

Liposomes or Extracellular Vesicles: A Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery

 , , , and
, , , and 
Abstract
:1. Introduction
2. Structure and Composition of Liposomes and Extracellular Vesicles
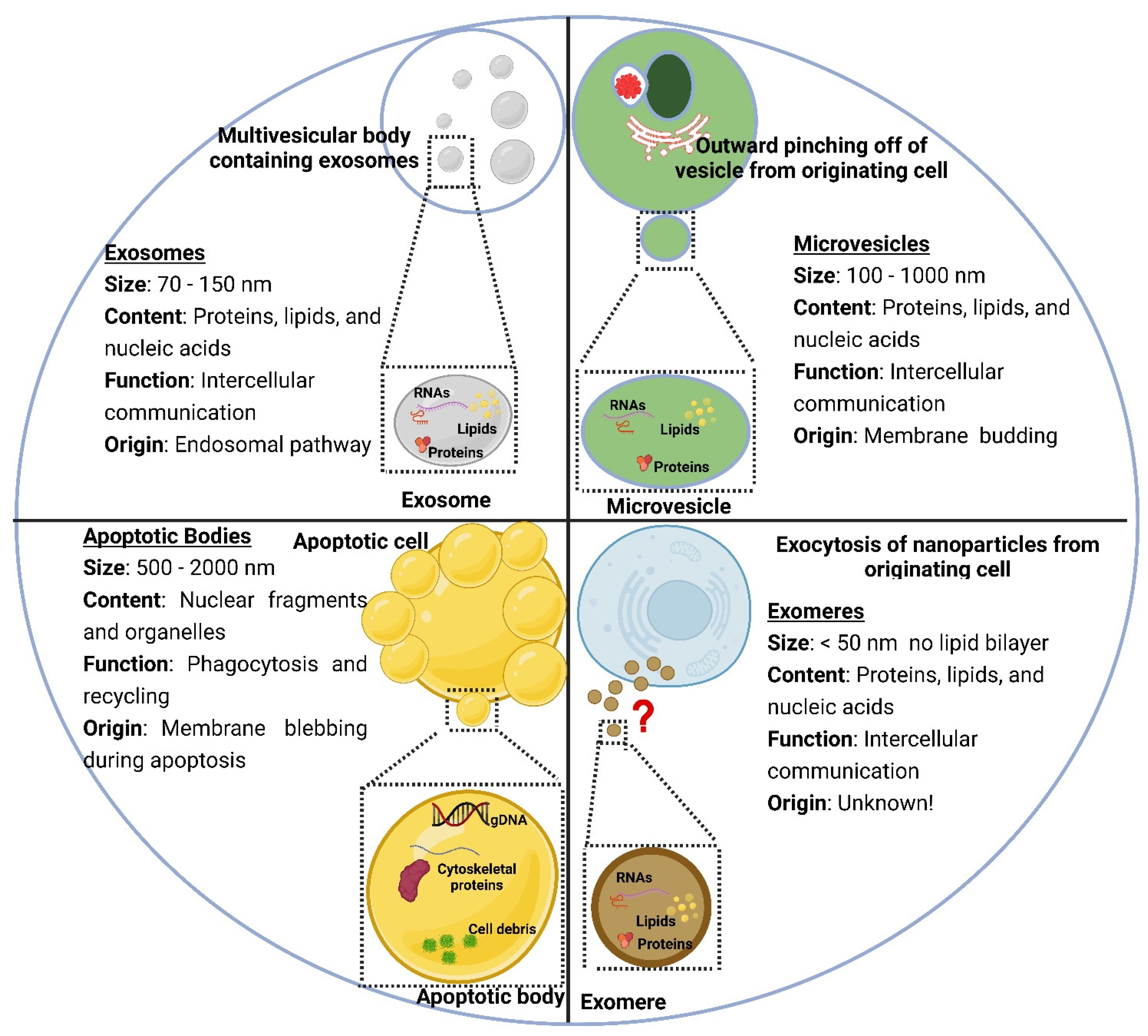
2.1. Structure of Liposomes and Extracellular Vesicles
2.2. Lipid Composition
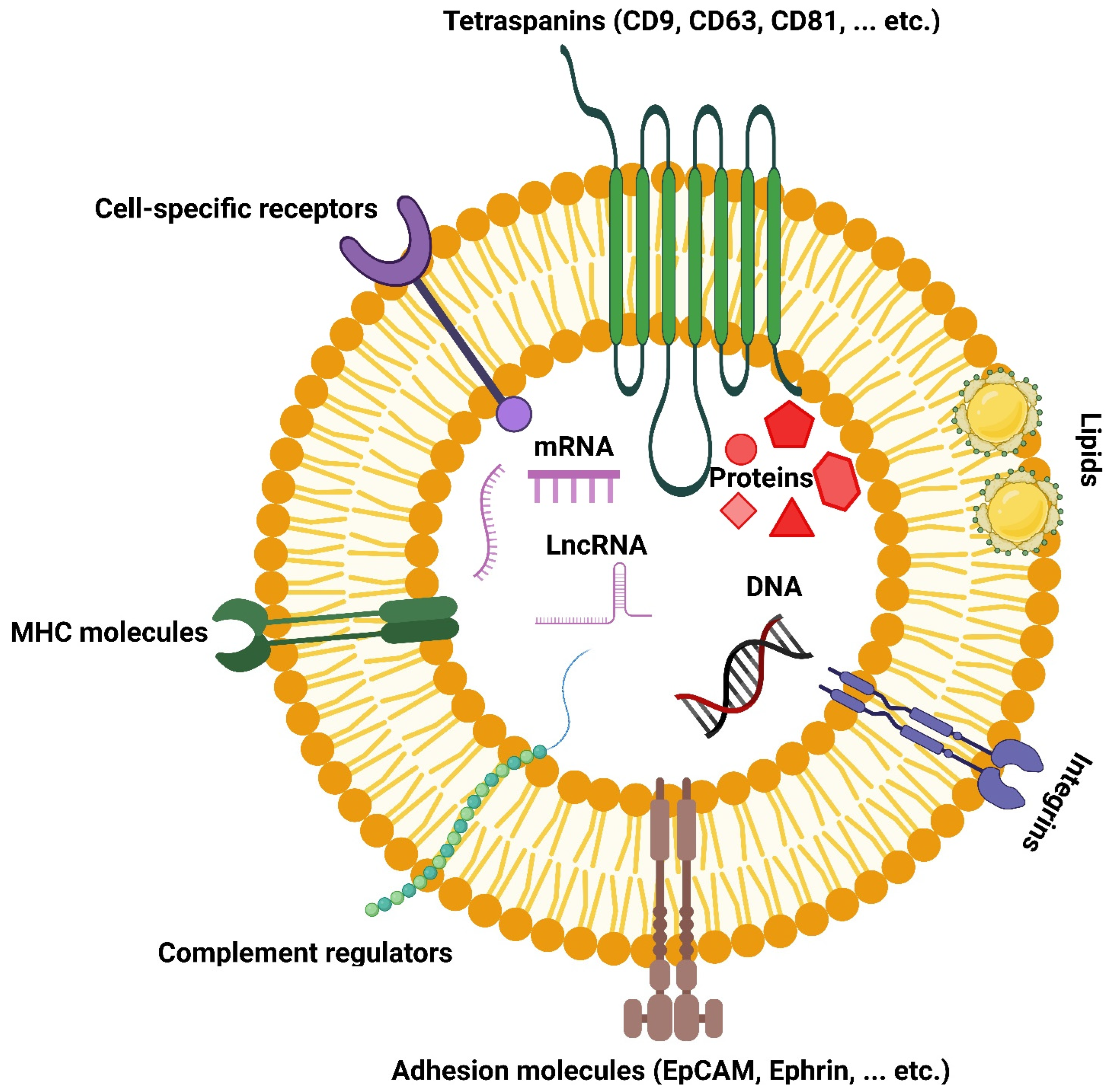
2.3. Protein Composition
2.4. Carbohydrate Composition
2.5. Polymer Composition
3. Methods of Preparation/Isolation
3.1. Liposomes Preparation Techniques
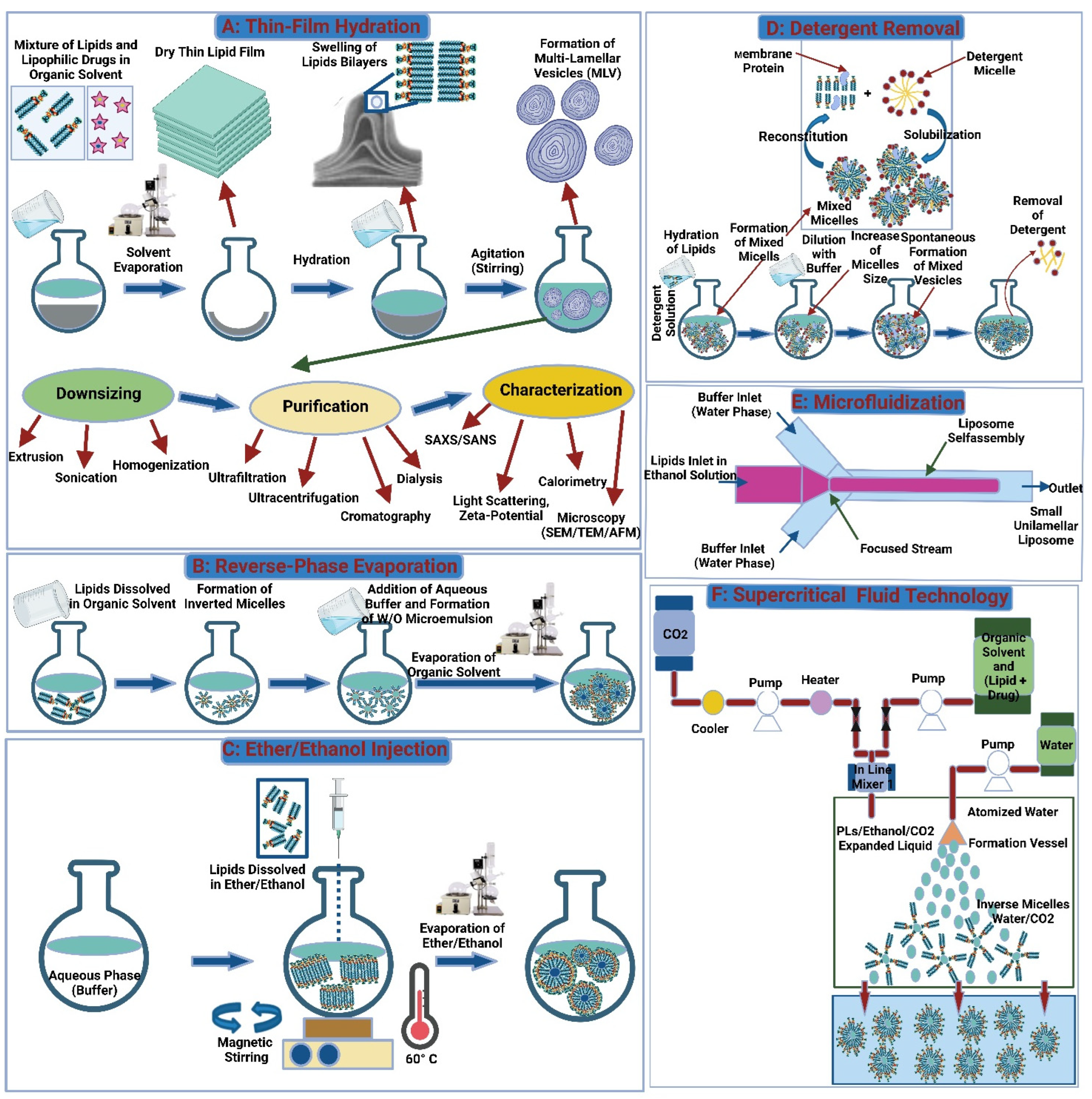
3.1.1. Conventional Methods
Thin-Film Hydration
Reverse-Phase Evaporation
Ether/Ethanol Injection
Detergent Removal Method
Freeze-Thaw Extrusion Method
Dehydration-Rehydration Method
Heating Method
3.1.2. Novel Methods
Microfluidization
Supercritical Fluid Technology
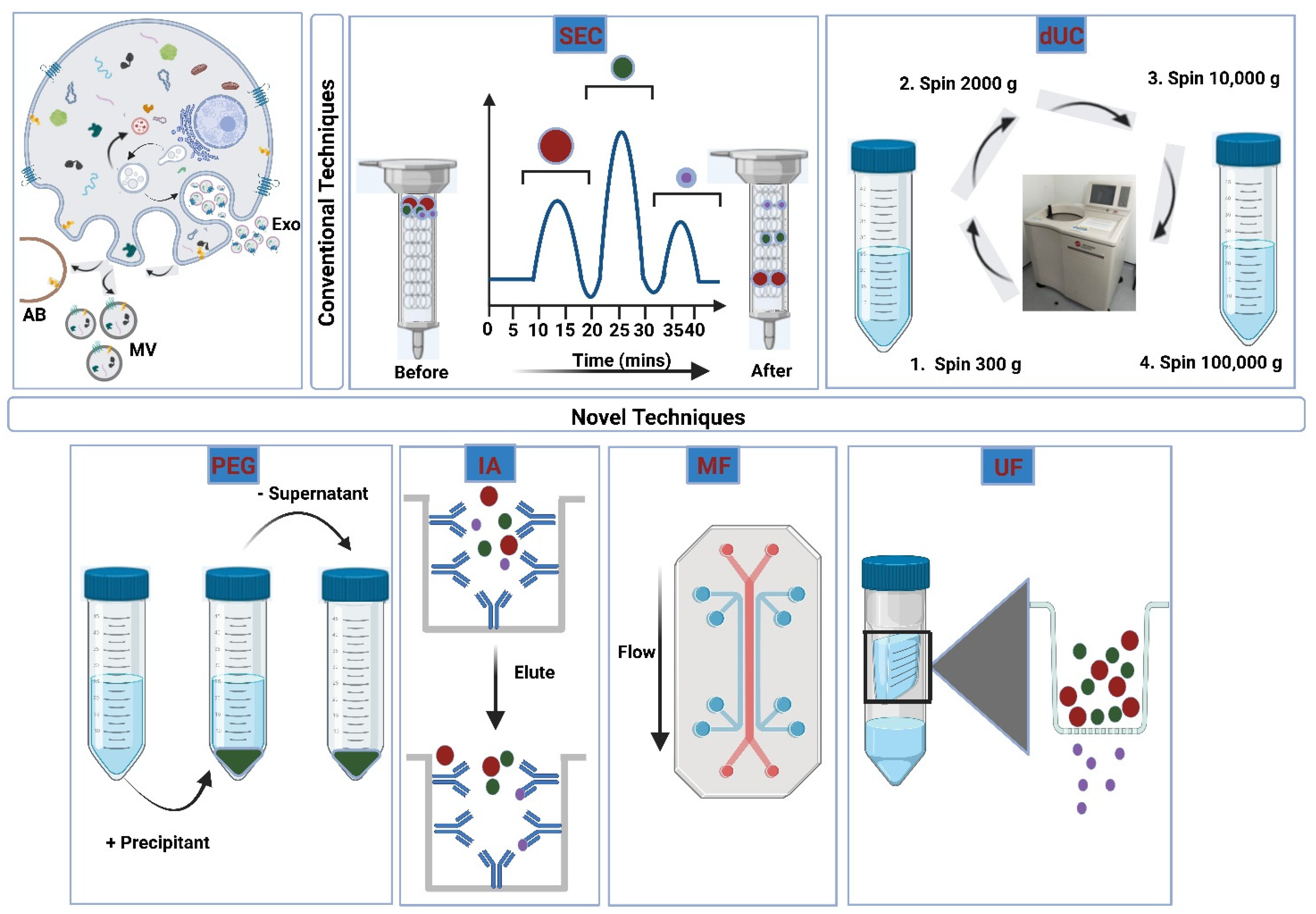
3.2. Isolation of Extracellular Vesicles
3.2.1. Ultracentrifugation
3.2.2. Size-Exclusion Chromatography
3.2.3. Ultrafiltration
3.2.4. Polymeric Precipitation
3.2.5. Immunoaffinity Isolation
3.2.6. Microfluidic Devices
4. Drug Loading into Liposomes and Extracellular Vesicles
4.1. Drug Loading into Liposomes
4.1.1. Passive Loading Approach
4.1.2. Active Loading Approach
4.2. Drug Loading into Extracellular Vesicles
4.2.1. Cell-Based Loading Approach
4.2.2. Non-Cell-Based Loading Approach
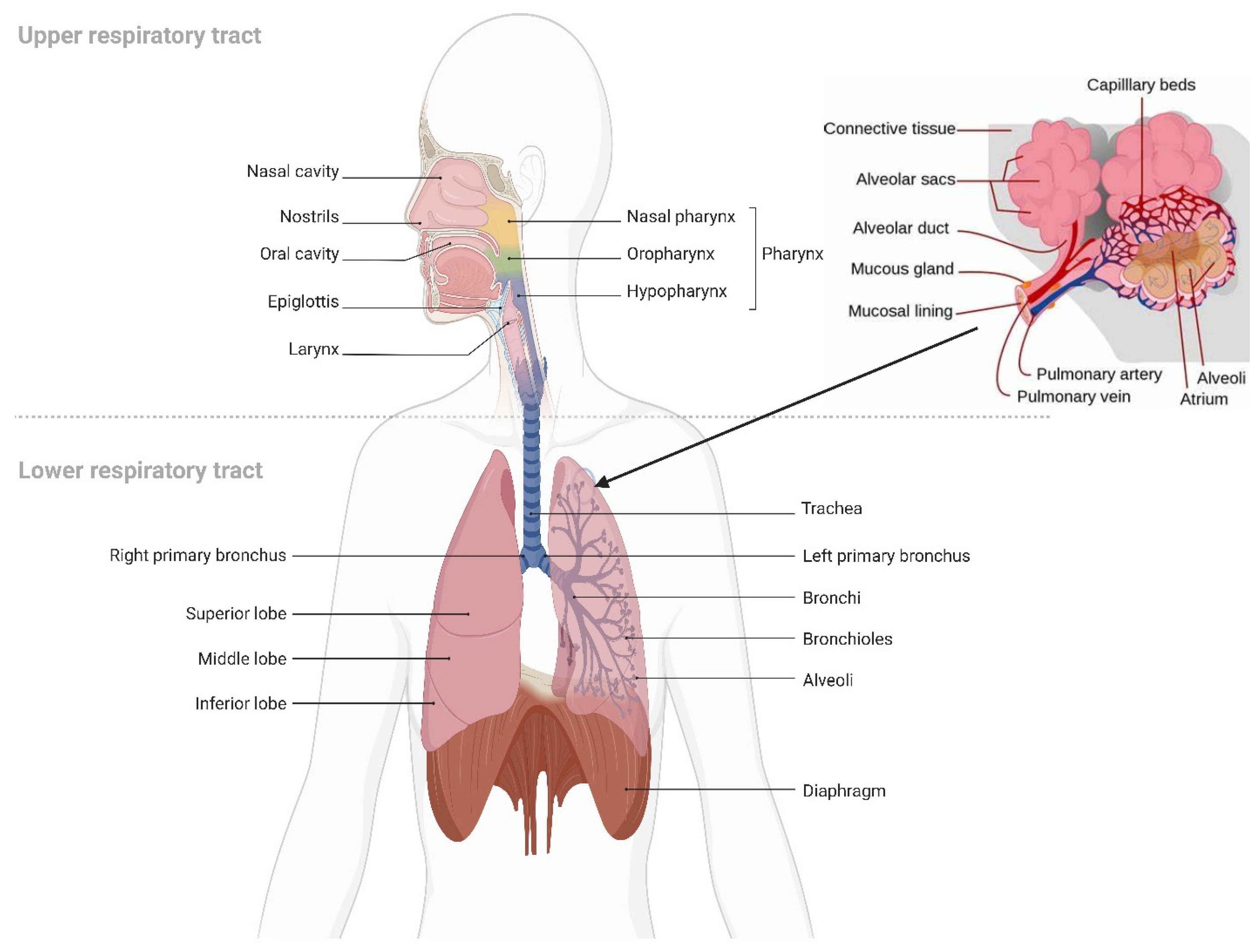
5. Pulmonary Drug Delivery
5.1. Overview of Pulmonary Drug Delivery
5.2. Requirements for Pulmonary Drug Deep Deposition
5.2.1. Inhaled Drug Formulation
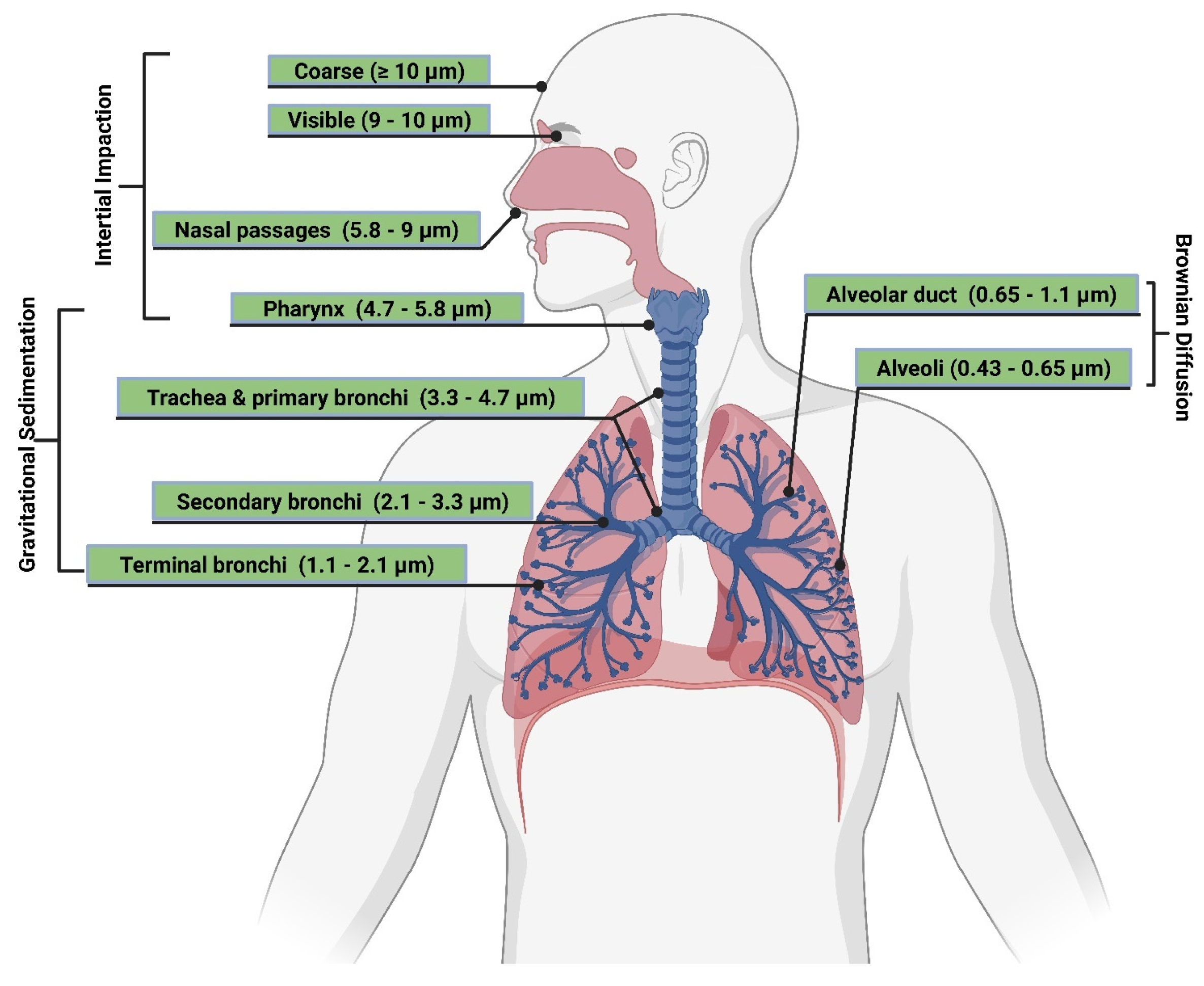
Particle Size and Deposition Pattern
Particle Shape and Surface Morphology
Particle Hygroscopicity
Particle Surface Charge
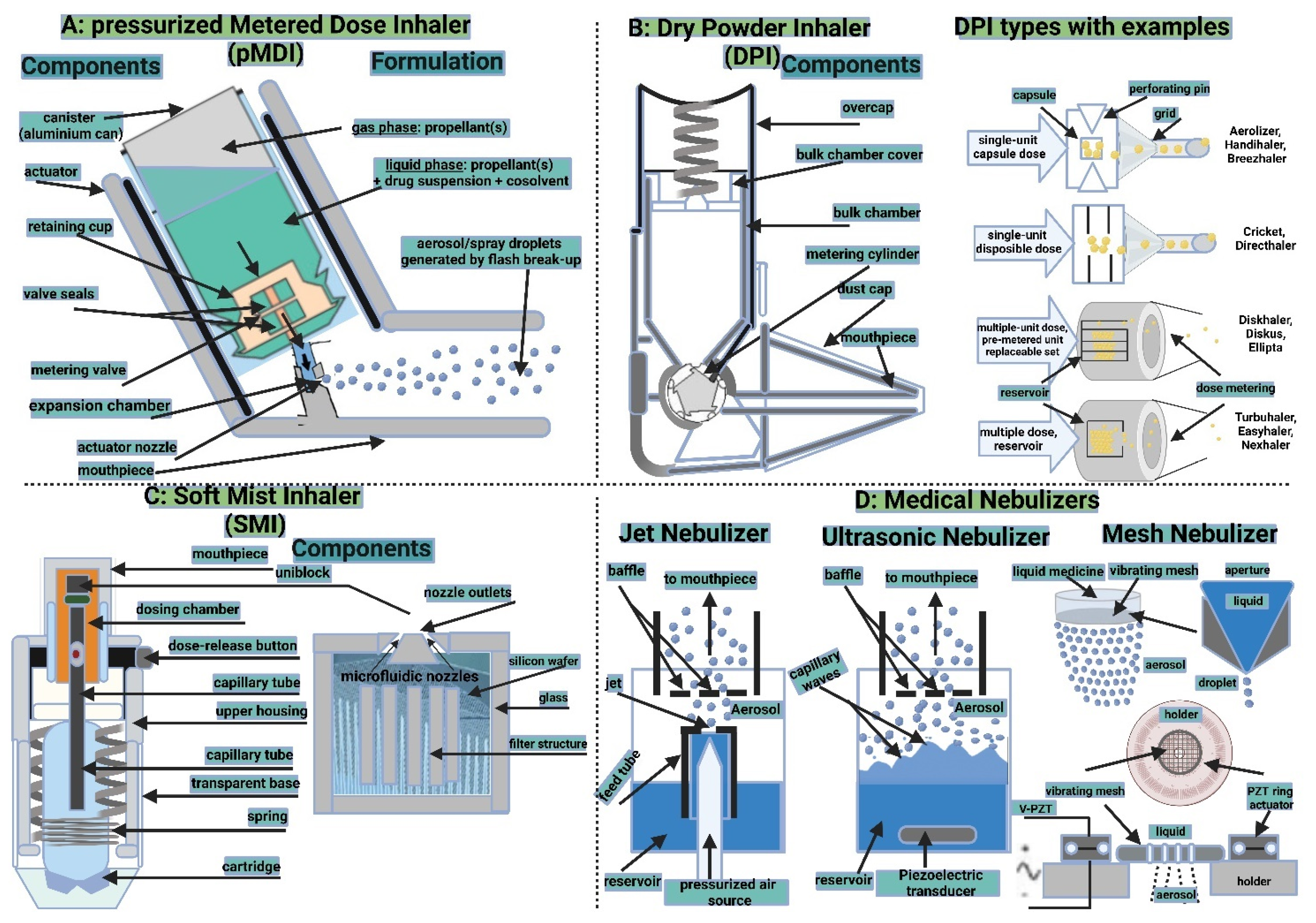
5.2.2. Inhalation Drug Delivery Devices
Pressurized Metered-Dose Inhalers
Dry Powder Inhalers
Soft Mist Inhalers
Medical Nebulizers
6. Applications of Liposomes and Extracellular Vesicles in Pulmonary Drug Delivery
6.1. Inhalable Liposomes

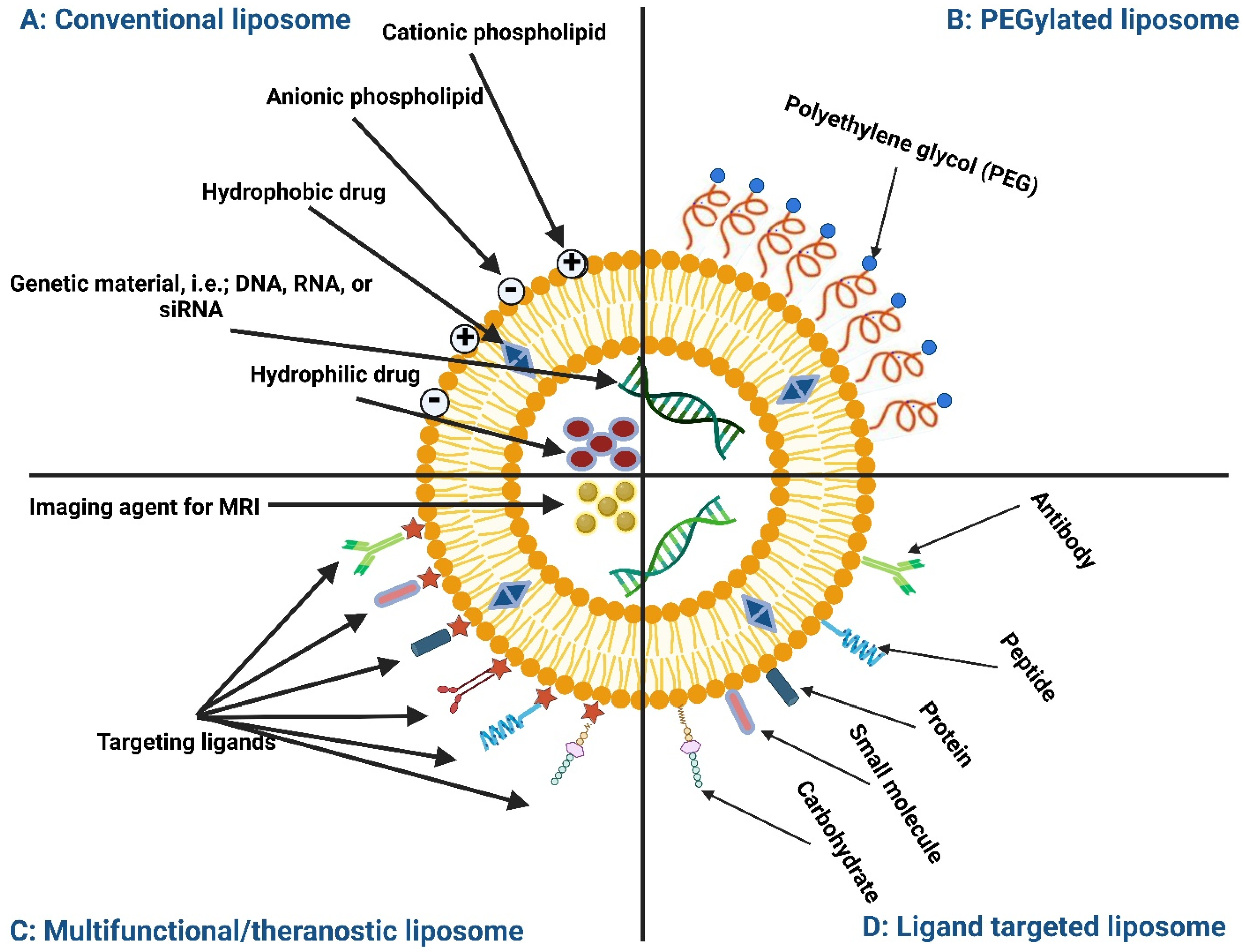{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Drug | Study Goals | Liposomes Composition | Drying Instrumentation | Reference |
|---|---|---|---|---|---|
| Cancer | Docetaxel-FA (DTX-FA) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between LPs-DTx-FA solution and co-spray dried LPs-DTx-FA | -Drug:lipid (1:25 (w/w)) -PC:Chol (6:1 (w/w)) -DSPE-PEG-FA: DSPE-PEG-COOH (1:2 (w/w)) | Spray Dryer | [260] |
| Cancer | Curcumin (CRC) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between CRC drug powder, GTB drug powder and freeze-dried LPs- CRC | -SPC:Chol (5:1 (w/w)) | Lyophilizer | [266] |
| Idiopathic pulmonary fibrosis (IPF) | Colchicine (COL) & Budesonide (BSD) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between LPs-COL/BSD solution and freeze-dried LPs-COL/BSD | Drug:lipid (composition) ratio -1:17.5 (w/w) ≈ 12 mg COL (DPPG:SPC:Chol 3:6:1 (w/w)) -1:17.5 (w/w) ≈ 10 mg BSD (DPPG:HSPC:Chol 4:5:1 (w/w)) | Lyophilizer | [270] |
| Infection | Moxifloxacin (MFX) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between mannosylated LPs-MXF solution and co-spray dried mannosylated LPs-MXF | -Drug:lipid (0.15:1 (w/w)) -PC:Chol (7:3 (w/w)) -DOTAP:PC:Chol (3.5:3.5:3 (w/w)) | Spray Dryer | [263] |
| Infection | Clarithromycin (CTM) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between LPs-CTM solution and ultrasonic spray freeze dried LPs- CTM | -Drug:SPC:Chol (2:4:1 (w/w)) | Lyophilizer | [265] |
| Infection | Ciprofloxacin (CFX) | Feasibility of converting LPs-CFX nanocrystals into a co-spray dried LPs-CFX | -SPC:Chol (7:3 (w/w)) -Sucrose:lipid (2:1 (w/w)) | Spray Dryer | [267] |
| Infection | Andrographolide (AGL) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between AGL drug powder and freeze-dried LPs- AGL | -SPC:Chol (6:1 (w/w)) | Lyophilizer | [268] |
| Infection | Licorice extract (LR-E) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between LPs-LR-E solution and freeze-dried LPs-LR-E | -Drug:lipid (1:6, 7, 8, & 9 (w/w)) -Lipid:trehalose (1:4 (w/w)) | Lyophilizer | [269] |
| Influenza | Oseltamivir phosphate (OTV-P) | Physicochemical, pharmacokinetics and pharmacodynamic properties comparison between LPs-OTV-P solution and co-spray dried LPs-OTV-P, OTV-carboxylate plasma concentration | -Drug:LPs (1:10 (w/w)) -Ovelecithin:Chol (6.7:1 (w/w)) | Spray Dryer | [262] |
6.2. Inhalable Extracellular Vesicles
| Disease | Drug Loading/Bioengineering | Study Goals | EVs Extraction | Reference |
|---|---|---|---|---|
| Cancer | A 10 mM solution of CRC powder dissolved in DMSO and added to the growth medium for intervals of 24 to 72 h. | -According to BCS, CRC belongs to a category IV drug (i.e., low solubility—low permeability) as well as CRC poor stability and rapid elimination paving the way for novel delivery systems [289]. Then, due to higher CRC concentrations in recipient cells, CRC, which is released by EVs, exerts a stronger anti-cancer effect. -Pharmacokinetics and pharmacodynamics comparison between CRC drug alone and EVs-CRC | Differential centrifugation | [278] |
| Cancer | After treating with MTX, tumor cells were exposed to UV light, 300 Jm-2 irradiations for various period of times for different cell types. After 24 h of incubation, supernatants were taken out and repeatedly centrifuged to remove cells, debris, and lastly to pellet ATCMPs. | -In vitro cells cytotoxicity assay and MPE mice model intrapleural injected with PBS, MTX drug alone, naïve empty ATCMPs, or ATCMPs-MTX. In contrast, 11 human MPE patients’ autologous tumor cells obtained via indwelling pleural catheter to produce ATCMPs to package MTX for individualized MPE therapy. -In vitro and in vivo comparing the pharmacokinetics and pharmacodynamics of the MTX alone and ATCMPs-MTX malignancy targeting. | Differential Centrifugation | [279] |
| Cancer | Loading Paclitaxel (PTX) into EVs released by autologous macrophages were followed three methods; incubation, electroporation, and sonication. | -Physicochemical comparison among different drug loading into EVs methods; incubation, electroporation, and sonication. The highest loading efficiency was achieved with mild EVs sonication in the presence of PTX. -In vitro and in vivo (intranasally (i.n.) administered in murine model of tumor lung metastases) pharmacokinetics and pharmacodynamics cytotoxicity comparison between PTX drug alone and EVs-PTX. | Polymer Precipitation (ExoQuick-TC™ Kit) | [164] |
| Cancer | First: PTX was added to EVs in PBS. Second: different amounts of aminoethylanisamide-polyethylene glycol-DSPE (AA-PEG-DSPE) were mixed with the EVs-PTX combination. Third: the final mixture was sonicated to obtain a solution of AA-vectorized EVs loaded with PTX (AA-PEG-EVs-PTX). | -Development and optimization of a formulation of AA-vectorized EVs superior structure loaded with PTX (AA-PEG-EVs-PTX) target the sigma receptor, which lung cancer cells overexpress. -In vitro and in vivo (intravenously (i.v.) administered in murine model of tumor lung metastases) pharmacokinetics and pharmacodynamics comparison between autologous vectorized labeled (DiL-AA-PEG-EVs-PTX) and non-vectorized labeled (Dil-PEG-EVs-PTX) concerning prolongation circulation time via PEGylation, targeting/accumulation via AA-vectorization, and bypassing Pgp (P-glycoprotein efflux pump)-mediated drug efflux in MDR cancer cells. | Polymer Precipitation (ExoQuick-TC™ Kit) | [161] |
| Idiopathic pulmonary fibrosis (IPF) | Human bone-marrow derived mesenchymal stem cells EVs (hBM-MSCs-EVs). | -Proving that normal and IPF lung fibroblasts’ TFG-1-induced myofibroblastic differentiation is suppressed by hBM-MSCs-EVs and not by fibroblast EVs. -Evaluating cellular EVs uptaking kinetics, hBM-MSCs-EVs exhibit higher time- and dose-dependent cellular uptake compared to fibroblast EVs. Contrarily, Thy-1 removing or blocking as well as Thy-1-beta integrin interactions inhibiting reduced the hBM-MSCs-EVs uptake and thereby avoided suppressing of myofibroblastic differentiation. | Differential Centrifugation | [284] |
| Idiopathic pulmonary fibrosis (IPF) | Human lung spheroid cells EVs (hLSCs-EVs) and Human bone-marrow derived mesenchymal stem cells EVs (hBM-MSCs-EVs). | -A viable way to mitigate cell-based therapy clinical challenges, is to substitute conditioned medium or secretome for real cells. Thus, stem cell’s regenerative ability via paracrine activity can be gained through their secretions, i.e., secretome and EVs. -Demonstrating that mice model of BLM/silica-induced fibrosis, nebulizer inhalation of hLSCs-EVs promotes lung repair superior to hBM-MSCs-EVs. | Differential Centrifugation | [285] |
| Idiopathic pulmonary fibrosis (IPF) | Human amnion epithelial cells derived EVs (hAECs-EVs) | -Although stem cell-derived EVs offer several therapeutic advantages over their parenteral cells, their therapeutic effects can be impaired by fibrosis. -In vivo treatment efficacy comparison on OVA/NA induced chronic AAD and BLM induced pulmonary fibrosis mice model between hAECs-EVs alone and hAECs-EVs + serelaxin (SLX). | Differential Centrifugation | [286] |
| Asthma | Human bone-marrow derived mesenchymal stem cells EVs (hBM-MSCs-EVs). | -Examining the role of hBM-MSCs-EVs paracrine effects in immune modulation that mimics paternal MSCs and hence therapeutic potential for asthma. -hBM-MSCs-EVs promote Tregs propagation and immunological suppression capacity by upregulating PBMCs cytokines IL-10 and TFG-β1 of asthmatic patient. | Differential Centrifugation | [280] |
6.3. Inhalable Hybrid Vesicles
7. Models for Testing Inhaled Aerosolized Mist/Dried Lipid Bilayer Vesicles
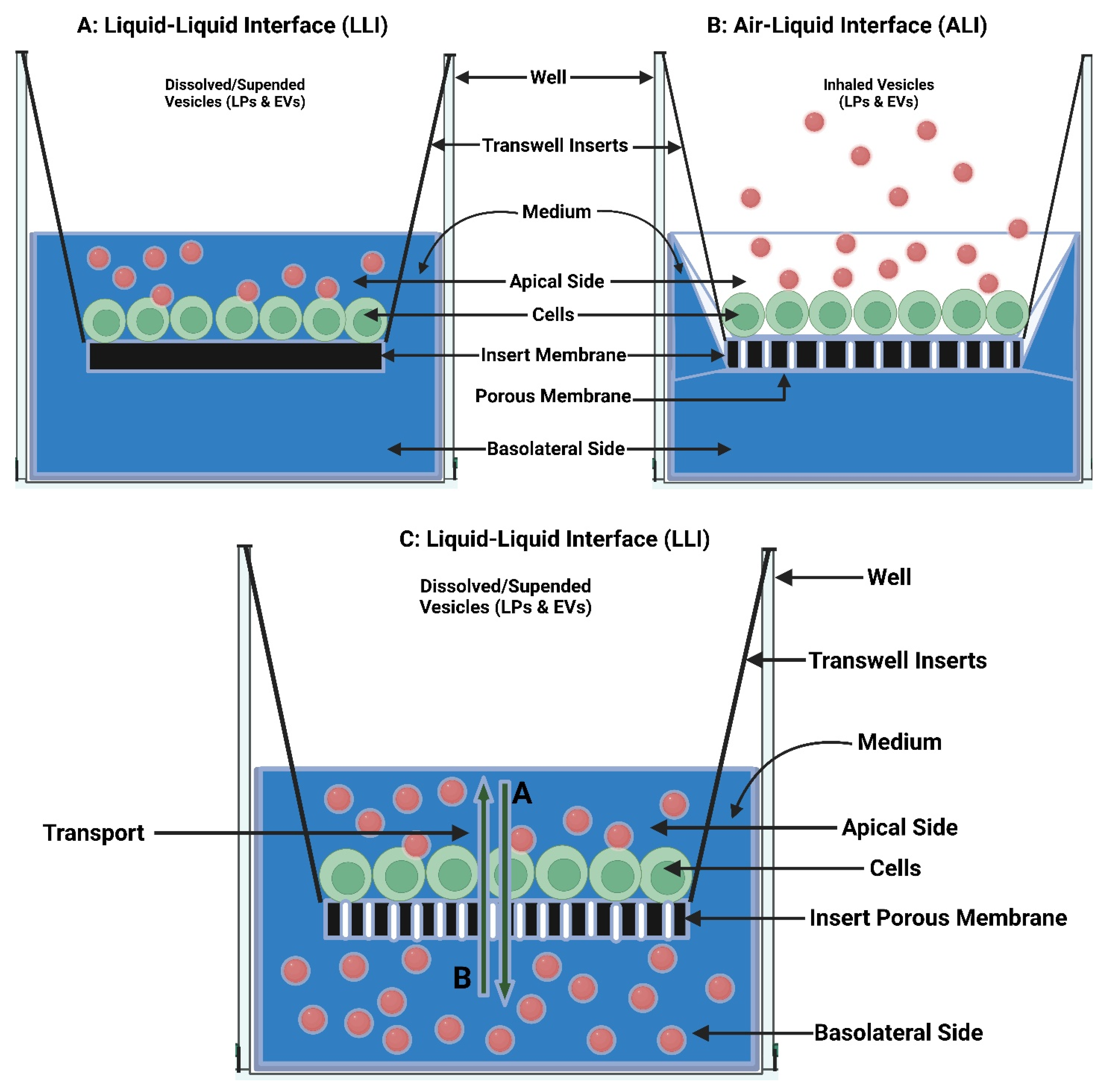
7.1. In Vitro Pulmonary Cell-Based Models
7.2. Ex Vivo Lung Tissue/Organ-Based Models
7.3. In Vivo Whole Animal-Based Models
8. Challenges in Clinical Translation of Liposomes and Extracellular Vesicles
8.1. Pharmacological/PKPD Challenges
8.2. Manufacturing and Administration Challenges
8.2.1. Drug Loading and Release
8.2.2. Identification and Purity
8.2.3. Potency
8.2.4. Large-Scale Production
8.2.5. Process Validation
8.2.6. Stability
9. Opinion: Suitability of Liposomes and Extracellular Vesicles for Pulmonary Drug Delivery, Comparison and Proposed Solutions to Overcome the Challenges
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Obaidi, H.; Granger, A.; Hibbard, T.; Opesanwo, S. Pulmonary drug delivery of antimicrobials and anticancer drugs using solid dispersions. Pharmaceutics 2021, 13, 1056. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Bera, H.; Shi, C.; Zhang, L.; Cun, D.; Yang, M. Pharmaceutical strategies to extend pulmonary exposure of inhaled medicines. Acta Pharm. Sin. B 2021, 11, 2565–2584. [Google Scholar] [PubMed]
