
Biobased Copolyamides 56/66: Synthesis, Characterization and Crystallization Kinetics
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Method
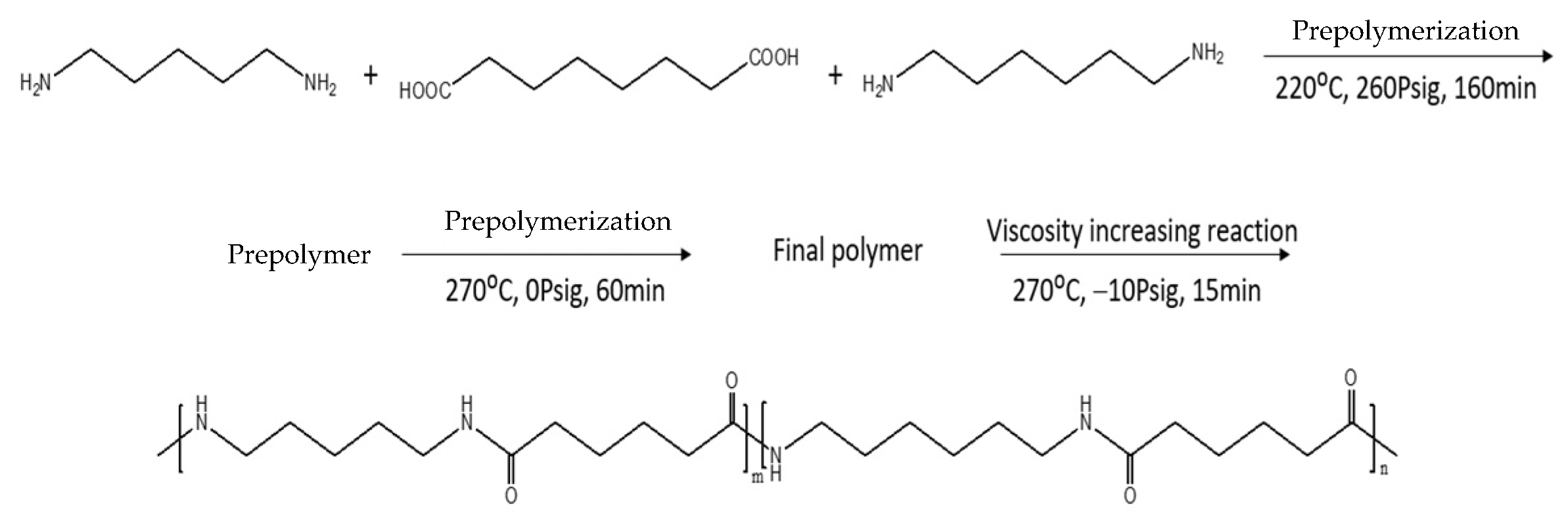
2.2.1. Synthesis of PA56/PA66 Co-PA Salt
2.2.2. Synthesis of PA56/PA66 Co-PAs
2.3. Characterization
2.3.1. DSC Measurements
2.3.2. FT-IR and 1H-NMR
2.3.3. Gel Permeation Chromatography (GPC)
2.3.4. TGA Measurements
2.3.5. Polarized Optical Microscopy (POM)
2.3.6. X-ray Diffraction (XRD)
2.3.7. Mechanical Properties
2.3.8. Nonisothermal Crystallization Process
3. Results and Discussion
3.1. Characterization of PA56/66
3.2. Thermal Properties of PA56/PA66
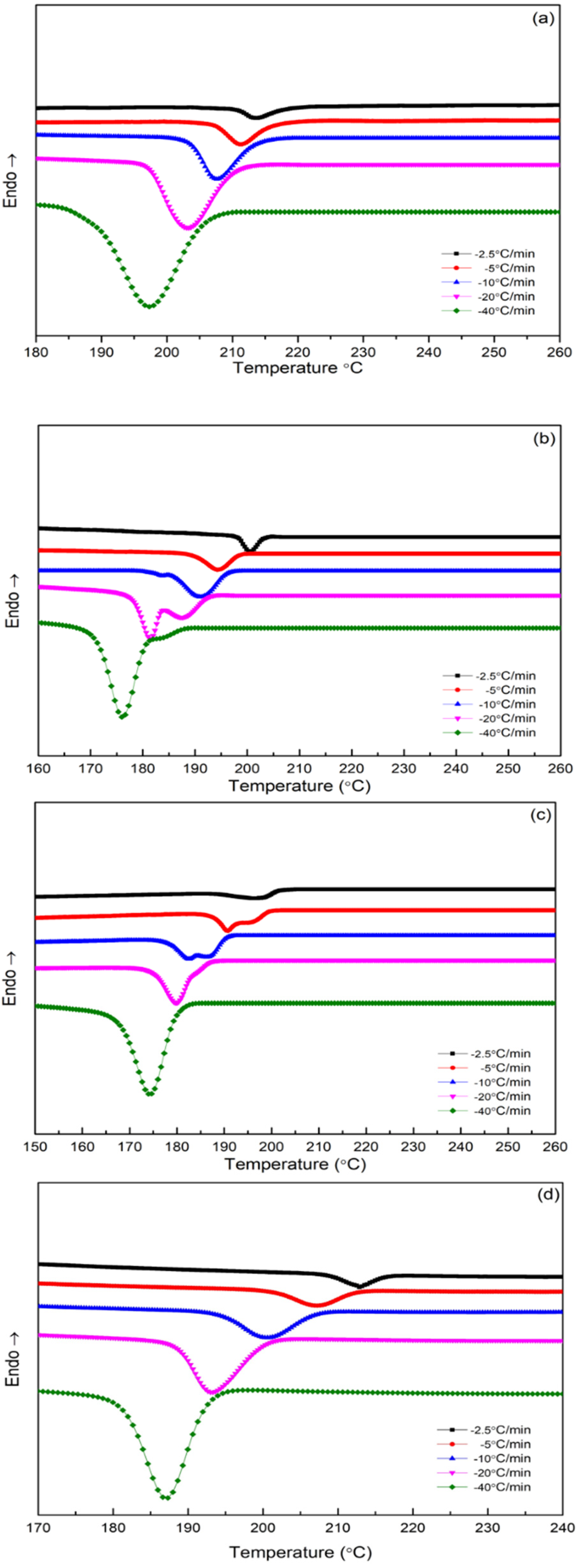
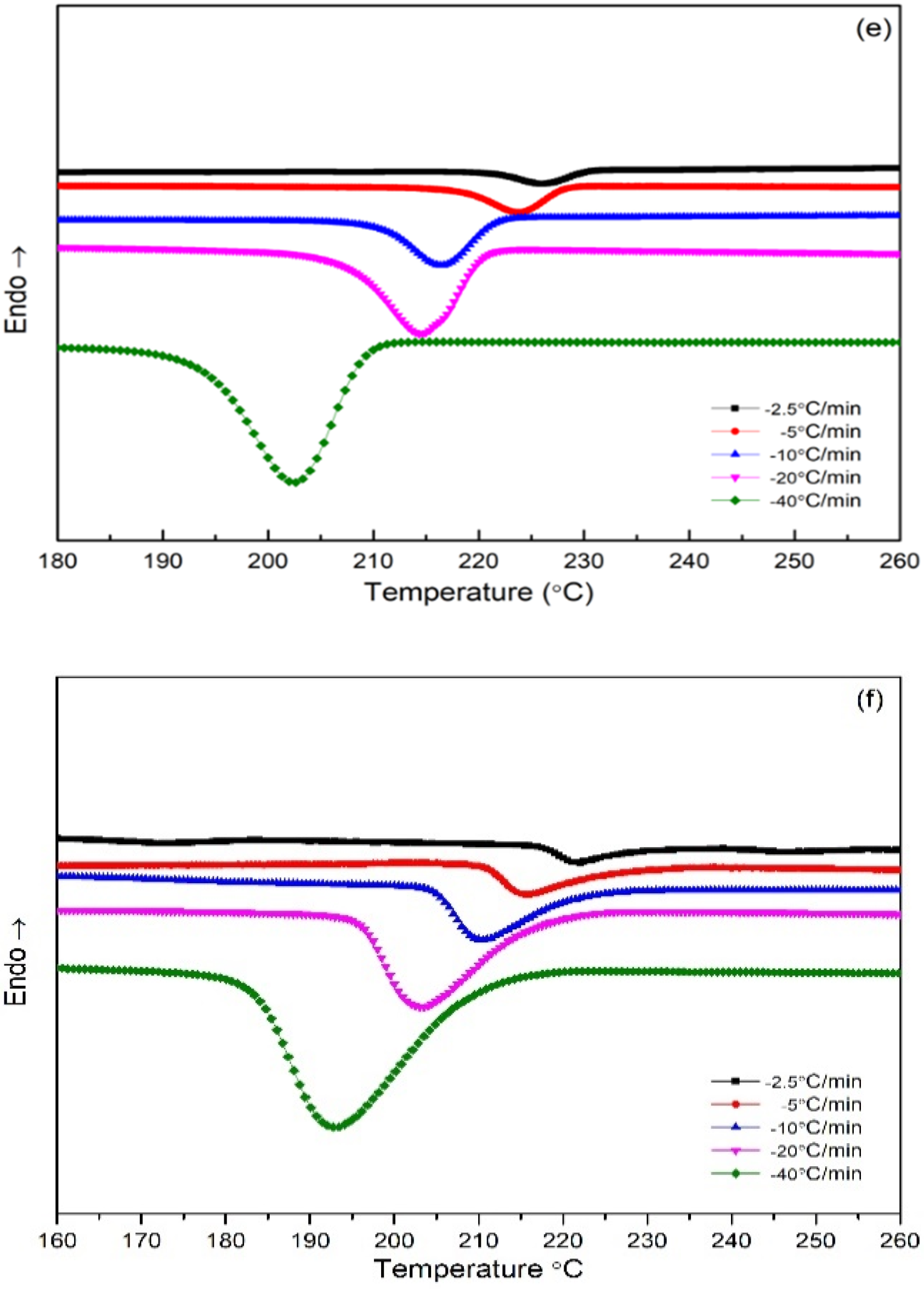
3.3. Differential Scanning Calorimetry (DSC)
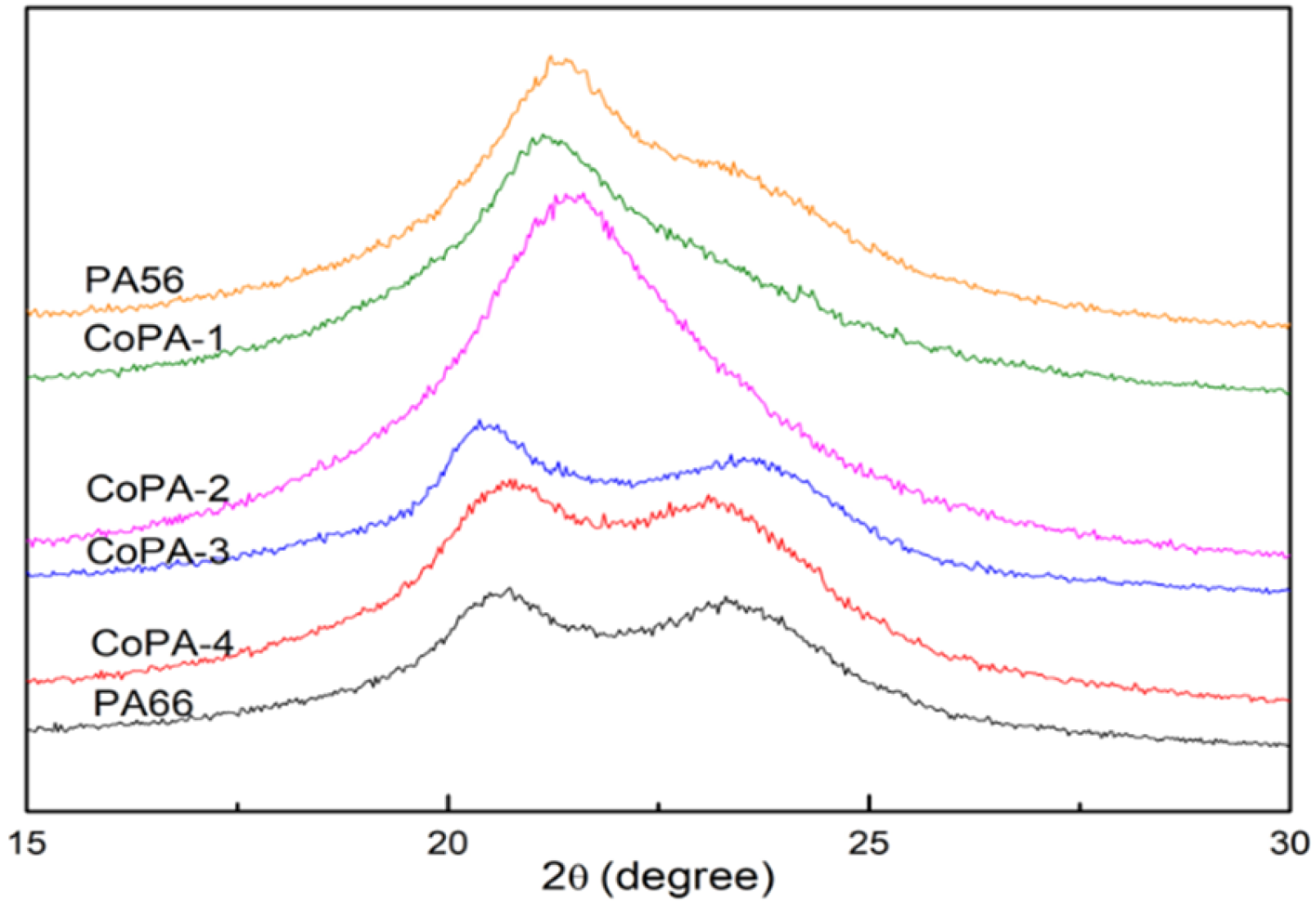
3.4. Crystal Structure of PA56/PA66
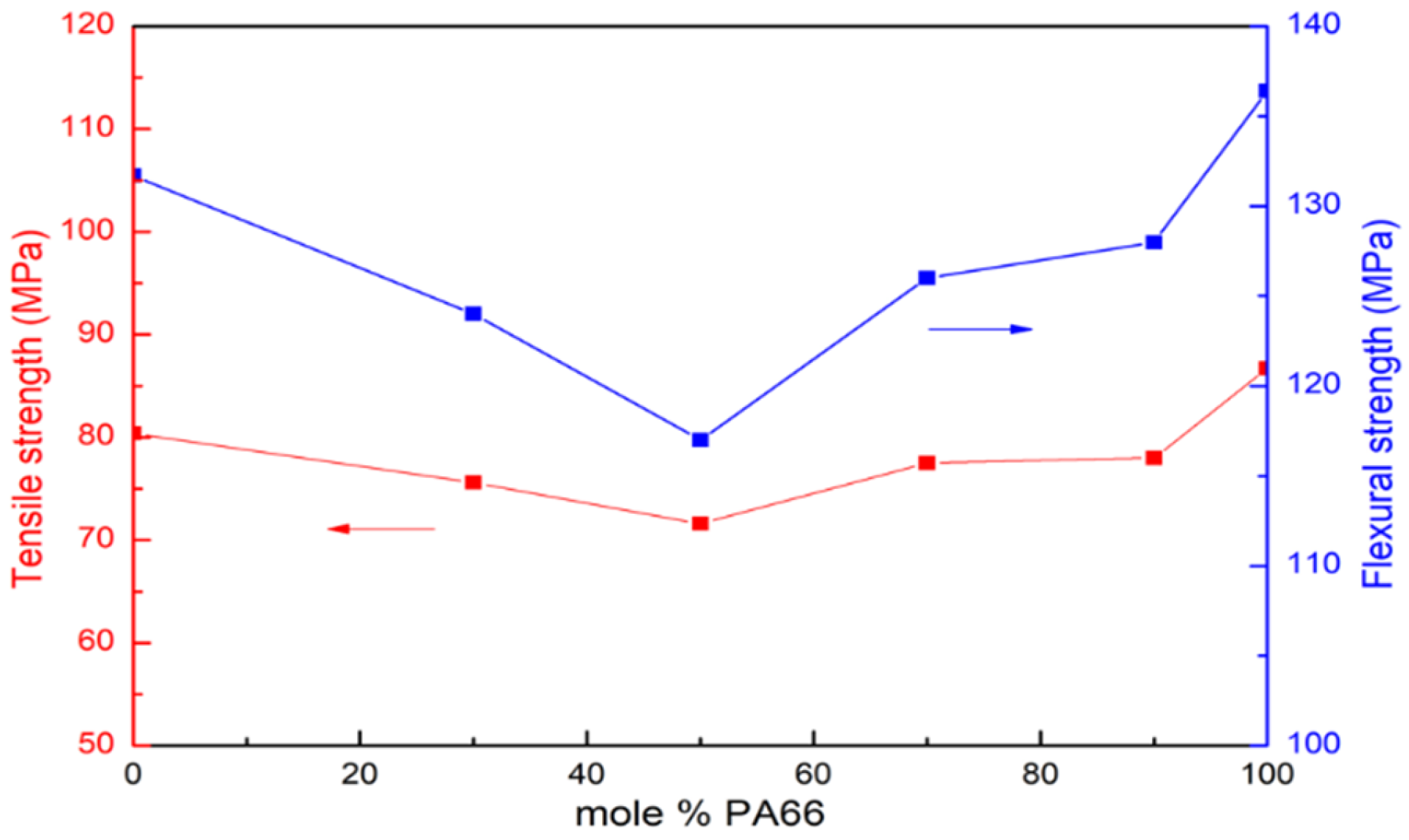
3.5. Mechanical Properties
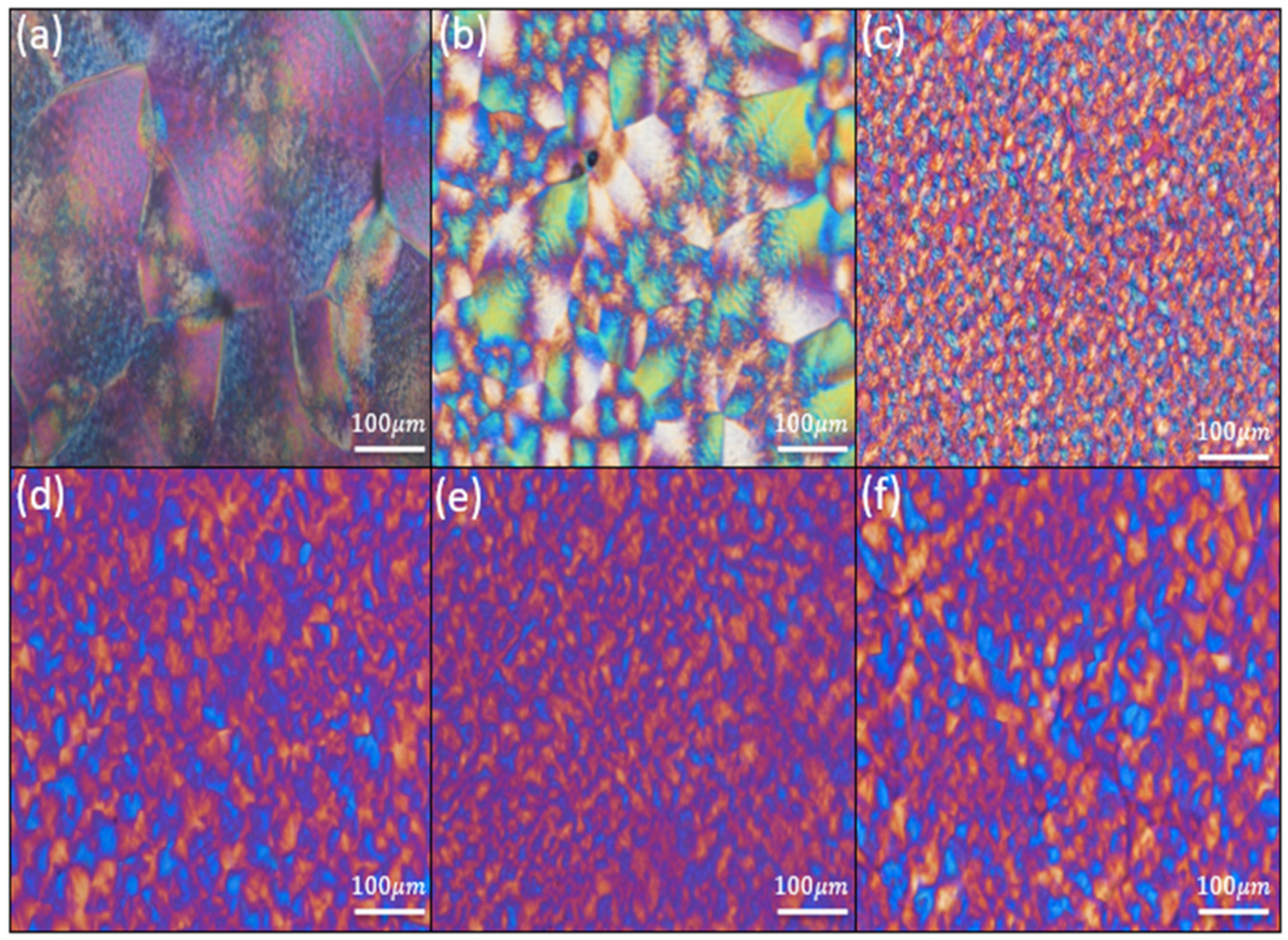
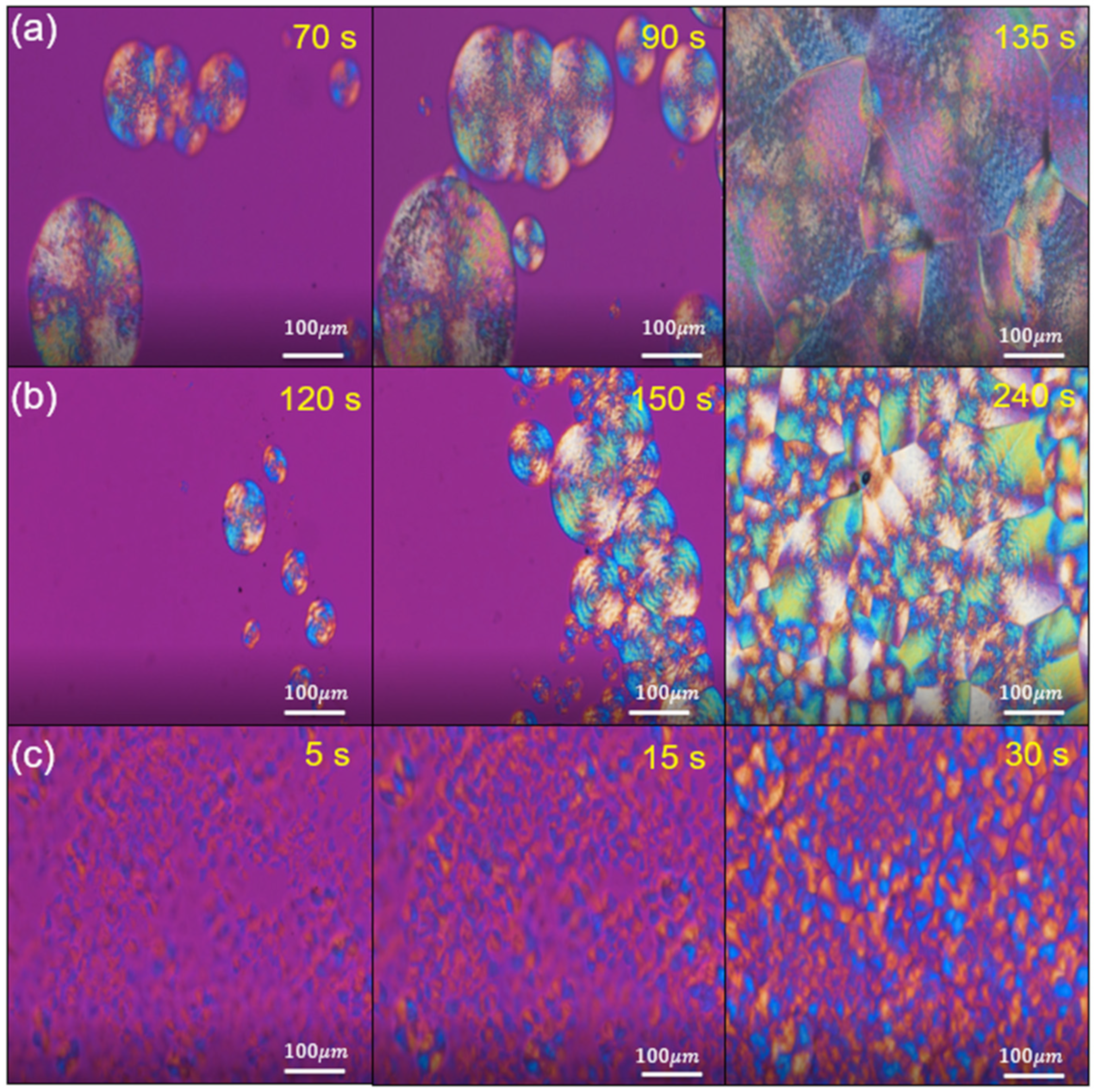
3.6. Crystallite Morphology
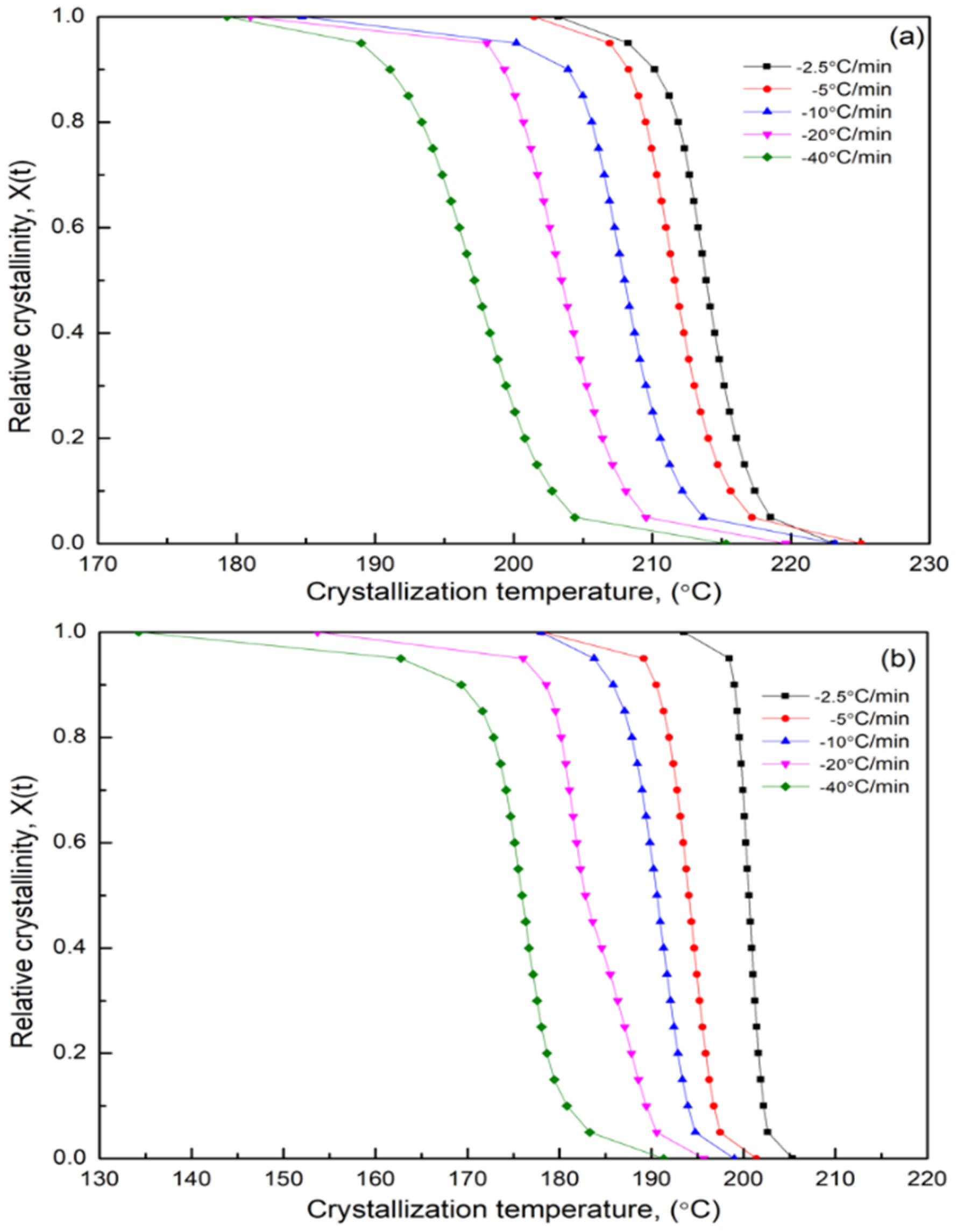
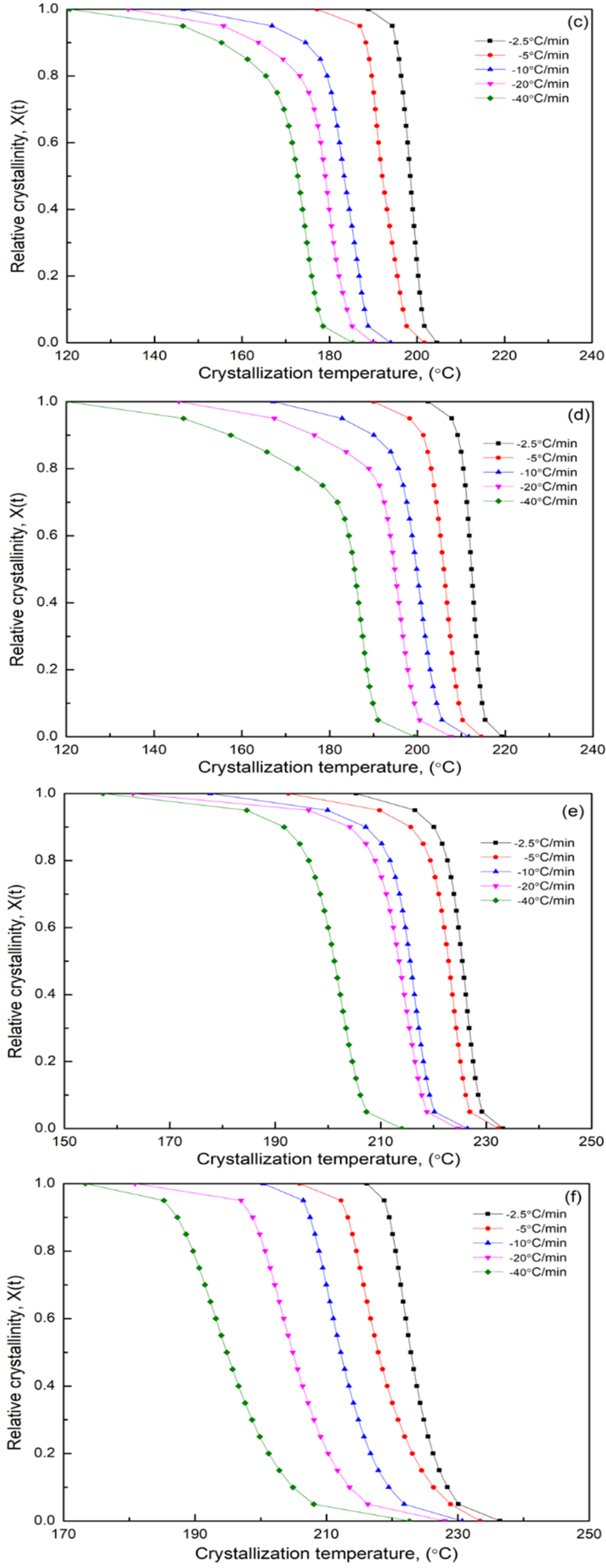
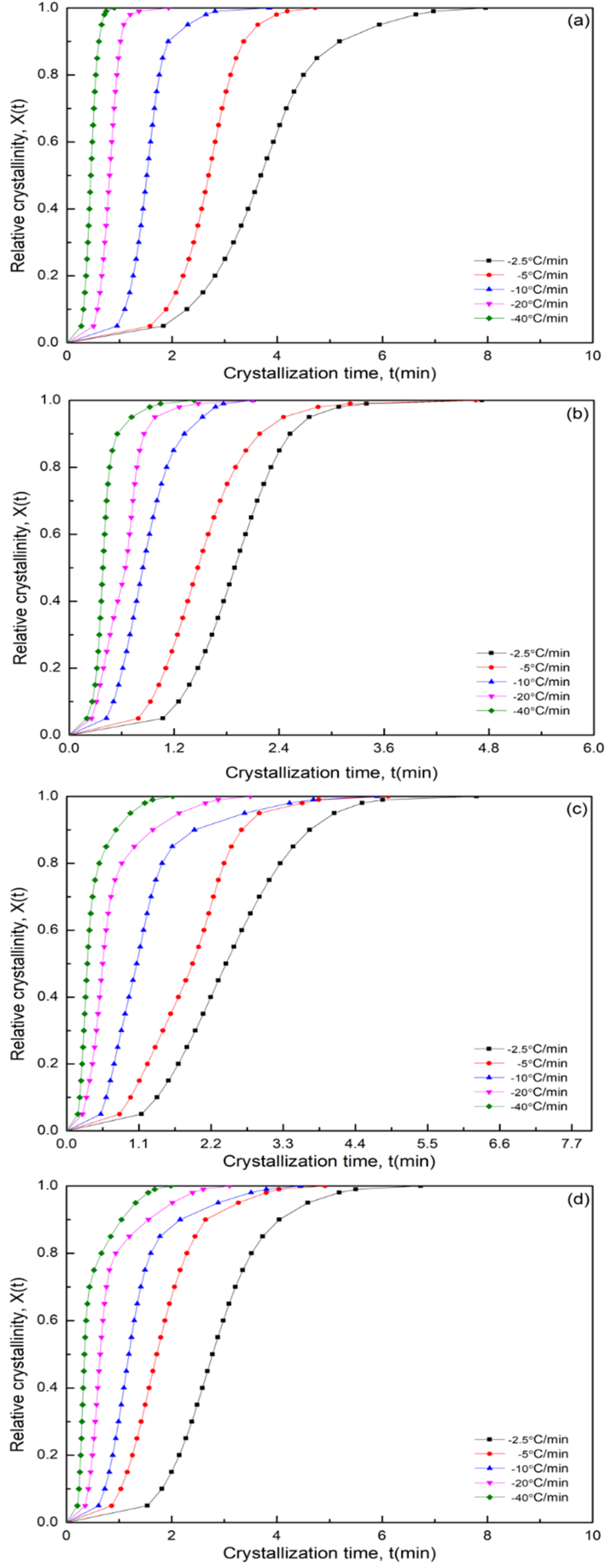
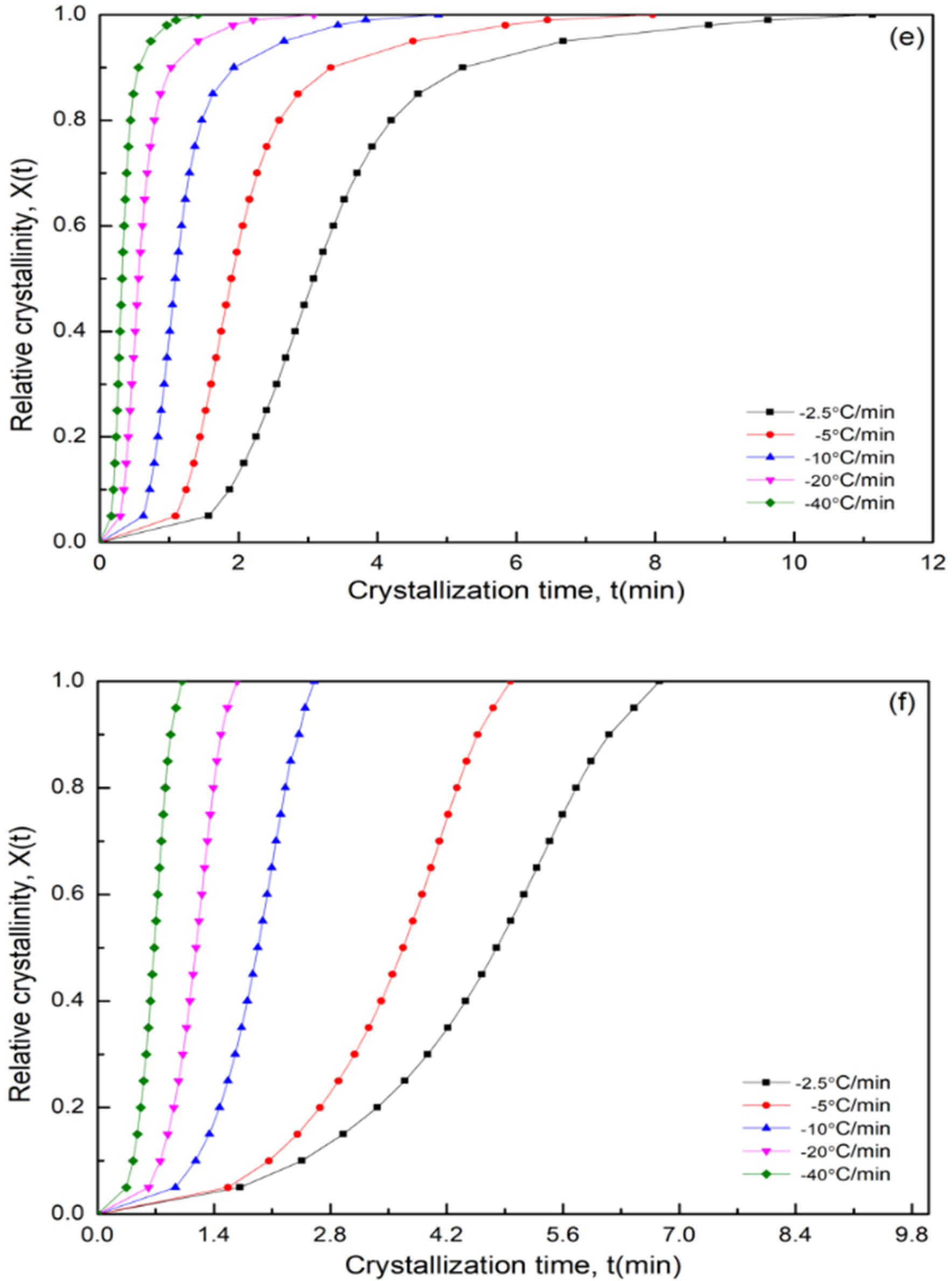
3.7. Nonisothermal Crystallization Kinetics
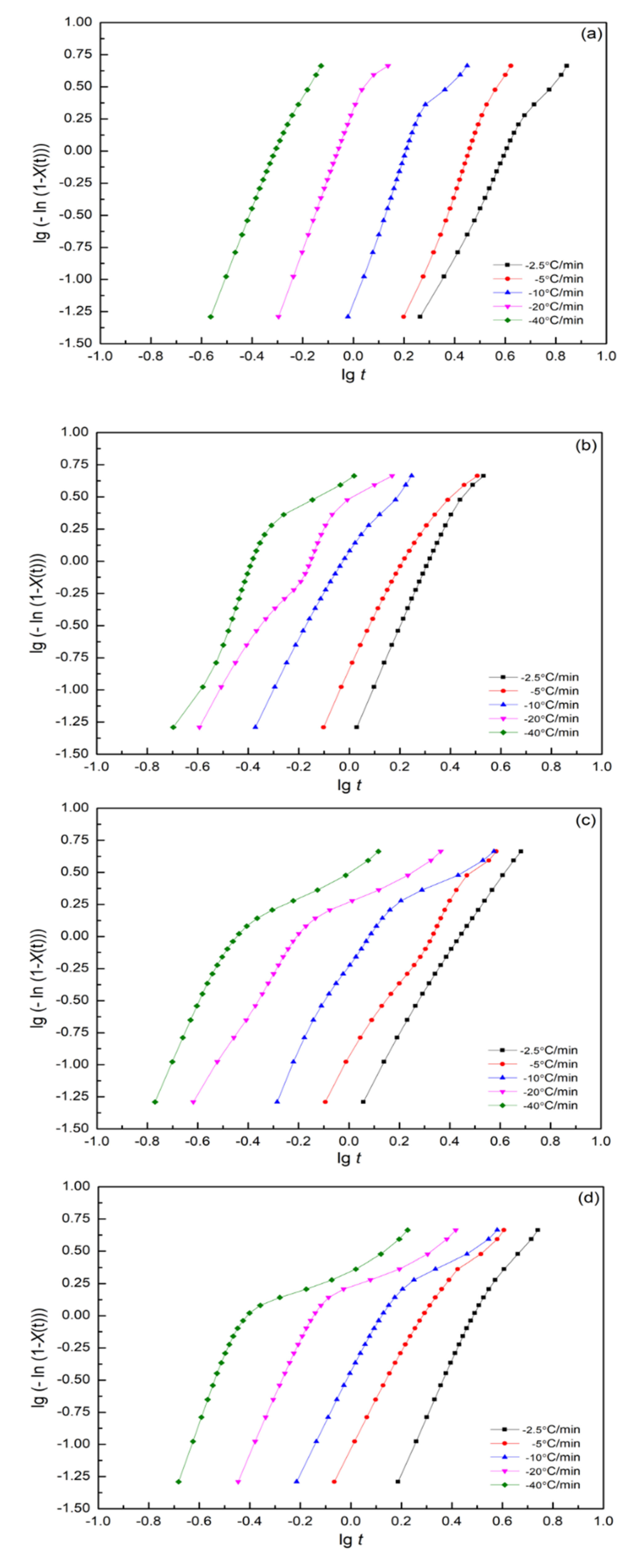
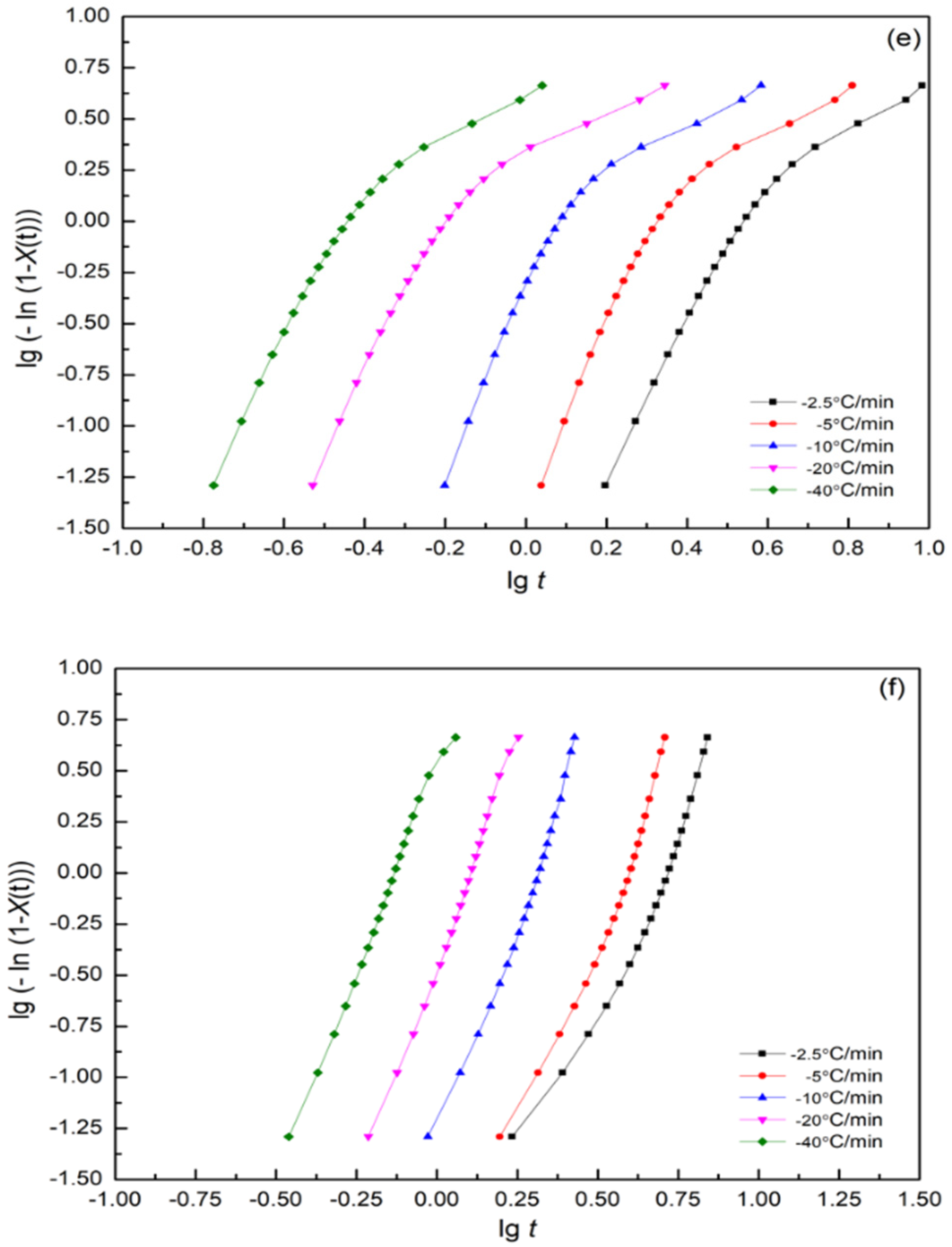
3.7.1. Modified Avrami Equation by Jeziorny
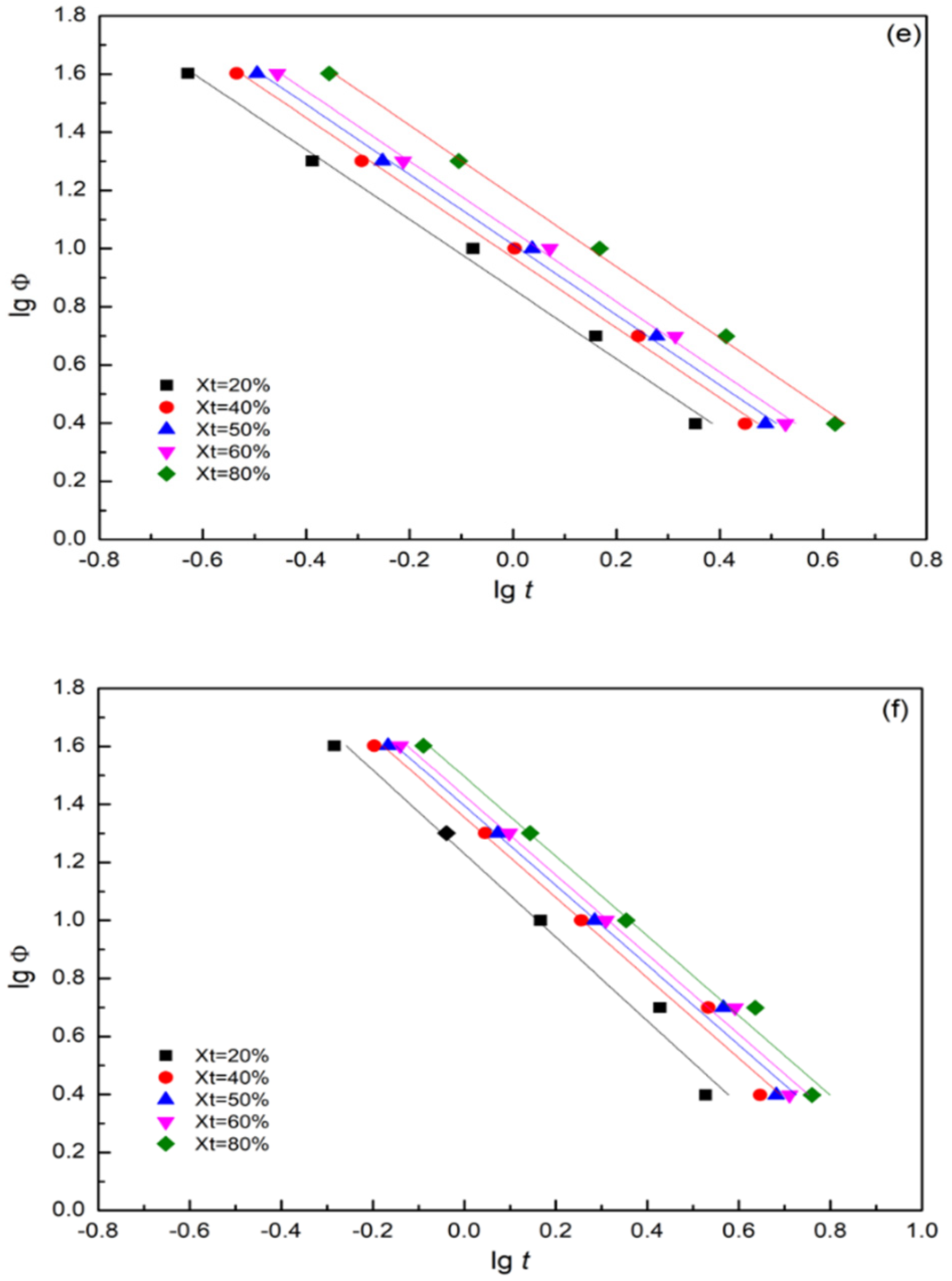
3.7.2. Mo Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carothers, W.H. Synthetic Fiber. U.S. Patent 2,130,948A, 20 September 1938. [Google Scholar]
- Kohan, M.I. Nylon Plastics Handbook; Hanser Publishers: New York, NY, USA, 1995. [Google Scholar]
- Liu, B.; Hu, G.; Zhang, J.; Yan, W. Non-isothermal crystallization, yellowing resistance and mechanical properties of heat resistance nylon 10T/66/titania dioxide/glass fibre composites. RSC Adv. 2019, 9, 7057–7064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, L.; Li, L.; Huang, B.; Wang, T.; Wang, Y. Direct injection molding and mechanical properties of high strength steel/composite hybrids. Compos. Struct. 2019, 210, 70–81. [Google Scholar] [CrossRef]
- Biricik, G.D.; Celebi, H.; Seyhan, A.T.; Ates, F. Thermal and mechanical properties of flax char/carbon fiber reinforced polyamide 66 hybrid composites. Polym. Compos. 2022, 43, 503–516. [Google Scholar] [CrossRef]
- Cho, B.G.; Lee, J.E.; Hwang, S.H.; Han, J.H.; Chae, H.G.; Park, Y.B. Enhancement in mechanical properties of polyamide 66-carbon fiber composites containing graphene oxide-carbon nanotube hybrid nanofillers synthesized through in situ interfacial polymerization. Compos. Part A Appl. Sci. Manuf. 2020, 135, 105938. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, T.; Xue, X.; He, M.; Xue, J.; Song, M.; Wu, S.; Kang, H.; Zhang, L.; Jia, Q. Synthesis of fully bio-based polyamides with tunable properties by employing itaconic acid. Polymer 2014, 55, 4846–4856. [Google Scholar] [CrossRef]
