
Self-Healing of Polymers and Polymer Composites


Abstract
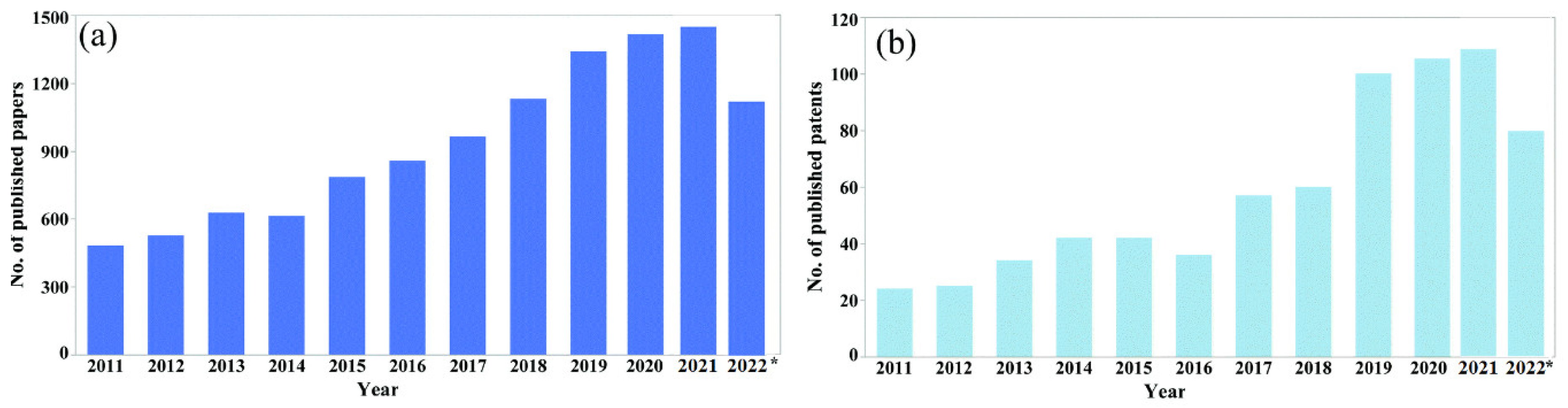
:1. Introduction
2. Methods of Self-Healing of Polymeric Materials
3. Intrinsic Self-Healing
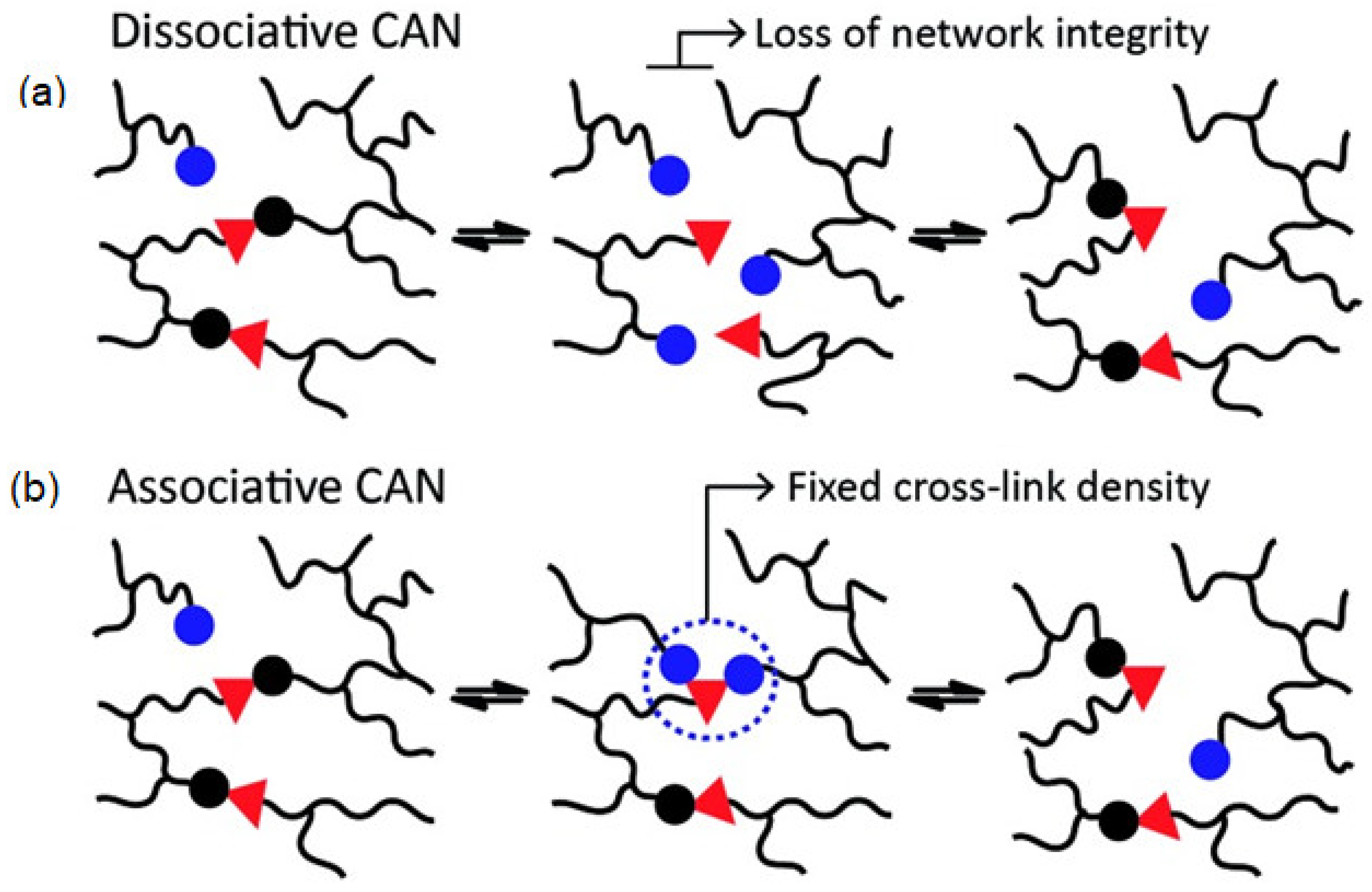
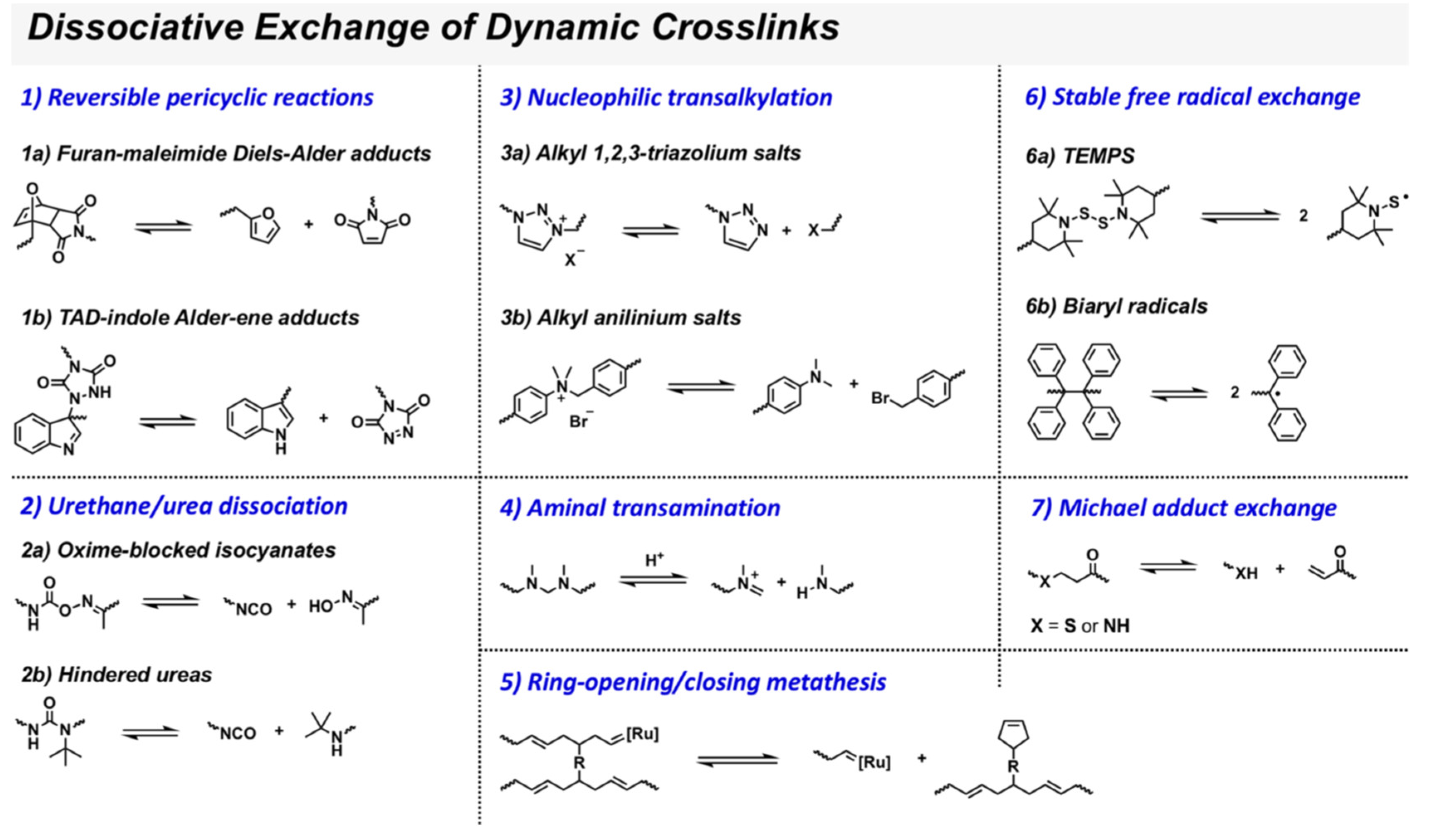
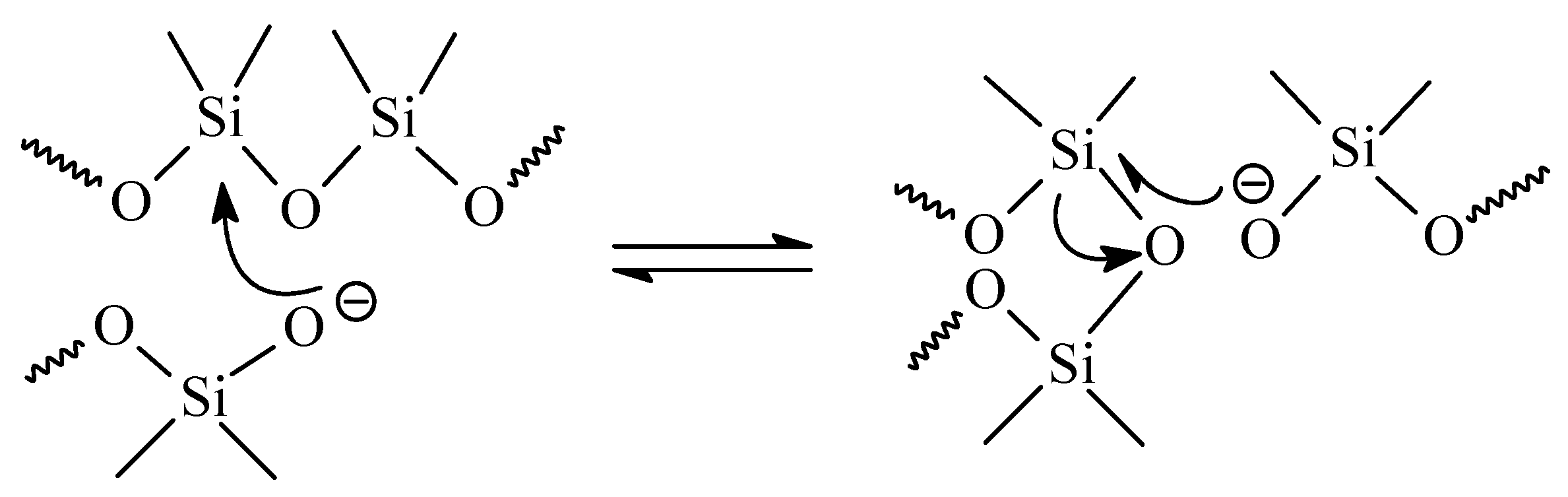
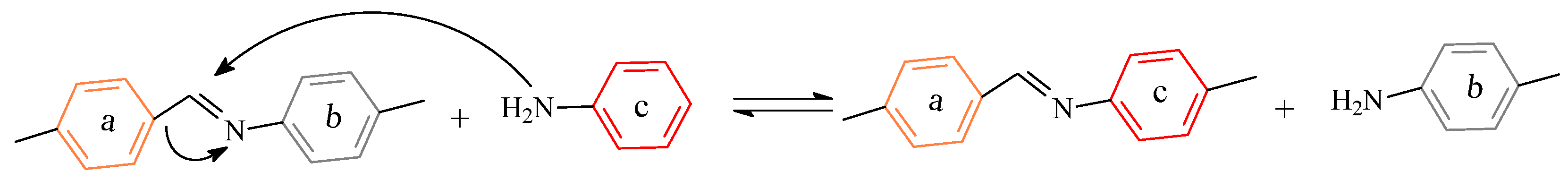
3.1. Dissociative Approach
3.1.1. Covalent Bonds
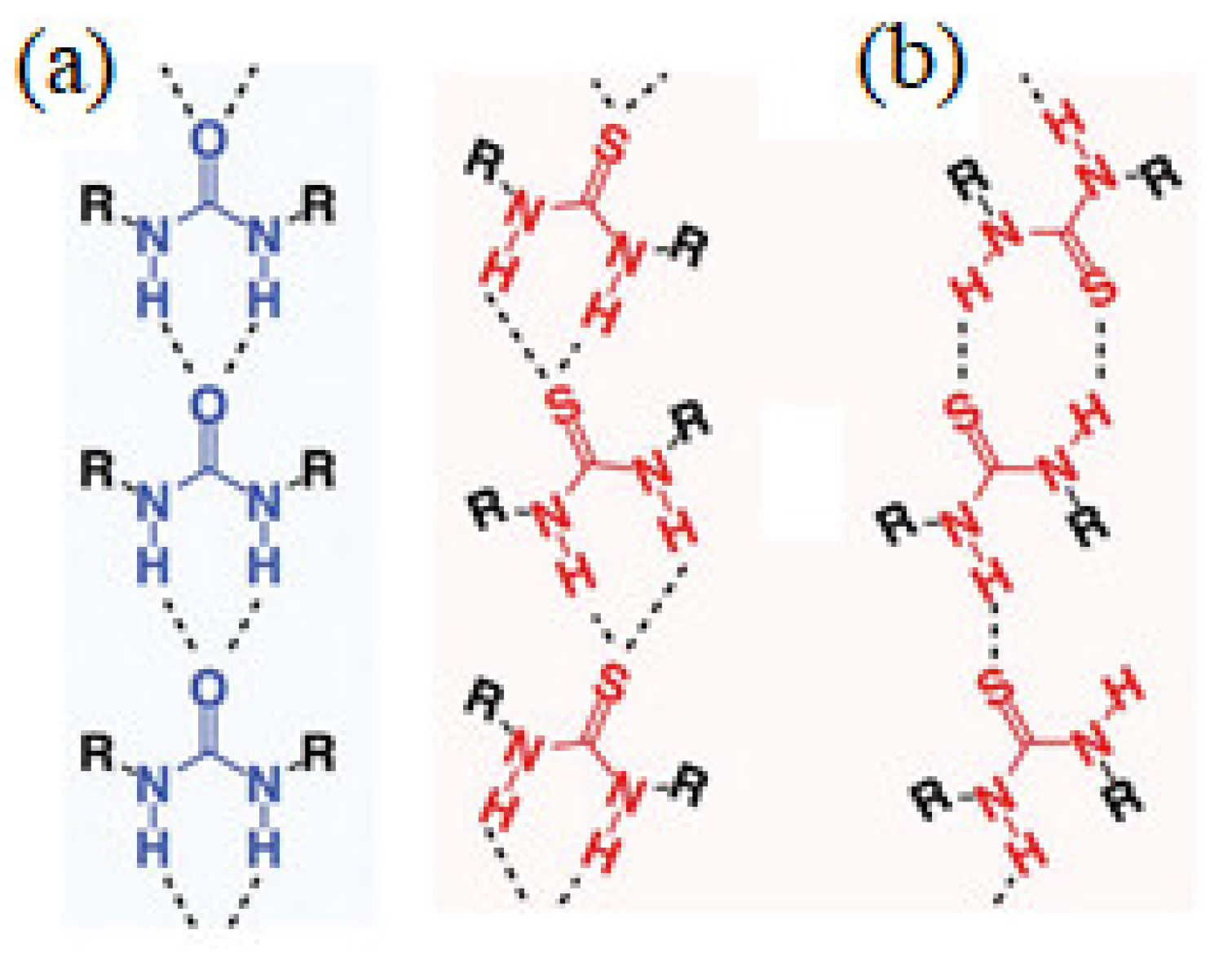
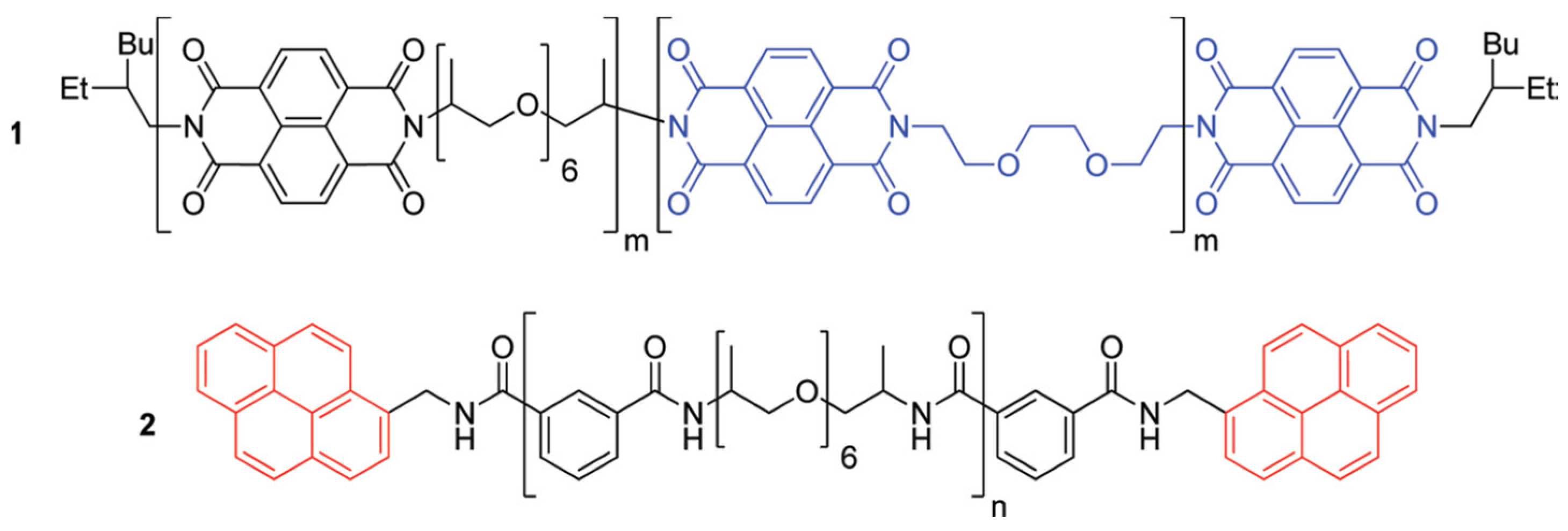
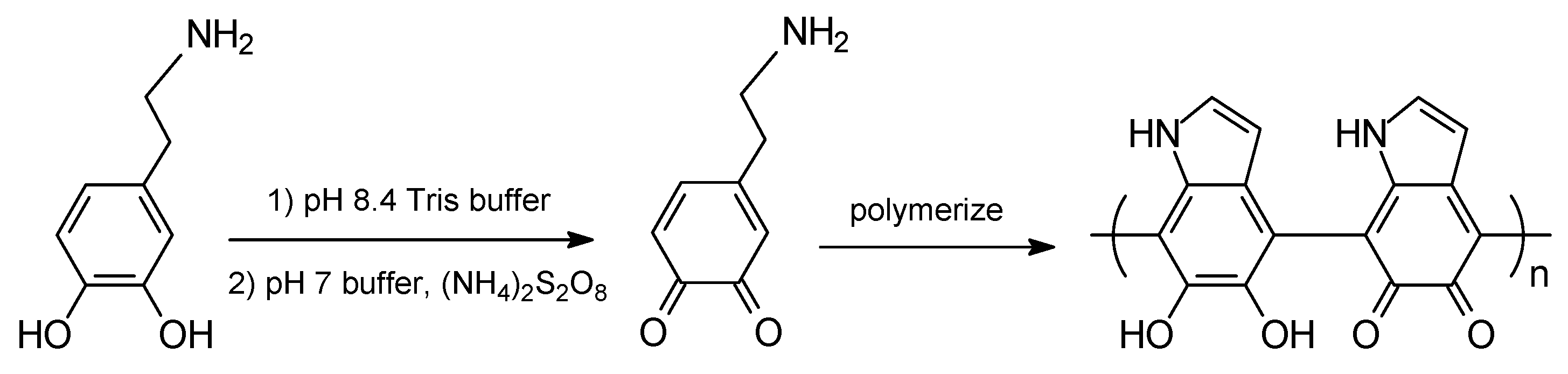
3.1.2. Non-Covalent Interactions
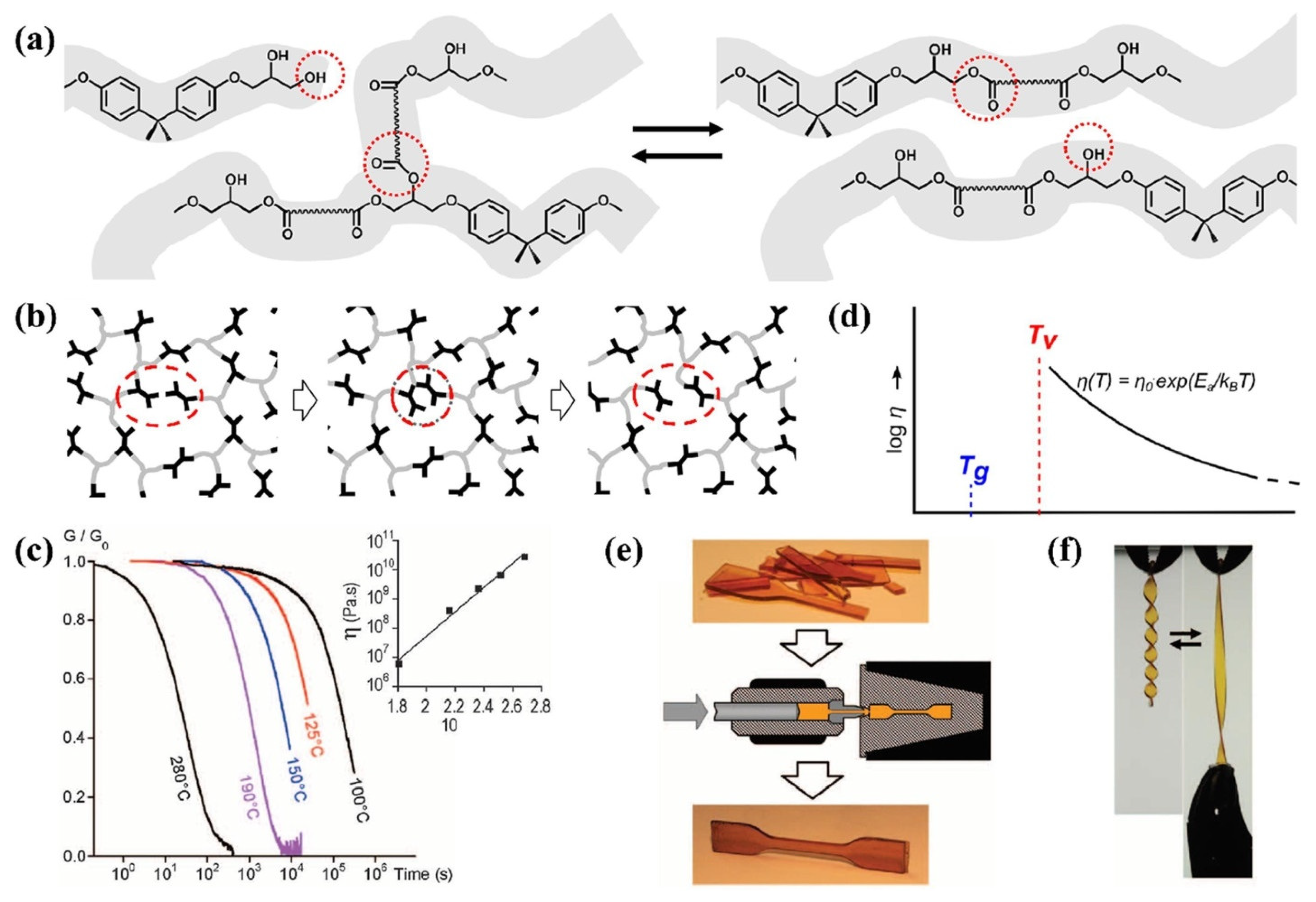
3.2. Associative Approach: Vitrimers
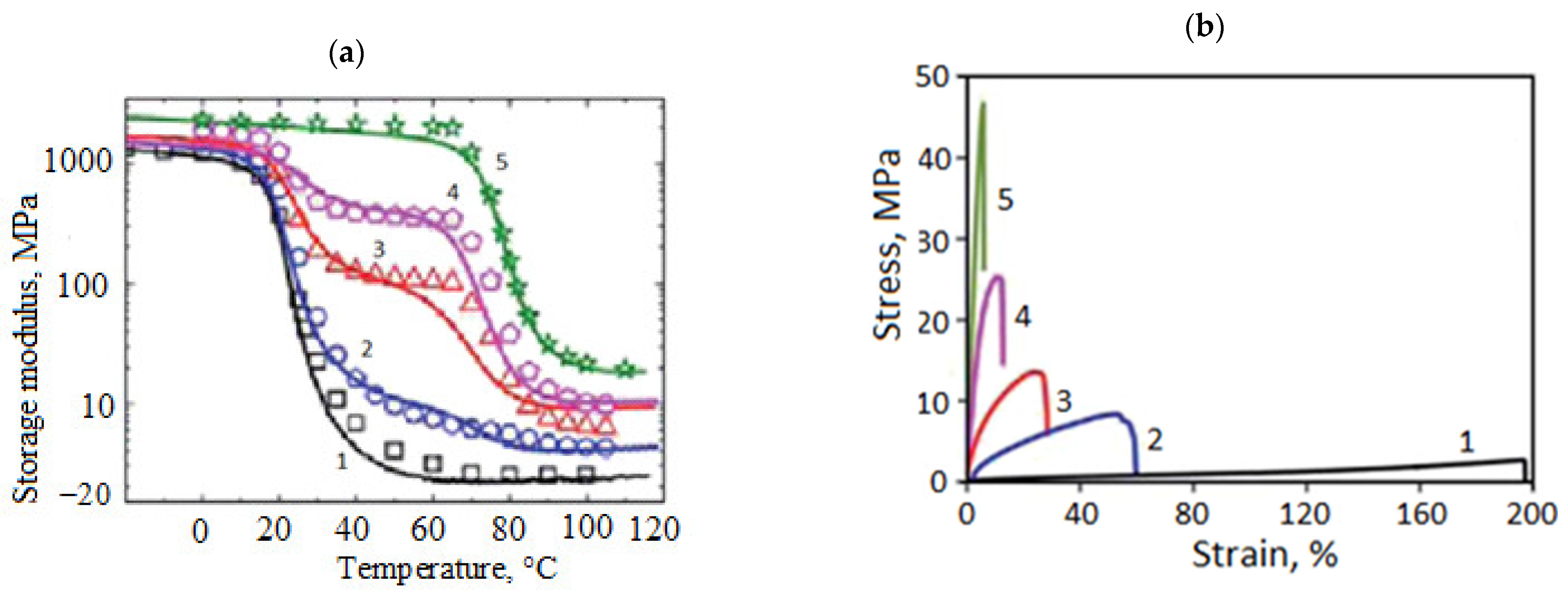
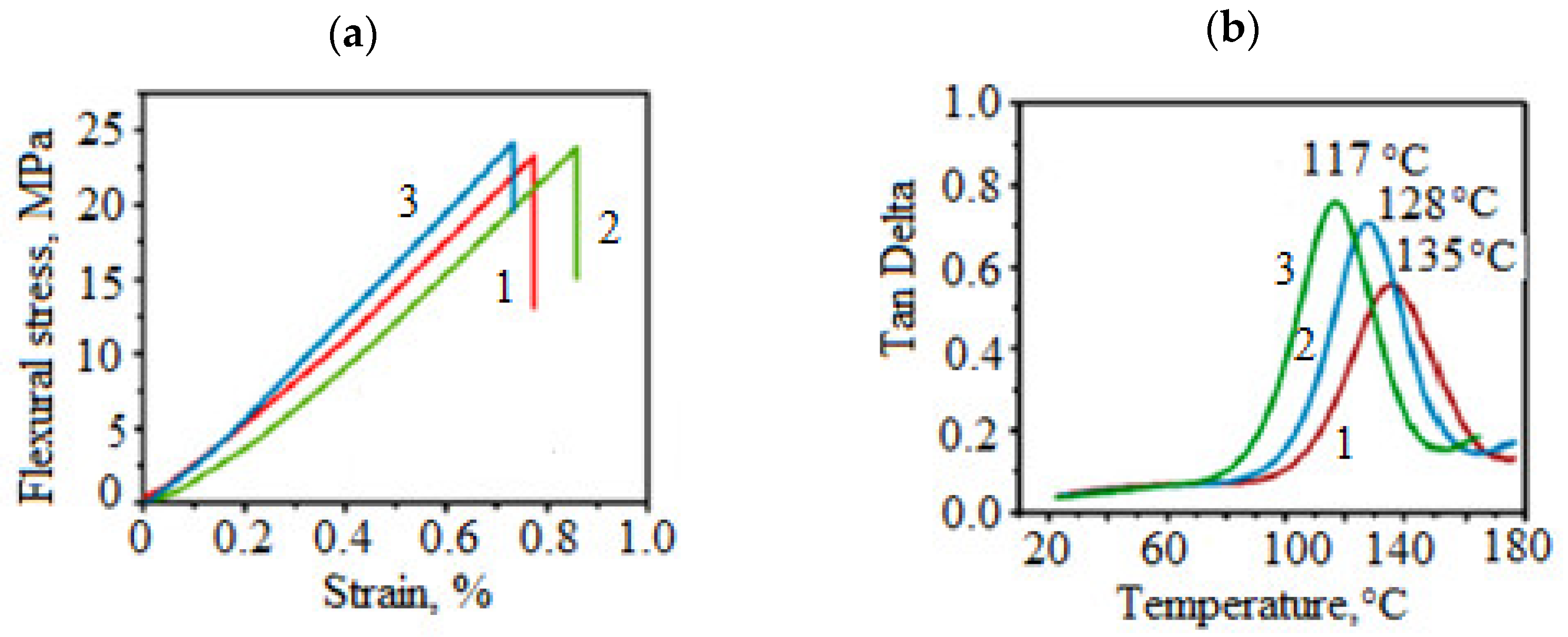
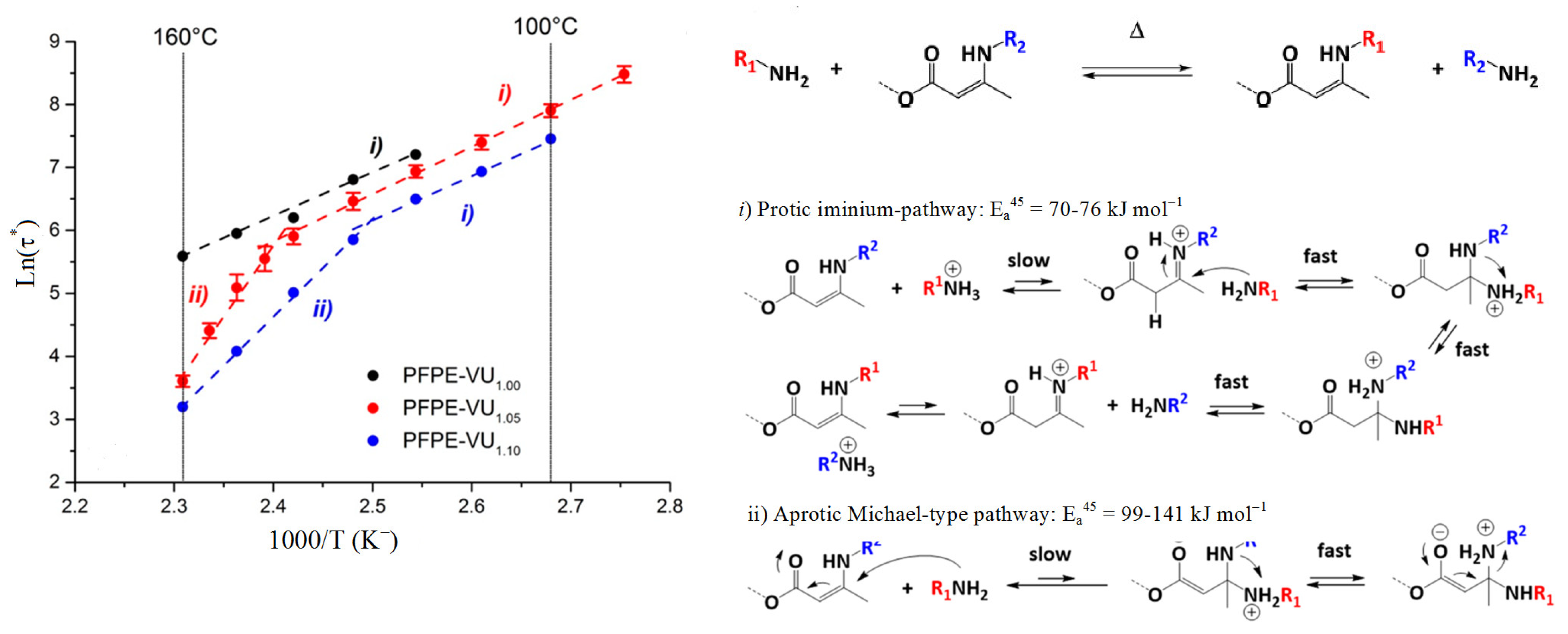
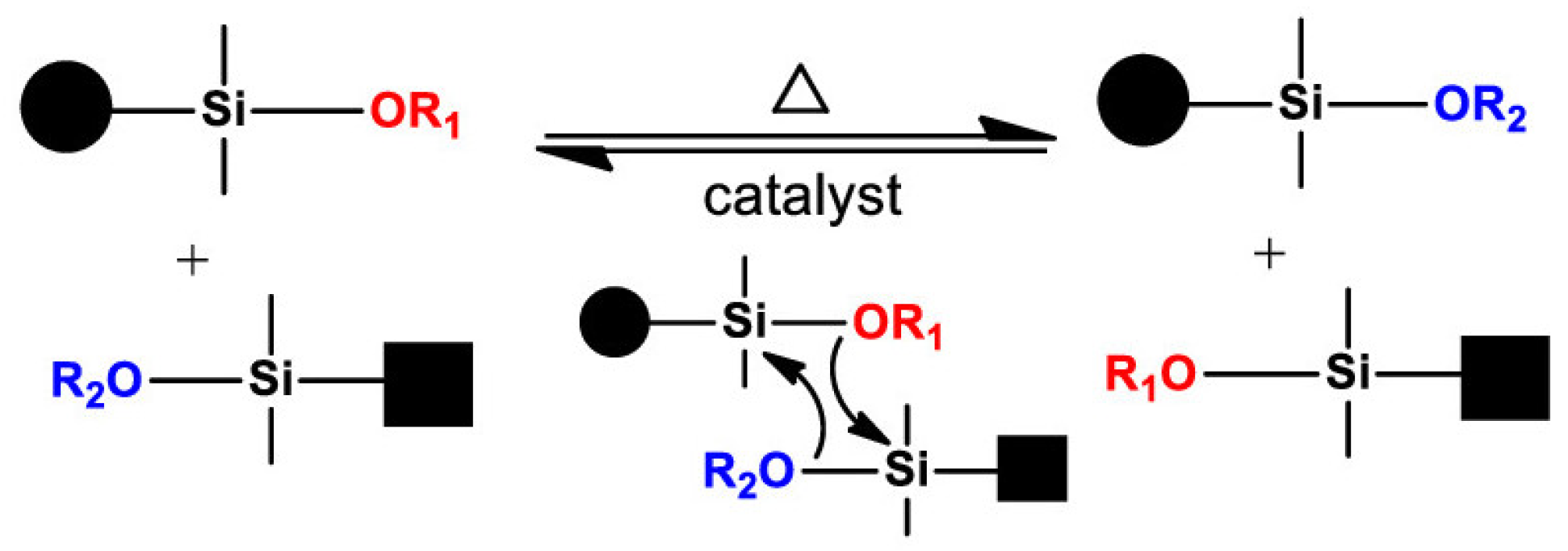
3.2.1. Vitrimers Based on the Transesterification Reaction
3.2.2. Vitrimers Based on Disulfide Exchange
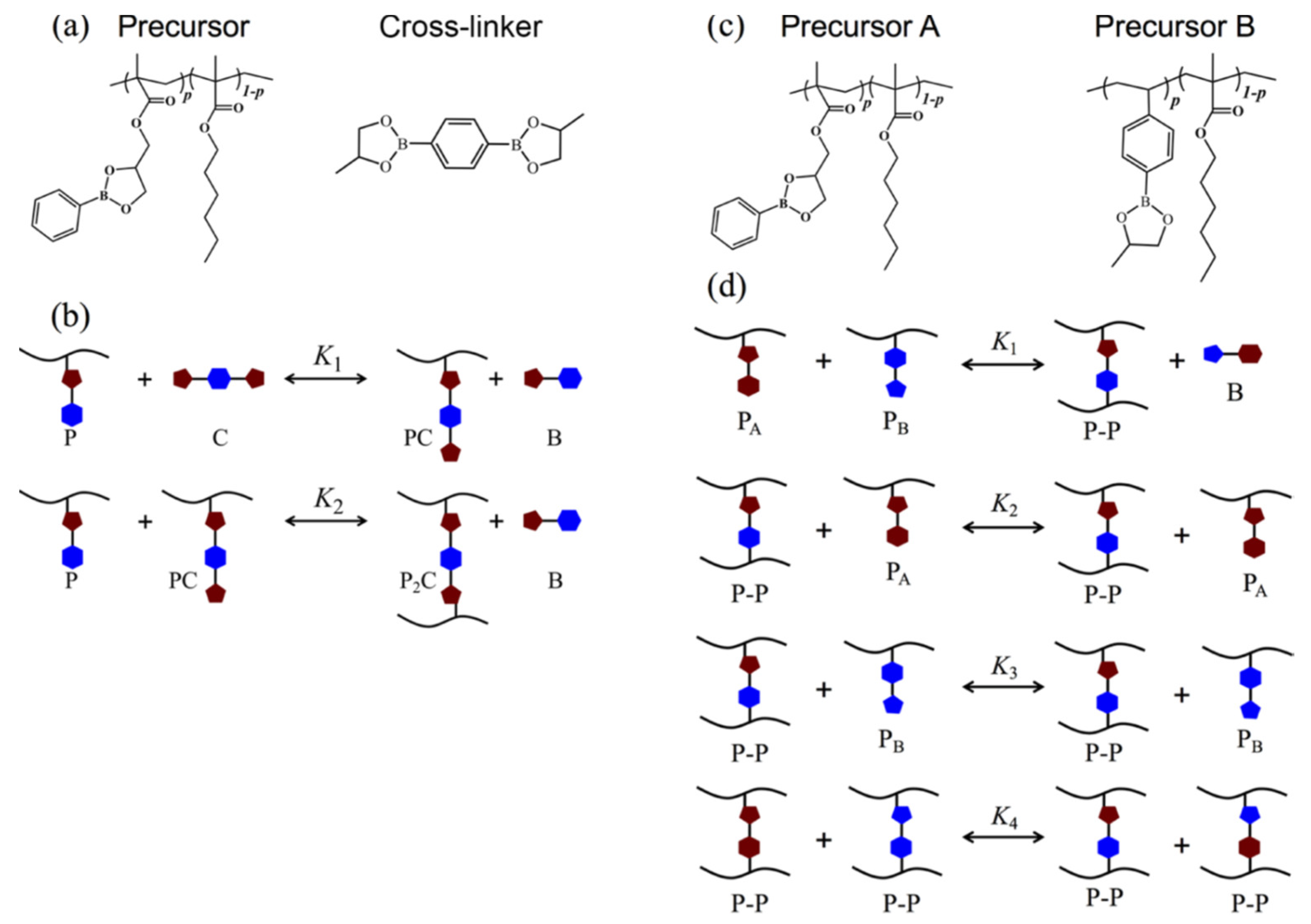
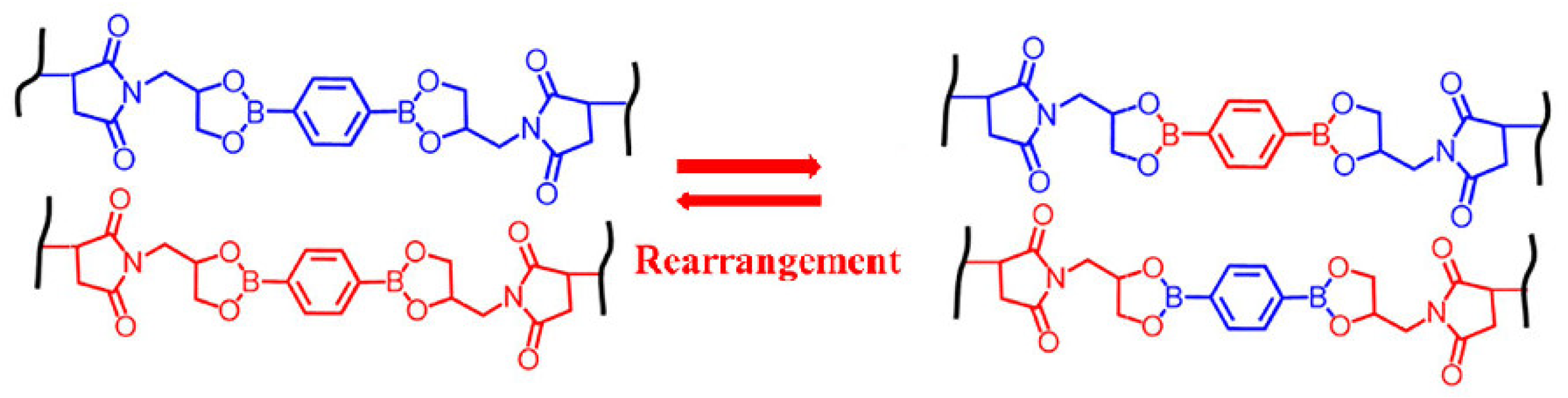
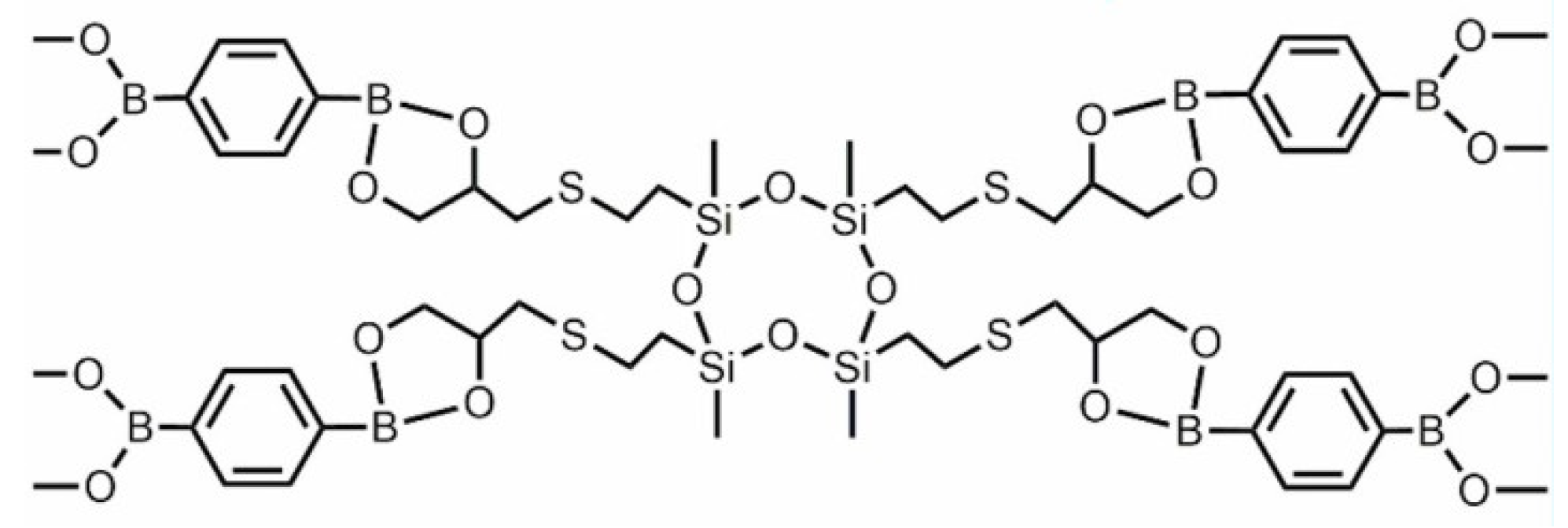
3.2.3. Vitrimers Based on Dioxaborolanes
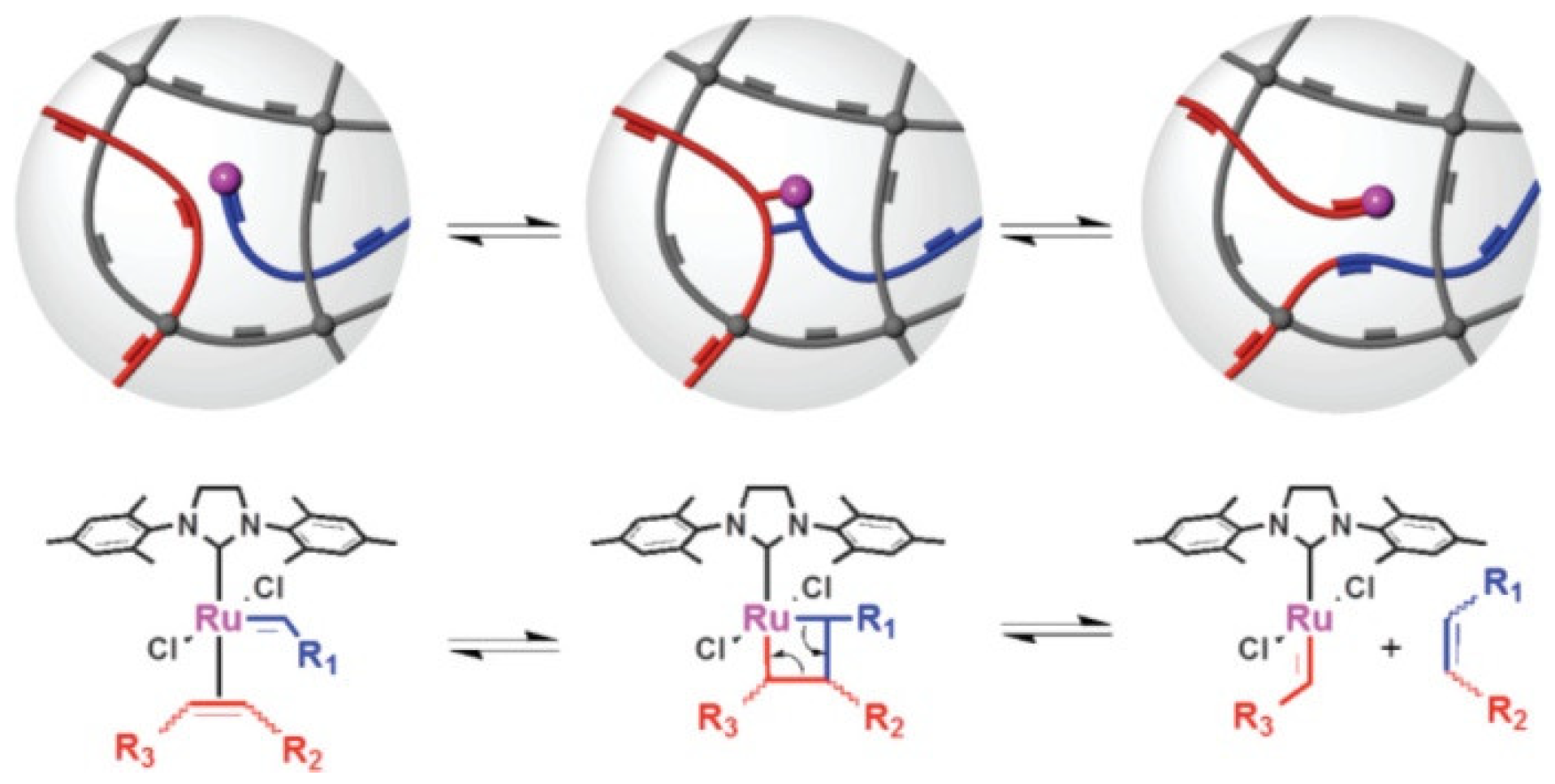
3.2.4. Vitrimers of Other Structures
4. External Self-Healing
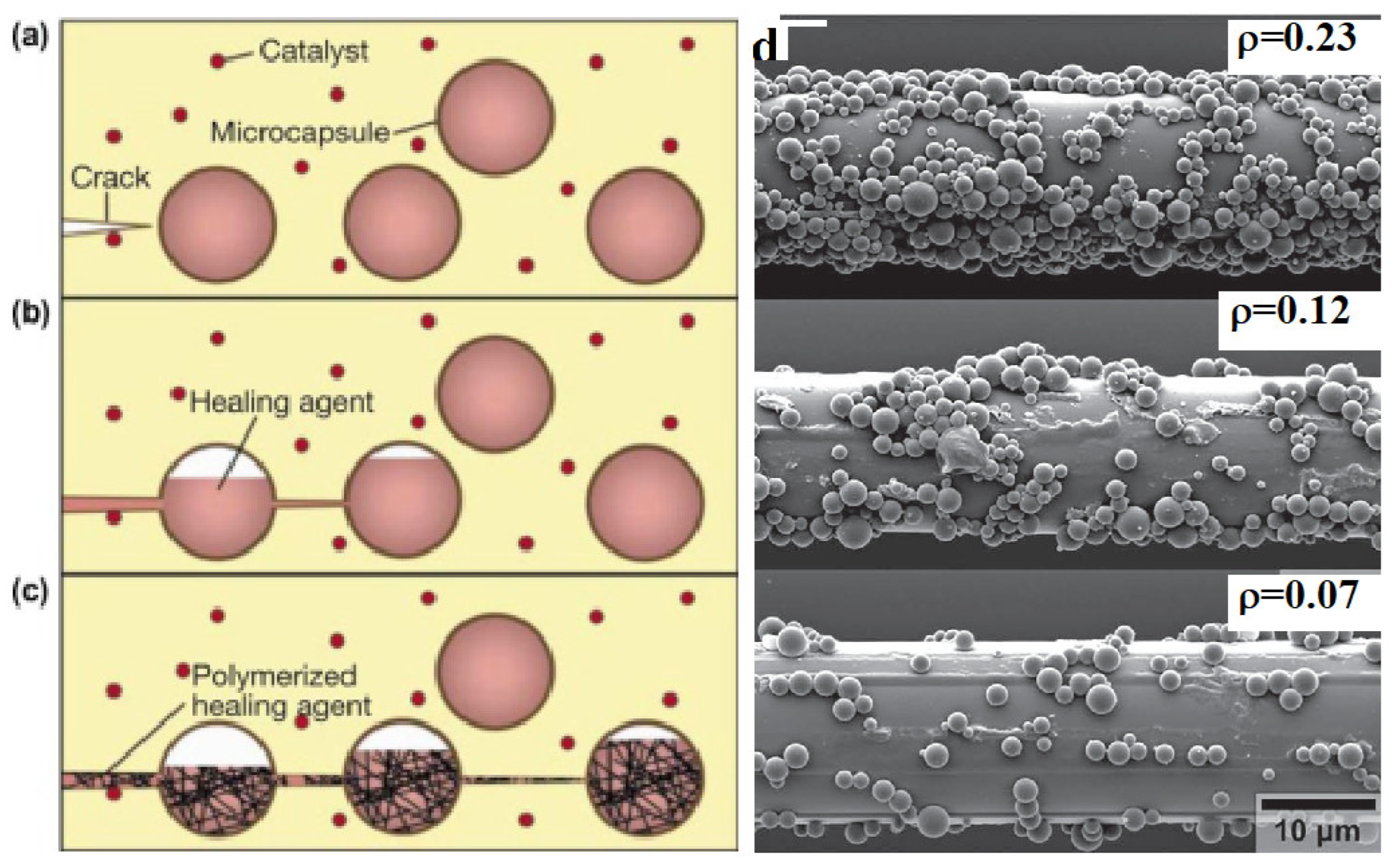
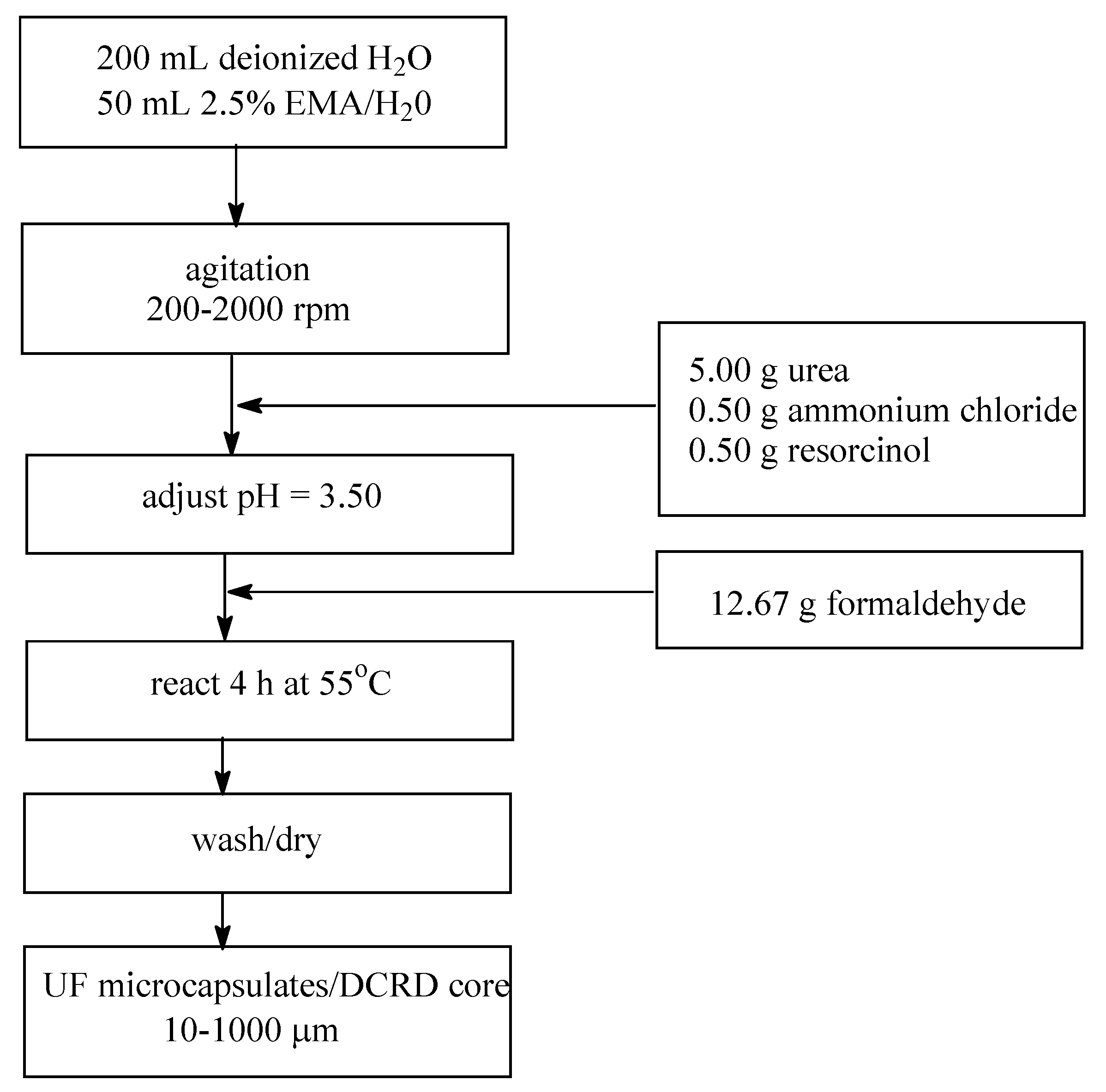
4.1. Use of Microcapsules

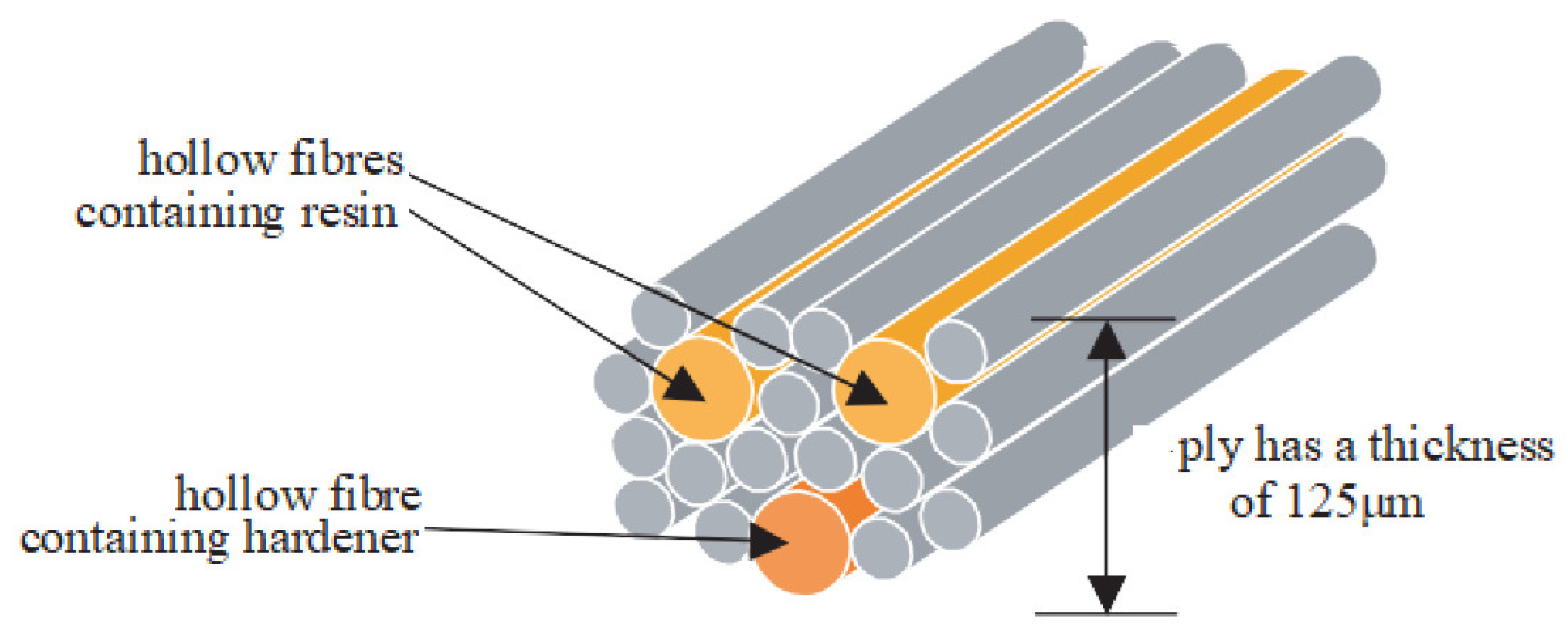
4.2. Use of Hollow Glass Fibers
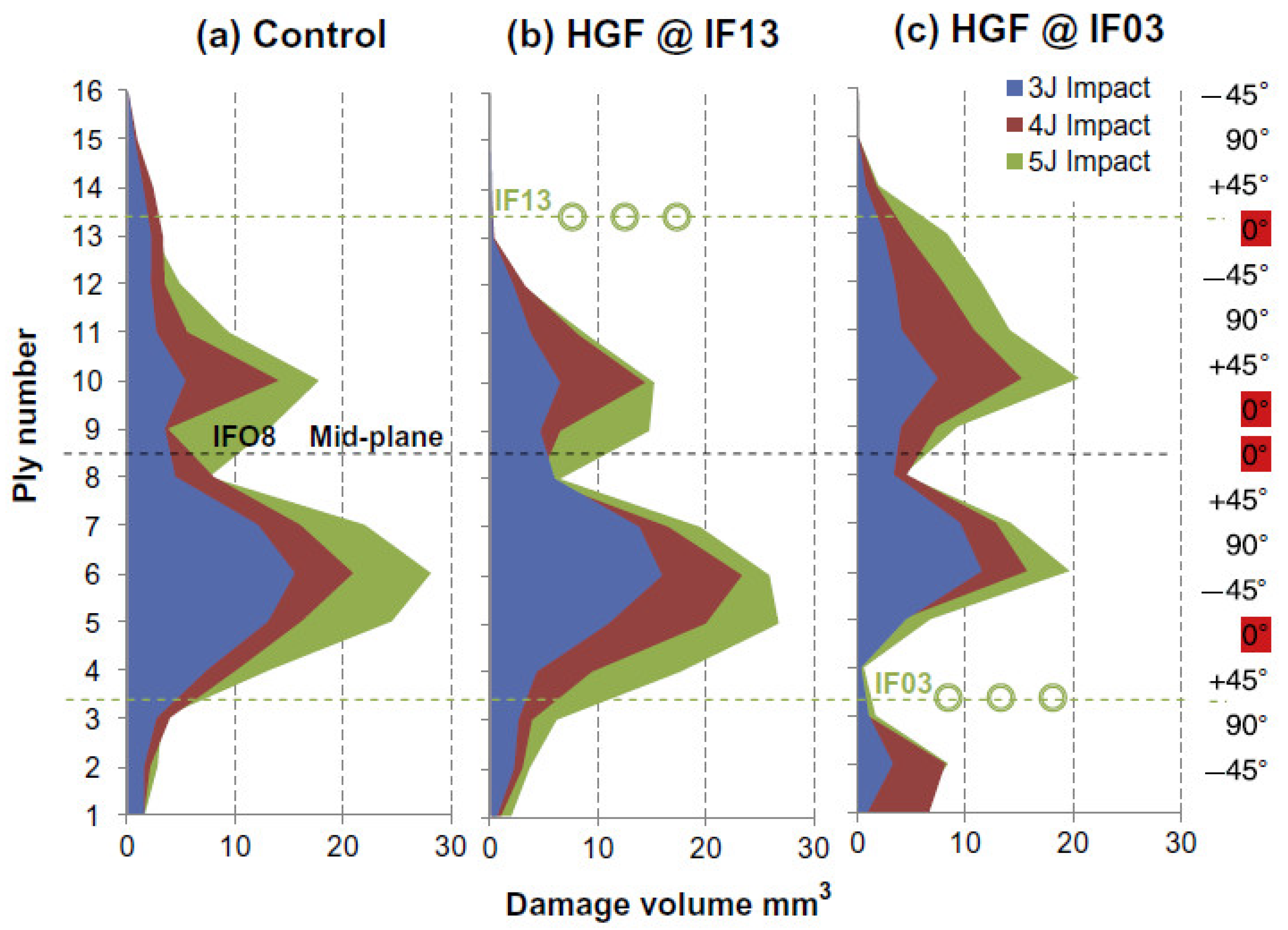
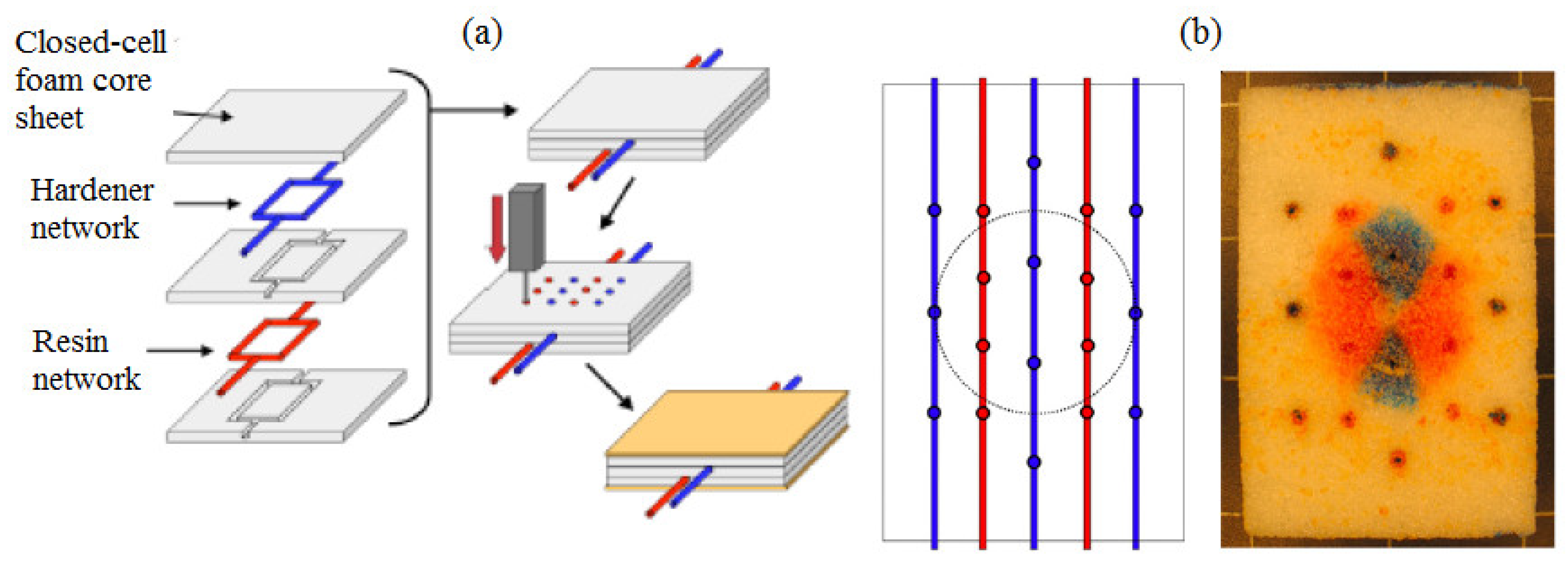
4.3. Materials with Microvasculature
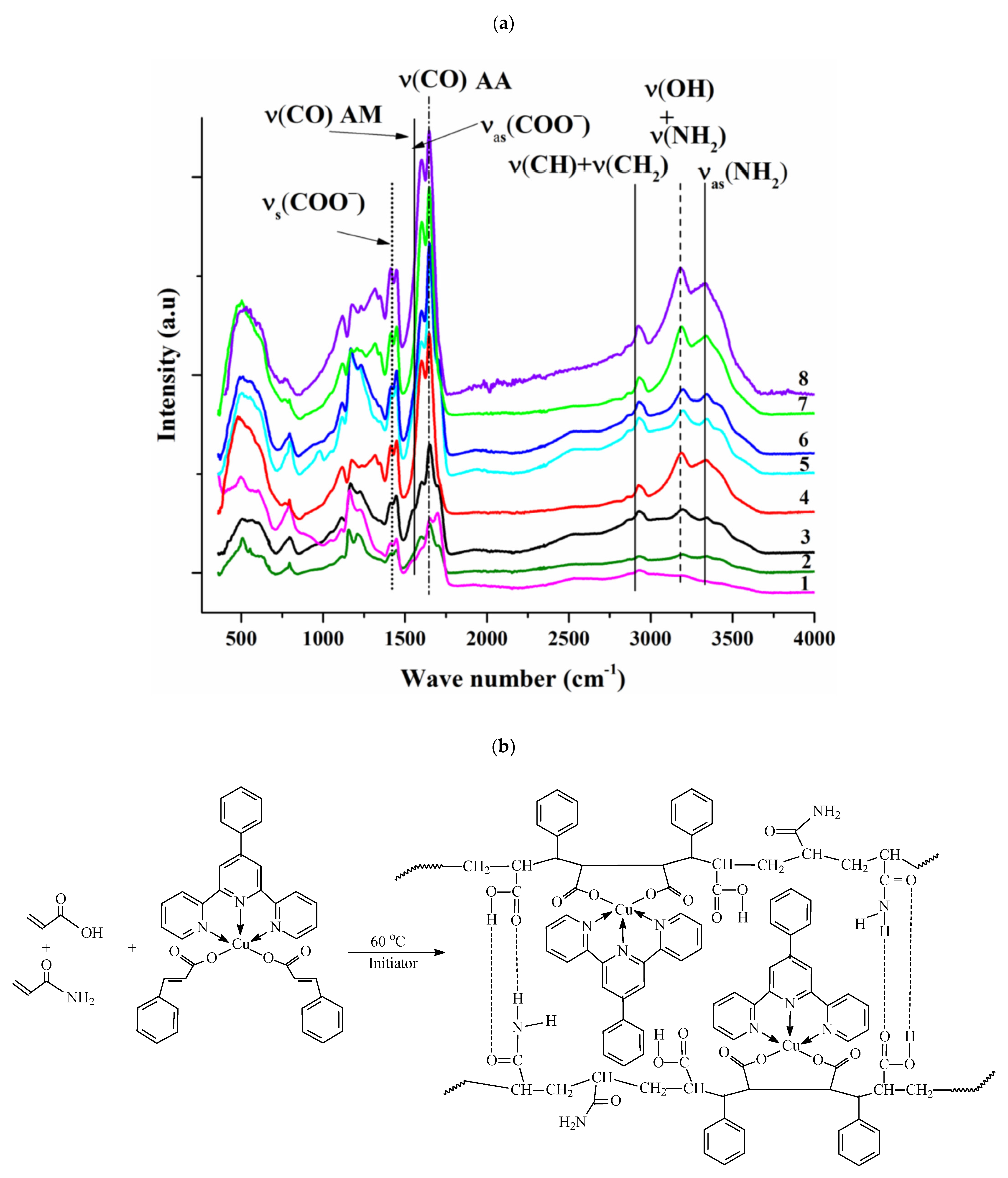
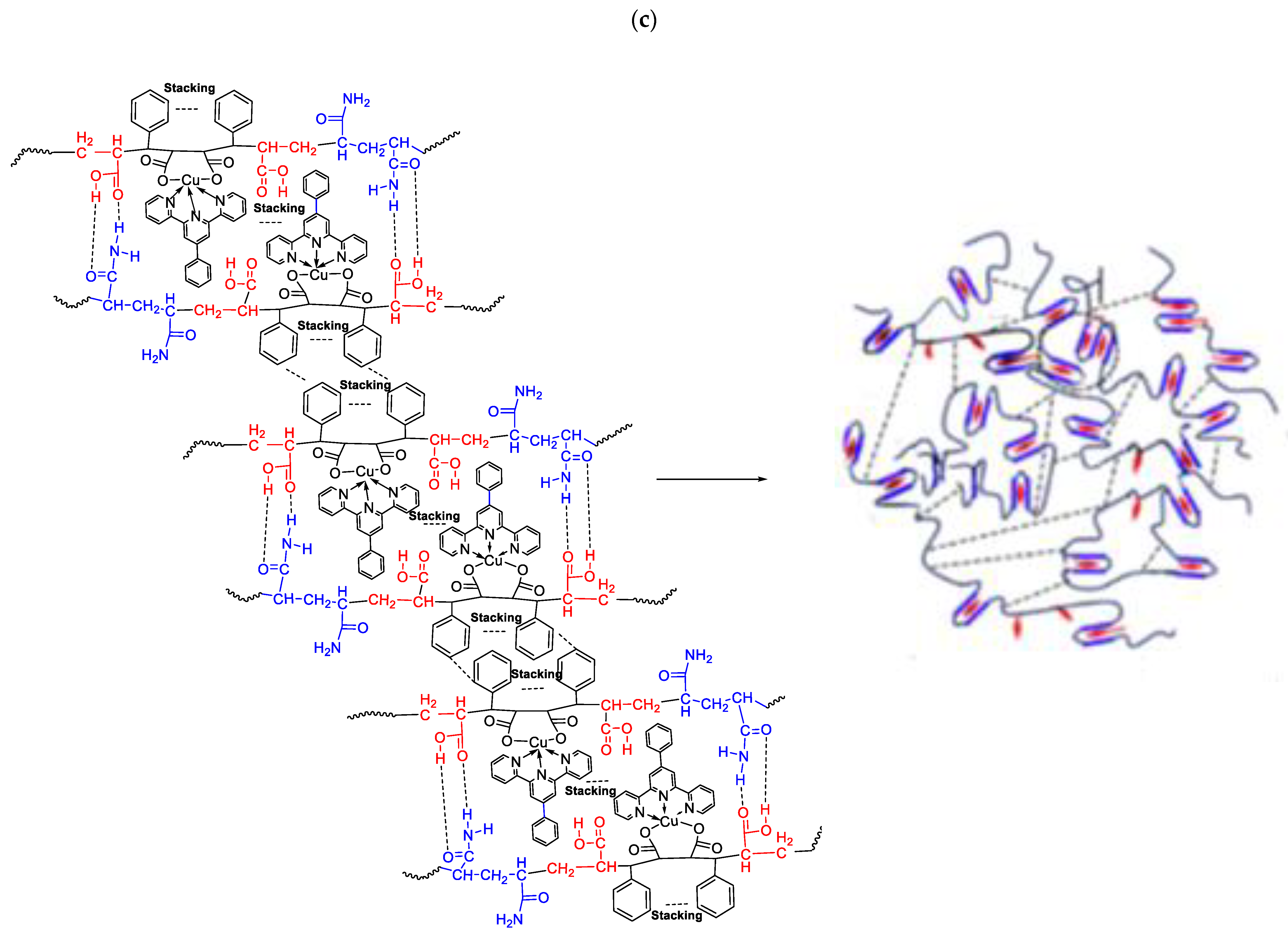
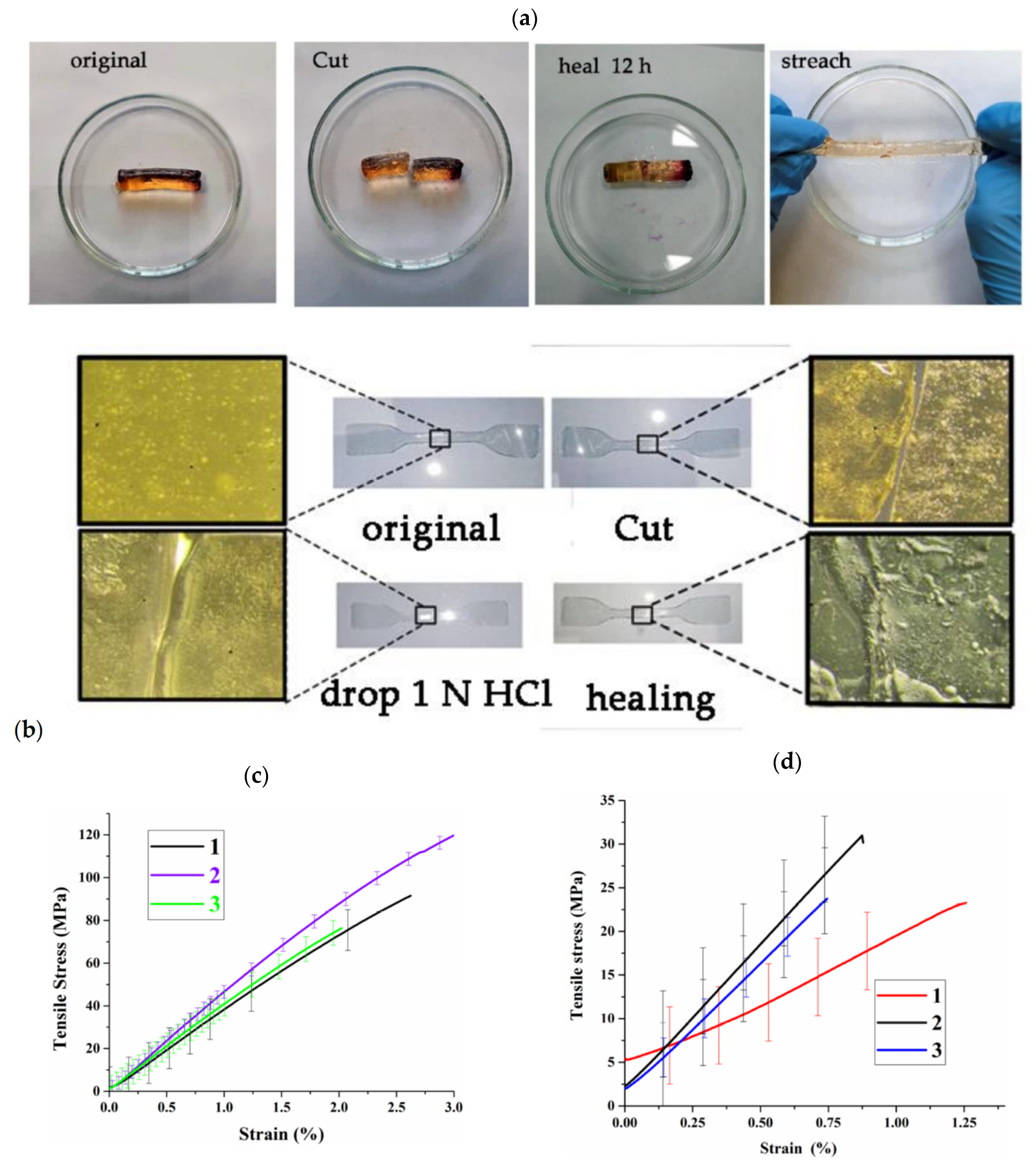
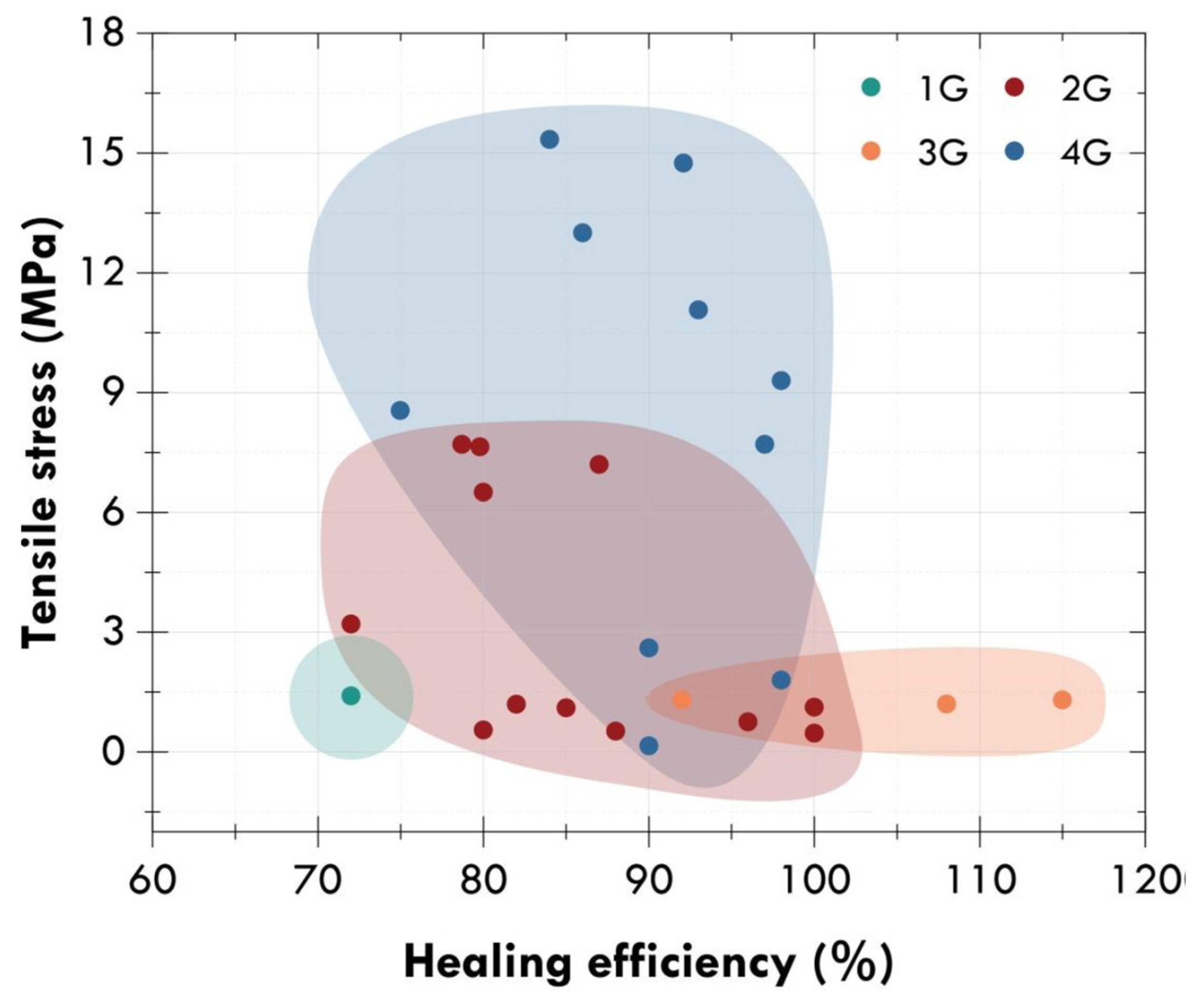
5. Self-Healing Metallopolymers
5.1. With Terpyridine Ligands
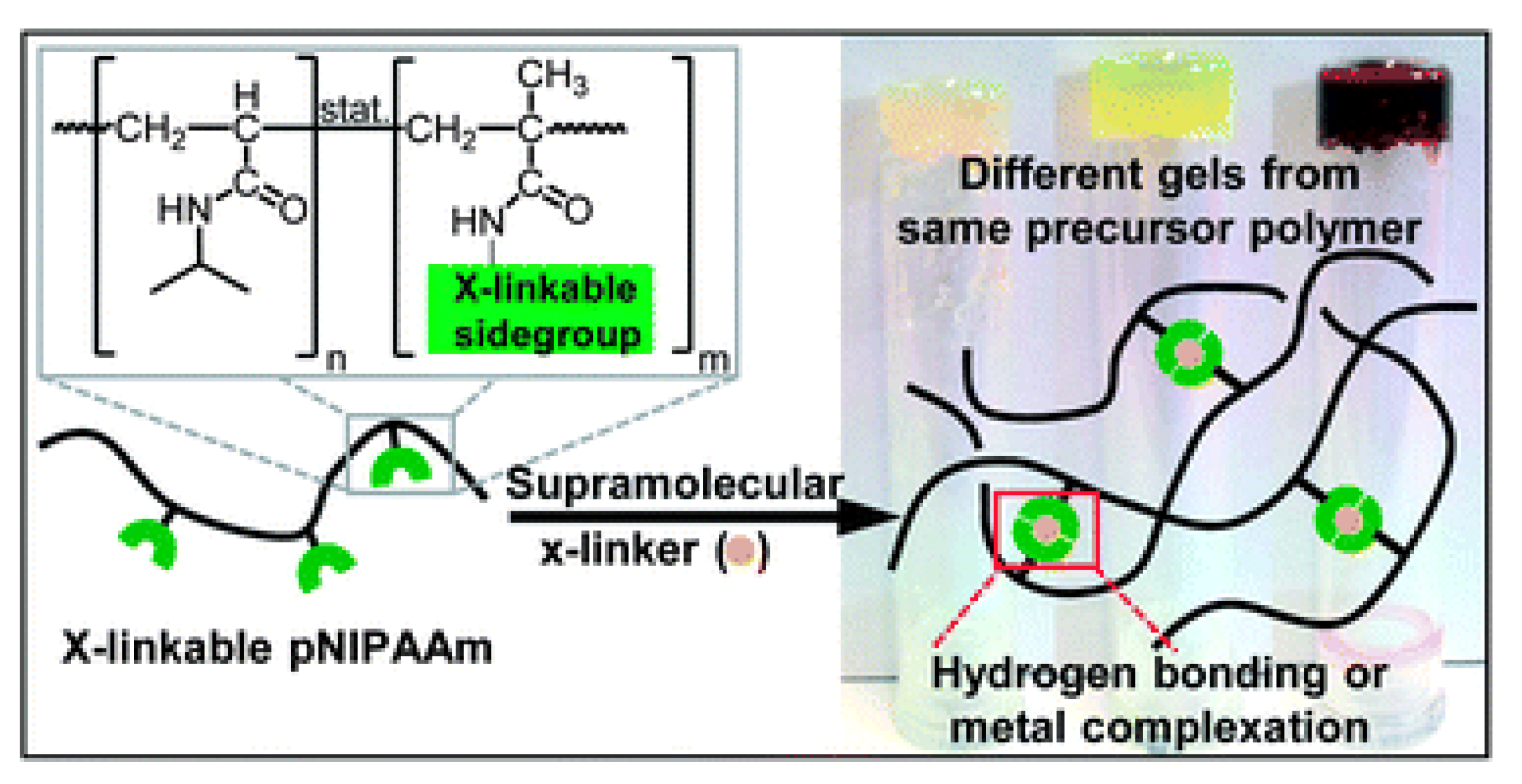
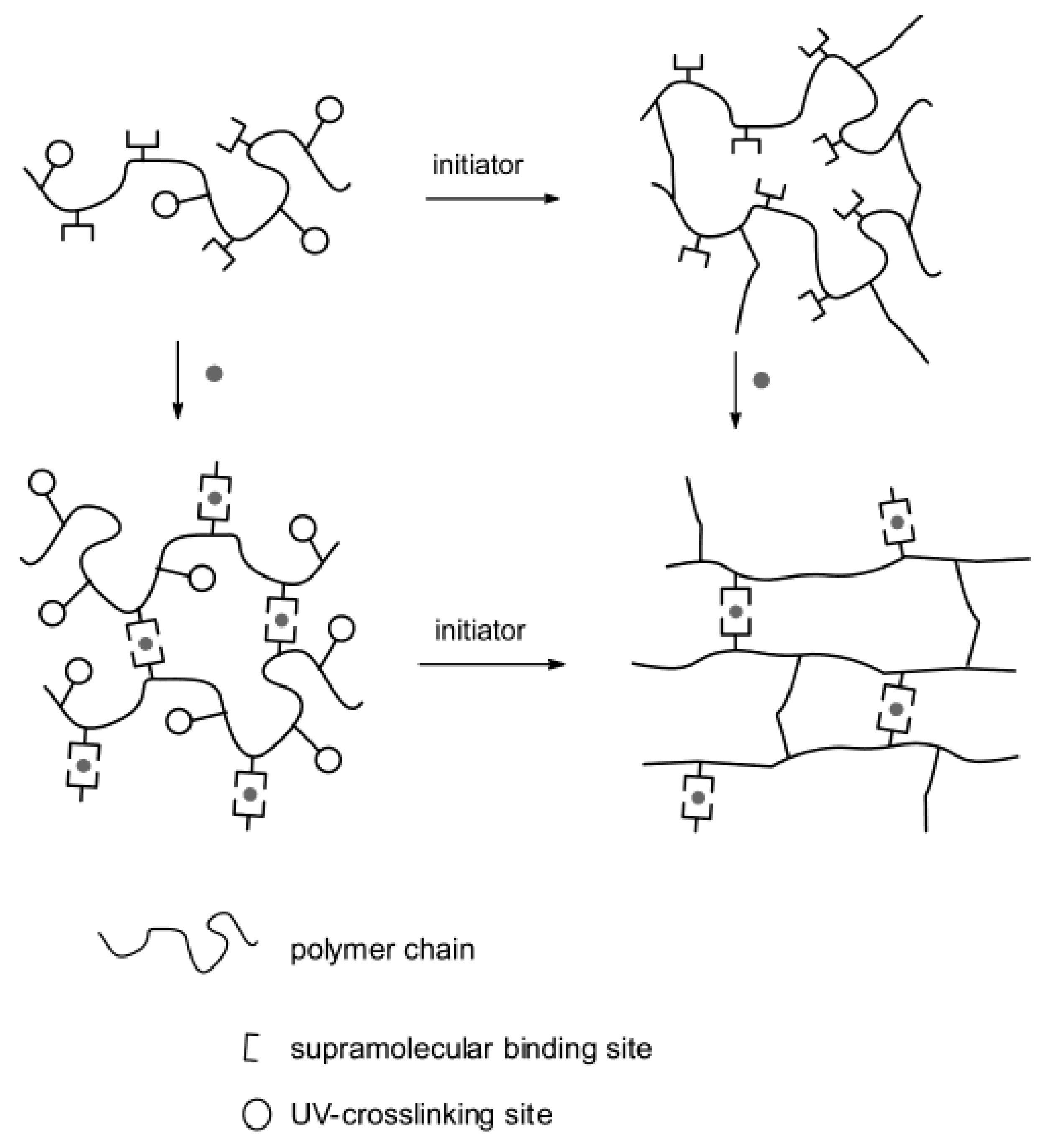
5.2. Llocopolymer Hydrogels
6. Summary and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, M.Q.; Rong, M.Z. Self-Healing Polymers and Polymer Composites; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Madara, S.R.; Sarath Raj, N.S.; Selvan, C.P. Review of research and developments in self healing composite materials. IOP Conf. Ser. Mater. Sci. Eng. 2018, 346, 012011. [Google Scholar] [CrossRef]
- Islam, S.; Bhat, G. Progress and challenges in self-healing composite materials. Mater. Adv. 2021, 2, 1896–1926. [Google Scholar] [CrossRef]
- Kenari, M.A.; Verma, D. Sustainable Biopolymer Composites: Biocompatibility, Self-Healing, Modeling, Repair and Recyclability; Verma, D., Sharma, M., Goh, K., Jain, S., Sharma, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; p. 155. [Google Scholar]
- Cioffi, M.O.H.; Bomfim, A.S.C.; Ambrogi, V.; Advani, S.G. A review on self-healing polymers and polymer composites for structural applications. Polym. Compos. 2022, 43, 7643–7668. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Rong, M.Z. Extrinsic and Intrinsic Approaches to Self-Healing Polymers and Polymer Composites; Wiley: Hoboken, NJ, USA, 2022. [Google Scholar]
- van der Zwaag, S. (Ed.) Self-Healing Materials: An Alternative Approach to 20 Centuries of Materials Science; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Nagori, I.; Goud, M.; Rajput, V. Self-Healing Composite Materials: A Review on Preceding and Perspective Research. Int. J. Technol. Innov. Modern Eng. Sci. 2019, 5, 687–697. [Google Scholar]
- Wool, R.P. Self-healing materials: A review. Soft Matter 2008, 4, 400–418. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, L.; Raimondo, M.; Naddeo, C.; Longo, P.; Mariconda, A. Self-healing materials for structural applications. Polym. Eng. Sci. 2013, 54, 777–784. [Google Scholar] [CrossRef]
- Blaiszik, B.J.; Kramer, S.L.B.; Olugebefola, S.C.; Moore, J.S.; Sottos, N.R.; White, S.R. Self-Healing Polymers and Composites. Annu. Rev. Mater. Res. 2010, 40, 179–211. [Google Scholar] [CrossRef]
- Williams, G.; Trask, R.; Bond, I. A self-healing carbon fibre reinforced polymer for aerospace applications. Compos. A 2007, 38, 1525–1532. [Google Scholar] [CrossRef]
- Toohey, K.S.; Sottos, N.R.; White, S.R. Characterization of Microvascular-Based Self-healing Coatings. Experim. Mech. 2008, 49, 707–717. [Google Scholar] [CrossRef]
- Fan, P.; Xue, C.; Zhou, X.; Yang, Z.; Ji, H. Dynamic Covalent Bonds of Si-OR and Si-OSi Enabled A Stiff Polymer to Heal and Recycle at Room Temperature. Materials 2021, 14, 2680. [Google Scholar] [CrossRef]
- Samadzadeh, M.; Boura, S.H.; Peikari, M.; Kasiriha, S.M.; Ashrafi, A. A review on self-healing coatings based on micro/nanocapsules. Prog. Org. Coat. 2010, 68, 159–164. [Google Scholar] [CrossRef]
- Stankiewicz, A.; Szczygieł, I.; Szczygieł, B. Self-healing coatings in anti-corrosion applications. J. Mater. Sci. 2013, 48, 8041–8051. [Google Scholar] [CrossRef] [Green Version]
- Sitnikov, N.N.; Shelyakov, A.V.; Khabibullina, I.A.; Mitina, N.A.; Resnina, N.N. Effect of Copper Content on the Structure and Phase Transformations in Melt-Spun TiNi–TiCu Alloys. Inorg. Mater. Appl. Res. 2018, 9, 279–285. [Google Scholar] [CrossRef]
- Yamuna, J.; Siva, T.; Kumari, S.S.S.; Sathiyanarayanan, S. A smart poly(aniline-formaldehyde) microcapsule based self-healing anticorrosive coating. RSC Adv. 2016, 6, 79–86. [Google Scholar] [CrossRef]
- Wei, H.; Wang, Y.; Guo, J.; Shen, N.Z.; Jiang, D.; Zhang, X.; Guo, Z. Advanced micro/nanocapsules for self-healing smart anticorrosion coatings. J. Mater. Chem. A 2015, 3, 469–480. [Google Scholar] [CrossRef]
- Guin, A.K.; Nayak, S.; Bhadu, M.K.; Singh, V.; Rout, T.K. Development and Performance Evaluation of Corrosion Resistance Self-Healing Coating. ISRN Corros. 2014, 2014, 979323. [Google Scholar] [CrossRef]
- Sadrabadi, T.E.; Allahkaram, S.R.; Staab, T.; Towhidi, N. Preparation and characterization of durable micro/nanocapsules for use in self-healing anticorrosive coatings. Polym. Sci. Ser. B 2017, 59, 281–291. [Google Scholar] [CrossRef]
- Caruso, M.M.; Davis, D.A.; Shen, Q.; Odom, S.A.; Sottos, N.R.; White, S.R.; Moore, J.S. Mechanically-Induced Chemical Changes in Polymeric Materials. Chem. Rev. 2009, 109, 5755–5798. [Google Scholar] [CrossRef]