- Cidem, A.; Bradbury, P.; Traini, D.; Ong, H.X. Modifying and Integrating in vitro and ex vivo Respiratory Models for Inhalation Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 581995. [Google Scholar] [CrossRef]
- Valent, P.; Groner, B.; Schumacher, U.; Superti-Furga, G.; Busslinger, M.; Kralovics, R.; Zielinski, C.; Penninger, J.M.; Kerjaschki, D.; Stingl, G. Paul Ehrlich (1854–1915) and his contributions to the foundation and birth of translational medicine. J. Innate Immun. 2016, 8, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Garello, F.; Circosta, P.; Stefania, R.; Aime, S.; Saglio, G.; Giachino, C. Nanocarriers as magic bullets in the treatment of leukemia. Nanomaterials 2020, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Trams, E.G.; Lauter, C.J.; Salem, J.N.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta Biomembr. 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Pan, B.-T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Gill, S.; Catchpole, R.; Forterre, P. Extracellular membrane vesicles in the three domains of life and beyond. FEMS Microbiol. Rev. 2019, 43, 273–303. [Google Scholar]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Abreu, S.C.; Lopes-Pacheco, M.; Weiss, D.J.; Rocco, P.R. Mesenchymal stromal cell-derived extracellular vesicles in lung diseases: Current status and perspectives. Front. Cell Dev. Biol. 2021, 9, 600711. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Dembek, A.; Linnenberger, R.; Dahlem, C.; Barghash, A.; Fecher-Trost, C.; Fuhrmann, G.; Koch, M.; Kraegeloh, A.; Huwer, H. Toll-like receptor 2 release by macrophages: An anti-inflammatory program induced by glucocorticoids and lipopolysaccharide. Front. Immunol. 2019, 10, 1634. [Google Scholar] [CrossRef] [Green Version]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.H.A.M.; Heijnen, H.F.G.; van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles-based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [Green Version]
- Börger, V.; Bremer, M.; Ferrer-Tur, R.; Gockeln, L.; Stambouli, O.; Becic, A.; Giebel, B. Mesenchymal stem/stromal cell-derived extracellular vesicles and their potential as novel immunomodulatory therapeutic agents. Int. J. Mol. Sci. 2017, 18, 1450. [Google Scholar] [CrossRef] [Green Version]
- Ferrati, S.; Wu, T.; Kanapuram, S.R.; Smyth, H.D. Dosing considerations for inhaled biologics. Int. J. Pharm. 2018, 549, 58–66. [Google Scholar] [CrossRef]
- Liang, W.; Pan, H.W.; Vllasaliu, D.; Lam, J.K. Pulmonary delivery of biological drugs. Pharmaceutics 2020, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-R.; Wei, X.-Q.; Zhang, S.; Fu, N.; Lin, Y.-F.; Cai, X.-X.; Peng, Q. Effects of micro-environmental pH of liposome on chemical stability of loaded drug. Nanoscale Res. Lett. 2017, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drescher, S.; van Hoogevest, P. The phospholipid research center: Current research in phospholipids and their use in drug delivery. Pharmaceutics 2020, 12, 1235. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; Chuesiang, P.; Shin, G.H.; Park, H.J. Post-processing techniques for the improvement of liposome stability. Pharmaceutics 2021, 13, 1023. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Salar-Behzadi, S. Oral inhalation for delivery of proteins and peptides to the lungs. Eur. J. Pharm. Biopharm. 2021, 163, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Tang, Q.D.; Doan, D.C.T.; Dang, M.C. Micro and nano liposome vesicles containing curcumin for a drug delivery system. Adv. Nat. Sci. Nanosci. Nanotechnol. 2016, 7, 035003. [Google Scholar] [CrossRef] [Green Version]
- Drazenovic, J.; Wang, H.; Roth, K.; Zhang, J.; Ahmed, S.; Chen, Y.; Bothun, G.; Wunder, S.L. Effect of lamellarity and size on calorimetric phase transitions in single component phosphatidylcholine vesicles. Biochim. Biophys. Acta Biomembr. 2015, 1848, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supramaniam, P.; Ces, O.; Salehi-Reyhani, A. Microfluidics for artificial life: Techniques for bottom-up synthetic biology. Micromachines 2019, 10, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Deatherage, B.L.; Cookson, B.T. Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.G.; Ding, Y.; Jiang, L. Unconventional protein secretion in plants: A critical assessment. Protoplasma 2016, 253, 31–43. [Google Scholar] [CrossRef]
- Kim, D.-K.; Kang, B.; Kim, O.Y.; Choi, D.-S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C. EVpedia: An integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20384. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinger, S.A.; Cha, D.J.; Franklin, J.L.; Higginbotham, J.N.; Dou, Y.; Ping, J.; Shu, L.; Prasad, N.; Levy, S.; Zhang, B. Diverse long RNAs are differentially sorted into extracellular vesicles secreted by colorectal cancer cells. Cell Rep. 2018, 25, 715–725.e4. [Google Scholar] [CrossRef] [Green Version]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging (Albany N. Y.) 2018, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct 2013, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.H.; Korma, S.A.; Lipeng, Q.; Jinghua, C. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2019, 27, 742–761. [Google Scholar] [CrossRef]
- Tsuji, T.; Morita, S.-Y.; Ikeda, Y.; Terada, T. Enzymatic fluorometric assays for quantifying all major phospholipid classes in cells and intracellular organelles. Sci. Rep. 2019, 9, 8607. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11, 20140459. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Lee, K.Y. Characteristics and Clinical Application of Extracellular Vesicle-Derived DNA. Cancers 2021, 13, 3827. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratta, S.; Shimanaka, Y.; Costanzi, E.; Ni, S.; Urbanelli, L.; Kono, N.; Morena, F.; Sagini, K.; Giovagnoli, S.; Romani, R. Lipotoxic stress alters the membrane lipid profile of extracellular vesicles released by Huh-7 hepatocarcinoma cells. Sci. Rep. 2021, 11, 4613. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.-C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S. Zeta potential of extracellular vesicles: Toward understanding the attributes that determine colloidal stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Yasue, Y.; Takahashi, Y.; Takakura, Y. Determining the role of surface glycans in the pharmacokinetics of small extracellular vesicles. J. Pharm. Sci. 2021, 110, 3261–3267. [Google Scholar] [CrossRef] [PubMed]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 22, 204–220. [Google Scholar] [CrossRef]
- Srivatsav, A.T.; Kapoor, S. The emerging world of membrane vesicles: Functional relevance, theranostic avenues and tools for investigating membrane function. Front. Mol. Biosci. 2021, 8, 640355. [Google Scholar] [CrossRef]
- Piffoux, M.; Silva, A.K.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of extracellular vesicles by fusion with liposomes for the design of personalized biogenic drug delivery systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef]
- Abdelrehim, A.; Shaltiel, L.; Zhang, L.; Barenholz, Y.; High, S.; Harris, L.K. The use of tail-anchored protein chimeras to enhance liposomal cargo delivery. PLoS ONE 2019, 14, e0212701. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Sasaki, Y.; Akiyoshi, K. Preparation of cationic proteoliposomes using cell-free membrane protein synthesis: The chaperoning effect of cationic liposomes. RSC Adv. 2020, 10, 28741–28745. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.; Schwieger, C.; Schmidt, C. Liposomes as Carriers of Membrane-Associated Proteins and Peptides for Mass Spectrometric Analysis. Angew. Chem. Int. Ed. Engl. 2021, 60, 11523–11530. [Google Scholar] [CrossRef]
- Majeed, S.; Ahmad, A.B.; Sehar, U.; Georgieva, E.R. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes 2021, 11, 685. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Lee, C.-S.; Lee, M. Bioactive Scaffolds Integrated with Liposomal or Extracellular Vesicles for Bone Regeneration. Bioengineering 2021, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Goers, R.; Thoma, J.; Ritzmann, N.; Di Silvestro, A.; Alter, C.; Gunkel-Grabole, G.; Fotiadis, D.; Müller, D.J.; Meier, W. Optimized reconstitution of membrane proteins into synthetic membranes. Commun. Chem. 2018, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Edwards, K.; Johnsson, M.; Karlsson, G.; Silvander, M. Effect of polyethyleneglycol-phospholipids on aggregate structure in preparations of small unilamellar liposomes. Biophys. J. 1997, 73, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Ghazarian, H.; Idoni, B.; Oppenheimer, S.B. A glycobiology review: Carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem. 2011, 113, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta Gen. Subj. 1999, 1473, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [PubMed]
- Maja, L.; Željko, K.; Mateja, P. Sustainable technologies for liposome preparation. J. Supercrit. Fluids 2020, 165, 104984. [Google Scholar]
- AL-Japairai, K.A.S.; Mahmood, S. Systemic Delivery of Calcium Channel Blockers for Hypertension through Transdermal Delivery—A Review. In Materials Science Forum; Trans Tech Publications Ltd.: Wollerau, Switzerland, 2021; pp. 204–208. [Google Scholar]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New developments in liposomal drug delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [PubMed]
- Nkanga, C.I.; Bapolisi, A.M.; Okafor, N.I.; Krause, R.W.M. General perception of liposomes: Formation, manufacturing and applications. In Liposomes: Advances and Perspectives; Intech Open: London, UK, 2019. [Google Scholar]
- Xiang, B.; Cao, D.-Y. Preparation of drug liposomes by thin-film hydration and homogenization. In Liposome-Based Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2021; pp. 25–35. [Google Scholar]
- Sipai, A.; Vandana, Y.; Mamatha, Y.; Prasanth, V. Liposomes: An overview. J. Pharm. Sci. Innov. 2012, 1, 13–21. [Google Scholar]
- Hong, S.-S.; Lim, S.-J. Laboratory scale production of injectable liposomes by using cell disruptor to avoid the probe sonication process. J. Pharm. Investig. 2015, 45, 73–78. [Google Scholar] [CrossRef]
- Sharma, D.; Ali, A.A.E.; Trivedi, L.R. An Updated Review on: Liposomes as drug delivery system. PharmaTutor 2018, 6, 50–62. [Google Scholar] [CrossRef]
