
Thermally Degradable Poly(n-butyl acrylate) Model Networks Prepared by PhotoATRP and Radical Trap-Assisted Atom Transfer Radical Coupling

 , , ,
, , ,  and
and
Abstract
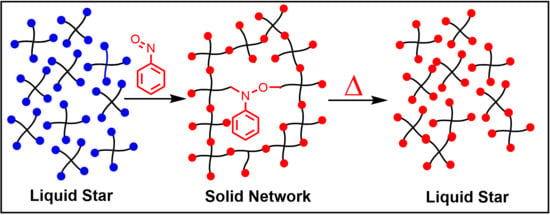
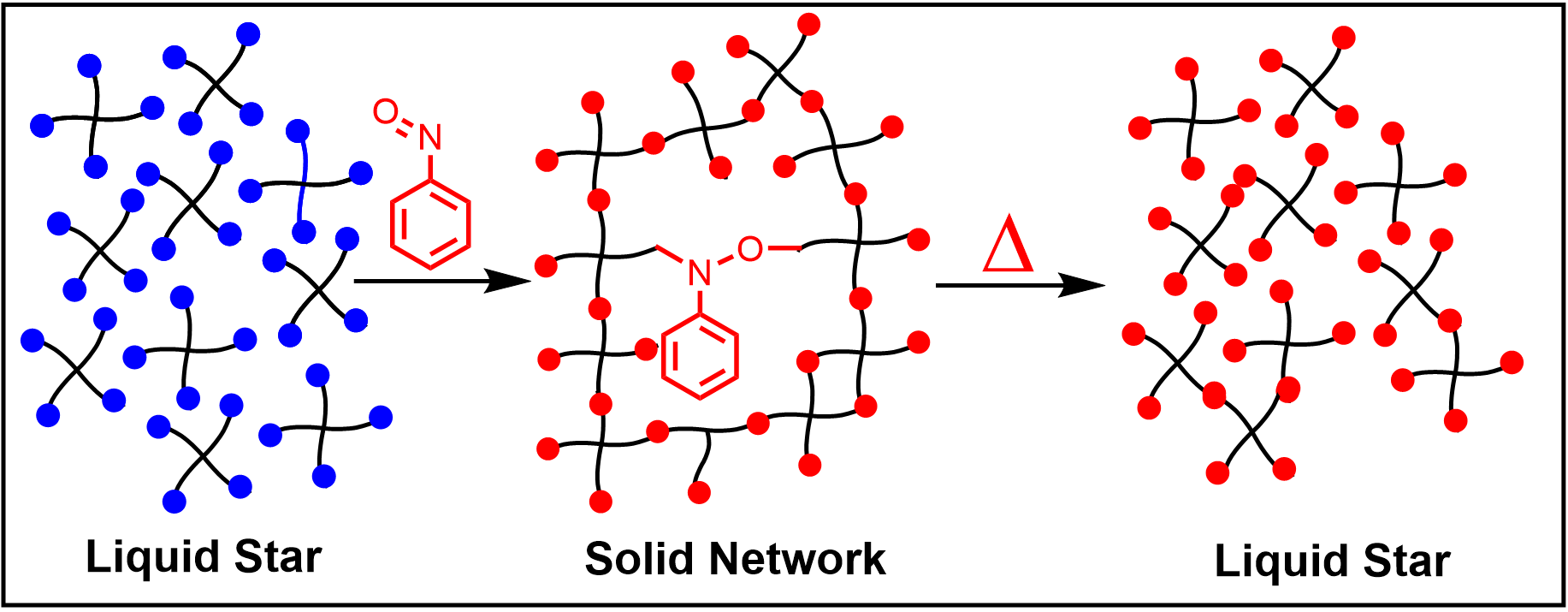
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instrumentation
2.2.1. Gel Permeation Chromatography (GPC)
2.2.2. Nuclear Magnetic Resonance (1H NMR)
2.2.3. Thermogravimetric Analysis (TGA)
2.2.4. Dynamic Mechanical Analysis (DMA)
2.3. Synthesis
2.3.1. Synthesis of Tetrafunctional ATRP Initiator (4f-BiB)
2.3.2. Synthesis of Poly(n-butyl acrylate) Star Polymer (PBA4-Br) by PhotoATRP
2.3.3. Curing of PBA Networks by RTA-ATRC
3. Results
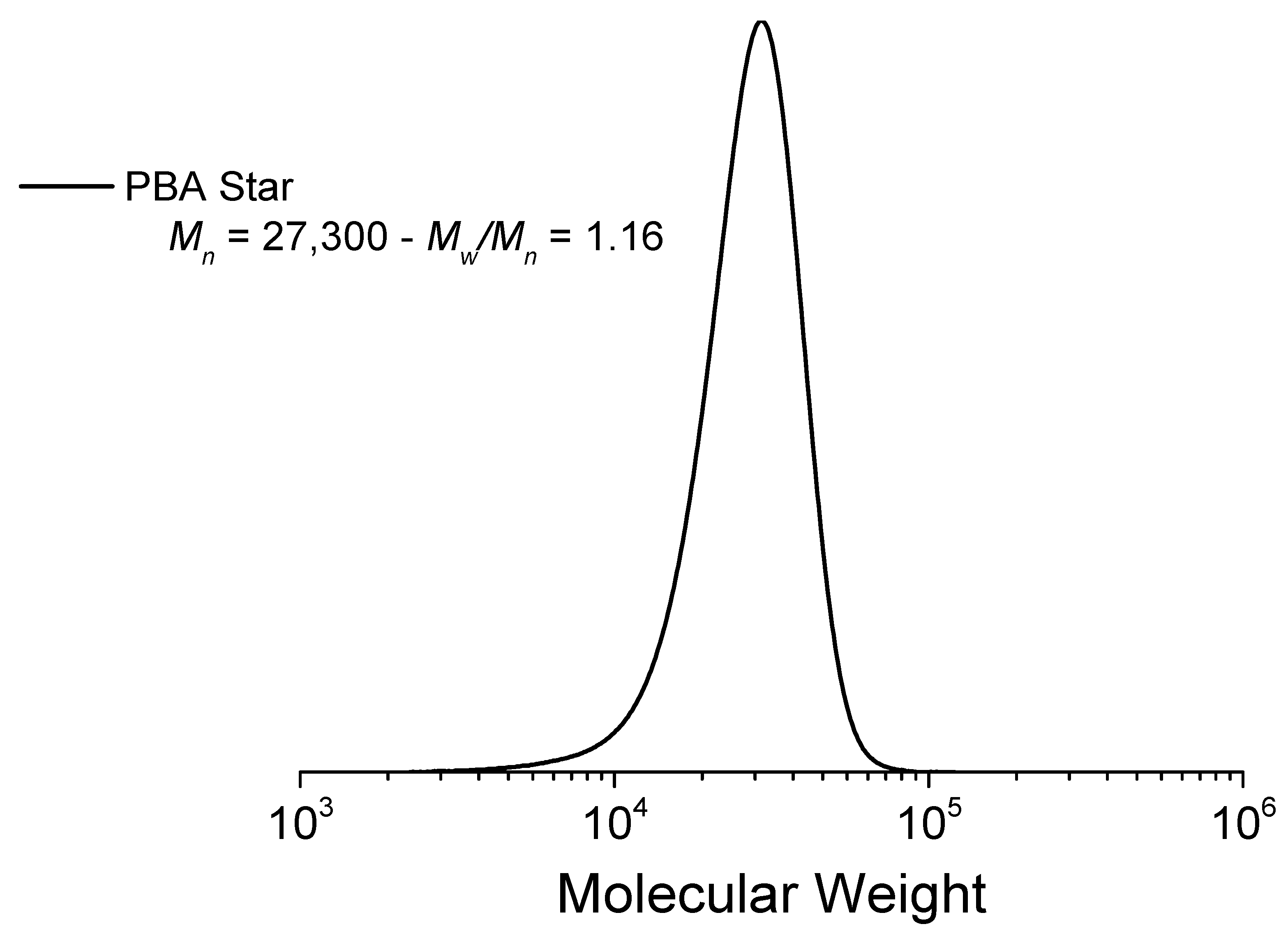
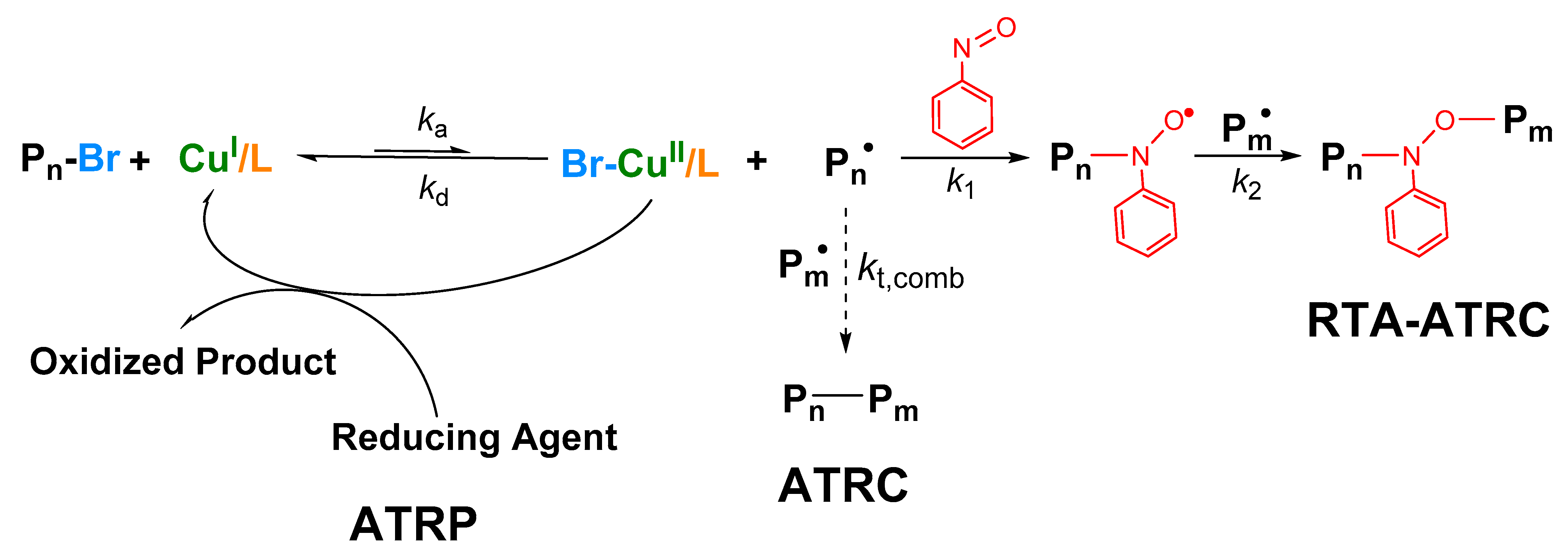
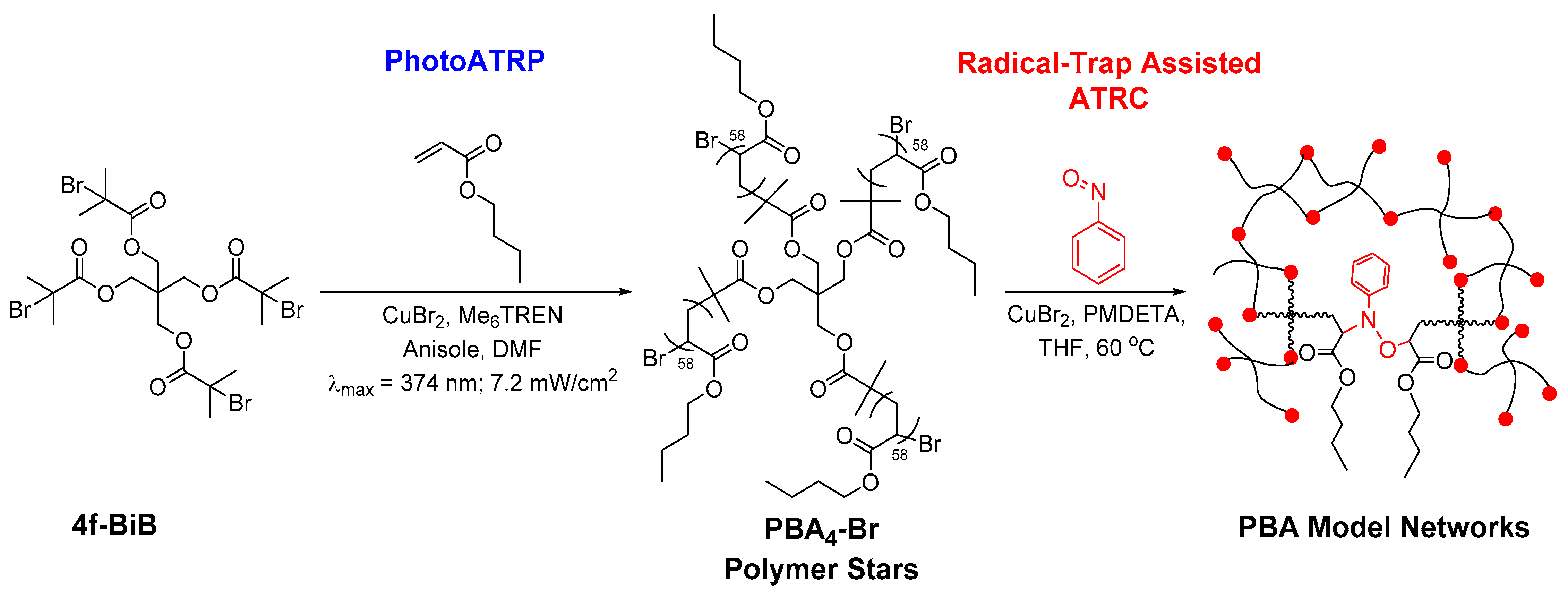
3.1. Network Synthesis
3.2. Network Characterization
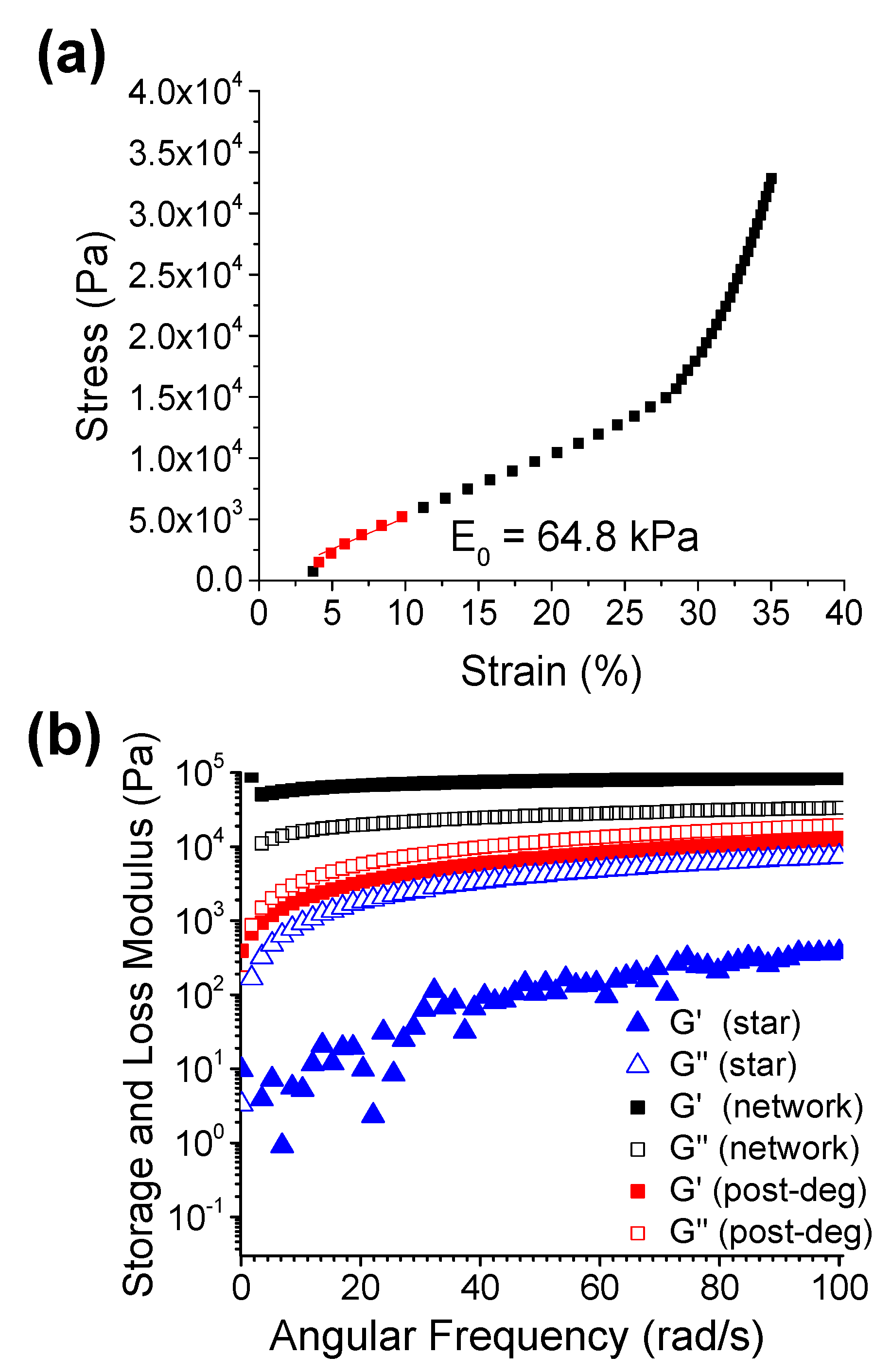
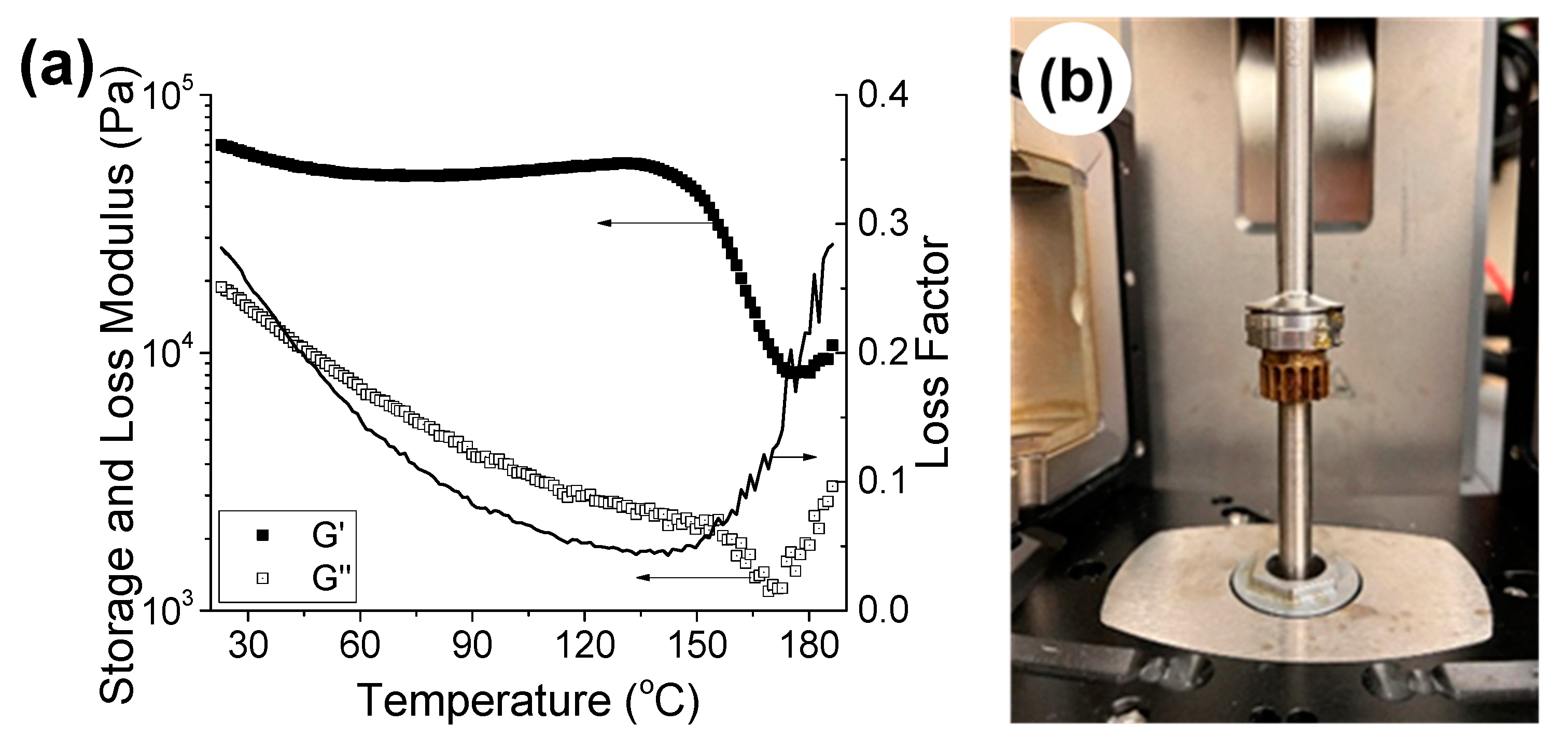
3.2.1. Mechanical Properties of the Networks
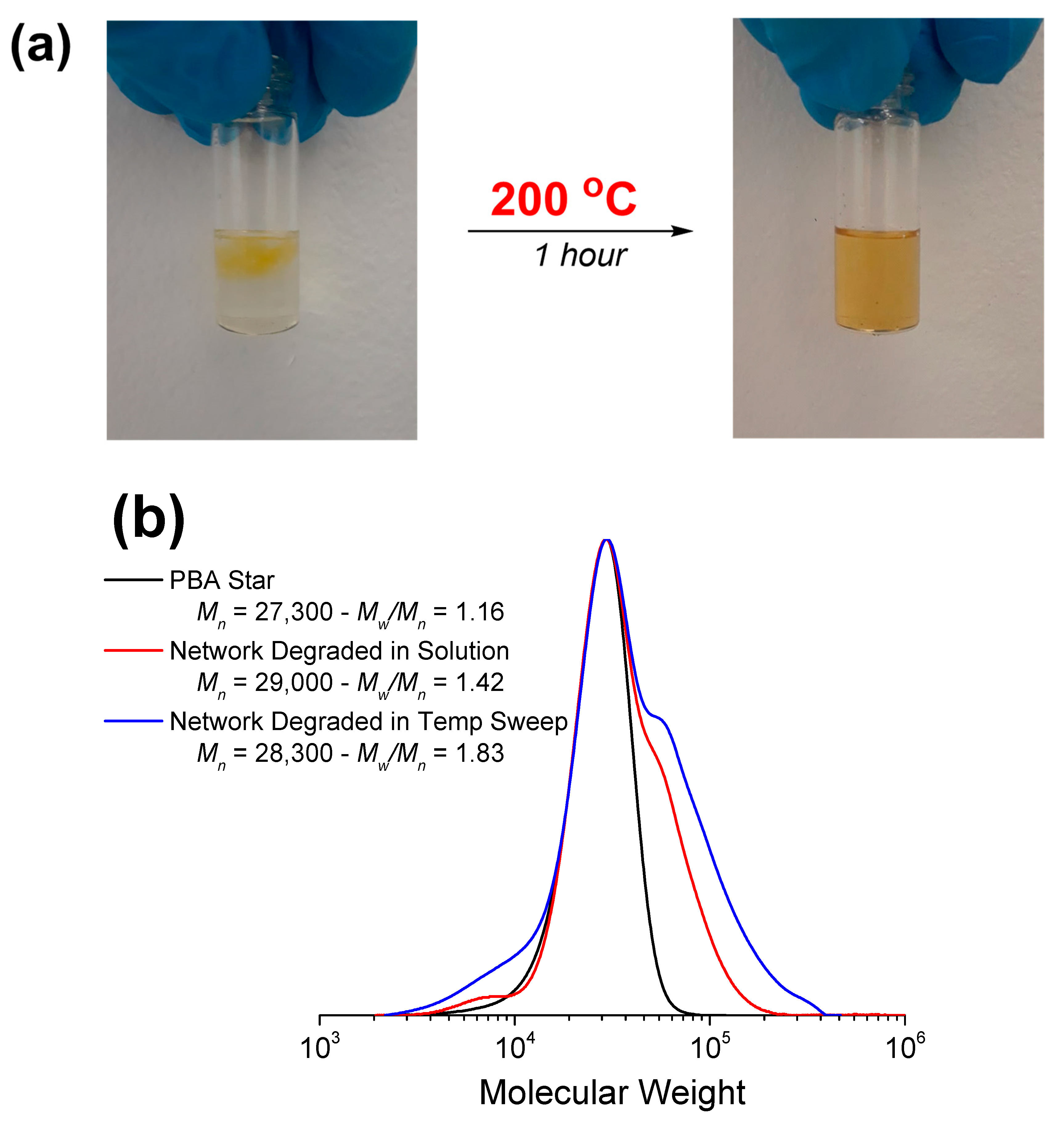
3.2.2. Degradation in Solution
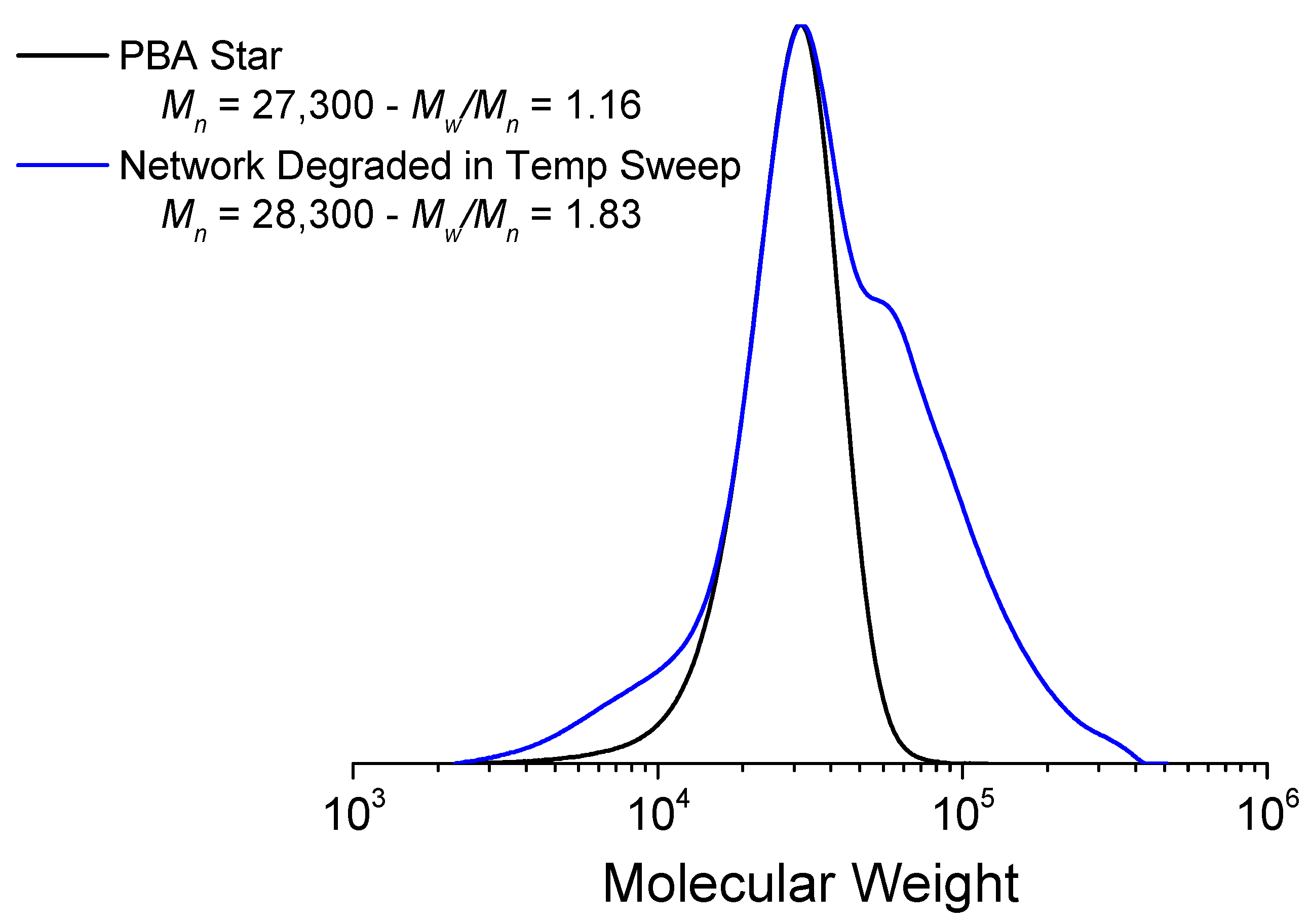
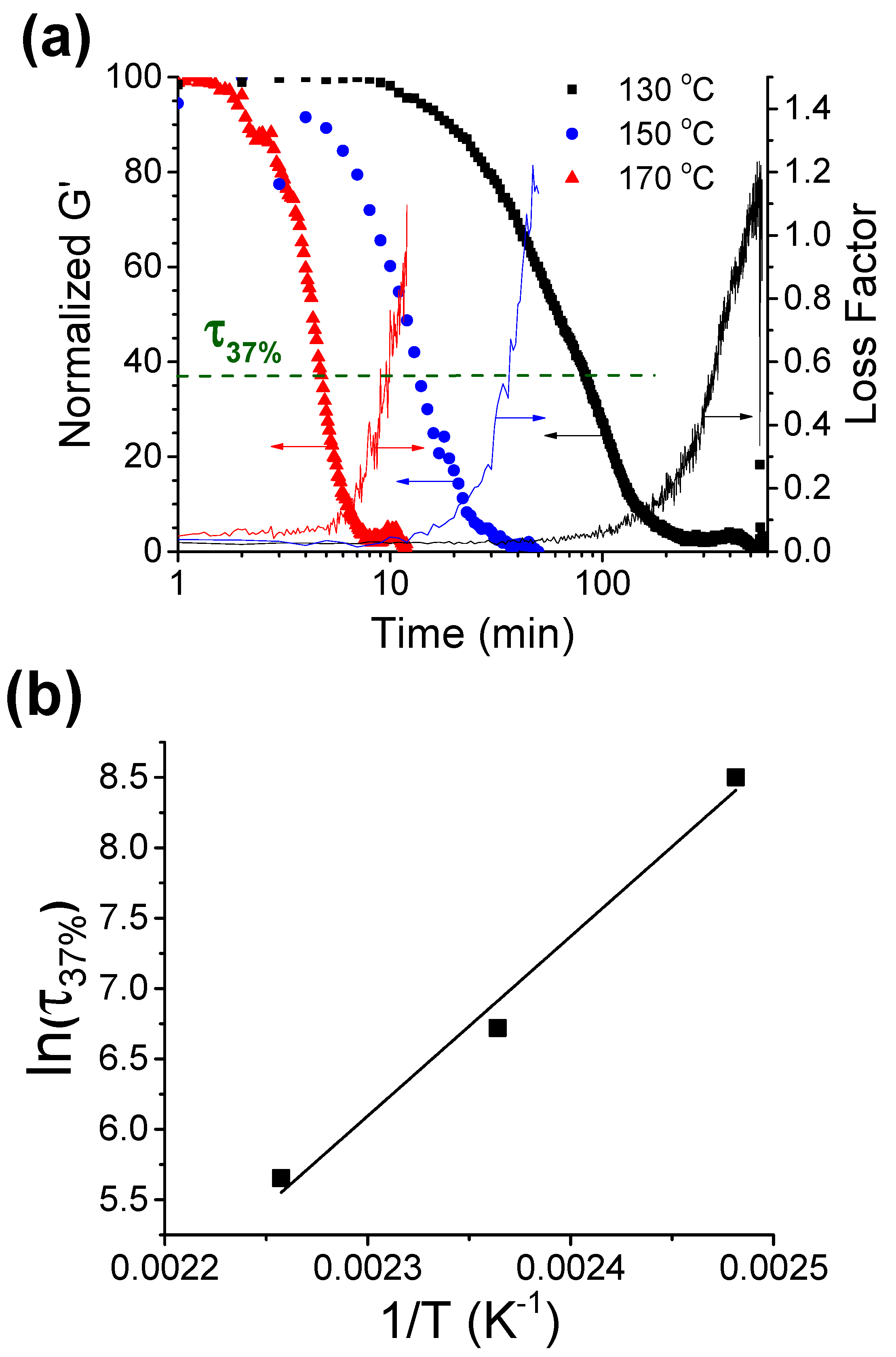
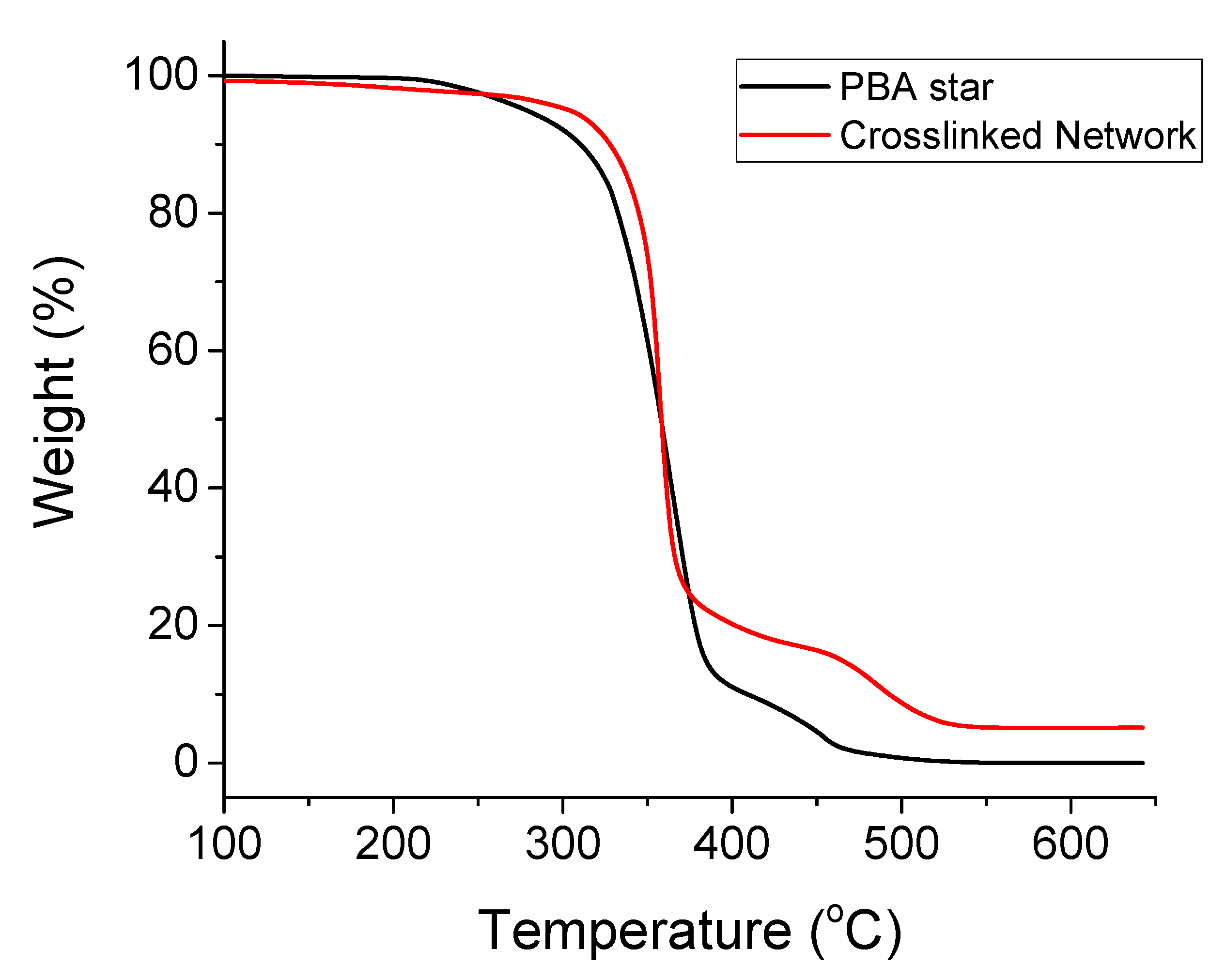
3.2.3. Thermogravimetric Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, Y.; Zhao, J.; Johnson, J.A. A (Macro) Molecular-Level Understanding of Polymer Network Topology. Trends Chem. 2019, 1, 318–334. [Google Scholar] [CrossRef]
- Daniel, W.F.; Burdyńska, J.; Vatankhah-Varnoosfaderani, M.; Matyjaszewski, K.; Paturej, J.; Rubinstein, M.; Dobrynin, A.V.; Sheiko, S.S. Solvent-free, supersoft and superelastic bottlebrush melts and networks. Nat. Mater. 2016, 15, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, V.G.; Mukherjee, S.; Xie, R.; Levi, A.E.; Atassi, A.; Uchiyama, T.; Wang, H.; Chabinyc, M.L.; Bates, C.M. Super-soft solvent-free bottlebrush elastomers for touch sensing. Mater. Horiz. 2020, 7, 181–187. [Google Scholar] [CrossRef]
- Liang, H.; Sheiko, S.S.; Dobrynin, A.V. Supersoft and hyperelastic polymer networks with brushlike strands. Macromolecules 2018, 51, 638–645. [Google Scholar] [CrossRef]