- Hart, L.R.; Harries, J.L.; Greenland, B.W.; Colquhoun, H.M.; Hayes, W. Healable supramolecular polymers. Polym. Chem. 2013, 4, 4860–4870. [Google Scholar]
- Yang, Y.; Xu, Y.; Ji, Y.; Wei, Y. Functional epoxy vitrimers and composites. Prog. Mater. Sci. 2020, 120, 100710. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, X.; Urban, M.W. Chemical and physical aspects of self-healing materials. Prog. Polym. Sci. 2015, 49–50, 34–59. [Google Scholar] [CrossRef] [Green Version]
- Bekas, D.G.; Tsirka, K.; Baltzis, D.; Paipetis, A.S. Self-healing materials: A review of advances in materials, evaluation, characterization and monitoring techniques. Compos. B 2016, 87, 92–119. [Google Scholar] [CrossRef]
- Fischer, H. Self-repairing material systems—A dream or a reality? Nat. Sci. 2010, 2, 873–901. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.J.; Bond, I.P.; Trask, R.S. Compression after impact assessment of self-healing CFRP. Compos. A 2009, 40, 1399–1406. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, S.; Toendepi, I.; Jiang, Q.; Wei, Y.; Qiu, Y.; Liu, W. Reprocessable, Reworkable, and Mechanochromic Polyhexahydrotriazine Thermoset with Multiple Stimulus Responsiveness. Polymers 2020, 12, 2375. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Taynton, P.; Zhang, W.; Dunn, M.L.; Qi, H.J. Reprocessing and recycling of thermosetting polymers based on bond exchange reactions. RSC Adv. 2014, 4, 10108–10117. [Google Scholar] [CrossRef]
- Sánchez-Silva, L.; Gutiérrez, N.; Sánchez, P.; Romero, A.; Valverde, J.L. Smart microcapsules containing nonpolar chemical compounds and carbon nanofibers. Chem. Eng. J. 2012, 181–182, 813–822. [Google Scholar] [CrossRef]
- Trask, R.S.; Williams, G.J.; Bond, I.P. Bioinspired self-healing of advanced composite structures using hollow glass fibres. J. Roy. Soc. Interface 2006, 4, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Blaiszik, B.J.; Baginska, M.; White, S.R.; Sottos, N.R. Autonomic Recovery of Fiber/Matrix Interfacial Bond Strength in a Model Composite. Adv. Funct. Mater. 2010, 20, 3547–3554. [Google Scholar] [CrossRef]
- Wang, Y.; Pham, D.T.; Ji, C. Self-healing composites: A review. Cogent. Eng. 2015, 2, 1075686. [Google Scholar] [CrossRef] [Green Version]
- Esser-Kahn, A.P.; Thakre, P.R.; Dong, H.; Patrick, J.F.; Vlasko-Vlasov, V.K.; Sottos, N.R.; White, S.R. Three-dimensional microvascular fiber-reinforced composites. Adv. Mater. 2011, 23, 3654–3658. [Google Scholar] [CrossRef] [PubMed]
- Trask, R.S.; Bond, I.P. Bioinspired engineering study of Plantae vascules for self-healing composite structures. J. Roy. Soc. Interface 2009, 7, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Luterbacher, R.; Trask, R.S.; Bond, I.P. Static and fatigue tensile properties of cross-ply laminates containing vascules for self-healing applications. Smart Mater. Struct. 2015, 25, 015003. [Google Scholar] [CrossRef] [Green Version]
- Shields, Y.; De Belie, N.; Jefferson, A.; Van Tittelboom, K. A review of vascular networks for self-healing applications. Smart Mater. Struct. 2021, 30, 063001. [Google Scholar] [CrossRef]
- Kostopoulos, V.; Kotrotsos, A.; Baltopoulos, A.; Tsantzalis, S.; Tsokanas, P.; Loutas, T.; Bosman, A.W. Mode II fracture toughening and healing of composites using supramolecular polymer interlayers. Express Polym. Lett. 2016, 10, 914–926. [Google Scholar] [CrossRef]
- Swait, T.J.; Rauf, A.; Grainger, R.; Bailey, P.B.S.; Lafferty, A.D.; Fleet, E.J.; Hayes, S.A. Smart composite materials for self-sensing and self-healing. Plast. Rubb. Compos. 2012, 41, 215–224. [Google Scholar] [CrossRef]
- Kostopoulos, V.; Kotrotsos, A.; Tsantzalis, S.; Tsokanas, P.; Loutas, T.; Bosman, A.W. Toughening and healing of continuous fibre reinforced composites by supramolecular polymers. Compos. Sci. Technol. 2016, 128, 84–93. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; De Hoe, G.X.; Snyder, R.L.; Dichtel, W.R.; Hillmyer, M.A. Approaches to Sustainable and Continually Recyclable Cross-Linked Polymers. ACS Sustain. Chem. Eng. 2018, 6, 11145–11159. [Google Scholar] [CrossRef] [Green Version]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Wojtecki, R.; Meador, M.; Rowan, S. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [Google Scholar] [CrossRef]
- Rozenberg, B.A.; Irzhak, V.I.; Enikolopyan, N.S. Interchain Exchange in Polymers; Khimiya: Moscow, Russia, 1975. (In Russian) [Google Scholar]
- Liu, T.; Zhao, B.; Zhang, J. Recent development of repairable, malleable and recyclable thermosetting polymers through dynamic transesterification. Polymer 2020, 194, 122392. [Google Scholar] [CrossRef]
- Korshak, V.V.; Vinogradova, S.V. Heterochain Polyesters and Polyethers; Nauka: Moscow, Russia, 1958. (In Russian) [Google Scholar]
- Tretbar, C.A.; Neal, J.A.; Guan, Z. Direct Silyl Ether Metathesis for Vitrimers with Exceptional Thermal Stability. J. Am. Chem. Soc. 2019, 141, 16595–16599. [Google Scholar] [CrossRef] [PubMed]
- Handbook of Metathesis; Grubbs, R.H.; Wenzel, A.G.; O’Leary, D.J.; Khosravi, E. (Eds.) Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Hayashi, M. Implantation of Recyclability and Healability into Cross-Linked Commercial Polymers by Applying the Vitrimer Concept. Polymers 2020, 12, 1322. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Xin, A.; Wang, Q. Mechanics of self-healing polymer networks crosslinked by dynamic bonds. J. Mech. Phys. Solids 2018, 121, 409–431. [Google Scholar] [CrossRef]
- Roy, N.; Bruchmann, B.; Lehn, J.-M. DYNAMERS: Dynamic polymers as self-healing materials. Chem. Soc. Rev. 2015, 44, 3786–3807. [Google Scholar] [CrossRef] [Green Version]
- Lehn, J.M. Dynamers: Dynamic Molecular and Supramolecular Polymers. Austr. J. Chem. 2010, 63, 611–623. [Google Scholar] [CrossRef]
- Lehn, J.M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Fox, J.; Wie, J.J.; Greenland, B.W.; Burattini, S.; Hayes, W.; Colquhoun, H.M.; Rowan, S.J. High-strength, healable, supramolecular polymer nanocomposites. J. Am. Chem. Soc. 2012, 134, 5362–5368. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, Z.; Wu, S.; Guo, B. Integrating Sacrificial Bonds into Dynamic Covalent Networks toward Mechanically Robust and Malleable Elastomers. ACS Macro Lett. 2019, 8, 193–199. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.; Zhou, Q. Preparation and characterization of microcapsules based self-healing coatings containing epoxy ester as healing agent. Prog. Org. Coat. 2018, 125, 403–410. [Google Scholar] [CrossRef]