- Yu, H.; Park, J.-Y.; Kwon, C.W.; Hong, S.-C.; Park, K.-M.; Chang, P.-S. An overview of nanotechnology in food science: Preparative methods, practical applications, and safety. J. Chem. Chem. Eng. 2018, 2018, 5427978. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Qi, X.-R. Preparation of drug liposomes by reverse-phase evaporation. In Liposome-Based Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2021; pp. 37–46. [Google Scholar]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv. 2011, 2011, 591325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouda, A.; Sakr, O.S.; Nasr, M.; Sammour, O. Ethanol injection technique for liposomes formulation: An insight into development, influencing factors, challenges and applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102174. [Google Scholar] [CrossRef]
- Charcosset, C.; Juban, A.; Valour, J.-P.; Urbaniak, S.; Fessi, H. Liposome-Based Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2015; Volume 94, pp. 508–515. [Google Scholar]
- Shaker, S.; Gardouh, A.R.; Ghorab, M.M. Factors affecting liposomes particle size prepared by ethanol injection method. Res. Pharm. Sci. 2017, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Laouini, A.; Jaafar-Maalej, C.; Limayem-Blouza, I.; Sfar, S.; Charcosset, C.; Fessi, H. Preparation, characterization and applications of liposomes: State of the art. J. Colloid Sci. Biotechnol. 2012, 1, 147–168. [Google Scholar] [CrossRef]
- Mukherjee, F.; Prasad, A.; Bahekar, V.S.; Rana, S.K.; Rajendra, L.; Sharma, G.K.; Srinivasan, V.A. Evaluation of immunogenicity and protective efficacy of a liposome containing Brucella abortus S19 outer membrane protein in BALB/c mice. Iran. J. Vet. Med. 2016, 17, 1. [Google Scholar]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Maherani, B.; Arab-Tehrany, E.; R Mozafari, M.; Gaiani, C.; Linder, M. Liposomes: A review of manufacturing techniques and targeting strategies. Curr. Nanosci. 2011, 7, 436–452. [Google Scholar] [CrossRef]
- Costa, A.P.; Xu, X.; Burgess, D.J.J.P.r. Freeze-anneal-thaw cycling of unilamellar liposomes: Effect on encapsulation efficiency. Pharm. Res. 2014, 31, 97–103. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. A quality by design (QbD) case study on liposomes containing hydrophilic API: I. Formulation, processing design and risk assessment. Int. J. Pharm. 2011, 419, 52–59. [Google Scholar] [CrossRef]
- Sanarova, E.; Lantsova, A.; Oborotova, N.; Orlova, O.; Polozkova, A.; Dmitrieva, M.; Nikolaeva, N. Liposome drug delivery. J. Pharm. Sci. Res. 2019, 11, 1148–1155. [Google Scholar]
- Cliff, L.; Chadda, R.; Robertson, J.L. Occupancy distributions of membrane proteins in heterogeneous liposome populations. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183033. [Google Scholar] [CrossRef] [PubMed]
- Karami, N.; Moghimipour, E.; Salimi, A. Liposomes as a novel drug delivery system: Fundamental and pharmaceutical application. Asian J. Pharm. 2018, 12, S31–S41. [Google Scholar]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Has, C.; Sunthar, P. A comprehensive review on recent preparation techniques of liposomes. J. Liposome Res. 2020, 30, 336–365. [Google Scholar] [CrossRef]
- Khorasani, S.; Danaei, M.; Mozafari, M.R. Nanoliposome technology for the food and nutraceutical industries. Trends Food Sci. Technol. 2018, 79, 106–115. [Google Scholar] [CrossRef]
- Shah, V.M.; Nguyen, D.X.; Patel, P.; Cote, B.; Al-Fatease, A.; Pham, Y.; Huynh, M.G.; Woo, Y.; Alani, A.W. Liposomes produced by microfluidics and extrusion: A comparison for scale-up purposes. Nanomedicine 2019, 18, 146–156. [Google Scholar] [CrossRef]
- Garg, S.; Heuck, G.; Ip, S.; Ramsay, E. Microfluidics: A transformational tool for nanomedicine development and production. J. Drug Target. 2016, 24, 821–835. [Google Scholar] [CrossRef]
- Kastner, E.; Kaur, R.; Lowry, D.; Moghaddam, B.; Wilkinson, A.; Perrie, Y. High-throughput manufacturing of size-tuned liposomes by a new microfluidics method using enhanced statistical tools for characterization. Int. J. Pharm. 2014, 477, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Mazumder, R.; Padhi, S. Glycerosomes: Advanced liposomal drug delivery system. Indian J. Pharm. Sci. 2020, 82, 385–397. [Google Scholar] [CrossRef]
- Patil, Y.P.; Jadhav, S.J.C. Novel methods for liposome preparation. Chem. Phys. Lipids 2014, 177, 8–18. [Google Scholar] [PubMed]
- Penoy, N.; Grignard, B.; Evrard, B.; Piel, G. A supercritical fluid technology for liposome production and comparison with the film hydration method. Int. J. Pharm. 2021, 592, 120093. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ansari, V.A.; Singh, K.; Rakib, M. Liposomal Cosmeceuticals: New Era of Anti-Ageing Formulations. World J. Pharm. Res. 2018, 7, 202–215. [Google Scholar]
- Kao, C.-Y.; Papoutsakis, E.T. Extracellular vesicles: Exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Curr. Opin. Biotechnol. 2019, 60, 89–98. [Google Scholar] [CrossRef]
- Kenari, A.N.; Cheng, L.; Hill, A.F. Methods for loading therapeutics into extracellular vesicles and generating extracellular vesicles mimetic-nanovesicles. Methods 2020, 177, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.A.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F. Methodological guidelines to study extracellular vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Martin, K.; FitzGerald, S.; O’sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [Green Version]
- Szatanek, R.; Baran, J.; Siedlar, M.; Baj-Krzyworzeka, M. Isolation of extracellular vesicles: Determining the correct approach. Int. J. Mol. Med. 2015, 36, 11–17. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular vesicles: Composition, biological relevance, and methods of study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Gardiner, C.; Vizio, D.D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef]
- Livshits, M.A.; Khomyakova, E.; Evtushenko, E.G.; Lazarev, V.N.; Kulemin, N.A.; Semina, S.E.; Generozov, E.V.; Govorun, V.M. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 2015, 5, 17319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.D.; Shah, S.J.M. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Talebjedi, B.; Tasnim, N.; Hoorfar, M.; Mastromonaco, G.F.; De Almeida Monteiro Melo Ferraz, M. Exploiting microfluidics for extracellular vesicle isolation and characterization: Potential use for standardized embryo quality assessment. Front. Vet. Sci. 2021, 7, 620809. [Google Scholar] [CrossRef] [PubMed]
- Lucchetti, D.; Fattorossi, A.; Sgambato, A. Extracellular vesicles in oncology: Progress and pitfalls in the methods of isolation and analysis. Biotechnol. J. 2019, 14, 1700716. [Google Scholar] [CrossRef] [Green Version]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [Green Version]
- Linares, R.; Tan, S.; Gounou, C.; Arraud, N.; Brisson, A.R. High-speed centrifugation induces aggregation of extracellular vesicles. J. Extracell. Vesicles 2015, 4, 29509. [Google Scholar] [CrossRef]
- Ma, C.; Jiang, F.; Ma, Y.; Wang, J.; Li, H.; Zhang, J. Isolation and detection technologies of extracellular vesicles and application on cancer diagnostic. Nanotechnol. Microtechnol. Drug Deliv. Syst. 2019, 17, 1559325819891004. [Google Scholar] [CrossRef]
- Liangsupree, T.; Multia, E.; Riekkola, M.-L. Modern isolation and separation techniques for extracellular vesicles. J. Chromatogr. A 2021, 1636, 461773. [Google Scholar]
- Böing, A.N.; Van Der Pol, E.; Grootemaat, A.E.; Coumans, F.A.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef]
- Lozano-Ramos, I.; Bancu, I.; Oliveira-Tercero, A.; Armengol, M.P.; Menezes-Neto, A.; Portillo, H.A.D.; Lauzurica-Valdemoros, R.; Borràs, F.E. Size-exclusion chromatography-based enrichment of extracellular vesicles from urine samples. J. Extracell. Vesicles 2015, 4, 27369. [Google Scholar] [CrossRef] [Green Version]
- Lobb, R.J.; Becker, M.; Wen Wen, S.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized cholesterol-siRNA chemistry improves productive loading onto extracellular vesicles. Mol. Ther. 2015, 4, 27031. [Google Scholar]
- Yoon, Y.; Kim, S.; Lee, J.; Choi, J.; Kim, R.-K.; Lee, S.-J.; Sul, O.; Lee, S.-B. Clogging-free microfluidics for continuous size-based separation of microparticles. Sci. Rep. 2016, 6, 26531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, W.; Liu, G.; Lu, W.; Fu, J. Continuous-flow microfluidic blood cell sorting for unprocessed whole blood using surface-micromachined microfiltration membranes. Lab Chip 2014, 14, 2565–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-Y.; Huang, C.-K.; Lu, Y.-W. Enhancement of microfluidic particle separation using cross-flow filters with hydrodynamic focusing. Biomicrofluidics 2016, 10, 011906. [Google Scholar] [CrossRef] [Green Version]
- Haraszti, R.A.; Miller, R.; Stoppato, M.; Sere, Y.Y.; Coles, A.; Didiot, M.-C.; Wollacott, R.; Sapp, E.; Dubuke, M.L.; Li, X.; et al. Exosomes produced from 3D cultures of MSCs by tangential flow filtration show higher yield and improved activity. Mol. Ther. 2018, 26, 2838–2847. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Park, J.; Jung, J.-H.; Lee, R.; Park, J.-H.; Yuk, J.M.; Hwang, H.; Yeon, J.H. Cyclic tangential flow filtration system for isolation of extracellular vesicles. APL Bioeng. 2021, 5, 016103. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.-Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef]
- Kordelas, L.; Rebmann, V.; Ludwig, A.; Radtke, S.; Ruesing, J.; Doeppner, T.; Epple, M.; Horn, P.; Beelen, D.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of extracellular vesicles: General methodologies and latest trends. Biomed. Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef]
- Zarovni, N.; Corrado, A.; Guazzi, P.; Zocco, D.; Lari, E.; Radano, G.; Muhhina, J.; Fondelli, C.; Gavrilova, J.; Chiesi, A. Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods 2015, 87, 46–58. [Google Scholar] [CrossRef]
- Alvarez, M.L.; Khosroheidari, M.; Ravi, R.K.; DiStefano, J.K. Comparison of protein, microRNA, and mRNA yields using different methods of urinary exosome isolation for the discovery of kidney disease biomarkers. Kidney Int. 2012, 82, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 2014, 3, 24858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.E.; Kim, G.; Kim, M.; Park, D.; Kang, H.J.; Lee, M.; Huh, N. A direct extraction method for microRNAs from exosomes captured by immunoaffinity beads. Anal. Biochem. 2012, 431, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Koliha, N.; Wiencek, Y.; Heider, U.; Jüngst, C.; Kladt, N.; Krauthäuser, S.; Johnston, I.C.; Bosio, A.; Schauss, A.; Wild, S. A novel multiplex bead-based platform highlights the diversity of extracellular vesicles. J. Extracell. Vesicles 2016, 5, 29975. [Google Scholar] [CrossRef] [PubMed]
- Boriachek, K.; Masud, M.K.; Palma, C.; Phan, H.-P.; Yamauchi, Y.; Hossain, M.S.A.; Nguyen, N.-T.; Salomon, C.; Shiddiky, M.J.A. Avoiding pre-isolation step in exosome analysis: Direct isolation and sensitive detection of exosomes using gold-loaded nanoporous ferric oxide nanozymes. Anal. Chem. 2019, 91, 3827–3834. [Google Scholar] [CrossRef] [Green Version]