- Mutua, F.N.; Cheng, C.; Dong, Y.; Zheng, C.; Zhu, B.; He, Y. Synthesis and properties of bio-based poly(pentamethylene oxamide). Polym. Eng. Sci. 2017, 58, 659–664. [Google Scholar] [CrossRef]
- Lee, J.A.; Ann, J.H.; Kim, I.; Li, S.; Lee, Y. Synthesis, characterization and application of fully bio-based and biodegradable nylon-4,4 and -5,4. ACS Sustain. Chem. Eng. 2020, 8, 5604–5614. [Google Scholar] [CrossRef]
- Lee, M.O.; Kim, J.H.; Park, J.Y.; Kim, S.Y. Bio-based poly(pentamethylene sebacamide) by solid-state polymerization from bio-based monomers. Green Chem. 2021, 23, 6469–6476. [Google Scholar] [CrossRef]
- Eltahir, Y.A.; Saeed, H.A.M.; Xia, Y.M.; Wang, Y.M. Preparation of polyamide 5,6 (PA56) fibers and its mechanical properties. Adv. Mater. Res. 2014, 937, 86–91. [Google Scholar] [CrossRef]
- Kim, H.T.; Baritugo, K.A.; Oh, Y.H.; Hyun, S.M.; Khang, T.U.; Kang, K.H.; Jung, S.H.; Song, B.K.; Park, K.; Kim, I.; et al. Metabolic engineering of Corynebacterium glutamicum for the high-level production of cadaverine that can be used for the synthesis of biopolyamide 510. ACS Sustain. Chem. Eng. 2018, 6, 5296–5305. [Google Scholar] [CrossRef]
- Meng, C.; Liu, X. Synthesis of bio-based semi aromatic high temperature polyamide PA5T/56 and effect of benzene ring on non-isothermal crystallization kinetics. J. Polym. Res. 2021, 28, 383. [Google Scholar] [CrossRef]
- Gale, E.F.; Epps, H.M. Studies on bacterial amino-acid decarboxylases: 1. l(+)-lysine decarboxylase. Biochem. J. 1944, 38, 232–242. [Google Scholar] [CrossRef]
- Buschke, N.; Schroeder, H.; Wittmann, C. Metabolic engineering of Corynebacterium glutamicum for production of 1,5-diaminopentane from hemicellulose. Biotechnol. J. 2011, 6, 306–307. [Google Scholar] [CrossRef]
- Kind, S.; Wittmann, C. Bio-based production of the platform chemical 1,5-diaminopentane. Appl. Microbiol. Biotechnol. 2011, 91, 1287–1296. [Google Scholar] [CrossRef]
- Takahashi, E.; Furui, M.; Seko, H.; Shibatani, T. d-lysine production from l-lysine by successive chemical racemization and microbial asymmetric degradation. Appl. Microbiol. Biotechnol. 1997, 47, 347–351. [Google Scholar] [CrossRef]
- Xue, C.; Hsu, K.M.; Chiu, C.Y.; Chang, Y.K.; Ng, I.S. Fabrication of bio-based polyamide 56 and antibacterial nanofiber membrane from cadaverine. Chemosphere 2021, 266, 128967. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, S.; Wang, X.; Yu, J.; Ding, B. Efficient and reusable polyamide-56 nanofiber/nets membrane with bimodal structures for air filtration. J. Colloid Interface Sci. 2015, 457, 203–211. [Google Scholar] [CrossRef]
- Gadomski, A. Improvement of color value of bio-based polyamide 56 fibers. e-Polymers 2018, 18, 91–95. [Google Scholar]