- Cuthbert, J.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. STEM gels by controlled radical polymerization. Trends Chem. 2020, 2, 341–353. [Google Scholar] [CrossRef]
- Pandey, P.; Chauhan, R. Membranes for gas separation. Prog. Polym. Sci. 2001, 26, 853–893. [Google Scholar] [CrossRef]
- Javaid, A. Membranes for solubility-based gas separation applications. Chem. Eng. J. 2005, 112, 219–226. [Google Scholar] [CrossRef]
- Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Porous polymer catalysts with hierarchical structures. Chem. Soc. Rev. 2015, 44, 6018–6034. [Google Scholar] [CrossRef]
- Huang, J.; Turner, S.R. Hypercrosslinked polymers: A review. Polym. Rev. 2018, 58, 1–41. [Google Scholar] [CrossRef]
- Rikkou, M.D.; Kolokasi, M.; Matyjaszewski, K.; Patrickios, C.S. End-linked amphiphilic polymer conetworks: Synthesis by sequential atom transfer radical polymerization and swelling characterization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1878–1886. [Google Scholar] [CrossRef]
- Patrickios, C.S.; Matyjaszewski, K. Amphiphilic polymer co-networks: 32 years old and growing stronge—A perspective. Polym. Int. 2021, 70, 10–13. [Google Scholar] [CrossRef]
- Cuthbert, J.; Wanasinghe, S.V.; Matyjaszewski, K.; Konkolewicz, D. Are RAFT and ATRP Universally Interchangeable Polymerization Methods in Network Formation? Macromolecules 2021, 54, 8331–8340. [Google Scholar] [CrossRef]
- Rikkou-Kalourkoti, M.; Loizou, E.; Porcar, L.; Matyjaszewski, K.; Patrickios, C.S. End-linked, amphiphilic, degradable polymer conetworks: Synthesis by sequential atom transfer radical polymerization using a bifunctional, cleavable initiator. Polym. Chem. 2012, 3, 105–116. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolaÿ, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-Healing Polymer Films Based on Thiol–Disulfide Exchange Reactions and Self-Healing Kinetics Measured Using Atomic Force Microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Kamada, J.; Koynov, K.; Corten, C.; Juhari, A.; Yoon, J.A.; Urban, M.W.; Balazs, A.C.; Matyjaszewski, K. Redox responsive behavior of thiol/disulfide-functionalized star polymers synthesized via atom transfer radical polymerization. Macromolecules 2010, 43, 4133–4139. [Google Scholar] [CrossRef]