- Cui, G.; Zhang, C.; Wang, A.; Zhou, X.; Xing, X.; Liu, J.; Lu, Q. Research progress on self-healing polymer/graphene anticorrosion coatings. Prog. Org. Coat. 2021, 155, 106231. [Google Scholar] [CrossRef]
- Balazs, A.C.; Emrick, T.; Russell, T.P. Nanoparticle polymer composites: Where two small worlds meet. Science 2006, 314, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.C. Modeling self-healing materials. Mater. Today 2007, 10, 18–23. [Google Scholar]
- Gupta, S.; Zhang, Q.; Emrick, T.; Balazs, A.; Russell, T. Entropy-driven segregation of nanoparticles to cracks in multilayered composite polymer structures. Nat. Mater. 2006, 5, 229–233. [Google Scholar] [CrossRef]
- Stukalin, E.B.; Cai, L.-H.; Kumar, N.A.; Leibler, L.; Rubinstein, M. Self-Healing of Unentangled Polymer Networks with Reversible Bonds. Macromolecules 2013, 46, 7525–7541. [Google Scholar] [CrossRef] [PubMed]
- Knipprath, C.; McCombe, G.P.; Trask, R.S.; Bond, I.P. Predicting self-healing strength recovery using a multi-objective genetic algorithm. Compos. Sci. Technol. 2012, 72, 752–759. [Google Scholar] [CrossRef]
- Yang, T.; Wang, C.H.; Zhang, J.; He, S.; Mouritz, A.P. Toughening and self-healing of epoxy matrix laminates using mendable polymer stitching. Compos. Sci. Technol. 2012, 72, 1396–1401. [Google Scholar] [CrossRef]
- Wang, C.H.; Sidhu, K.; Yang, T.; Zhang, J.; Shanks, R. Interlayer self-healing and toughening of carbon fibre/epoxy composites using copolymer films. Compos. A 2012, 43, 512–518. [Google Scholar] [CrossRef]
- Wu, A.S.; Coppola, A.M.; Sinnott, M.J.; Chou, T.-W.; Thostenson, E.T.; Byun, J.-H.; Kim, B.-S. Sensing of damage and healing in three-dimensional braided composites with vascular channels. Compos. Sci. Technol. 2012, 72, 1618–1626. [Google Scholar] [CrossRef]
- Podgórski, M.; Fairbanks, B.D.; Kirkpatrick, B.E.; McBride, M.; Martinez, A.; Dobson, A.; Bowman, C.N. Toward Stimuli-Responsive Dynamic Thermosets through Continuous Development and Improvements in Covalent Adaptable Networks (CANs). Adv. Mater. 2020, 32, 1906876. [Google Scholar] [CrossRef]
- Scheutz, G.M.; Lessard, J.J.; Sims, M.B.; Sumerlin, B.S. Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc. 2019, 141, 16181–16196. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lei, Y.; Xiao, Y.; Fu, X.; Kong, W.; Wang, Y.; Lei, J. Mechanically robust, exceptionally recyclable and shape memory cross-linked network based on reversible dynamic urea bonds. J. Mater. Chem. A 2020, 8, 22369–22378. [Google Scholar] [CrossRef]
- Lai, Y.; Kuang, X.; Zhu, P.; Huang, M.; Dong, X.; Wang, D. Colorless, Transparent, Robust, and Fast Scratch-Self-Healing Elastomers via a Phase-Locked Dynamic Bonds Design. Adv. Mater. 2018, 30, 1802556. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Goseki, R.; Ito, K.; Otsuka, H. Thermally Healable and Reprocessable Bis(hindered amino)disulfide-Cross-Linked Polymethacrylate Networks. ACS Macro Lett. 2017, 6, 1280–1284. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, H.; Zhang, D.; Lou, Y.; Huang, L.; Ma, L.; Hao, X.; Dong, L.; Rosei, F.; Lau, W.M. Ultrafast and high-efficient self-healing epoxy coatings with active multiple hydrogen bonds for corrosion protection. Corros. Sci. 2021, 187, 109485. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Nan, Y.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2017, 359, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef]
- Balkenende, D.W.R.; Olson, R.A.; Balog, S.; Weder, C.; Montero de Espinosa, L. Epoxy Resin-Inspired Reconfigurable Supramolecular Networks. Macromolecules 2016, 49, 7877–7885. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Y.; Xu, X.; Yuan, W.; Yang, L.; Shao, Q.; Huang, Y. Efficient intrinsic self-healing epoxy acrylate formed from host-guest chemistry. Polymer 2019, 164, 79–85. [Google Scholar] [CrossRef]
- Mohamadhoseini, M.; Mohamadnia, Z. Supramolecular self-healing materials via host-guest strategy between cyclodextrin and specific types of guest molecules. Coord. Chem. Rev. 2021, 432, 213711. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, D.; Lu, F.; Yuan, W.; Xu, X.; Zhang, Q.; Huang, Y. Multistimuli-Responsive Intrinsic Self-Healing Epoxy Resin Constructed by Host–Guest Interactions. Macromolecules 2016, 51, 5294–5303. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Winne, J.M.; Leibler, L.; Du Prez, F. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Nicolaÿ, R. Vitrimers: Permanently crosslinked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104, 101233. [Google Scholar] [CrossRef]
- Jourdain, A.; Asbai, R.; Anaya, O.; Chehimi, M.M.; Drockenmuller, E.; Montarnal, D. Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks. Macromolecules 2020, 53, 1884–1900. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Png, Z.M.; Ng, S.H.; Tham, G.X.; Ye, E.; Goh, S.S.; Li, Z. Vitrimers: Current research trends and their emerging applications. Mater. Today 2021, 51, 586–625. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, A.; Lin, Y. Reprocessability of dynamic polydioxaborolane networks activated by heat, moisture and mechanical force. Polymer 2020, 209, 123037. [Google Scholar] [CrossRef]
- Song, Z.; Wang, Z.; Cai, S. Mechanics of vitrimer with hybrid networks. Mech. Mater. 2021, 153, 103687. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Jin, K.; Torkelson, J.M. Vitrimers Designed Both to Strongly Suppress Creep and To Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments. Macromolecules 2018, 51, 5537–5546. [Google Scholar] [CrossRef]