- Biscans, A.; Haraszti, R.A.; Echeverria, D.; Miller, R.; Didiot, M.-C.; Nikan, M.; Roux, L.; Aronin, N.; Khvorova, A. Hydrophobicity of lipid-conjugated siRNAs predicts productive loading to small extracellular vesicles. Mol. Ther. 2018, 26, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.G.; Mohamadi, R.M.; Poudineh, M.; Kermanshah, L.; Ahmed, S.; Safaei, T.S.; Stojcic, J.; Nam, R.K.; Sargent, E.H.; Kelley, S.O. Interrogating circulating microsomes and exosomes using metal nanoparticles. Nano Micro Small 2016, 12, 727–732. [Google Scholar] [CrossRef]
- Ghosh, A.; Davey, M.; Chute, I.C.; Griffiths, S.G.; Lewis, S.; Chacko, S.; Barnett, D.; Crapoulet, N.; Fournier, S.; Joy, A. Rapid isolation of extracellular vesicles from cell culture and biological fluids using a synthetic peptide with specific affinity for heat shock proteins. PLoS ONE 2014, 9, e110443. [Google Scholar] [CrossRef] [Green Version]
- Balaj, L.; Atai, N.A.; Chen, W.; Mu, D.; Tannous, B.A.; Breakefield, X.O.; Skog, J.; Maguire, C.A. Heparin affinity purification of extracellular vesicles. Sci. Rep. 2015, 5, 10266. [Google Scholar] [CrossRef]
- Reiner, A.T.; Witwer, K.W.; Van Balkom, B.W.; De Beer, J.; Brodie, C.; Corteling, R.L.; Gabrielsson, S.; Gimona, M.; Ibrahim, A.G.; De Kleijn, D. Concise review: Developing best-practice models for the therapeutic use of extracellular vesicles. Stem Cells Transl. Med. 2017, 6, 1730–1739. [Google Scholar] [CrossRef] [Green Version]
- Sidhom, K.; Obi, P.O.; Saleem, A. A review of exosomal isolation methods: Is size exclusion chromatography the best option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Asghari, M.; Aslan, M.K.; Yilmaz, A.; Mateescu, B.; Stavrakis, S.; deMello, A.J. Microfluidics for extracellular vesicle separation and mimetic synthesis: Recent advances and future perspectives. Chem. Eng. J. 2021, 404, 126110. [Google Scholar] [CrossRef]
- Guo, S.-C.; Tao, S.-C.; Dawn, H. Microfluidics-based on-a-chip systems for isolating and analysing extracellular vesicles. J. Extracell. Vesicles 2018, 7, 1508271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Naranjo, J.C.; Wu, H.-J.; Ugaz, V.M. Microfluidics for exosome isolation and analysis: Enabling liquid biopsy for personalized medicine. Lab Chip 2017, 17, 3558–3577. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.T.; Kim, J.; Jang, S.C.; Choi, E.-J.; Gho, Y.S.; Park, J. Microfluidic filtration system to isolate extracellular vesicles from blood. Lab Chip 2012, 12, 5202–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Guo, J.; Tian, F.; Yang, N.; Yan, F.; Ding, Y.; Wei, J.; Hu, G.; Nie, G.; Sun, J. Field-free isolation of exosomes from extracellular vesicles by microfluidic viscoelastic flows. ACS Nano 2017, 11, 6968–6976. [Google Scholar] [CrossRef] [Green Version]
- Bruus, H. Acoustofluidics 7: The acoustic radiation force on small particles. Lab Chip 2012, 12, 1014–1021. [Google Scholar] [CrossRef]
- Destgeer, G.; Sung, H.J. Recent advances in microfluidic actuation and micro-object manipulation via surface acoustic waves. Lab. Chip. 2015, 15, 2722–2738. [Google Scholar] [CrossRef]
- El-Hammadi, M.M.; Arias, J.L. An update on liposomes in drug delivery: A patent review (2014–2018). Expert Opin. Ther. Pat. 2019, 29, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Margiana, R.; Bokov, D.O.; Abdelbasset, W.K.; Kouhbanani, M.A.J.; Varma, R.S.; Marofi, F.; Jarahian, M.; Beheshtkhoo, N. Liposomes: Structure, Biomedical Applications, and Stability Parameters with Emphasis on Cholesterol. Front. Bioeng. Biotechnol. 2021, 9, 705886. [Google Scholar]
- Garg, T.; K Goyal, A. Liposomes: Targeted and controlled delivery system. Drug Deliv. Lett. 2014, 4, 62–71. [Google Scholar] [CrossRef]
- Leigh, S.; Leigh, M.L.S.; Van Hoogevest, P. Method of Solubilizing Biologically Active Compounds. U.S. Patent US20110244028A1, 6 October 2011. [Google Scholar]
- Pauli, G.; Tang, W.-L.; Li, S.-D. Development and characterization of the solvent-assisted active loading technology (SALT) for liposomal loading of poorly water-soluble compounds. Pharmaceutics 2019, 11, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Zhao, Y.; May, J.P.; Chen, I.-W.; Undzys, E.; Li, S.-D. A study of liposomal formulations to improve the delivery of aquated cisplatin to a multidrug resistant tumor. Pharm. Res. 2015, 32, 3261–3268. [Google Scholar] [CrossRef]
- Kim, E.-M.; Jeong, H.-J. Liposomes: Biomedical Applications. Chonnam Med. J. 2021, 57, 27. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J. Extracell. Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef]
- Han, Y.; Jones, T.W.; Dutta, S.; Zhu, Y.; Wang, X.; Narayanan, S.P.; Fagan, S.C.; Zhang, D. Overview and update on methods for cargo loading into extracellular vesicles. Processes 2021, 9, 356. [Google Scholar] [CrossRef]
- Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Sutaria, D.S.; Badawi, M.; Phelps, M.A.; Schmittgen, T.D. Achieving the promise of therapeutic extracellular vesicles: The devil is in details of therapeutic loading. Pharm. Res. 2017, 34, 1053–1066. [Google Scholar]
- Marcus, M.E.; Leonard, J.N. FedExosomes: Engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals 2013, 6, 659–680. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Brozovich, A.; Pasto, A.; Sushnitha, M.; Martinez, J.O.; Evangelopoulos, M.; Boada, C.; Tasciotti, E.; Taraballi, F. Bioinspired extracellular vesicles: Lessons learned from nature for biomedicine and bioengineering. Nanomaterials 2020, 10, 2172. [Google Scholar] [CrossRef]
- Agrawal, A.K.; Aqil, F.; Jeyabalan, J.; Spencer, W.A.; Beck, J.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.; Munagala, R.; Bondada, S.; et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomedicine 2017, 13, 1627–1636. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Miller, R.; Didiot, M.-C.; Biscans, A.; Alterman, J.F.; Hassler, M.R.; Roux, L.; Echeverria, D.; Sapp, E.; DiFiglia, M. Optimized cholesterol-siRNA chemistry improves productive loading onto extracellular vesicles. Mol. Ther. 2018, 26, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Familtseva, A.; Jeremic, N.; Tyagi, S.C. Exosomes: Cell-created drug delivery systems. Mol. Cell. Biochem. 2019, 459, 1–6. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, S.; Almurisi, S.H.; Al-Japairai, K.; Hilles, A.R.; Alelwani, W.; Bannunah, A.M.; Alshammari, F.; Alheibshy, F. Ibuprofen-Loaded Chitosan–Lipid Nanoconjugate Hydrogel with Gum Arabic: Green Synthesis, Characterisation, In Vitro Kinetics Mechanistic Release Study and PGE2 Production Test. Gels 2021, 7, 254. [Google Scholar] [CrossRef]
- Ortega, A.; Martinez-Arroyo, O.; Forner, M.J.; Cortes, R. Exosomes as drug delivery systems: Endogenous nanovehicles for treatment of systemic lupus erythematosus. Pharmaceutics 2020, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wu, T.; Zhang, D.; Zhang, Z. Cell or cell membrane-based drug delivery systems. Theranostics 2015, 5, 863. [Google Scholar] [CrossRef] [Green Version]
- Al-Japairai, K.A.S.; Mahmood, S.; Almurisi, S.H.; Venugopal, J.R.; Hilles, A.R.; Azmana, M.; Raman, S. Current trends in polymer microneedle for transdermal drug delivery. Int. J. Pharm. 2020, 587, 119673. [Google Scholar] [CrossRef] [PubMed]
- Lennaárd, A.J.; Mamand, D.R.; Wiklander, R.J.; El Andaloussi, S.; Wiklander, O.P.B. Optimised Electroporation for Loading of Extracellular Vesicles with Doxorubicin. Pharmaceutics 2022, 14, 38. [Google Scholar] [CrossRef]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef]
- Jeyaram, A.; Lamichhane, T.N.; Wang, S.; Zou, L.; Dahal, E.; Kronstadt, S.M.; Levy, D.; Parajuli, B.; Knudsen, D.R.; Chao, W. Enhanced loading of functional miRNA cargo via pH gradient modification of extracellular vesicles. Mol. Ther. 2020, 28, 975–985. [Google Scholar] [CrossRef]
- Sibum, I.; Hagedoorn, P.; de Boer, A.H.; Frijlink, H.W.; Grasmeijer, F. Challenges for pulmonary delivery of high powder doses. Int. J. Pharm. 2018, 548, 325–336. [Google Scholar] [CrossRef]
- Chellappan, D.K.; Prasher, P.; Saravanan, V.; Yee, V.S.V.; Chi, W.C.W.; Wong, J.W.; Wong, J.K.; Wong, J.T.; Wan, W.; Chellian, J. Protein and peptide delivery to lungs by using advanced targeted drug delivery. Chem. Biol. Interact. 2022, 351, 109706. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2019, 18, 19–40. [Google Scholar]
- Movia, D.; Prina-Mello, A. Preclinical development of orally inhaled drugs (OIDs)—Are animal models predictive or shall we move towards in vitro non-animal models? Animals 2020, 10, 1259. [Google Scholar] [CrossRef]
- Takano, M.; Kawami, M.; Aoki, A.; Yumoto, R. Receptor-mediated endocytosis of macromolecules and strategy to enhance their transport in alveolar epithelial cells. Expert Opin. Drug Deliv. 2015, 12, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Lavorini, F.; Buttini, F.; Usmani, O.S. 100 years of drug delivery to the lungs. Concepts Princ. Pharmacol. 2019, 260, 143–159. [Google Scholar]
- Moon, C.; Smyth, H.D.; Watts, A.B.; Williams, R.O. Delivery technologies for orally inhaled products: An update. AAPS PharmSciTech 2019, 20, 117. [Google Scholar] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.d.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar]
- Claridge, B.; Lozano, J.; Poh, Q.H.; Greening, D.W. Development of extracellular vesicle therapeutics: Challenges, considerations, and opportunities. Front. Cell Dev. Biol. 2021, 9, 734720. [Google Scholar] [CrossRef]
- Murgia, X.; de Souza Carvalho, C.; Lehr, C.-M. Overcoming the pulmonary barrier: New insights to improve the efficiency of inhaled therapeutics. Eur. J. Nanomed. 2014, 6, 157–169. [Google Scholar] [CrossRef]
- Lechanteur, A.; Evrard, B. Influence of composition and spray-drying process parameters on carrier-free DPI properties and behaviors in the lung: A review. Pharmaceutics 2020, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, I.M.; El-Baz, N.M.; Yacoub, M.H. Inhaled nano-and microparticles for drug delivery. Glob. Cardiol. Sci. Pract. 2015, 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Tena, A.F.; Clarà, P.C. Deposition of inhaled particles in the lungs. Arch. Bronconeumol. (Engl. Ed.) 2012, 48, 240–246. [Google Scholar] [CrossRef]
- Newman, S.P. Drug delivery to the lungs: Challenges and opportunities. Ther. Deliv. 2017, 8, 647–661. [Google Scholar] [CrossRef]
- Abdelaziz, H.M.; Gaber, M.; Abd-Elwakil, M.M.; Mabrouk, M.T.; Elgohary, M.M.; Kamel, N.M.; Kabary, D.M.; Freag, M.S.; Samaha, M.W.; Mortada, S.M. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J. Control. Release 2018, 269, 374–392. [Google Scholar] [PubMed]