- Kolmakov, G.V.; Matyjaszewski, K.; Balazs, A.C. Harnessing labile bonds between nanogel particles to create self-healing materials. ACS Nano 2009, 3, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Self, J.L.; Sample, C.S.; Levi, A.E.; Li, K.; Xie, R.; de Alaniz, J.R.; Bates, C.M. Dynamic bottlebrush polymer networks: Self-healing in super-soft materials. J. Am. Chem. Soc. 2020, 142, 7567–7573. [Google Scholar] [CrossRef]
- Mark, J.; Sullivan, J. Model networks of end-linked polydimethylsiloxane chains. I. Comparisons between experimental and theoretical values of the elastic modulus and the equilibrium degree of swelling. J. Chem. Phys. 1977, 66, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Villar, M.A.; Vallés, E.M. Influence of the final extent of reaction on the structure of model polydimethylsiloxane networks obtained by the end-linking hydrosilation reaction. Polym. Bull. 1995, 35, 279–284. [Google Scholar] [CrossRef]
- Johnson, J.A.; Lewis, D.R.; Díaz, D.D.; Finn, M.; Koberstein, J.T.; Turro, N.J. Synthesis of degradable model networks via ATRP and click chemistry. J. Am. Chem. Soc. 2006, 128, 6564–6565. [Google Scholar] [CrossRef]
- Johnson, J.A.; Finn, M.G.; Koberstein, J.T.; Turro, N.J. Synthesis of Photocleavable Linear Macromonomers by ATRP and Star Macromonomers by a Tandem ATRP–Click Reaction: Precursors to Photodegradable Model Networks. Macromolecules 2007, 40, 3589–3598. [Google Scholar] [CrossRef]
- Shih, H.; Lin, C.-C. Cross-linking and degradation of step-growth hydrogels formed by thiol–ene photoclick chemistry. Biomacromolecules 2012, 13, 2003–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cok, A.M.; Zhou, H.; Johnson, J.A. Synthesis of Model Network Hydrogels via Tetrazine-O lefin Inverse Electron Demand Diels-A lder Cycloaddition. Macromol. Symp. 2013, 329, 108–112. [Google Scholar] [CrossRef]