- Gong, X.; Zhang, S.; Wang, J. Mixed versus networked bio-based PA56/Polyethylene terephthalate composite yarns: A study of two modification methods. J. Biobased Mater. Bioenergy 2021, 15, 90–96. [Google Scholar]
- Stouffer, J.M.; Howard, W.; Starkweather, J.; Hsiao, B.S.; Avakian, P.; Jones, G.A. Copolymer modification of nylon-6,6 with 2-methylpentamethylenediamine. Polymer 1996, 37, 1217–1228. [Google Scholar] [CrossRef]
- Chen, C.W.; Hsu, T.S.; Huang, K.W.; Rwei, S.P. Effect of 1,2,4,5-benzenetetracarboxylic acid on unsaturated poly(butylene adipate-co-butylene itaconate) copolyesters: Synthesis, non-isothermal crystallization kinetics, thermal and mechanical properties. Polymers 2020, 12, 1160. [Google Scholar] [CrossRef]
- Lin, C.; Zou, F.; Fernández-Ronco, M.P.; Yan, Y.; Hufenus, R. Melting behavior and non-isothermal crystallization kinetics of copolyimide 6/12. Polym. Cryst. 2019, 2, 10054. [Google Scholar]
- Qu, C.; Tang, Y.; Wang, D.; Fan, X.P.; Li, H.F.; Liu, C.W.; Su, K.; Zhao, D.X.; Jing, J.Q.; Zhang, X. Improved processability of PA66-polyimide copolymers with different polyimide contents. J. Appl. Polym. Sci. 2020, 138, 49640. [Google Scholar] [CrossRef]
- Xu, Q.; Guan, B.; Guo, W.; Liu, X. Effect of antioxidants on thermo-oxidative stability and aging of bio-based PA56T and fast characterization of anti-oxidation performance. Polymers 2022, 14, 1280. [Google Scholar] [CrossRef]
- Rwei, S.P.; Ranganathan, P.; Chiang, W.Y.; Lee, Y.H. Synthesis of low melting temperature aliphatic-aromatic copolyamides derived from novel bio-based semi aromatic monomer. Polymers 2018, 10, 793. [Google Scholar] [CrossRef]
- Feng, W.; Wang, P.; Zou, G.; Ren, Z.; Ji, J. Synthesis and characterization of semiaromatic copolyimide 10T/1014 with high performance and flexibility. Des. Monomers Polym. 2018, 21, 33–42. [Google Scholar] [CrossRef]
- Yu, A.J.; Evans, R.D. Isomorphous replacement in copolyamide systems: Adipic and terephthalic acids. J. Polym. Sci. 1960, 42, 249–257. [Google Scholar] [CrossRef]
- Garmans, R.J.; Aalto, S.; Maurer, E.H.J. Copolyamide of nylon-4,6 and nylon-4,T. J. Polym. Part A Polym. Chem. 1989, 27, 423–430. [Google Scholar] [CrossRef]
- Novitsky, T.F.; Lange, C.A.; Mathias, L.J.; Osborn, S.; Ayotte, R.; Manning, S. Eutectic melting behavior of polyamide 10,T-co-6,T and 12,T-co-6,T copolyterephthalamides. Polymer 2010, 51, 2417–2425. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Zhang, Y.; Yang, G. Melting behavior and nonisothermal crystallization kinetics of polyamide 6/polyamide 66 molecular composites via in situ polymerization. J. Appl. Polym. Sci. 2005, 98, 2172–2177. [Google Scholar] [CrossRef]