- Muralidharan, P.; Malapit, M.; Mallory, E.; Hayes, D., Jr.; Mansour, H.M. Inhalable nanoparticulate powders for respiratory delivery. Nanomedicine 2015, 11, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.S. Physiological Factors Affecting Lung Deposition. J. Aerosol Med. Pulm. Drug Deliv. 2021, 34, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Darquenne, C. Deposition mechanisms. J. Aerosol Med. Pulm. Drug Deliv. 2020, 33, 181–185. [Google Scholar] [CrossRef]
- Hofmann, W. Modelling inhaled particle deposition in the human lung—A review. J Aerosol Sci. 2011, 42, 693–724. [Google Scholar] [CrossRef]
- Janssen, W.J.; Stefanski, A.L.; Bochner, B.S.; Evans, C.M. Control of lung defence by mucins and macrophages: Ancient defence mechanisms with modern functions. Eur. Respir. J. 2016, 48, 1201–1214. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Gupta, N.; Ahsan, F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur. J. Pharm. Biopharm. 2015, 89, 163–174. [Google Scholar] [CrossRef]
- Malamatari, M.; Charisi, A.; Malamataris, S.; Kachrimanis, K.; Nikolakakis, I. Spray drying for the preparation of nanoparticle-based drug formulations as dry powders for inhalation. Processes 2020, 8, 788. [Google Scholar] [CrossRef]
- Ourique, A.F.; dos Santos Chaves, P.; Souto, G.D.; Pohlmann, A.R.; Guterres, S.S.; Beck, R.C.R. Redispersible liposomal-N-acetylcysteine powder for pulmonary administration: Development, in vitro characterization and antioxidant activity. Eur. J. Pharm. Sci. 2014, 65, 174–182. [Google Scholar] [CrossRef]
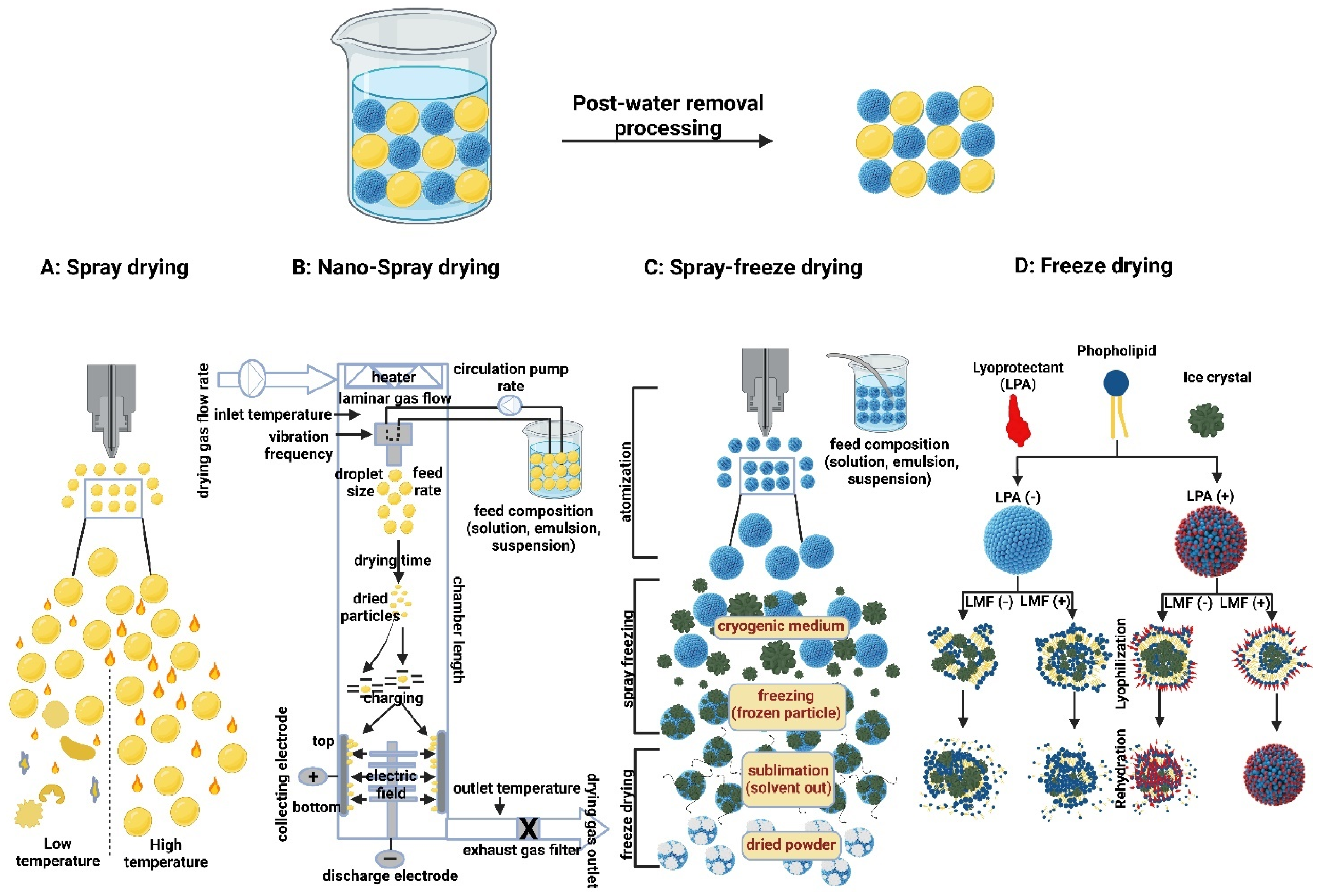
- Sharma, A.; Khamar, D.; Cullen, S.; Hayden, A.; Hughes, H. Innovative Drying Technologies for Biopharmaceuticals. Int. J. Pharm. 2021, 609, 121115. [Google Scholar]
- Kusuma, G.D.; Barabadi, M.; Tan, J.L.; Morton, D.A.; Frith, J.E.; Lim, R. To protect and to preserve: Novel preservation strategies for extracellular vesicles. Front. Pharmacol. 2018, 9, 1199. [Google Scholar] [CrossRef] [Green Version]
- Arpagaus, C. Pharmaceutical particle engineering via nano spray drying—Process parameters and application examples on the laboratory-scale. Int. J. Med. Nano Res 2018, 5, 026. [Google Scholar] [CrossRef]
- Yang, E.; Yu, H.; Choi, S.; Park, K.-M.; Jung, H.-S.; Chang, P.-S. An Advanced Lyophilization Toward Intact Lipid Nanovesicles: Liquid-mediated Freezing with Cryoprotectant to Retain the Integrity of Lipid Nanovesicles. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Kwon, Y.-B.; Kang, J.-H.; Han, C.-S.; Kim, D.-W.; Park, C.-W. The effect of particle size and surface roughness of spray-dried bosentan microparticles on aerodynamic performance for dry powder inhalation. Pharmaceutics 2020, 12, 765. [Google Scholar] [CrossRef]
- Garbuzenko, O.B.; Mainelis, G.; Taratula, O.; Minko, T. Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention. Cancer Biol. Med. 2014, 11, 44. [Google Scholar]
- Abdulbaqi, I.M.; Assi, R.A.; Yaghmur, A.; Darwis, Y.; Mohtar, N.; Parumasivam, T.; Saqallah, F.G.; Wahab, H.A. Pulmonary delivery of anticancer drugs via lipid-based nanocarriers for the treatment of lung cancer: An update. Pharmaceuticals 2021, 14, 725. [Google Scholar] [CrossRef]
- Bermeo, M.; El Hadri, N.; Ravaux, F.; Zaki, A.; Zou, L.; Jouiad, M. Adsorption capacities of hygroscopic materials based on NaCl-TiO2 and NaCl-SiO2 core/shell particles. J. Nanotechnol. 2020, 2020, 3683629. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Gu, Z.; Lu, W.; Zhang, L.; Okuda, T.; Fujioka, K.; Luo, H.; Yu, C.W. Atmospheric humidity and particle charging state on agglomeration of aerosol particles. Atmos. Environ. 2019, 197, 141–149. [Google Scholar] [CrossRef]
- Zhou, Q.T.; Gengenbach, T.; Denman, J.A.; Yu, H.H.; Li, J.; Chan, H.K. Synergistic antibiotic combination powders of colistin and rifampicin provide high aerosolization efficiency and moisture protection. AAPS J. 2014, 16, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Schattling, P.; Dreier, C.; Städler, B. Janus subcompartmentalized microreactors. Soft Matter 2015, 11, 5327–5335. [Google Scholar] [CrossRef] [Green Version]
- Georgatzakou, H.T.; Pavlou, E.G.; Papageorgiou, E.G.; Papassideri, I.S.; Kriebardis, A.G.; Antonelou, M.H. The Multi-Faced Extracellular Vesicles in the Plasma of Chronic Kidney Disease Patients. Front. Cell Dev. Biol. 2020, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Kaialy, W. A review of factors affecting electrostatic charging of pharmaceuticals and adhesive mixtures for inhalation. Int. J. Pharm. 2016, 503, 262–276. [Google Scholar] [PubMed]
- Sarangi, S.; Thalberg, K.; Frenning, G. Effect of fine particle shape on the stability and performance of adhesive mixtures intended for inhalation. Powder Technol. 2021, 385, 299–305. [Google Scholar] [CrossRef]
- Dabbagh, A.; Abu Kasim, N.H.; Yeong, C.H.; Wong, T.W.; Abdul Rahman, N. Critical parameters for particle-based pulmonary delivery of chemotherapeutics. J. Aerosol Med. Pulm. Drug Deliv. 2018, 31, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Jańczewski, D.; Zhu, X.; Quintana, R.; He, T.; Neoh, K.G. Surface charge control for zwitterionic polymer brushes: Tailoring surface properties to antifouling applications. J. Colloid Interface Sci. 2015, 452, 43–53. [Google Scholar] [CrossRef]
- Adel, I.M.; ElMeligy, M.F.; Abdelrahim, M.E.; Maged, A.; Abdelkhalek, A.A.; Abdelmoteleb, A.M.; Elkasabgy, N.A. Design and characterization of spray-dried proliposomes for the pulmonary delivery of curcumin. Int. J. Nanomed. 2021, 16, 2667. [Google Scholar] [CrossRef]
- Chen, J.; Li, P.; Zhang, T.; Xu, Z.; Huang, X.; Wang, R.; Du, L. Review on Strategies and Technologies for Exosome Isolation and Purification. Front. Bioeng. Biotechnol. 2022, 9, 811971. [Google Scholar] [CrossRef]
- Chaurasiya, B.; Zhao, Y.-Y. Dry powder for pulmonary delivery: A comprehensive review. Pharmaceutics 2020, 13, 31. [Google Scholar] [CrossRef]
- Anderson, S.; Atkins, P.; Bäckman, P.; Cipolla, D.; Clark, A.; Daviskas, E.; Disse, B.; Entcheva-Dimitrov, P.; Fuller, R.; Gonda, I. Inhaled Medicines: Past, Present, and Future. Pharmacol. Rev. 2022, 74, 48–118. [Google Scholar]
- Holtzman, J.; Lee, H. Emerging role of extracellular vesicles in the respiratory system. Exp. Mol. Med. 2020, 52, 887–895. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Araya, J.; Ochiya, T.; Kuwano, K. Extracellular vesicle-mediated cellular crosstalk in lung repair, remodelling and regeneration. Eur. Respir. Rev. 2022, 31, 210106. [Google Scholar] [CrossRef]
- Talaat, M.; Si, X.A.; Xi, J. Effect of MDI Actuation Timing on Inhalation Dosimetry in a Human Respiratory Tract Model. Pharmaceuticals 2022, 15, 61. [Google Scholar] [CrossRef]
- Stein, S.W.; Thiel, C.G. The history of therapeutic aerosols: A chronological review. J. Aerosol Med. Pulm. Drug Deliv. 2017, 30, 20–41. [Google Scholar] [CrossRef]
- Farr, S.J.; Kellaway, I.W.; Carman-Meakin, B. Assessing the potential of aerosol-generated liposomes from pressurised pack formulations. J. Control. Release 1987, 5, 119–127. [Google Scholar] [CrossRef]
- Vyas, S.; Sakthivel, T. Pressurized pack-based liposomes for pulmonary targeting of isoprenaline—Development and characterization. J. Microencapsul. 1994, 11, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Wauthoz, N.; Amighi, K. Phospholipids in pulmonary drug delivery. Eur. J. Lipid Sci. Technol. 2014, 116, 1114–1128. [Google Scholar] [CrossRef]
- Cipolla, D.; Wu, H.; Gonda, I.; Chan, H.-K. Aerosol performance and stability of liposomes containing ciprofloxacin nanocrystals. J. Aerosol Med. Pulm. Drug Deliv. 2015, 28, 411–422. [Google Scholar] [CrossRef]
- Weers, J. Comparison of phospholipid-based particles for sustained release of ciprofloxacin following pulmonary administration to bronchiectasis patients. Pulm. Ther. 2019, 5, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, D.P.; Vital, J.; Leiva, M.C.; Gonçalves, L.M.; Taboada, P.; Remuñán-López, C.; Vítor, J.; Almeida, A.J. Transfection of pulmonary cells by stable pDNA-polycationic hybrid nanostructured particles. Nanomedicine 2019, 14, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Yousaf, S.; Najlah, M.; Ahmed, W.; Elhissi, A. Proliposome powder or tablets for generating inhalable liposomes using a medical nebulizer. J. Pharm. Investig. 2021, 51, 61–73. [Google Scholar] [CrossRef]
- Khan, I.; Yousaf, S.; Subramanian, S.; Alhnan, M.A.; Ahmed, W.; Elhissi, A. Proliposome powders for the generation of liposomes: The influence of carbohydrate carrier and separation conditions on crystallinity and entrapment of a model antiasthma steroid. AAPS PharmSciTech 2018, 19, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Jain, M.; Wan, K.-W.; Ahmed, W.; Albed Alhnan, M.; Phoenix, D.A.; Taylor, K.M.; Elhissi, A. Ethanol-based proliposome delivery systems of paclitaxel for in vitro application against brain cancer cells. J. Liposome Res. 2018, 28, 74–85. [Google Scholar] [CrossRef]
- Bahr, M.M.; Amer, M.S.; Abo-El-Sooud, K.; Abdallah, A.N.; El-Tookhy, O.S. Preservation techniques of stem cells extracellular vesicles: A gate for manufacturing of clinical grade therapeutic extracellular vesicles and long-term clinical trials. Int. J. Vet. Sci. Med. 2020, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A review of its classification, isolation techniques, storage, diagnostic and targeted therapy applications. Int. J. Nanomed. 2020, 15, 6917. [Google Scholar] [CrossRef] [PubMed]
- Tamura, G. Comparison of the aerosol velocity of Respimat® soft mist inhaler and seven pressurized metered dose inhalers. Allergol. Int. 2015, 64, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.C.; McConville, J.T. The function and performance of aqueous aerosol devices for inhalation therapy. J. Pharm. Pharmacol. 2016, 68, 556–578. [Google Scholar] [PubMed]
- Ibrahim, M.; Verma, R.; Garcia-Contreras, L. Inhalation drug delivery devices: Technology update. Med Devices (Auckl) 2015, 8, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Khairnar, S.V.; Jain, D.D.; Tambe, S.M.; Chavan, Y.R.; Amin, P.D. Nebulizer systems: A new frontier for therapeutics and targeted delivery. Ther. Deliv. 2022, 13, 31–49. [Google Scholar] [CrossRef]
- Bassetti, M.; Vena, A.; Russo, A.; Peghin, M. Inhaled liposomal antimicrobial delivery in lung infections. Drugs 2020, 80, 1309–1318. [Google Scholar] [CrossRef]
- Wang, X.; Xie, Z.; Zhao, J.; Zhu, Z.; Yang, C.; Liu, Y. Prospects of Inhaled Phage Therapy for Combatting Pulmonary Infections. Front. Cell. Infect. Microbiol. 2021, 11, 758392. [Google Scholar] [CrossRef]
- Bianco, F.; Salomone, F.; Milesi, I.; Murgia, X.; Bonelli, S.; Pasini, E.; Dellacà, R.; Ventura, M.L.; Pillow, J. Aerosol drug delivery to spontaneously-breathing preterm neonates: Lessons learned. Respir. Res. 2021, 22, 1–31. [Google Scholar]