- Jia, Y.; Matt, Y.; An, Q.; Wessely, I.; Mutlu, H.; Theato, P.; Bräse, S.; Llevot, A.; Tsotsalas, M. Dynamic covalent polymer networks via combined nitroxide exchange reaction and nitroxide mediated polymerization. Polym. Chem. 2020, 11, 2502–2510. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, J. Versatility of radical coupling in construction of topological polymers. Polym. Chem. 2014, 5, 277–308. [Google Scholar] [CrossRef]
- Sarbu, T.; Lin, K.-Y.; Spanswick, J.; Gil, R.R.; Siegwart, D.J.; Matyjaszewski, K. Synthesis of hydroxy-telechelic poly (methyl acrylate) and polystyrene by atom transfer radical coupling. Macromolecules 2004, 37, 9694–9700. [Google Scholar] [CrossRef]
- Sarbu, T.; Lin, K.-Y.; Ell, J.; Siegwart, D.J.; Spanswick, J.; Matyjaszewski, K. Polystyrene with Designed Molecular Weight Distribution by Atom Transfer Radical Coupling. Macromolecules 2004, 37, 3120–3127. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, K.; Chen, Y.; Xi, F. Isomeric Dicyclic Polymers via Atom Transfer Radical Polymerization and Atom Transfer Radical Coupling Cyclization. Macromolecules 2014, 47, 1993–1998. [Google Scholar] [CrossRef]
- Luo, X.; Wang, G.; Huang, J. Preparation of H-shaped ABCAB terpolymers by atom transfer radical coupling. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 59–68. [Google Scholar] [CrossRef]
- Nicolaÿ, R.; Marx, L.; Hémery, P.; Matyjaszewski, K. Synthesis of multisegmented degradable polymers by atom transfer radical cross-coupling. Macromolecules 2007, 40, 9217–9223. [Google Scholar] [CrossRef]
- Li, I.; Howell, B.A.; Matyjaszewski, K.; Shigemoto, T.; Smith, P.B.; Priddy, D.B. Kinetics of decomposition of 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidine and its implications on nitroxyl-mediated styrene polymerization. Macromolecules 1995, 28, 6692–6693. [Google Scholar] [CrossRef]
- Voter, A.F.; Tillman, E.S.; Findeis, P.M.; Radzinski, S.C. Synthesis of macrocyclic polymers formed via intramolecular radical trap-assisted atom transfer radical coupling. ACS Macro Lett. 2012, 1, 1066–1070. [Google Scholar] [CrossRef]