- Ridgway, J.S. Structure-property relationships of ring-containing nylon 66 copolyamides. J. Polym. Sci. Part A-1 Polym. Chem. 1970, 8, 3089–3111. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computation on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Chrissafis, K.; Lorenzo, M.L.D.; Koga, N.; Pijolat, M.; Roduit, B.; Sbirrazzuoli, N.; Suñol, J.J. ICTAC Kinetics Committee recommendations for collecting experimental thermal analysis data for kinetic computations. Thermochim. Acta 2014, 590, 1–23. [Google Scholar] [CrossRef]
- Bradbury, E.M.; Brown, L.; Elliott, A.; Parry, D.A.D. The structure of the gamma form of polycaproamide (Nylon 6). Polymer 1965, 6, 465–482. [Google Scholar] [CrossRef]
- Wang, Y.; Kang, H.L.; Guo, Y.F.; Liu, R.G.; Hao, X.M.; Qiao, R.R.; Yan, J.L. The structure and properties of bio-based polyamide 56 fibers prepared by high-speed spinning. J. Appl. Polym. Sci. 2020, 137, 49344. [Google Scholar] [CrossRef]
- Cooper, S.J.; Coogan, M.; Everall, N.; Priestnall, I. A polarized μ-FTIR study on a model system for nylon 66: Implication for the nylon Brill structure. Polymer 2001, 42, 10119–10132. [Google Scholar] [CrossRef]
- Cui, X.; Liu, Z.; Yan, D. Synthesis and characterization of novel even-odd nylons based on undecanedioic acid. Eur. Polym. J. 2004, 40, 1111–1118. [Google Scholar] [CrossRef]
- Pepin, J.; Gaucher, V.; Lefebvre, J.M.; Stroeks, A. Biaxial stretching behavior as a probe of H-bond organization in semi-crystalline polyamides. Polymer 2016, 101, 217–224. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Zhang, J.; Cao, X.; Wu, P.F.; Ye, G.; Fu, Y.; Zhuang, Y.F.; Zhang, A.; Zheng, K.; Ma, Y.M. High heat resistance and good melt spinnability of a polyamide 66 containing benzene structure. Mater. Adv. 2021, 2, 6647–6654. [Google Scholar] [CrossRef]
- Ge, H.; Wang, W.; Pan, Y.; Yu, X.; Hu, W.; Hu, Y. An inherently flame-retardant polyamide containing a phosphorus pendent group prepared by interfacial polymerization. RSC Adv. 2016, 6, 81802–81808. [Google Scholar] [CrossRef]
- Rwei, S.P.; Tseng, Y.C.; Chiu, K.C.; Chang, S.M.; Chen, Y.M. The crystallization kinetics of nylon 6/6T and nylon 66/6T copolymers. Thermochim. Acta 2013, 555, 37–45. [Google Scholar] [CrossRef]
- Rwei, S.P.; Ranganathan, P.; Lee, Y.H. Isothermal crystallization kinetics study of fully aliphatic PA6 copolyamides: Effect of novel long-chain polyamide salt a comonomer. Polymers 2019, 11, 472. [Google Scholar] [CrossRef]
- Jia, F.; Mao, J.L.; Ma, Y.; Yao, C. Mechanical and thermal properties of dimer acid based nylon 636/nylon 66 copolymers. J. Appl. Polym. Sci. 2014, 131, 39845. [Google Scholar] [CrossRef]
- Moran, C.S.; Barthelon, A.; Pearsall, A.; Mittal, V.; Dorgan, J.R. Biorenewable blends of polyamide-4,10 and polyamide-6,10. J. Appl. Polym. Sci. 2016, 133, 43626. [Google Scholar] [CrossRef]