- Lessard, J.J.; Scheutz, G.M.; Sung, S.H.; Lantz, K.A.; Epps, T.H., III; Sumerlin, B.S. Block Copolymer Vitrimers. J. Am. Chem. Soc. 2020, 142, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chabert, E.; Vial, J.; Cauchois, J.-P.; Mihaluta, M.; Tournilhac, F. Multiple welding of long fiber epoxy vitrimer composites. Soft Matter. 2016, 12, 4838–4845. [Google Scholar] [CrossRef] [PubMed]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Amirkhosravi, M.; Ke, K.; Gray, T.G.; Manas-Zloczower, I. Cellulose Nanocrystals: Accelerator and Reinforcing Filler for Epoxy Vitrimerization. ACS Appl. Mater. Interfaces 2021, 13, 3419–3425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; An, L.; Wang, S. Recyclable High-Performance Epoxy-Anhydride Resins with DMP-30 as the Catalyst of Transesterification Reactions. Polymers 2021, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yuan, C.; Zhang, W.; Dunn, M.L.; Qi, H.J.; Liu, Z.; Ge, Q. Recycling of vitrimer blends with tunable thermomechanical properties. RSC Adv. 2019, 9, 5431–5437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, L.; Amirkhosravi, M.; Gong, X.; Gray, T.G.; Manas-Zloczower, I. Recycling Epoxy by Vitrimerization: Influence of an Initial Thermoset Chemical Structure. ACS Sustain. Chem. Eng. 2020, 8, 12706–12712. [Google Scholar] [CrossRef]
- Yue, L.; Ke, K.; Amirkhosravi, M.; Gray, T.G.; Manas-Zloczower, I. Catalyst-Free Mechanochemical Recycling of Biobased Epoxy with Cellulose Nanocrystals. ACS Appl. Bio Mater. 2021, 4, 4176–4183. [Google Scholar] [CrossRef]
- Jouyandeh, M.; Tikhani, F.; Hampp, N.; Akbarzadeh Yazdi, D.; Zarrintaj, P.; Reza Ganjali, M.; Reza Saeb, M. Highly curable self-healing vitrimer-like cellulose-modified halloysite nanotube/epoxy nanocomposite coatings. Chem. Eng. J. 2020, 396, 125196. [Google Scholar] [CrossRef]
- Fang, H.; Ye, W.; Yang, K.; Song, K.; Wei, H.; Ding, Y. Vitrimer chemistry enables epoxy nanocomposites with mechanical robustness and integrated conductive segregated structure for high performance electromagnetic interference shielding. Compos. B 2021, 215, 108782. [Google Scholar] [CrossRef]
- Barabanova, A.I.; Afanasyev, E.S.; Askadskii, A.A.; Khokhlov, A.R.; Philippova, O.E. Synthesis and Properties of Epoxy Networks with a Tunable Matrix. Polym. Sci. Ser. A 2019, 61, 375–381. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Black, S.P.; Sanders, J.K.M.; Stefankiewicz, A.R. Disulfide exchange: Exposing supramolecular reactivity through dynamic covalent chemistry. Chem. Soc. Rev. 2014, 43, 1861–1872. [Google Scholar] [CrossRef]
- Azcune, I.; Odriozola, I. Aromatic disulfide crosslinks in polymer systems: Self-healing, reprocessability, recyclability and more. Eur. Polym. J. 2016, 84, 147–160. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekond, A.; Cabanero, G.; Rodriguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Compos. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Matxain, J.M.; Asua, J.M.; Ruipérez, F. Design of new disulfide-based organic compounds for the improvement of self-healing materials. Phys. Chem. Chem. Phys. 2016, 18, 1758–1770. [Google Scholar] [CrossRef]
- Tran, T.-N.; Di Mauro, C.; Malburet, S.; Graillot, A.; Mija, A. Dual Cross-linking of Epoxidized Linseed Oil with Combined Aliphatic/Aromatic Diacids Containing Dynamic S−S Bonds Generating Recyclable Thermosets. ACS Appl. Bio Mater. 2020, 3, 7550–7561. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, L.; Wu, Y.; Zhao, X.; Zhang, Y. Rapid Stress Relaxation and Moderate Temperature of Malleability Enabled by the Synergy of Disulfide Metathesis and Carboxylate Transesterification in Epoxy Vitrimers. ACS Macro Lett. 2019, 8, 255–260. [Google Scholar] [CrossRef]
- Röttger, M.; Domenech, T.; van der Weegen, R.; Breuillac, A.; Nicolaÿ, R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65. [Google Scholar] [CrossRef]
- Lei, Q.-L.; Xia, X.; Yang, J.; Pica Ciamarra, M.; Ni, R. Entropy-controlled cross-linking in linker-mediated vitrimers. Proc. Natl. Acad. Sci. USA 2020, 117, 27111–27115. [Google Scholar] [CrossRef]
- Caffy, F.; Nicolaÿ, R. Transformation of polyethylene into a vitrimer by nitroxide radical coupling of a bis-dioxaborolane. Polym. Chem. 2019, 10, 3107–3115. [Google Scholar] [CrossRef]
- Wu, S.; Yang, H.; Xu, W.-S.; Chen, Q. Relationship between Reaction Kinetics and Chain Dynamics of Vitrimers Based on Dioxaborolane Metathesis. Macromolecules 2020, 53, 1180–1190. [Google Scholar] [CrossRef]
- Wu, S.; Yang, H.; Xu, W.-S.; Chen, Q. Thermodynamics and Reaction Kinetics of Symmetric Vitrimers Based on Dioxaborolane Metathesis. Macromolecules 2021, 54, 6799–6809. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Zha, X.-J.; Bao, R.-Y.; Ke, K.; Liu, Z.-Y.; Yang, M.-B.; Yang, W. Vitrimers of polyolefin elastomer with physically cross-linked network. J. Polym. Res. 2021, 28, 210. [Google Scholar] [CrossRef]
- Schmolke, W.; Perner, N.; Seiffert, S. Dynamically Cross-Linked Polydimethylsiloxane Networks with Ambient-Temperature Self-Healing. Macromolecules 2015, 48, 8781–8788. [Google Scholar] [CrossRef]
- Nishimura, Y.; Chung, J.; Muradyan, H.; Guan, Z. Silyl Ether as a Robust and Thermally Stable Dynamic Covalent Motif for Malleable Polymer Design. J. Am. Chem. Soc. 2017, 139, 14881–14884. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, L.; Zhao, Y.; Wang, Y.; Lei, J. Reprocessable and Shape Memory Thermosetting Epoxy Resins Based on Silyl Ether Equilibration. Macromol. Chem. Phys. 2019, 220, 1900149. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Yu, R.; Zhao, X.-J.; Zhang, Y.; Huang, W. A facile access to stiff epoxy vitrimers with excellent mechanical properties via siloxane equilibration. J. Mater. Chem. A 2018, 6, 10184–10188. [Google Scholar] [CrossRef]