- Valente, C.J.; Schellenberger, A.M.; Tillman, E.S. Dimerization of poly (methyl methacrylate) chains using radical trap-assisted atom transfer radical coupling. Macromolecules 2014, 47, 2226–2232. [Google Scholar] [CrossRef]
- Arce, M.M.; Pan, C.W.; Thursby, M.M.; Wu, J.P.; Carnicom, E.M.; Tillman, E.S. Influence of solvent on radical trap-assisted dimerization and cyclization of polystyrene radicals. Macromolecules 2016, 49, 7804–7813. [Google Scholar] [CrossRef]
- Butcher, W.E.; Radzinski, S.C.; Tillman, E.S. Selective formation of diblock copolymers using radical trap-assisted atom transfer radical coupling. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3619–3626. [Google Scholar] [CrossRef]
- Blackburn, S.C.; Myers, K.D.; Tillman, E.S. Macrocyclic poly (methyl acrylate) and macrocyclic poly (methyl acrylate-block-styrene) synthesized by radical trap-assisted atom transfer radical coupling. Polymer 2015, 68, 284–292. [Google Scholar] [CrossRef] [Green Version]
- McFadden, B.D.; Arce, M.M.; Carnicom, E.M.; Herman, J.; Abrusezze, J.; Tillman, E.S. Radical Trap-Assisted Atom Transfer Radical Coupling of Diblock Copolymers as a Method of Forming Triblock Copolymers. Macromol. Chem. Phys. 2016, 217, 2473–2482. [Google Scholar] [CrossRef]
- Andry, J.J.; Lee, J.J.; Wu, J.; Xia, K.; Tillman, E.S. Universal chain-end coupling conditions for brominated polystyrenes, polyacrylates, and polymethacrylates. Processes 2021, 9, 1001. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, G.; Huang, J. Synthesis of H-shaped A3BA3 copolymer by methyl-2-nitrosopropane induced single electron transfer nitroxide radical coupling. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2811–2817. [Google Scholar] [CrossRef]
- Telitel, S.; Amamoto, Y.; Poly, J.; Morlet-Savary, F.; Soppera, O.; Lalevee, J.; Matyjaszewski, K. Introduction of self-healing properties into covalent polymer networks via the photodissociation of alkoxyamine junctions. Polym. Chem. 2014, 5, 921–930. [Google Scholar] [CrossRef]