- Chandel, A.; Goyal, A.K.; Ghosh, G.; Rath, G. Recent advances in aerosolised drug delivery. Biomed. Pharmacother. 2019, 112, 108601. [Google Scholar] [CrossRef] [PubMed]
- Park, H.M.; Chang, K.H.; Moon, S.-H.; Park, B.J.; Yoo, S.K.; Nam, K.C. In vitro delivery efficiencies of nebulizers for different breathing patterns. Biomed. Eng. Online 2021, 20, 1–14. [Google Scholar] [CrossRef]
- Shi, M.M.; Yang, Q.Y.; Monsel, A.; Yan, J.Y.; Dai, C.X.; Zhao, J.Y.; Shi, G.C.; Zhou, M.; Zhu, X.M.; Li, S.K.; et al. Preclinical efficacy and clinical safety of clinical-grade nebulized allogenic adipose mesenchymal stromal cells-derived extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12134. [Google Scholar] [CrossRef]
- Dong, L.; Wang, Y.; Zheng, T.; Pu, Y.; Ma, Y.; Qi, X.; Zhang, W.; Xue, F.; Shan, Z.; Liu, J. Hypoxic hUCMSC-derived extracellular vesicles attenuate allergic airway inflammation and airway remodeling in chronic asthma mice. Stem Cell. Res. Ther. 2021, 12, 4. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart nanoparticles for drug delivery application: Development of versatile nanocarrier platforms in biotechnology and nanomedicine. J. Nanomater. 2019, 2019, e3702518. [Google Scholar] [CrossRef]
- Witika, B.A.; Mweetwa, L.L.; Tshiamo, K.O.; Edler, K.; Matafwali, S.K.; Ntemi, P.V.; Chikukwa, M.T.; Makoni, P.A. Vesicular drug delivery for the treatment of topical disorders: Current and future perspectives. J. Pharm. Pharmacol. 2021, 73, 1427–1441. [Google Scholar] [CrossRef]
- Rodríguez, D.A.; Vader, P. Extracellular Vesicle-Based Hybrid Systems for Advanced Drug Delivery. Pharmaceutics 2022, 14, 267. [Google Scholar] [CrossRef]
- Tang, T.-T.; Wang, B.; Lv, L.-L.; Liu, B.-C. Extracellular vesicle-based Nanotherapeutics: Emerging frontiers in anti-inflammatory therapy. Theranostics 2020, 10, 8111. [Google Scholar] [CrossRef]
- Patras, L.; Ionescu, A.E.; Munteanu, C.; Hajdu, R.; Kosa, A.; Porfire, A.; Licarete, E.; Rauca, V.F.; Sesarman, A.; Luput, L. Trojan horse treatment based on PEG-coated extracellular vesicles to deliver doxorubicin to melanoma in vitro and in vivo. Cancer Biol. Ther. 2022, 23, 1–16. [Google Scholar] [CrossRef]
- Mukherjee, D.; Paul, D.; Sarker, S.; Hasan, M.N.; Ghosh, R.; Prasad, S.E.; Vemula, P.K.; Das, R.; Adhikary, A.; Pal, S.K. Polyethylene Glycol-Mediated Fusion of Extracellular Vesicles with Cationic Liposomes for the Design of Hybrid Delivery Systems. ACS Appl. Bio. Mater. 2021, 4, 8259–8266. [Google Scholar] [CrossRef]
- Mohan, A.; Agarwal, S.; Clauss, M.; Britt, N.S.; Dhillon, N.K. Extracellular vesicles: Novel communicators in lung diseases. Respir. Res. 2020, 21, 175. [Google Scholar]
- Šutić, M.; Vukić, A.; Baranašić, J.; Försti, A.; Džubur, F.; Samaržija, M.; Jakopović, M.; Brčić, L.; Knežević, J. Diagnostic, predictive, and prognostic biomarkers in non-small cell lung cancer (NSCLC) management. J. Pers. Med. 2021, 11, 1102. [Google Scholar] [CrossRef]
- Gao, Y.; Qin, Y.; Wan, C.; Sun, Y.; Meng, J.; Huang, J.; Hu, Y.; Jin, H.; Yang, K. Small extracellular vesicles: A novel avenue for cancer management. Front. Oncol. 2021, 11, 441. [Google Scholar]
- Mehta, P.P.; Ghoshal, D.; Pawar, A.P.; Kadam, S.S.; Dhapte-Pawar, V.S. Recent advances in inhalable liposomes for treatment of pulmonary diseases: Concept to clinical stance. J. Drug Deliv. Sci. Technol. 2020, 56, 101509. [Google Scholar] [CrossRef]
- Pastor, L.; Vera, E.; Marin, J.M.; Sanz-Rubio, D. Extracellular Vesicles from Airway Secretions: New Insights in Lung Diseases. Int. J. Mol. Sci. 2021, 22, 583. [Google Scholar] [CrossRef]
- Zareba, L.; Szymanski, J.; Homoncik, Z.; Czystowska-Kuzmicz, M. Evs from balf—Mediators of inflammation and potential biomarkers in lung diseases. Int. J. Mol. Sci. 2021, 22, 3651. [Google Scholar] [CrossRef]
- Xiroudaki, S.; Schoubben, A.; Giovagnoli, S.; Rekkas, D.M. Dry Powder Inhalers in the Digitalization Era: Current Status and Future Perspectives. Pharmaceutics 2021, 13, 1455. [Google Scholar] [CrossRef]
- Sou, T.; Bergström, C.A. Contemporary formulation development for inhaled pharmaceuticals. J. Pharm. Sci. 2021, 110, 66–86. [Google Scholar]
- Ye, Y.; Ma, Y.; Zhu, J. The future of dry powder inhaled therapy: Promising or Discouraging for systemic disorders? Int. J. Pharm. 2022, 614, 121457. [Google Scholar] [CrossRef]
- Plaunt, A.J.; Nguyen, T.L.; Corboz, M.R.; Malinin, V.S.; Cipolla, D.C. Strategies to Overcome Biological Barriers Associated with Pulmonary Drug Delivery. Pharmaceutics 2022, 14, 302. [Google Scholar] [CrossRef]
- Ponkshe, P.; Feng, S.; Tan, C. Inhalable liposomes for treating lung diseases: Clinical development and challenges. Biomed. Mater. 2021, 16, 054101. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Cai, L.; Xu, C.; Sun, S.; Liu, Y.; Rosenecker, J.; Guan, S. Nanotechnologies in Delivery of DNA and mRNA Vaccines to the Nasal and Pulmonary Mucosa. Nanomaterials 2022, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Kong, Y.; Liu, Q.; Lu, Y.; Xing, H.; Lu, X.; Yang, Y.; Xu, J.; Li, N.; Zhao, D. Inhalable dry powder prepared from folic acid-conjugated docetaxel liposomes alters pharmacodynamic and pharmacokinetic properties relevant to lung cancer chemotherapy. Pulm. Pharmacol. Ther. 2019, 55, 50–61. [Google Scholar] [CrossRef]
- Gandhi, M.; Pandya, T.; Gandhi, R.; Patel, S.; Mashru, R.; Misra, A.; Tandel, H. Inhalable liposomal dry powder of gemcitabine-HCl: Formulation, in vitro characterization and in vivo studies. Int. J. Pharm. 2015, 496, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, H.; Lu, X.; Jiang, L.; Xi, X.; Liu, J.; Zhu, J. Development and evaluation of a dry powder formulation of liposome-encapsulated oseltamivir phosphate for inhalation. Drug Deliv. 2015, 22, 608–618. [Google Scholar] [CrossRef]
- Hamed, A.; Osman, R.; Al-Jamal, K.T.; Holayel, S.M.; Geneidi, A.-S. Enhanced antitubercular activity, alveolar deposition and macrophages uptake of mannosylated stable nanoliposomes. J. Drug Deliv. Sci. Technol. 2019, 51, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Honmane, S.; Hajare, A.; More, H.; Osmani, R.A.M.; Salunkhe, S. Lung delivery of nanoliposomal salbutamol sulfate dry powder inhalation for facilitated asthma therapy. J. Liposome Res. 2019, 29, 332–342. [Google Scholar] [CrossRef]
- Ye, T.; Yu, J.; Luo, Q.; Wang, S.; Chan, H.-K. Inhalable clarithromycin liposomal dry powders using ultrasonic spray freeze drying. Powder Technol. 2017, 305, 63–70. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, Y.; Ge, Y.; Hu, Y.; Li, M.; Jin, Y. Inhalation treatment of primary lung cancer using liposomal curcumin dry powder inhalers. Acta Pharm. Sin. B 2018, 8, 440–448. [Google Scholar] [CrossRef]
- Khatib, I.; Khanal, D.; Ruan, J.; Cipolla, D.; Dayton, F.; Blanchard, J.D.; Chan, H.-K. Ciprofloxacin nanocrystals liposomal powders for controlled drug release via inhalation. Int. J. Pharm. 2019, 566, 641–651. [Google Scholar] [CrossRef]
- Li, M.; Zhang, T.; Zhu, L.; Wang, R.; Jin, Y. Liposomal andrographolide dry powder inhalers for treatment of bacterial pneumonia via anti-inflammatory pathway. Int. J. Pharm. 2017, 528, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.; Pharande, R.; Bannalikar, A.; Gupta, P.; Gupta, U.; Mukne, A. Inhalable liposomes of Glycyrrhiza glabra extract for use in tuberculosis: Formulation, in vitro characterization, in vivo lung deposition, and in vivo pharmacodynamic studies. Drug Dev. Ind. Pharm. 2019, 45, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chennakesavulu, S.; Mishra, A.; Sudheer, A.; Sowmya, C.; Reddy, C.S.; Bhargav, E. Pulmonary delivery of liposomal dry powder inhaler formulation for effective treatment of idiopathic pulmonary fibrosis. Asian J. Pharm. Sci. 2018, 13, 91–100. [Google Scholar] [CrossRef]
- Zillen, D.; Beugeling, M.; Hinrichs, W.L.; Frijlink, H.W.; Grasmeijer, F. Natural and bioinspired excipients for dry powder inhalation formulations. Curr. Opin. Colloid Interface Sci. 2021, 56, 101497. [Google Scholar] [CrossRef]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Roda, M.A.; Xu, X.; Rezonzew, G.; Viera, L. Activated PMN exosomes: Pathogenic entities causing matrix destruction and disease in the lung. Cell 2019, 176, 113–126.e5. [Google Scholar] [CrossRef] [Green Version]
- Yuan, K.; Shamskhou, E.A.; Orcholski, M.E.; Nathan, A.; Reddy, S.; Honda, H.; Mani, V.; Zeng, Y.; Ozen, M.O.; Wang, L. Loss of endothelium-derived Wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation 2019, 139, 1710–1724. [Google Scholar] [CrossRef]
- Zhao, L.; Luo, H.; Li, X.; Li, T.; He, J.; Qi, Q.; Liu, Y.; Yu, Z. Exosomes derived from human pulmonary artery endothelial cells shift the balance between proliferation and apoptosis of smooth muscle cells. Cardiology 2017, 137, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3, 23743. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Nguyen, J. Exosomes as therapeutics: The implications of molecular composition and exosomal heterogeneity. J. Control. Release 2016, 228, 179–190. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Wang, X.; Rai, A.; Groot, M.; Jin, Y. Exosome-mediated small RNA delivery: A novel therapeutic approach for inflammatory lung responses. Mol. Ther. 2018, 26, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, J.; Zeng, C.; Wu, D.; Mu, Z.; Chen, B.; Xie, Y.; Ye, Y.; Liu, J. Curcumin increases exosomal TCF21 thus suppressing exosome-induced lung cancer. Oncotarget 2016, 7, 87081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Wu, F.; Hu, G.; Chen, L.; Xu, J.; Xu, P.; Wang, X.; Li, Y.; Liu, S.; Zhang, S. Autologous tumor cell–derived microparticle-based targeted chemotherapy in lung cancer patients with malignant pleural effusion. Sci. Transl. Med. 2019, 11, eaat5690. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.-M.; Zhuansun, Y.-X.; Chen, R.; Lin, L.; Lin, Y.; Li, J.-G. Mesenchymal stem cell exosomes promote immunosuppression of regulatory T cells in asthma. Exp. Cell Res. 2018, 363, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.F.; Borg, Z.D.; Goodwin, M.; Sokocevic, D.; Wagner, D.E.; Coffey, A.; Antunes, M.; Robinson, K.L.; Mitsialis, S.A.; Kourembanas, S. Systemic administration of human bone marrow-derived mesenchymal stromal cell extracellular vesicles ameliorates aspergillus hyphal extract-induced allergic airway inflammation in immunocompetent mice. Stem Cells Transl. Med. 2015, 4, 1302–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Takehara, K.; Koga, Y.; Hachisu, Y.; Utsugi, M.; Sawada, Y.; Saito, Y.; Yoshimi, S.; Yatomi, M.; Shin, Y.; Wakamatsu, I. Differential Discontinuation Profiles between Pirfenidone and Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 143. [Google Scholar] [CrossRef]