- Zhao, S.; Abu-Omar, M.M. Recyclable and Malleable Epoxy Thermoset Bearing Aromatic Imine Bonds. Macromolecules 2018, 51, 9816–9824. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Nicolay, R.; Winne, J.M.; Du Prez, F.E. Fluorinated Vitrimer Elastomers with a Dual Temperature Response. J. Am. Chem. Soc. 2018, 140, 13272–13284. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Winne, J.M.; Du Prez, F.E. Vitrimers: Directing chemical reactivity to control material properties. Chem. Sci. 2020, 11, 4855–4870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-X.; Guan, Z. Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of Strong Carbon–Carbon Double Bonds. J. Am. Chem. Soc. 2012, 134, 14226–14231. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-X.; Tournilhac, F.; Leibler, L.; Guan, Z. Making Insoluble Polymer Networks Malleable via Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 8424–8427. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Brutman, J.P.; Delgado, P.A.; Hillmyer, M.A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Guadagno, L.; Raimondo, M.; Naddeo, C.; Longo, P.; Mariconda, A.; Binder, W.H. Healing efficiency and dynamic mechanical properties of self-healing epoxy systems. Smart Mater. Struct. 2014, 23, 045001. [Google Scholar] [CrossRef]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. [Google Scholar] [CrossRef]
- Patel, A.J.; Sottos, N.R.; Wetzel, E.D.; White, S.R. Autonomic healing of low-velocity impact damage in fiber-reinforced composites. Compos. A 2010, 41, 360–368. [Google Scholar] [CrossRef]
- Jones, A.S.; Rule, J.D.; Moore, J.S.; White, S.R.; Sottos, N.R. Catalyst morphology and dissolution kinetics for self-healing polymers. Chem. Mater. 2006, 18, 1312–1317. [Google Scholar] [CrossRef]
- Gould, P. Self-help for ailing structures. Mater. Today 2003, 6, 44–49. [Google Scholar] [CrossRef]
- Pratama, P.A.; Sharifi, M.; Peterson, A.M.; Palmese, G.R. Room Temperature Self-Healing Thermoset Based on the Diels–Alder Reaction. ACS Appl. Mater. Interfaces 2013, 5, 12425–12431. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; White, S.R.; Braun, P.V. Room-Temperature Polydimethylsiloxane-Based Self-Healing Polymers. Chem. Mater. 2012, 24, 4209–4214. [Google Scholar] [CrossRef]
- Brown, E.N.; Kessler, M.R.; Sottos, N.R.; White, S.R. In situ poly(urea-formaldehyde) microencapsulation of dicyclopentadiene. J. Microencapsul. 2003, 20, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Rahamtullah; Kumar, D.; Rajagopal, C.; Kumar Roy, P. Influence of microcapsule shell material on the mechanical behavior of epoxy composites for self-healing applications. J. Appl. Polym. Sci. 2014, 131, 40572. [Google Scholar] [CrossRef]
- Caruso, M.M.; Blaiszik, B.J.; Jin, H.; Schelkopf, S.R.; Stradley, D.S.; Sottos, N.R.; Moore, J.S. Robust, Double-Walled Microcapsules for Self-Healing Polymeric Materials. ACS Appl. Mater. Interfaces 2010, 2, 1195–1199. [Google Scholar] [CrossRef]
- Kang, S.; Baginska, M.; White, S.R.; Sottos, N.R. Core-shell polymeric microcapsules with superior thermal and solvent stability. ACS Appl. Mater. Interfaces 2015, 7, 10952–10956. [Google Scholar] [CrossRef]
- Li, Q.; Mishra, A.K.; Kim, N.H.; Kuila, T.; Lau, K.; Lee, J.H. Effects of processing conditions of poly (methylmethacrylate) encapsulated liquid curing agent on the properties of self-healing composites. Compos. B 2013, 49, 6–15. [Google Scholar] [CrossRef]
- Ning, K.; Loomans, B.; Yeung, C.; Li, J.; Yang, F.; Leeuwenburgh, S. Influence of microcapsule parameters and initiator concentration on the self-healing capacity of resin-based dental composites. Dental Mater. 2021, 37, 403–412. [Google Scholar] [CrossRef]
- Odom, S.A.; Jackson, A.C.; Prokup, A.M.; Chayanupatkul, S.; Sottos, N.R.; White, S.R.; Moore, J.S. Visual Indication of Mechanical Damage Using Core–Shell Microcapsules. ACS Appl. Mater. Interfaces 2011, 3, 4547–4551. [Google Scholar] [CrossRef]
- Sadrabadi, T.E.; Allahkaram, S.R.; Towhidi, N. Preparation and Property Investigation of Epoxy/Amine Micro/Nanocapsule Based Self-healing Coatings. Int. Polym. Process. 2018, 33, 721–730. [Google Scholar] [CrossRef]
- Zainuddin, S.; Arefin, T.; Fahim, A.; Hosur, M.V.; Tyson, J.D.; Kumar, A.; Jeelani, S. Recovery and improvement in low-velocity impact properties of e-glass/epoxy composites through novel self-healing technique. Compos. Struct. 2014, 108, 277–286. [Google Scholar] [CrossRef]
- McCombe, G.P.; Rouse, J.; Trask, R.S.; Withers, P.J.; Bond, I.P. X-ray damage characterisation in self-healing fibre reinforced polymers. Compos. A 2012, 43, 613–620. [Google Scholar] [CrossRef]
- Trask, R.S.; Williams, H.R.; Bond, I.P. Biomimetic planar and branched selfhealing networks in composite laminates. In Proceedings of the First International Conference on Self Healing Materials, Noordwijk Aan Zee, The Netherlands, 18–20 April 2007. [Google Scholar]
- Williams, H.R.; Trask, R.S.; Bond, I.P. Self-healing sandwich panels: Restoration of compressive strength after impact. Compos. Sci. Technol. 2008, 68, 3171–3177. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Trask, R.S.; Bond, I.P. Characterization and analysis of carbon fibre-reinforced polymer composite laminates with embedded circular vasculature. J. Roy. Soc. Interface 2010, 7, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.S.; White, S.R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. [Google Scholar] [CrossRef]
- Patrick, J.F.; Hart, K.R.; Krull, B.P.; Diesendruck, C.E.; Moore, J.S.; White, S.R.; Sottos, N.R. Continuous Self-Healing Life Cycle in Vascularized Structural Composites. Adv. Mater. 2014, 26, 4302–4308. [Google Scholar] [CrossRef]
- Norris, C.J.; Bond, I.P.; Trask, R.S. The role of embedded bioinspired vasculature on damage formation in self-healing carbon fibre reinforced composites. Compos. A 2011, 42, 639–648. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Trask, R.S.; Bond, I.P. Analytical study of vascular networks for self-healing composite laminates. In Proceedings of the 17th International Conference on Composite Materials, ICCM-17, Edinburgh, UK, 27−31 July 2009. [Google Scholar]
- Hamilton, A.R.; Sottos, N.R.; White, S.R. Local Strain Concentrations in a Microvascular Network. Exper. Mech. 2009, 50, 255–263. [Google Scholar] [CrossRef]
- Norris, C.J.; White, J.A.P.; McCombe, G.; Chatterjee, P.; Bond, I.P.; Trask, R.S. Autonomous stimulus triggered self-healing in smart structural composites. Smart Mater. Struct. 2012, 21, 094027. [Google Scholar] [CrossRef]
- Tsilimigkra, X.; Baltopoulos, A.; Tsantzalis, S.; Kotrotsos, A.; Siakavellas, N.; Kostopoulos, V.; Flórez, S. Strategies on implementing a potential self-healing functionality in a composite structure. Cienc. Tecnol. Mater. 2016, 28, 147–154. [Google Scholar] [CrossRef]