- Su, J.; Amamoto, Y.; Nishihara, M.; Takahara, A.; Otsuka, H. Reversible cross-linking of hydrophilic dynamic covalent polymers with radically exchangeable alkoxyamines in aqueous media. Polym. Chem. 2011, 2, 2021–2026. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Jin, K.; Rusayyis, M.B.; Torkelson, J.M. Arresting Elevated-Temperature Creep and Achieving Full Cross-Link Density Recovery in Reprocessable Polymer Networks and Network Composites via Nitroxide-Mediated Dynamic Chemistry. Macromolecules 2021, 54, 1452–1464. [Google Scholar] [CrossRef]
- van Ravensteijn, B.G.P.; Bou Zerdan, R.; Helgeson, M.E.; Hawker, C.J. Minimizing Star–Star Coupling in Cu(0)-Mediated Controlled Radical Polymerizations. Macromolecules 2019, 52, 601–609. [Google Scholar] [CrossRef]
- Martinez, M.R.; Sobieski, J.; Lorandi, F.; Fantin, M.; Dadashi-Silab, S.; Xie, G.; Olszewski, M.; Pan, X.; Ribelli, T.G.; Matyjaszewski, K. Understanding the Relationship between Catalytic Activity and Termination in photoATRP: Synthesis of Linear and Bottlebrush Polyacrylates. Macromolecules 2020, 53, 59–67. [Google Scholar] [CrossRef]
- Pan, X.; Tasdelen, M.A.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated controlled radical polymerization. Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar] [CrossRef] [Green Version]
- Konkolewicz, D.; Schröder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. Visible light and sunlight photoinduced ATRP with ppm of Cu catalyst. ACS Macro Lett. 2012, 1, 1219–1223. [Google Scholar] [CrossRef]
- Cuthbert, J.; Yerneni, S.S.; Sun, M.; Fu, T.; Matyjaszewski, K. Degradable polymer stars based on tannic acid cores by ATRP. Polymers 2019, 11, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyjaszewski, K.; Miller, P.J.; Pyun, J.; Kickelbick, G.; Diamanti, S. Synthesis and Characterization of Star Polymers with Varying Arm Number, Length, and Composition from Organic and Hybrid Inorganic/Organic Multifunctional Initiators. Macromolecules 1999, 32, 6526–6535. [Google Scholar] [CrossRef]