- Shentu, T.-P.; Huang, T.-S.; Cernelc-Kohan, M.; Chan, J.; Wong, S.S.; Espinoza, C.R.; Tan, C.; Gramaglia, I.; van der Heyde, H.; Chien, S. Thy-1 dependent uptake of mesenchymal stem cell-derived extracellular vesicles blocks myofibroblastic differentiation. Sci. Rep. 2017, 7, 18052. [Google Scholar] [CrossRef] [Green Version]
- Dinh, P.-U.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun. 2020, 11, 1064. [Google Scholar] [CrossRef] [Green Version]
- Royce, S.G.; Patel, K.P.; Mao, W.; Zhu, D.; Lim, R.; Samuel, C.S. Serelaxin enhances the therapeutic effects of human amnion epithelial cell-derived exosomes in experimental models of lung disease. Br. J. Pharmacol. 2019, 176, 2195–2208. [Google Scholar] [CrossRef]
- Krishnan, A.; Turner, A.M. Chronic Obstructive Pulmonary Disease: The Present and Future. Biomedicines 2022, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Stolk, J.; Broekman, W.; Mauad, T.; Zwaginga, J.J.; Roelofs, H.; Fibbe, W.E.; Oostendorp, J.; Bajema, I.; Versteegh, M.I.; Taube, C. A phase I study for intravenous autologous mesenchymal stromal cell administration to patients with severe emphysema. QJM 2016, 109, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truzzi, F.; Tibaldi, C.; Zhang, Y.; Dinelli, G. An overview on dietary polyphenols and their biopharmaceutical classification system (BCS). Int. J. Mol. Sci. 2021, 22, 5514. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Matoori, S.; Leroux, J.-C. Twenty-five years of polymersomes: Lost in translation? Mater. Horiz. 2020, 7, 1297–1309. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Graf, I.; Kuang, Y.; Zheng, X.; Haupt, M.; Majid, A.; Kilic, E.; Hermann, D.M.; Psychogios, M.-N.; Weber, M.S. Neural progenitor cell-derived extracellular vesicles enhance blood-brain barrier integrity by NF-κB (Nuclear Factor-κB)-Dependent regulation of ABCB1 (ATP-Binding Cassette Transporter B1) in Stroke Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1127–1145. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.-E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-derived extracellular vesicles breach the intact blood–brain barrier via transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Kooijmans, S.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Rankin-Turner, S.; Vader, P.; O’Driscoll, L.; Giebel, B.; Heaney, L.M.; Davies, O.G. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Adv. Drug Deliv. Rev. 2021, 173, 479–491. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome–liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, R.; Sasaki, Y.; Kawasaki, R.; Katagiri, K.; Sawada, S.-I.; Mukai, S.-A.; Akiyoshi, K. Magnetically navigated intracellular delivery of extracellular vesicles using amphiphilic nanogels. Bioconjug. Chem. 2019, 30, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Sawada, S.-I.; Sato, Y.T.; Kawasaki, R.; Yasuoka, J.-I.; Mizuta, R.; Sasaki, Y.; Akiyoshi, K. Nanogel hybrid assembly for exosome intracellular delivery: Effects on endocytosis and fusion by exosome surface polymer engineering. Biomater. Sci. 2020, 8, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.-A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Cheng, L.; Lu, Y.; Zhang, X.; Wang, Y.; Deng, J.; Zhou, J.; Liu, B.; Liu, J. Thermosensitive Exosome–Liposome Hybrid Nanoparticle-Mediated Chemoimmunotherapy for Improved Treatment of Metastatic Peritoneal Cancer. Adv. Sci. 2020, 7, 2000515. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, X.; Tang, J.; Lv, Q.; Liu, J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials 2021, 275, 120964. [Google Scholar] [CrossRef]
- Jhan, Y.-Y.; Prasca-Chamorro, D.; Zuniga, G.P.; Moore, D.M.; Kumar, S.A.; Gaharwar, A.K.; Bishop, C.J. Engineered extracellular vesicles with synthetic lipids via membrane fusion to establish efficient gene delivery. Int. J. Pharm. 2020, 573, 118802. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.; van de Wakker, S.I.; de Groot, E.M.; de Jong, O.G.; Gitz-François, J.J.; Seinen, C.S.; Sluijter, J.P.; Schiffelers, R.M.; Vader, P. Functional siRNA Delivery by Extracellular Vesicle–Liposome Hybrid Nanoparticles. Adv. Healthc. Mater. 2021, 11, 2101202. [Google Scholar] [CrossRef]
- Sun, L.; Fan, M.; Huang, D.; Li, B.; Xu, R.; Gao, F.; Chen, Y. Clodronate-loaded liposomal and fibroblast-derived exosomal hybrid system for enhanced drug delivery to pulmonary fibrosis. Biomaterials 2021, 271, 120761. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Z.; Tian, H.; Chen, X. Production and clinical development of nanoparticles for gene delivery. Mol. Ther. Methods Clin. Dev. 2016, 3, 16023. [Google Scholar] [CrossRef]
- Lu, M.; Xing, H.; Xun, Z.; Yang, T.; Zhao, X.; Cai, C.; Wang, D.; Ding, P. Functionalized extracellular vesicles as advanced therapeutic nanodelivery systems. Eur. J. Pharm. Sci. 2018, 121, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in anti-tumor treatments targeting the CD47/SIRPα axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.O.; He, M. Unlocking the power of exosomes for crossing biological barriers in drug delivery. Pharmaceutics 2021, 13, 122. [Google Scholar] [CrossRef]
- Ferraris, C.; Cavalli, R.; Panciani, P.P.; Battaglia, L. Overcoming the Blood–Brain Barrier: Successes and Challenges in Developing Nanoparticle-Mediated Drug Delivery Systems for the Treatment of Brain Tumours. Int. J. Nanomed. 2020, 15, 2999. [Google Scholar] [CrossRef]
- Joshi, B.S.; de Beer, M.A.; Giepmans, B.N.; Zuhorn, I.S. Endocytosis of extracellular vesicles and release of their cargo from endosomes. ACS Nano 2020, 14, 4444–4455. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, W.; Chen, X.; Wang, Q.; Li, C.; Chen, Q.; Zhang, Y.; Lu, Y.; Ding, X.; Jiang, C. Bone marrow mesenchymal stem cells-derived exosomes for penetrating and targeted chemotherapy of pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 1563–1575. [Google Scholar] [CrossRef]
- Soininen, S.K.; Vellonen, K.-S.; Heikkinen, A.T.; Auriola, S.; Ranta, V.-P.; Urtti, A.; Ruponen, M. Intracellular PK/PD relationships of free and liposomal doxorubicin: Quantitative analyses and PK/PD modeling. Mol. Pharm. 2016, 13, 1358–1365. [Google Scholar] [CrossRef]
- Schindler, C.; Collinson, A.; Matthews, C.; Pointon, A.; Jenkinson, L.; Minter, R.R.; Vaughan, T.J.; Tigue, N.J. Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. PLoS ONE 2019, 14, e0214545. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Brigstock, D.R. Integrins and heparan sulfate proteoglycans on hepatic stellate cells (HSC) are novel receptors for HSC-derived exosomes. FEBS Lett. 2016, 590, 4263–4274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.E.; de Jong, O.G.; Evers, M.J.; Nurazizah, M.; Schiffelers, R.M.; Vader, P. Natural or synthetic RNA delivery: A stoichiometric comparison of extracellular vesicles and synthetic nanoparticles. Nano Lett. 2021, 21, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.-U.; Allen, T.; Shen, D. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Zehentmeier, S.; Pereira, J.P. Cell circuits and niches controlling B cell development. Immunol. Rev. 2019, 289, 142–157. [Google Scholar]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Borghardt, J.M.; Weber, B.; Staab, A.; Kloft, C. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 2015, 17, 853–870. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Thörn, H.; Lennernäs, H.; Sjögren, E. Pulmonary drug absorption and systemic exposure in human: Predictions using physiologically based biopharmaceutics modeling. Eur. J. Pharm. Biopharm. 2020, 156, 191–202. [Google Scholar] [CrossRef]
- Nahar, K.; Gupta, N.; Gauvin, R.; Absar, S.; Patel, B.; Gupta, V.; Khademhosseini, A.; Ahsan, F. In vitro, in vivo and ex vivo models for studying particle deposition and drug absorption of inhaled pharmaceuticals. Eur. J. Pharm. Sci. 2013, 49, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.G.; Hennen, J.; Serchi, T.; Blömeke, B.; Gutleb, A.C. Potential of coculture in vitro models to study inflammatory and sensitizing effects of particles on the lung. Toxicol. Vitr. 2011, 25, 1516–1534. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.J.; Endter, S.; Tachon, G.; Falson, F.; Buckley, S.T.; Ehrhardt, C. Transport of the fluorescent organic cation 4-(4-(dimethylamino) styryl)-N-methylpyridinium iodide (ASP+) in human respiratory epithelial cells. Eur. J. Pharm. Biopharm. 2012, 81, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Daneshian, M.; Bouwstra, J.; Caloni, F.; Constant, S.; Davies, D.E.; Dandekar, G.; Guzman, C.A.; Fabian, E.; Haltner, E. Non-animal models of epithelial barriers (skin, intestine and lung) in research, industrial applications and regulatory toxicology. Altex 2015, 32, 327–378. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Daum, N.; Carius, P.; Horstmann, J.C.; Lehr, C.-M. Reconstituted 2D Cell and Tissue Models. In Pharmaceutical Inhalation Aerosol Technology; CRC Press: Boca Raton, FL, USA, 2019; pp. 627–651. [Google Scholar]
- Lenz, A.-G.; Karg, E.; Brendel, E.; Hinze-Heyn, H.; Maier, K.L.; Eickelberg, O.; Stoeger, T.; Schmid, O. Inflammatory and oxidative stress responses of an alveolar epithelial cell line to airborne zinc oxide nanoparticles at the air-liquid interface: A comparison with conventional, submerged cell-culture conditions. Biomed. Res. Int. 2013, 2013, 652632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, F.; Clift, M.J.; Piccapietra, F.; Behra, R.; Schmid, O.; Petri-Fink, A.; Rothen-Rutishauser, B. Exposure of silver-nanoparticles and silver-ions to lung cells in vitro at the air-liquid interface. Part. Fibre Toxicol. 2013, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Schmid, O.; Jud, C.; Umehara, Y.; Mueller, D.; Bucholski, A.; Gruber, F.; Denk, O.; Egle, R.; Petri-Fink, A.; Rothen-Rutishauser, B. Biokinetics of aerosolized liposomal ciclosporin a in human lung cells in vitro using an air-liquid cell interface exposure system. J. Aerosol Med. Pulm. Drug Deliv. 2017, 30, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Lenz, A.-G.; Stoeger, T.; Cei, D.; Schmidmeir, M.; Semren, N.; Burgstaller, G.; Lentner, B.; Eickelberg, O.; Meiners, S.; Schmid, O. Efficient bioactive delivery of aerosolized drugs to human pulmonary epithelial cells cultured in air–liquid interface conditions. Am. J. Respir. Cell Mol. Biol. 2014, 51, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Lenz, A.; Stoeger, T.; Cei, D.; Schmidmeir, M.; Pfister, N.; Burgstaller, G.; Lentner, B.; Eickelberg, O.; Meiners, S.; Schmid, O. Drug Delivery to the Lungs. Available online: https://ddl-conference.com/ddl24-2013/conference-papers/validation-alice-cloud-technology-functional-efficacy-studies-aerosolized-drugs-delivered-cells-air-liquid-interface/ (accessed on 31 December 2022).