- Schubert, U.S.; Newkome, G.R.; Manners, I. (Eds.) Metal-Containing and Metallosupramolecular Polymers and Materials; American Chemical Society: Washington, DC, USA, 2006; 575p. [Google Scholar]
- Whittell, G.R.; Manners, I. Metallopolymers: New Multifunctional Materials. Adv. Mater. 2007, 19, 3439–3468. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Manners, I. Frontiers in Transition-Metal-Containing Polymers; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2007; 704p. [Google Scholar]
- Wang, X.S.; McHale, R. Metal-Containing Polymers: Building Blocks for Functional (Nano)Materials. Macromol. Rapid Commun. 2010, 31, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.G.; Zhang, J.; Yan, Y.; Ren, L.; Tang, C. Metallopolymers with transition metals in the side-chain by living and controlled polymerization techniques. Prog. Polym. Sci. 2014, 39, 1742–1796. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Agatemor, C.; Wong, W.-Y. Macromolecules Incorporating Transition Metals: Tackling Global Challenges; Royal Society of Chemistry: London, UK, 2018; 231p. [Google Scholar]
- Zechel, S.; Hager, M.D.; Schubert, U.S. Self-Healing Materials. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Yan, J.; Zheng, X.; Yao, J.; Xu, P.; Miao, Z.; Li, J.; Lv, Z.; Zhang, Q.; Yan, Y. Metallopolymers from organically modified polyoxometalates (MOMPs): A review. J. Organomet. Chem. 2019, 884, 1–16. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.I.; Yadav, B.C.; Uflyand, I.E.; Oliva González, C.M.; Kharisov, B.I.; Kharissova, O.V.; Ortega García, B. A review on the polymers with shape memory assisted self-healing properties for triboelectric nanogenerators. J. Mater. Res. 2021, 36, 1225–1240. [Google Scholar] [CrossRef]
- Huang, Z.; Yu, H.; Wang, L.; Liu, X.; Lin, T.; Haq, F.; Vatsadze, S.Z.; Lemenovskiy, D.A. Ferrocene-contained metal organic frameworks: From synthesis to applications. Coord. Chem. Rev. 2021, 430, 213737. [Google Scholar] [CrossRef]
- Shevchenko, V.G.; Stepanov, G.V.; Kramarenko, E.Y. Dielectric Spectroscopy of Hybrid Magnetoactive Elastomers. Polymers 2021, 13, 2002. [Google Scholar] [CrossRef]
- Ivakha, N.B.; Savchenko, I.O.; Berezhnytska, O.S.; Rusakova, N.V.; Trunova, O.K. Ytterbium metal polymers as precursors of luminescent materials emitting in the near infrared region. Appl. Nanosci. 2020, 10, 4983–4990. [Google Scholar] [CrossRef]
- Stanley, J.M.; Holliday, B.J. Luminescent lanthanide-containing metallopolymers. Coord. Chem. Rev. 2012, 256, 1520–1530. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.I.; Yadav, B.C.; Kudaibergenov, S.E.; Uflyand, I.E. Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators. Polymers 2020, 12, 2594. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.I.; Yadav, B.C.; Singh, S.; Uflyand, I.E. Self-healing and shape memory metallopolymers: State-of-the-art and future perspectives. Dalton Trans. 2020, 49, 3042–3087. [Google Scholar] [CrossRef] [PubMed]
- Zechel, S.; Hager, M.D.; Schubert, U.S. Self-Healing Polymers: From Biological Systems to Highly Functional Polymers. In Functional Polymers (Polymers and Polymeric Composites: A Reference Series); Jafar Mazumder, M.A., Sheardown, H., Al-Ahmed, A., Eds.; Springer: Cham, Switzerland, 2019; pp. 665–717. [Google Scholar]
- Sandmann, B.; Bode, S.; Hager, M.D.; Schubert, D. Metallopolymers as an emerging class of self-healing materials. Adv. Polym. Sci. 2013, 262, 239–257. [Google Scholar]
- Yuan, J.; Fang, X.; Zhang, L.; Hong, G.; Lin, Y.; Zheng, Q.; Xu, Y.; Ruan, Y.; Weng, W.; Xia, H.; et al. Multi-responsive self-healing metallo-supramolecular gels based on ‘‘click’’ ligand. J. Mater. Chem. 2012, 22, 11515–11522. [Google Scholar] [CrossRef]
- Lee, J.; Choi, E.J.; Varga, I.; Claesson, P.M.; Yun, S.-H.; Song, C. Terpyridine-functionalized stimuli-responsive microgels and their assembly through metal–ligand interactions. Polym. Chem. 2018, 9, 1032–1039. [Google Scholar] [CrossRef]
- Sezer, S.; Köytepe, S.; Gültek, A.; Seçkin, T. Synthesis and Stimuli-Responsive Properties of Metallo-Supramolecular Phosphazene Polymers Based on Terpyridine Metal Complexes. J. Inorg. Organomet. Polym. Mater. 2021, 31, 3389–3405. [Google Scholar] [CrossRef]
- Rossow, T.; Hackelbusch, S.; van Assenbergh, P.; Seiffert, S. A modular construction kit for supramolecular polymer gels. Polym. Chem. 2013, 4, 2515–2527. [Google Scholar] [CrossRef]
- Götz, S.; Abend, M.; Zechel, S.; Hager, M.D.; Schubert, U.S. Platinum-terpyridine complexes in polymers: A novel approach for the synthesis of self-healing metallopolymers. J. Appl. Polym. Sci. 2019, 136, 47064. [Google Scholar] [CrossRef]
- Vega, J.M.; Grande, A.M.; van der Zwaag, S.; Garcia, S.J. On the role of free carboxylic groups and cluster conformation on the surface scratch healing behaviour of ionomers. Eur. Polym. J. 2014, 57, 121–126. [Google Scholar] [CrossRef]
- Dahlke, J.; Kimmig, J.; Abend, M.; Zechel, S.; Vitz, J.; Schubert, U.S.; Hager, M.D. Quantification of the scratch-healing efficiency for novel zwitterionic polymers. NPG Asia Mater. 2020, 12, 13. [Google Scholar] [CrossRef]
- Gubarev, A.S.; Lezov, A.A.; Mikusheva, N.G.; Perevyazko, I.; Senchukova, A.S.; Lezova, A.A.; Podsevalnikova, A.N.; Rogozhin, V.B.; Enke, M.; Winter, A.; et al. Hydrodynamic Characteristics and Conformational Parameters of Ferrocene-Terpyridine-Based Polymers. Polymers 2022, 14, 1776. [Google Scholar] [CrossRef]
- Zanardi, C.; Zanfrognini, B.; Morandi, S.; Terzi, F.; Pigani, L.; Pasquali, L.; Seeber, R. Synthesis, spectroscopic and electrochemical characterization of Co(II)-terpyridine based metallopolymer. Electrochim. Acta 2018, 260, 314–323. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Z.; Li, Y.; Geng, L.; Ren, J.; Feng, G. Fluorescent and Electrochemical Supramolecular Coordination Polymer Hydrogels Formed from Ion-Tuned Self-Assembly of Small Bis-Terpyridine Monomer. Inorg. Chem. 2017, 56, 7512–7518. [Google Scholar] [CrossRef] [PubMed]