- Sweet, G.E.; Bell, J.P. Multiple endotherm melting behavior in relation to polymer morphology. J. Polym. Sci. Part A-2 Polym. Phys. 1972, 10, 1273–1283. [Google Scholar] [CrossRef]
- Harvey, E.D.; Hybart, F.J. Rates of crystallization of copolyamides. II. Random copolymers of nylon 66 and 6. J. Appl. Polym. Sci. 1970, 14, 2133–2143. [Google Scholar] [CrossRef]
- Bunn, C.W.; Garner, E.V. The crystal structures of two polyamides (‘Nylon’). Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1947, 189, 39–68. [Google Scholar]
- Yang, T.; Gao, Y.; Wang, X.; Ma, B.; He, Y. Hydrogen bonding and crystalline structure of bio-based PA56. Polymer 2021, 237, 124356. [Google Scholar] [CrossRef]
- Puiggalí, J.; Franco, L.; Alemán, C.; Subirana, J.A. Crystal structure of nylon 5,6. A model with two hydrogen bond directions for derived from odd diamines. Macromolecules 1998, 31, 8540–8548. [Google Scholar] [CrossRef]
- Morales-Gámez, L.; Soto, D.; Franco, L.; Puiggalí, J. Brill transition and melt crystallization of nylon 56: An odd-even polyamide with two hydrogen-bonding directions. Polymer 2010, 51, 5788–5798. [Google Scholar] [CrossRef]
- Magill, J.H. Spherulitic crystallization. Part I. “Odd-even” polyamides: Nylon 56 and nylon 96. J. Polym. Sci. Part A 1965, 3, 1195–1219. [Google Scholar]
- Tseng, C.H.; Tsai, P.S. The isothermal and nonisothermal crystallization kinetics and morphology of solvent-precipitated nylon 66. Polymers 2022, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Taib, R.M.; Tham, C.Y. Isothermal crystallization kinetics and spherulite morphologies of poly(lactic acid)/ethylene acrylate copolymer blends. J. Appl. Polym. Sci. 2017, 134, 45487. [Google Scholar]
- Song, J.; Ren, M.; Chen, Q.; Sun, X.; Zhang, H.; Song, C.; Zhang, H.; Mo, Z. Isothermal and nonisothermal crystallization kinetics of irradiated nylon 1212. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2326–2333. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II Transformation tome relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change. III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Wunderlich, B. Macromolecular Physics; Academic Press: New York, NY, USA, 1977; Volume 2. [Google Scholar]
- Jeziorny, A. Parameters characterizing the kinetics of the non-isothermal crystallization of poly(ethylene terephthalate) determined by d.s.c. Polymer 1978, 19, 1142–1144. [Google Scholar] [CrossRef]
- Liu, S.; Yu, Y.; Cui, Y.; Zhang, H.; Mo, Z. Isothermal and nonisothermal crystallization kinetics of nylon-11. J. Appl. Polym. Sci. 1998, 70, 2371–2380. [Google Scholar] [CrossRef]
- McFerran, N.L.A.; Armstrong, C.G.; McNally, T. Nonisothermal and isothermal crystallization kinetics of nylon-12. J. Appl. Polym. Sci. 2008, 110, 1043–1058. [Google Scholar] [CrossRef]