- Ding, Y.; Weindl, P.; Lenz, A.-G.; Mayer, P.; Krebs, T.; Schmid, O. Quartz crystal microbalances (QCM) are suitable for real-time dosimetry in nanotoxicological studies using VITROCELL® Cloud cell exposure systems. Part. Fibre Toxicol. 2020, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, G.; Koch, W.; Ritter, D.; Gutleb, A.C.; Larsen, S.T.; Loret, T.; Zanetti, F.; Constant, S.; Chortarea, S.; Rothen-Rutishauser, B. Air–liquid Interface in vitro models for respiratory toxicology research: Consensus workshop and recommendations. Appl. Vitr. Toxicol. 2018, 4, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Byron, P.R.; Roberts, S.R.; Clark, A.R. An isolated perfused rat lung preparation for the study of aerosolized drug deposition and absorption. J. Pharm. Sci. 1986, 75, 168–171. [Google Scholar] [CrossRef]
- Byron, P.R.; Niven, R.W. A novel dosing method for drug administration to the airways of the isolated perfused rat lung. J. Pharm. Sci. 1988, 77, 693–695. [Google Scholar] [CrossRef]
- Li, M.; Byron, P.R. Tobramycin disposition in the rat lung following airway administration. J. Pharmacol. Exp. Ther. 2013, 347, 318–324. [Google Scholar] [CrossRef]
- Price, D.F.; Luscombe, C.N.; Eddershaw, P.J.; Edwards, C.D.; Gumbleton, M. The differential absorption of a series of P-glycoprotein substrates in isolated perfused lungs from Mdr1a/1b genetic knockout mice can be attributed to distinct physico-chemical properties: An insight into predicting transporter-mediated, pulmonary specific disposition. Pharm. Res. 2017, 34, 2498–2516. [Google Scholar]
- Bosquillon, C.; Madlova, M.; Patel, N.; Clear, N.; Forbes, B. A comparison of drug transport in pulmonary absorption models: Isolated perfused rat lungs, respiratory epithelial cell lines and primary cell culture. Pharm. Res. 2017, 34, 2532–2540. [Google Scholar] [CrossRef]
- Selg, E.; Ewing, P.; Acevedo, F.; Sjöberg, C.-O.; Ryrfeldt, Å.; Gerde, P. Dry powder inhalation exposures of the endotracheally intubated rat lung, ex vivo and in vivo: The pulmonary pharmacokinetics of fluticasone furoate. J. Aerosol Med. Pulm. Drug Deliv. 2013, 26, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.X.; Benaouda, F.; Traini, D.; Cipolla, D.; Gonda, I.; Bebawy, M.; Forbes, B.; Young, P.M. In vitro and ex vivo methods predict the enhanced lung residence time of liposomal ciprofloxacin formulations for nebulisation. Eur. J. Pharm. Biopharm. 2014, 86, 83–89. [Google Scholar] [CrossRef]
- Edwards, C.D.; Luscombe, C.; Eddershaw, P.; Hessel, E.M. Development of a novel quantitative structure-activity relationship model to accurately predict pulmonary absorption and replace routine use of the isolated perfused respiring rat lung model. Pharm. Res. 2016, 33, 2604–2616. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Sjögren, E.; Thörn, H.; Rubin, K.; Bäckman, P.; Lennernäs, H. Pulmonary absorption–estimation of effective pulmonary permeability and tissue retention of ten drugs using an ex vivo rat model and computational analysis. Eur. J. Pharm. Biopharm. 2018, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Thörn, H.; Sjögren, E.; Holmstén, L.; Rubin, K.; Lennernäs, H. Pulmonary dissolution of poorly soluble compounds studied in an ex vivo rat lung model. Mol. Pharm. 2019, 16, 3053–3064. [Google Scholar] [CrossRef]
- Eriksson, J.; Sjögren, E.; Lennernäs, H.; Thörn, H. Drug absorption parameters obtained using the isolated perfused rat lung model are predictive of rat in vivo lung absorption. AAPS J. 2020, 22, 71. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, L. In vivo animal models for controlled-release pulmonary drug delivery. In Controlled Pulmonary Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2011; pp. 443–474. [Google Scholar]
- Jones, R.M.; Harrison, A. A new methodology for predicting human pharmacokinetics for inhaled drugs from oratracheal pharmacokinetic data in rats. Xenobiotica 2012, 42, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Lamm Bergström, E.; Janzén, D.L.; Fridén, M.; Eriksson, U.; Grime, K.; Ferguson, D. Translational model to predict pulmonary pharmacokinetics and efficacy in man for inhaled bronchodilators. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Matei, A.C.; Antounians, L.; Zani, A. Extracellular Vesicles as a Potential Therapy for Neonatal Conditions: State of the Art and Challenges in Clinical Translation. Pharmaceutics 2019, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zaro, J.; Shen, Y. Advances in exosome-based drug delivery and tumor targeting: From tissue distribution to intracellular fate. Int. J. Nanomed. 2020, 15, 9355–9371. [Google Scholar] [CrossRef]
- Large, D.E.; Abdelmessih, R.G.; Fink, E.A.; Auguste, D.T. Liposome composition in drug delivery design, synthesis, characterization, and clinical application. Adv. Drug Deliv. Rev. 2021, 176, 113851. [Google Scholar]
- Matsumoto, A.; Takahashi, Y.; Chang, H.-Y.; Wu, Y.-W.; Yamamoto, A.; Ishihama, Y.; Takakura, Y. Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. J. Extracell. Vesicles 2020, 9, 1696517. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Liang, X.; Pavlova, S.; Wiklander, O.P.B.; Corso, G.; Zhao, Y.; Saher, O.; Bost, J.; Zickler, A.M.; Piffko, A.; et al. Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. J. Extracell. Vesicles 2020, 9, 1800222. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Duroux, M.; Moos, T.; Andresen, T.L.; Simonsen, J.B. On the use of liposome controls in studies investigating the clinical potential of extracellular vesicle-based drug delivery systems—A commentary. J. Control. Release 2018, 269, 10–14. [Google Scholar] [CrossRef]
- Silva, A.K.A.; Morille, M.; Piffoux, M.; Arumugam, S.; Mauduit, P.; Larghero, J.; Bianchi, A.; Aubertin, K.; Blanc-Brude, O.; Noël, D.; et al. Development of extracellular vesicle-based medicinal products: A position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs—EVOLVE France”. Adv. Drug Deliv. Rev. 2021, 179, 114001. [Google Scholar] [CrossRef]
- Sutaria, D.S.; Jiang, J.; Elgamal, O.A.; Pomeroy, S.M.; Badawi, M.; Zhu, X.; Pavlovicz, R.; Azevedo-Pouly, A.C.P.; Chalmers, J.; Li, C.; et al. Low active loading of cargo into engineered extracellular vesicles results in inefficient miRNA mimic delivery. J. Extracell. Vesicles 2017, 6, 1333882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Alyane, M.; Barratt, G.; Lahouel, M. Remote loading of doxorubicin into liposomes by transmembrane pH gradient to reduce toxicity toward H9c2 cells. Saudi Pharm. J. 2016, 24, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavian, S.A.; Bianconi, V.; Pirro, M.; Sahebkar, A. Challenges and pitfalls in the development of liposomal delivery systems for cancer therapy. Semin. Cancer Biol. 2021, 69, 337–348. [Google Scholar] [CrossRef]
- Seetharamu, N.; Kim, E.; Hochster, H.; Martin, F.; Muggia, F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. Anticancer Res. 2010, 30, 541–545. [Google Scholar]
- Burnouf, T.; Agrahari, V.; Agrahari, V. Extracellular Vesicles As Nanomedicine: Hopes And Hurdles in Clinical Translation. Int. J. Nanomed. 2019, 14, 8847–8859. [Google Scholar] [CrossRef]
- Webber, J.; Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2013, 2, 19861. [Google Scholar] [CrossRef]
- Knopp, J.L.; Holder-Pearson, L.; Chase, J.G. Insulin Units and Conversion Factors: A Story of Truth, Boots, and Faster Half-Truths. J. Diabetes Sci. Technol. 2019, 13, 597–600. [Google Scholar] [CrossRef]
- Willis, G.R.; Kourembanas, S.; Mitsialis, S.A. Toward exosome-based therapeutics: Isolation, heterogeneity, and fit-for-purpose potency. Front. Cardiovasc. Med. 2017, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Zickler, A.M.; El Andaloussi, S. Dosing extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 178, 113961. [Google Scholar] [CrossRef]
- Caron, W.P.; Clewell, H.; Dedrick, R.; Ramanathan, R.K.; Davis, W.L.; Yu, N.; Tonda, M.; Schellens, J.H.; Beijnen, J.H.; Zamboni, W.C. Allometric scaling of pegylated liposomal anticancer drugs. J. Pharmacokinet. Pharmacodyn. 2011, 38, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Takov, K.; Yellon, D.M.; Davidson, S.M. Comparison of small extracellular vesicles isolated from plasma by ultracentrifugation or size-exclusion chromatography: Yield, purity and functional potential. J. Extracell. Vesicles 2019, 8, 1560809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, L.; Parkin, C.G. Rethinking the Viability and Utility of Inhaled Insulin in Clinical Practice. J. Diabetes Res. 2018, 2018, 4568903. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Pittas, A.G. Inhaled insulin—What went wrong. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 24–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaram, A.; Jay, S.M. Preservation and Storage Stability of Extracellular Vesicles for Therapeutic Applications. AAPS J. 2017, 20, 1. [Google Scholar] [CrossRef]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrügger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 2019, 8, 1684862. [Google Scholar] [CrossRef]
















| In Vitro Model | Model Characteristic | Human Physiological Realism-In Vivo Correlation |
|---|---|---|
| LLI: liquid-liquid interface | Submerged cultured cells exposure to vesicles dissolution or suspension in the culture medium | Unrealistic artificial alveolar environment incapable of real-time cell-delivered vesicles dosimetry |
| ALI: air-liquid interface | Vesicles deposition onto the cultured cells exposed to the inhaled air | Realistic physiologically alveolar relevant environment capable of real-time cell-delivered vesicles dosimetry |
| ALICE-CLOUD: air-liquid interface cell exposure-cloud (e.g., VITROCELL®) empowered with quartz crystal microbalances (QCMs) [337] | Aerosols cloud generates by nebulization gently deposits onto the cultured cells due to single particle sedimentation and cloud setting | Highly realistic physiologically alveolar relevant environment capable of real-time cell-delivered vesicles dosimetry |
| Ex Vivo Model | Model Characteristic | Human Physiological Realism-In Vivo Correlation |
|---|---|---|
| IPRL: isolated perfused rat lung | Preserves lung architecture and function without confounding whole-body complications | Allow the in vivo-relevant lung tissue/organ-level kinetic assessment of absorption and deposition |
| IPRL-QSAR: isolated perfused rat lung-quantitative structure activity relationship (In Silico, i.e., Computer Simulation) | Improve compounds design via predicting pulmonary absorption | Replace the routine generation of IPRL model data for ranking and classifying compounds prior to synthesis |
| IPRL-PBBP: isolated perfused rat lung-physiologically based biopharmaceutical dissolution data (In Silico, i.e., Computer Simulation) | Improve pulmonary drug absorption understanding via comparing absorption rates of poorly soluble inhaled drugs from suspensions and dry powders with solutions | A useful tool for investigating and improving pulmonary dissolution of poorly soluble inhaled drugs |
| IPRL-IV PK: isolated perfused rat lung-intravenous pharmacokinetics (In Silico, i.e., Computer Simulation) | Improves extent predictability using input parameters from IPRL ex vivo model | IPRL ex vivo (not in vitro) data are better to understand how different drug properties and formulation might affect in vivo behavior of inhaled compounds |
| In Vivo Model | Model Characteristic | Human Physiological Realism-In Vivo Correlation |
|---|---|---|
| Non-surgical orotracheal administration | Spray-instilled as solution or suspension or insufflated as powders | Predict inhaled drugs human PK profiles from in vivo rodent PK data |
| Intratracheal powder insufflation | Spray-insufflated as powders | Predicted human PK profiles by kinetic modeling requires further systematic refinement |
| Intratracheal solution delivery | Spray- instilled as solution | Predicted human PK profiles by translational compartmental modeling are remarkably identical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Jipouri, A.; Almurisi, S.H.; Al-Japairai, K.; Bakar, L.M.; Doolaanea, A.A. Liposomes or Extracellular Vesicles: A Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery. Polymers 2023, 15, 318. https://doi.org/10.3390/polym15020318
Al-Jipouri A, Almurisi SH, Al-Japairai K, Bakar LM, Doolaanea AA. Liposomes or Extracellular Vesicles: A Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery. Polymers. 2023; 15(2):318. https://doi.org/10.3390/polym15020318
Chicago/Turabian StyleAl-Jipouri, Ali, Samah Hamed Almurisi, Khater Al-Japairai, Latifah Munirah Bakar, and Abd Almonem Doolaanea. 2023. "Liposomes or Extracellular Vesicles: A Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery" Polymers 15, no. 2: 318. https://doi.org/10.3390/polym15020318