- Miller, P.J.; Matyjaszewski, K. Atom transfer radical polymerization of (meth) acrylates from poly (dimethylsiloxane) macroinitiators. Macromolecules 1999, 32, 8760–8767. [Google Scholar] [CrossRef]
- Zhu, Y.; Egap, E. Light-Mediated Polymerization Induced by Semiconducting Nanomaterials: State-of-the-Art and Future Perspectives. ACS Polym. Au 2021, 1, 76–99. [Google Scholar] [CrossRef]
- Zhu, Y.; Jin, T.; Lian, T.; Egap, E. Enhancing the efficiency of semiconducting quantum dot photocatalyzed atom transfer radical polymerization by ligand shell engineering. J. Chem. Phys. 2021, 154, 204903. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yuan, L.; Wang, Z.; Rahman, M.A.; Huang, Y.; Zhu, T.; Wang, R.; Cheng, J.; Wang, C.; Chu, F.; et al. Photoinduced Metal-Free Atom Transfer Radical Polymerization of Biomass-Based Monomers. Macromolecules 2016, 49, 7709–7717. [Google Scholar] [CrossRef]
- Theriot, J.C.; McCarthy, B.G.; Lim, C.-H.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization: Perspectives on Catalyst Design and Performance. Macromol. Rapid Commun. 2017, 38, 1700040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Discekici, E.H.; Anastasaki, A.; Read de Alaniz, J.; Hawker, C.J. Evolution and Future Directions of Metal-Free Atom Transfer Radical Polymerization. Macromolecules 2018, 51, 7421–7434. [Google Scholar] [CrossRef] [Green Version]
- Ribelli, T.G.; Konkolewicz, D.; Bernhard, S.; Matyjaszewski, K. How are Radicals (Re)Generated in Photochemical ATRP? J. Am. Chem. Soc. 2014, 136, 13303–13312. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, A.; Willenbacher, J.; Fleischmann, C.; Gutekunst, W.R.; Hawker, C.J. End group modification of poly (acrylates) obtained via ATRP: A user guide. Polym. Chem. 2017, 8, 689–697. [Google Scholar] [CrossRef]
- Min, K.; Gao, H.; Matyjaszewski, K. Preparation of homopolymers and block copolymers in miniemulsion by ATRP using activators generated by electron transfer (AGET). J. Am. Chem. Soc. 2005, 127, 3825–3830. [Google Scholar] [CrossRef]