- Bosq, N.; Aht-Ong, D. Nonisothermal crystallization behavior of poly (butylene succinate)/NaY zeolite nanocomposites. Macromol. Res. 2018, 26, 13–21. [Google Scholar] [CrossRef]
- Addonizio, M.L.; Martuscelli, E.; Silvestre, C. Study of the non-isothermal crystallization of poly(ethylene oxide)/poly(methyl methacrylate) blends. Polymer 1987, 28, 183–188. [Google Scholar] [CrossRef]
- Liu, T.; Mo, Z.; Zhang, H. Isothermal and nonisothermal melt crystallization kinetic behavior of Poly(Aryl Ether Biphenyl Ether Ketone Ketone): PEDEKK. J. Polym. Eng. 1998, 18, 283–299. [Google Scholar] [CrossRef]
- Zhang, Q.; Mo, Z. Melting crystallization behavior of nylon 66. Chin. J. Polym. Sci. 2001, 19, 237–246. [Google Scholar]
- Sun, Z.; Wang, X.; Guo, F.; Jiang, C.; Pan, Q. Isothermal and nonisothermal crystallization kinetics of bio-sourced nylon 69. Chin. J. Chem. Eng. 2016, 24, 638–645. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Zhang, H.; Mo, Z. Isothermal and nonisothermal crystallization kinetics of nylon 46. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 1784–1793. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Q.; Wang, Y.; Zhang, C.; Mo, Z.; Cao, S. Melting behaviors, isothermal and non-isothermal crystallization kinetics of nylon 1212. Polymer 2003, 44, 2537–2545. [Google Scholar] [CrossRef]
- Wang, Y.; Kang, H.L.; Wang, R.; Liu, R.G.; Hao, X.M. Crystallization of polyamide 56/polyamide 66 blends: Nonisothermal crystallization kinetics. J. Appl. Polym. Sci. 2018, 135, 46409. [Google Scholar] [CrossRef]
- Ai, T.; Zou, G.; Feng, W.; Ren, Z.; Li, F.; Wang, P.; Lu, B.; Ji, J. Synthesis and properties of biobased copolyamides based on polyamide 10T and polyamide 56 through one-pot polymerization. New J. Chem. 2021, 45, 14677–14686. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Nylon 56/66 (mole%) | PMD (mole) | HMD (mole) | AA (mole) | SHP (ppm) |
|---|---|---|---|---|---|
| PA56 | 100/0 | 1 | 0 | 1 | 80 |
| CoPA-1 | 70/30 | 0.7 | 0.3 | 1 | 80 |
| CoPA-2 | 50/50 | 0.5 | 0.5 | 1 | 80 |
| CoPA-3 | 30/70 | 0.3 | 0.7 | 1 | 80 |
| CoPA-4 | 10/90 | 0.9 | 0.1 | 1 | 80 |
| PA66 | 0/100 | 0 | 1 | 1 | 80 |
| Sample | Feed Ratio a | Calculated b | Mn, g/mol | Mw, g/mol | PDI |
|---|---|---|---|---|---|
| PA56 | 100 | - | 31,500 | 52,300 | 1.66 |
| CoPA-1 | 70/30 | 72.53/27.46 | 24,600 | 43,800 | 1.78 |
| CoPA-2 | 50/50 | 51.35/48.64 | 19,700 | 43,600 | 2.21 |
| CoPA-3 | 30/70 | 25.44/74.56 | 25,700 | 47,600 | 1.85 |
| CoPA-4 | 10/90 | 11.53/88.46 | 32,200 | 52,300 | 1.63 |
| PA66 | 100 | - | 33,400 | 53,600 | 1.61 |
| Sample | Td~5% (°C) | Tonset (°C) | T50% (°C) | Tmax (°C) | Residue at 600 °C (%) |
|---|---|---|---|---|---|
| PA56 | 348 | 413 | 440 | 445 | 3.02 |
| CoPA-1 | 333 | 415 | 441 | 452 | 1.98 |
| CoPA-2 | 298 | 415 | 442 | 443 | 1.77 |
| CoPA-3 | 319 | 416 | 443 | 479 | 2.83 |
