight Techniques. Nuc. Instr. Methods 1975, 126, 87–92. [Google Scholar] [CrossRef]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Species | H | D | C | O | K+ | I− |
|---|---|---|---|---|---|---|
| Number of electrons | 1 | 1 | 6 | 8 | 18 | 54 |
| Coherent neutron scattering amplitude (10−13 cm) | −3.74 | 6.67 | 6.65 | 5.80 | 3.67 | 5.28 |
| Coherent neutron cross-sectional area (10−24 cm2) | 1.76 | 5.59 | 5.55 | 4.23 | 1.69 | 3.50 |
| Incoherent neutron cross-sectional area (10−24 cm2) | 80.27 | 2.05 | 0.00 | 0.00 | 0.27 | 0.31 |
| X-ray scattering factor at 2θ = 0° | 1 | 1 | 6 | 8 | 18 | 54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashiro, K.; Kusaka, K.; Yamamoto, H.; Hosoya, T.; Okada, S.; Ohhara, T. Hybridization of Wide-Angle X-ray and Neutron Diffraction Techniques in the Crystal Structure Analyses of Synthetic Polymers. Polymers 2023, 15, 465. https://doi.org/10.3390/polym15020465
Tashiro K, Kusaka K, Yamamoto H, Hosoya T, Okada S, Ohhara T. Hybridization of Wide-Angle X-ray and Neutron Diffraction Techniques in the Crystal Structure Analyses of Synthetic Polymers. Polymers. 2023; 15(2):465. https://doi.org/10.3390/polym15020465
Chicago/Turabian StyleTashiro, Kohji, Katsuhiro Kusaka, Hiroko Yamamoto, Takaaki Hosoya, Shuji Okada, and Takashi Ohhara. 2023. "Hybridization of Wide-Angle X-ray and Neutron Diffraction Techniques in the Crystal Structure Analyses of Synthetic Polymers" Polymers 15, no. 2: 465. https://doi.org/10.3390/polym15020465