- Bode, S.; Zedler, L.; Schacher, F.H.; Dietzek, B.; Schmitt, M.; Popp, J.; Hager, M.D.; Schubert, U.S. Self-healing polymer coatings based on crosslinked metallosupramolecular copolymers. Adv. Mater. 2013, 25, 1634–1638. [Google Scholar] [CrossRef]
- El-ghayoury, A.; Hofmeier, H.; de Ruiter, B.; Schubert, U.S. Combining Covalent and Noncovalent Cross-Linking: A Novel Terpolymer for Two-Step Curing Applications. Macromolecules 2003, 36, 3955–3959. [Google Scholar] [CrossRef]
- Jeong, D.-C.; Lee, J.; Ro, Y.H.; Song, C. Repairable photoactive polymer systems via metal-terpyridine-based self-assembly. Polym. Chem. 2017, 8, 1923–1931. [Google Scholar] [CrossRef]
- Baimuratova, R.K.; Dzhardimalieva, G.I.; Vaganov, E.V.; Lesnichaya, V.A.; Kugabaeva, G.D.; Kydralieva, K.A.; Zhinzhilo, V.A.; Uflyand, I.E. Novel Self-Healing Metallocopolymers with Pendent 4-Phenyl-2,2′:6′,2″-terpyridine Ligand: Kinetic Studies and Mechanical Properties. Polymers 2021, 13, 1760. [Google Scholar] [CrossRef] [PubMed]
- Dzhardimalieva, G.I.; Yadav, B.C.; Lifintseva, T.V.; Uflyand, I.E. Polymer chemistry underpinning materials for triboelectric nanogenerators (TENGs): Recent trends. Eur. Polym. J. 2021, 142, 110163. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.; Golubeva, N.; Bogdanova, L.; Irzhak, V.; Baimuratova, R.; Shershnev, V.; Kugabaeva, G.; Kydralieva, K.; Zarelli, M. Metallopolymer hybrid nanocomposites: Preparation and structures. Mater. Today Proc. 2021, 34, 366–369. [Google Scholar] [CrossRef]
- Sorin, E.S.; Baimuratova, R.K.; Chernyayev, D.A.; Korchagin, D.V.; Uflyand, I.E.; Dzhardimalieva, G.I. New Mixed-Ligand Metal-Containing Monomer Based on Cobalt Acrylate and 4-Phenyl-2,2′:6′,2″-Terpyridine Ligand: Synthesis, Characteristics and Thermal Properties. Key Eng. Mater. 2021, 899, 37–44. [Google Scholar] [CrossRef]
- Uflyand, I.E.; Zhinzhilo, V.A.; Dzhardimalieva, G.I. Coordination Polymer Based on Nickel(II) Maleate and 4’-Phenyl-2,2′:6′,2″-Terpyridine: Synthesis, Crystal Structure and Conjugated Thermolysis. J. Inorg. Organomet. Polym. Mater. 2020, 30, 965–975. [Google Scholar] [CrossRef]
- Uflyand, I.E.; Zhinzhilo, V.A.; Mukhanova, E.A.; Karyukov, E.V.; Tautieva, M.A.; Ostapenko, D.A.; Burlakova, V.E.; Dzhardimalieva, G.I. Metal Chelate Monomers Based on Nickel Maleate and Chelating N-Heterocycles as Precursors of Core-shell Nanomaterials with Advanced Tribological Properties. Z. Anorg. Allg. Chem. 2019, 645, 758–767. [Google Scholar] [CrossRef]
- Baimuratova, R.K.; Dzhardimalieva, G.I.; Zhinzhilo, V.A.; Kydralieva, K.A.; Uflyand, I.E. Metal-containing monomers based on copper and zinc salts of unsaturated acids and pendent 4-phenyl-2,2′:6′,2″-terpyridine ligands: Synthesis, characterization and thermal properties. Key Eng. Mater. 2020, 869, 119–128. [Google Scholar] [CrossRef]
- Uflyand, I.E.; Tkachev, V.V.; Zhinzhilo, V.A.; Drogan, E.G.; Burlakova, V.E.; Sokolov, M.E.; Panyushkin, V.T.; Baimuratova, R.K.; Dzhardimalieva, G.I. Synthesis, Crystal Structure, Thermal Properties of Copper(II) Acrylate Complex with 4′-Phenyl-2,2′:6′,2′′-Terpyridine and Its Use in Nanomaterials Science. J. Mol. Struct. 2022, 1250, 131909. [Google Scholar] [CrossRef]
- Li, J.; Suo, Z.; Vlassak, J.J. Stiff, Strong, and Tough Hydrogels with Good Chemical Stability. J. Mater. Chem. B 2014, 2, 6708–6713. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Ding, H.; Qian, J.; Yin, J.; Wu, Z.L.; Song, Y.; Zheng, Q. Metal-coordination complexes mediated physical hydrogels with high toughness, stick−slip tearing behavior, and good processability. Macromolecules 2016, 49, 9637–9646. [Google Scholar] [CrossRef]
- Tang, Q.; Zhao, D.; Yang, H.; Wang, L.; Zhang, X. A pH-responsive self-healing hydrogel based on multivalent coordination of Ni2+ with polyhistidine-terminated PEG and IDA-modified oligochitosan. J. Mater. Chem. B 2019, 7, 30–42. [Google Scholar] [CrossRef]
- Tunn, I.; Harrington, M.J.; Blank, K.G. Bioinspired histidine–Zn2+ coordination for tuning the mechanical properties of self-healing coiled coil cross-linked hydrogels. Biomimetics 2019, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, J.; Chen, Y.; Zhang, L.; Zhou, J. Dual Physical Crosslinking Strategy to Construct Moldable Hydrogels with Ultrahigh Strength and Toughness. Adv. Funct. Mater. 2018, 28, 1800739. [Google Scholar] [CrossRef]
- Jing, Z.; Xu, A.; Liang, Y.-Q.; Zhang, Z.; Yu, C.; Hong, P.; Li, Y. Biodegradable poly(acrylic acid-co-acrylamide)/poly(vinyl alcohol) double network hydrogels with tunable mechanics and high self-healing performance. Polymers 2019, 11, 952. [Google Scholar] [CrossRef] [Green Version]
- Al-Tayyar, N.A.; Youssef, A.M.; Al-hindi, R. Antimicrobial food packaging based on sustainable Bio-based materials for reducing foodborne Pathogens: A review. Food Chem. 2020, 310, 125915. [Google Scholar] [CrossRef]
- Xu, Y.S.; Li, Y.S.; Chen, Q.M.; Fu, L.H.; Tao, L.; Wei, Y. Injectable and Self-Healing Chitosan Hydrogel Based on Imine Bonds: Design and Therapeutic Applications. Int. J. Mol. Sci. 2018, 19, 2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenite, A.; Chaput, C.; Wang, D.; Combes, C.; Buschmann, M.D.; Hoemann, C.D.; Leroux, J.C.; Atkinson, B.L.; Binette, F.; Selmani, A. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials 2000, 21, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.F.; Du, Y.M.; Hu, X.W.; Shi, X.W.; Kennedy, J.F. Rheological characterisation of a novel thermosensitive chitosan/poly(vinyl alcohol) blend hydrogel. Carbohydr. Polym. 2007, 67, 491–499. [Google Scholar] [CrossRef]
- Biswas, S.; Datta, L.P.; Roy, S. A Stimuli-Responsive Supramolecular Hydrogel for Controlled Release of Drug. J. Mol. Eng. Mater. 2017, 5, 1750011. [Google Scholar] [CrossRef]
- Ren, Y.; Lou, R.Y.; Liu, X.C.; Gao, M.; Zheng, H.Z.; Yang, T.; Xie, H.; Yu, W.; Ma, X. A self-healing hydrogel formation strategy via exploiting endothermic interactions between polyelectrolytes. Chem. Commun. 2016, 52, 6273–6276. [Google Scholar] [CrossRef]