| CoPA-4 | 383 | 417 | 444 | 444 | 1.93 |
| PA66 | 379 | 423 | 450 | 457 | 4.64 |
| Sample | Tg (°C) a | Tm1 (°C) b | ΔHm1 (J·g−1) b | Tm2 (°C) b | ΔHm2 (J·g−1) b | Tc (°C) c | ΔHc (J·g−1) c |
|---|---|---|---|---|---|---|---|
| PA56 | 60.3 | 252.8 | 57.6 | - | - | 207.8 | −55.5 |
| CoPA-1 | 56.4 | 222.2 | 90.9 | 236.5 | 53.8 | 194 | −53.5 |
| CoPA-2 | 54.2 | 218.2 | 50.6 | 205.2 | 1.3 | 188.1 | −46.9 |
| CoPA-3 | 56.8 | 234.7 | 52.1 | 206.7 | 1.0 | 203.0 | −48.2 |
| CoPA-4 | 59.3 | 249.9 | 55.7 | 207.5 | 1.1 | 217.8 | −51.0 |
| PA66 | 64.0 | 260.9 | 60.6 | - | - | 228.7 | −63.0 |
| Sample | Φ (°Cmin−1) | T* (°C) | Tmax (min) | X (tmax) (%) | t1/2 (min) | τ1/2 (min−1) |
|---|---|---|---|---|---|---|
| PA56 | 2.5 | 213.63 | 3.800 | 54.66 | 3.691 | 0.2709 |
| 5 | 211.25 | 2.767 | 55.87 | 2.694 | 0.3712 | |
| 10 | 207.67 | 1.547 | 54.31 | 1.519 | 0.6583 | |
| 20 | 203.34 | 0.813 | 51.19 | 0.811 | 1.2330 | |
| 40 | 197.32 | 0.453 | 48.57 | 0.454 | 2.2026 | |
| CoPA-1 | 2.5 | 200.50 | 1.933 | 53.23 | 1.893 | 0.5283 |
| 5 | 194.34 | 1.413 | 45.26 | 1.470 | 0.6803 | |
| 10 | 191.00 | 0.800 | 40.01 | 0.842 | 1.1876 | |
| 20 | 181.34 | 0.713 | 66.75 | 0.642 | 1.5576 | |
| 40 | 176.00 | 0.383 | 49.00 | 0.385 | 2.5974 | |
| CoPA-2 | 2.5 | 198.96 | 2.237 | 41.64 | 2.430 | 0.4415 |
| 5 | 190.76 | 2.203 | 67.39 | 1.919 | 0.5211 | |
| 10 | 183.34 | 1.070 | 56.27 | 1.066 | 0.9381 | |
| 20 | 179.66 | 0.520 | 43.26 | 0.548 | 1.8248 | |
| 40 | 172.76 | 0.313 | 38.46 | 0.314 | 3.1847 | |
| CoPA-3 | 2.5 | 212.35 | 2.777 | 45.15 | 2.778 | 0.3600 |
| 5 | 206.59 | 1.597 | 42.05 | 1.716 | 0.5828 | |
| 10 | 199.00 | 1.267 | 48.55 | 1.186 | 0.8432 | |
| 20 | 195.66 | 0.603 | 41.09 | 0.902 | 1.1086 | |
| 40 | 186.68 | 0.313 | 39.09 | 0.342 | 2.9240 | |
| CoPA-4 | 2.5 | 226.04 | 2.847 | 41.37 | 3.079 | 0.3248 |
| 5 | 223.75 | 1.717 | 37.85 | 1.895 | 0.5277 | |
| 10 | 216.50 | 1.005 | 38.50 | 1.089 | 0.9183 | |
| 20 | 214.66 | 0.503 | 37.83 | 0.559 | 1.7889 | |
| 40 | 202.68 | 0.280 | 36.54 | 0.320 | 3.1250 | |
| PA66 | 2.5 | 236.33 | 1.687 | 48.75 | 1.701 | 0.5879 |
| 5 | 231.34 | 1.497 | 49.25 | 1.509 | 0.6627 | |
| 10 | 225.33 | 0.987 | 49.47 | 0.988 | 1.0121 | |
| 20 | 218.34 | 0.663 | 47.83 | 0.676 | 1.4793 | |
| 40 | 209.32 | 0.420 | 52.69 | 0.410 | 2.4390 |
| Primary Crystallization Stage | Secondary Crystallization Stage | ||||||
|---|---|---|---|---|---|---|---|
| Sample | (°Cmin−1) | n1 | Zt1 | Zc1 | n2 | Zt2 | Zc2 |
| PA56 | 2.5 | 4.110 | 0.003 | 0.101 | 2.304 | 0.051 | 0.304 |
| 5 | 5.576 | 0.003 | 0.308 | 2.981 | 0.064 | 0.577 | |
| 10 | 5.787 | 0.006 | 0.755 | 2.084 | 0.526 | 0.938 | |
| 20 | 5.455 | 2.136 | 1.039 | 1.262 | 3.098 | 1.058 | |
| 40 | 4.755 | 28.78 | 1.088 | 3.454 | 12.70 | 1.066 | |
| CoPA-1 | 2.5 | 4.491 | 0.039 | 0.273 | 2.713 | 0.178 | 0.502 |
| 5 | 3.875 | 0.151 | 0.685 | 2.010 | 0.466 | 0.858 | |