- Domingues, K.M.; Tillman, E.S. Radical–radical coupling of polystyrene chains using AGET ATRC. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 5737–5745. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Augustine, K.F.; Fantin, M.; Krys, P.; Poli, R.; Matyjaszewski, K. Disproportionation or Combination? The Termination of Acrylate Radicals in ATRP. Macromolecules 2017, 50, 7920–7929. [Google Scholar] [CrossRef]
- Xie, G.; Martinez, M.R.; Daniel, W.F.M.; Keith, A.N.; Ribelli, T.G.; Fantin, M.; Sheiko, S.S.; Matyjaszewski, K. Benefits of Catalyzed Radical Termination: High-Yield Synthesis of Polyacrylate Molecular Bottlebrushes without Gelation. Macromolecules 2018, 51, 6218–6225. [Google Scholar] [CrossRef]
- Audran, G.; Blyth, M.T.; Coote, M.L.; Gescheidt, G.; Hardy, M.; Havot, J.; Holzritter, M.; Jacoutot, S.; Joly, J.-P.; Marque, S.R. Homolysis/mesolysis of alkoxyamines activated by chemical oxidation and photochemical-triggered radical reactions at room temperature. Org. Chem. Front. 2021, 8, 6561–6576. [Google Scholar] [CrossRef]
- Yu-Hsiang, H.; Chen, C.; Wang, C. Thermal degradation kinetics of poly (n-butyl acrylate) initiated by lactams and thiols. Polym. Degrad. Stab. 2004, 84, 505–514. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, M.R.; Zhuang, Z.; Treichel, M.; Cuthbert, J.; Sun, M.; Pietrasik, J.; Matyjaszewski, K. Thermally Degradable Poly(n-butyl acrylate) Model Networks Prepared by PhotoATRP and Radical Trap-Assisted Atom Transfer Radical Coupling. Polymers 2022, 14, 713. https://doi.org/10.3390/polym14040713
Martinez MR, Zhuang Z, Treichel M, Cuthbert J, Sun M, Pietrasik J, Matyjaszewski K. Thermally Degradable Poly(n-butyl acrylate) Model Networks Prepared by PhotoATRP and Radical Trap-Assisted Atom Transfer Radical Coupling. Polymers. 2022; 14(4):713. https://doi.org/10.3390/polym14040713
Chicago/Turabian StyleMartinez, Michael R., Ziye Zhuang, Megan Treichel, Julia Cuthbert, Mingkang Sun, Joanna Pietrasik, and Krzysztof Matyjaszewski. 2022. "Thermally Degradable Poly(n-butyl acrylate) Model Networks Prepared by PhotoATRP and Radical Trap-Assisted Atom Transfer Radical Coupling" Polymers 14, no. 4: 713. https://doi.org/10.3390/polym14040713