- Chang, G.; Chen, Y.; Li, Y.; Li, S.; Huang, F.; Shen, Y.; Xie, A. Self-healable hydrogel on tumor cell as drug delivery system for localized and effective therapy. Carbohydr. Polym. 2015, 122, 336–342. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, T.T.; Liu, Y.; Shang, F.; Chen, B.; Hu, Y.; Wang, S.; Guo, Z. Supramolecular hydrogel of poly(vinyl alcohol)/chitosan: A dual cross-link design. Adv. Polym. Technol. 2018, 37, 2186–2194. [Google Scholar] [CrossRef]
- Kang, M.M.; Liu, S.L.; Oderinde, O.; Yao, F.; Fu, G.D.; Zhang, Z.H. Template method for dual network self-healing hydrogel with conductive property. Mater. Des. 2018, 148, 96–103. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, J.; Wang, Z.; Wu, J.; Meng, G.; Liu, Z.; Guo, X. One-pot fabrication of triple-network structure hydrogels with high-strength and self-healing properties. Mater. Lett. 2017, 207, 53–56. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, L.; Huang, L.; Chen, H.; Xu, K.; Tan, Y.; Wang, P.; Zheng, J. Fracture of the Physically Cross-Linked First Network in Hybrid Double Network Hydrogels. Macromolecules 2014, 47, 2140–2148. [Google Scholar] [CrossRef]
- You, J.; Xie, S.; Cao, J.; Ge, H.; Xu, M.; Zhang, L.; Zhou, J. Quaternized Chitosan/Poly(acrylic acid) Polyelectrolyte Complex Hydrogels with Tough, Self-Recovery, and Tunable Mechanical Properties. Macromolecules 2016, 49, 1049–1059. [Google Scholar] [CrossRef]
- Song, G.; Zhang, L.; He, C.; Fang, D.-C.; Whitten, P.G.; Wang, H. Facile fabrication of tough hydrogels physically cross-linked by strong cooperative hydrogen bonding. Macromolecules 2013, 46, 7423–7435. [Google Scholar] [CrossRef]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Moon, H.; Paeng, K.; Song, C. Reversible Assembly of Terpyridine Incorporated Norbornene-Based Polymer via a Ring-Opening Metathesis Polymerization and Its Self-Healing Property. Polymers 2018, 10, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, F.J.; Pyckhout-Hintzen, W.; Schumers, J.M.; Fustin, C.A.; Gohy, J.F.; Bailly, C. Linear viscoelastic rheology of moderately entangled telechelic polybutadiene temporary networks. Macromolecules 2009, 42, 6181–6192. [Google Scholar] [CrossRef]
- Yan, Y.; de Keizer, A.; Stuart, M.; Besseling, N. From Coordination Polymers to Hierarchical Self-Assembled Structures. Adv. Polym. Sci. 2011, 242, 91–115. [Google Scholar]
- Beck, J.B.; Rowan, S.J. Multistimuli, Multiresponsive Metallo-Supramolecular Polymers. J. Am. Chem. Soc. 2003, 125, 13922–13923. [Google Scholar] [CrossRef]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Small-molecule dynamics and mechanisms underlying the macroscopic mechanical properties of coordinatively cross-linked polymer networks. J. Am. Chem. Soc. 2005, 127, 14488–14496. [Google Scholar] [CrossRef]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Strong Means Slow: Dynamic Contributions to the Bulk Mechanical Properties of Supramolecular Networks. Angew. Chem. Int. Ed. 2005, 44, 2746–2748. [Google Scholar] [CrossRef]
- Van der Zwaag, S.; Brinkman, E. Self-Healing Materials: Pioneering Research in the Netherlands; Van der Zwaag, S., Brinkman, E., Eds.; IOS Press: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Utrera-Barrios, S.I.; Verdejo, R.; Lopez-Manchado, M.A.; Hernandez Santana, M. Evolution of self-healing elastomers, from extrinsic to combined intrinsic mechanisms: A review. Mater. Horiz. 2020, 7, 2882–2902. [Google Scholar] [CrossRef]
- Riedl, G.; Pugstaller, R.; Wallner, G.M. Development and implementation of a simultaneous fatigue crack growth test setup for polymeric hybrid laminates. Eng. Fract. Mech. 2022, 267, 108468. [Google Scholar] [CrossRef]
- Shinde, V.V.; Taylor, G.; Celestine, A.-D.N.; Beckingham, B.S. Fused Filament Fabrication 3D Printing of Self-Healing High-Impact Polystyrene Thermoplastic Polymer Composites Utilizing Eco-friendly Solvent-Filled Microcapsules. ACS Appl. Polym. Mater. 2022, 4, 3324–3332. [Google Scholar] [CrossRef]
- Wan, Z.; Xu, Y.; Zhang, Y.; He, S.; Šavija, B. Mechanical properties and healing efficiency of 3D-printed ABS vascular based self-healing cementitious composite: Experiments and modelling. Eng. Fract. Mech. 2022, 267, 108471. [Google Scholar] [CrossRef]
- OuYang, Q.; Wang, X.; Liu, L. High crack self-healing efficiency and enhanced free-edge delamination resistance of carbon fibrous composites with hierarchical interleaves. Compos. Sci. Technol. 2022, 217, 109115. [Google Scholar] [CrossRef]
- Chaudhary, K.; Kandasubramanian, B. Self-Healing Nanofibers for Engineering Applications. Ind. Eng. Chem. Res. 2022, 61, 3789–3816. [Google Scholar] [CrossRef]
- Michal, B.T.; McKenzie, B.M.; Felder, S.E.; Rowan, S.J. Metallo-, Thermo-, and Photoresponsive Shape Memory and Actuating Liquid Crystalline Elastomers. Macromolecules 2015, 48, 3239–3246. [Google Scholar] [CrossRef]
- Lee, H.I.; Vahedi, V.; Pasbakhsh, P. Self-healing polymer composites: Prospects, challenges, and applications. Polym. Rev. 2016, 56, 225–261. [Google Scholar]
- Zhao, P.-C.; Li, W.; Huang, W.; Li, C.-H. A Self-Healing Polymer with Fast Elastic Recovery upon Stretching. Molecules 2020, 25, 597. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Fu, Q.; Tan, B.; Ma, Y.; Fang, L.; Lu, C.; Xu, Z. Build a bridge from polymeric structure design to engineering application of self-healing coatings: A review. Prog. Org. Coat. 2022, 167, 106790. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Module | Strength |
|---|---|---|
| Microcapsules 5 wt.% | 72 | 88 |
| Microcapsules 10 wt.% | 57 | 87 |
| Vasculatures (wax) | 70 | 70 |
| Vasculatures (wire) | 74 | 76 |
| Polyethylene terephthalate | 52 | 49 |
| Nylon doped with 10% multiwalled carbon nanotubes | 38 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irzhak, V.I.; Uflyand, I.E.; Dzhardimalieva, G.I. Self-Healing of Polymers and Polymer Composites. Polymers 2022, 14, 5404. https://doi.org/10.3390/polym14245404
Irzhak VI, Uflyand IE, Dzhardimalieva GI. Self-Healing of Polymers and Polymer Composites. Polymers. 2022; 14(24):5404. https://doi.org/10.3390/polym14245404
Chicago/Turabian StyleIrzhak, Vadim I., Igor E. Uflyand, and Gulzhian I. Dzhardimalieva. 2022. "Self-Healing of Polymers and Polymer Composites" Polymers 14, no. 24: 5404. https://doi.org/10.3390/polym14245404