| 10 | 3.511 | 1.235 | 1.021 | 2.206 | 10.09 | 1.260 | |
| 20 | 2.679 | 2.584 | 1.049 | 1.391 | 2.800 | 1.053 | |
| 40 | 5.257 | 102.0 | 1.123 | 1.209 | 4.413 | 1.038 | |
| CoPA-2 | 2.5 | 3.219 | 0.040 | 0.275 | 2.727 | 0.064 | 0.333 |
| 5 | 2.739 | 0.121 | 0.656 | 2.102 | 0.278 | 0.774 | |
| 10 | 3.095 | 0.583 | 0.948 | 1.083 | 1.067 | 1.000 | |
| 20 | 3.169 | 4.533 | 1.079 | 1.015 | 1.841 | 1.031 | |
| 40 | 3.808 | 55.17 | 1.107 | 1.054 | 3.264 | 1.030 | |
| CoPA-3 | 2.5 | 4.350 | 0.009 | 0.145 | 2.409 | 0.067 | 0.340 |
| 5 | 3.611 | 0.098 | 0.628 | 1.728 | 0.401 | 0.833 | |
| 10 | 3.774 | 0.360 | 0.903 | 1.183 | 0.909 | 0.991 | |
| 20 | 4.241 | 4.461 | 1.078 | 0.992 | 1.621 | 1.042 | |
| 40 | 4.564 | 86.70 | 1.118 | 0.958 | 2.458 | 1.028 | |
| CoPA-4 | 2.5 | 3.491 | 0.013 | 0.177 | 1.195 | 0.304 | 0.621 |
| 5 | 3.922 | 0.053 | 0.555 | 1.080 | 0.600 | 0.903 | |
| 10 | 4.042 | 0.412 | 0.915 | 1.035 | 1.120 | 1.011 | |
| 20 | 3.457 | 4.866 | 0.968 | 0.967 | 2.134 | 1.039 | |
| 40 | 3.517 | 36.15 | 1.094 | 1.099 | 4.159 | 1.036 | |
| PA66 | 2.5 | 3.609 | 0.101 | 0.399 | 3.007 | 0.149 | 0.467 |
| 5 | 3.698 | 0.150 | 0.684 | 2.409 | 0.351 | 0.881 | |
| 10 | 3.715 | 0.721 | 0.968 | 1.965 | 1.1153 | 1.001 | |
| 20 | 4.240 | 3.597 | 1.066 | 1.066 | 2.321 | 1.043 | |
| 40 | 4.579 | 49.64 | 1.097 | 1.097 | 8.447 | 1.055 | |
| Sample | Xt(%) | a | F(T) |
|---|---|---|---|
| PA56 | 20 | 1.143 | 12.62 |
| 40 | 1.152 | 14.94 | |
| 50 | 1.161 | 15.93 | |
| 60 | 1.169 | 16.90 | |
| 80 | 1.190 | 19.30 | |
| CoPA-1 | 20 | 1.588 | 5.294 |
| 40 | 1.608 | 7.575 | |
| 50 | 1.591 | 8.831 | |
| 60 | 1.558 | 9.972 | |
| 80 | 1.496 | 12.75 | |
| CoPA-2 | 20 | 1.240 | 6.503 |
| 40 | 1.159 | 9.268 | |
| 50 | 1.136 | 10.40 | |
| 60 | 1.141 | 11.66 | |
| 80 | 1.313 | 15.98 | |
| CoPA-3 | 20 | 1.323 | 7.509 |
| 40 | 1.268 | 9.892 | |
| 50 | 1.255 | 10.99 | |
| 60 | 1.256 | 12.20 | |
| 80 | 1.621 | 19.77 | |
| CoPA-4 | 20 | 1.121 | 7.739 |
| 40 | 1.144 | 9.647 | |
| 50 | 1.153 | 10.61 | |
| 60 | 1.160 | 11.71 | |
| 80 | 1.168 | 15.33 | |
| PA66 | 20 | 1.696 | 6.062 |
| 40 | 1.631 | 8.712 | |
| 50 | 1.611 | 9.906 | |
| 60 | 1.591 | 11.12 | |
| 80 | 1.558 | 14.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, C.-H.; Tsai, P.-S. Biobased Copolyamides 56/66: Synthesis, Characterization and Crystallization Kinetics. Polymers 2022, 14, 3879. https://doi.org/10.3390/polym14183879
Tseng C-H, Tsai P-S. Biobased Copolyamides 56/66: Synthesis, Characterization and Crystallization Kinetics. Polymers. 2022; 14(18):3879. https://doi.org/10.3390/polym14183879
Chicago/Turabian StyleTseng, Chia-Hsiung, and Ping-Szu Tsai. 2022. "Biobased Copolyamides 56/66: Synthesis, Characterization and Crystallization Kinetics" Polymers 14, no. 18: 3879. https://doi.org/10.3390/polym14183879