
Photocatalytic CO2 Conversion Using Metal-Containing Coordination Polymers and Networks: Recent Developments in Material Design and Mechanistic Details


Abstract
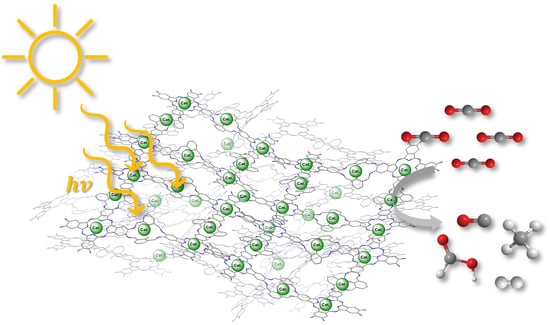
:
1. Introduction
2. 1D-Coordination Polymers
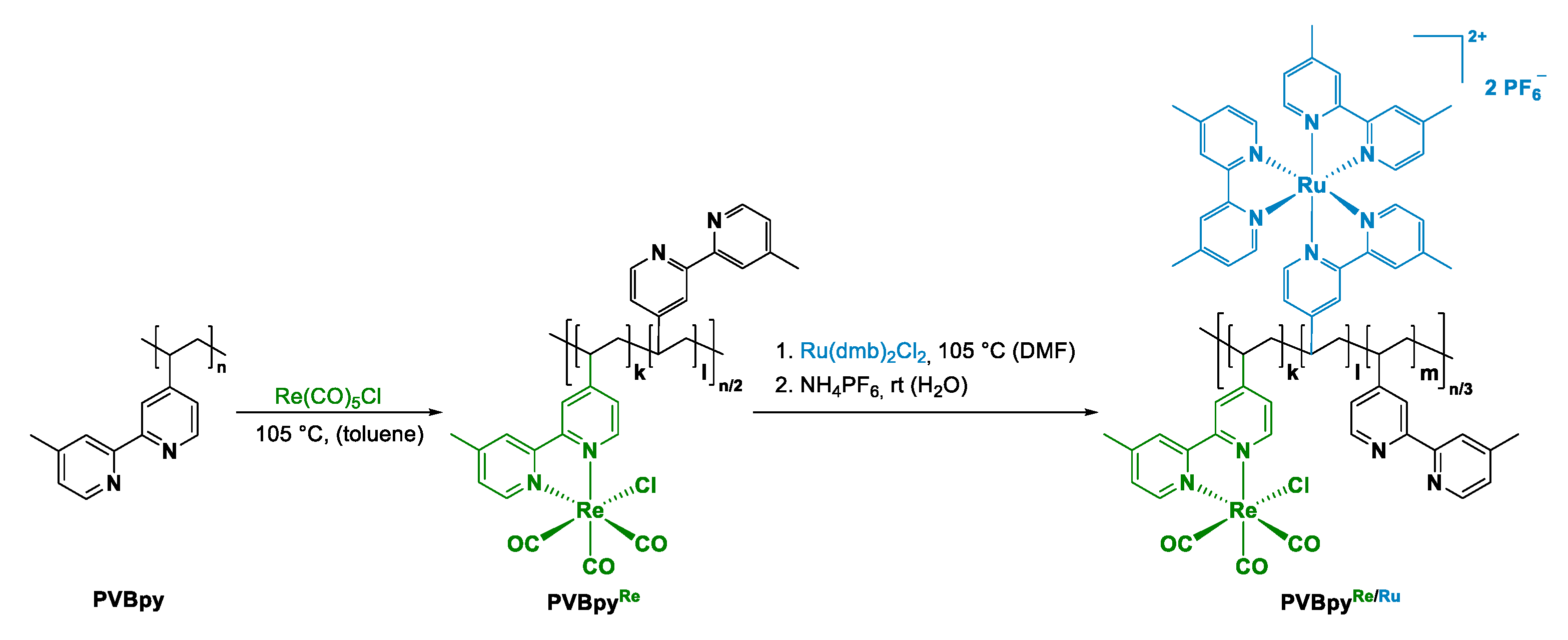
2.1. Re/Ru-Systems
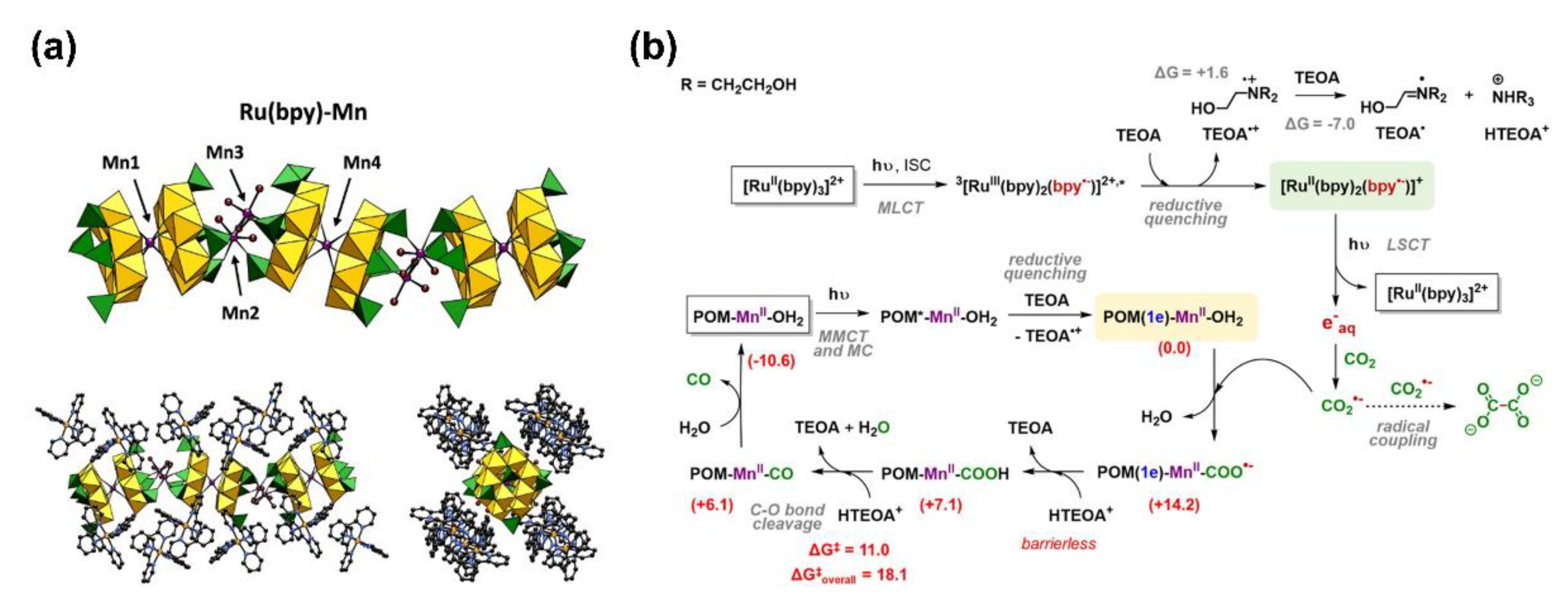
2.2. Ni, Fe, Mn-Systems
3. 2D- or 3D-Coordination Networks
3.1. Supramolecular Polymers and Polymer Gels
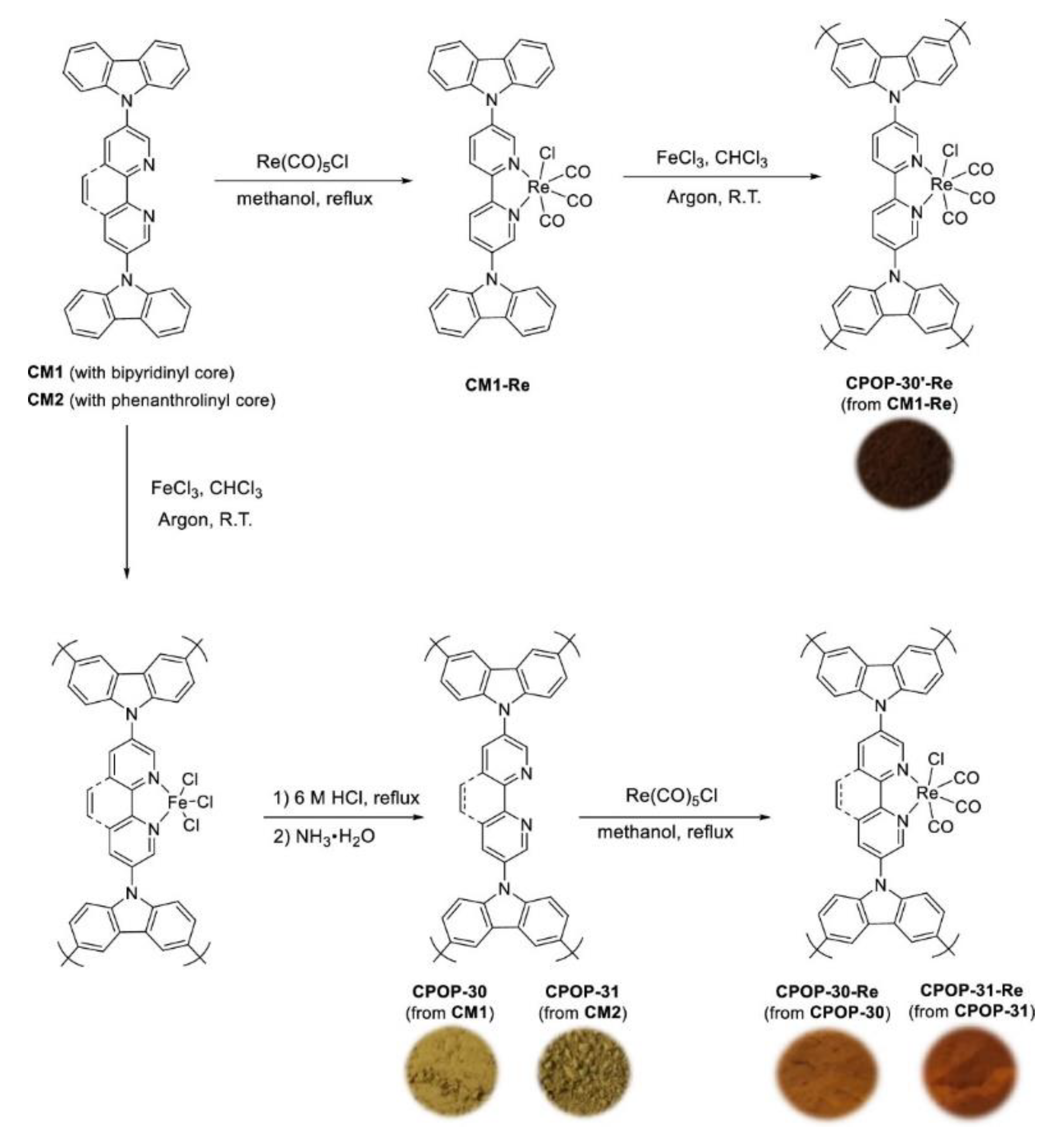
3.2. Porous Organic Polymers
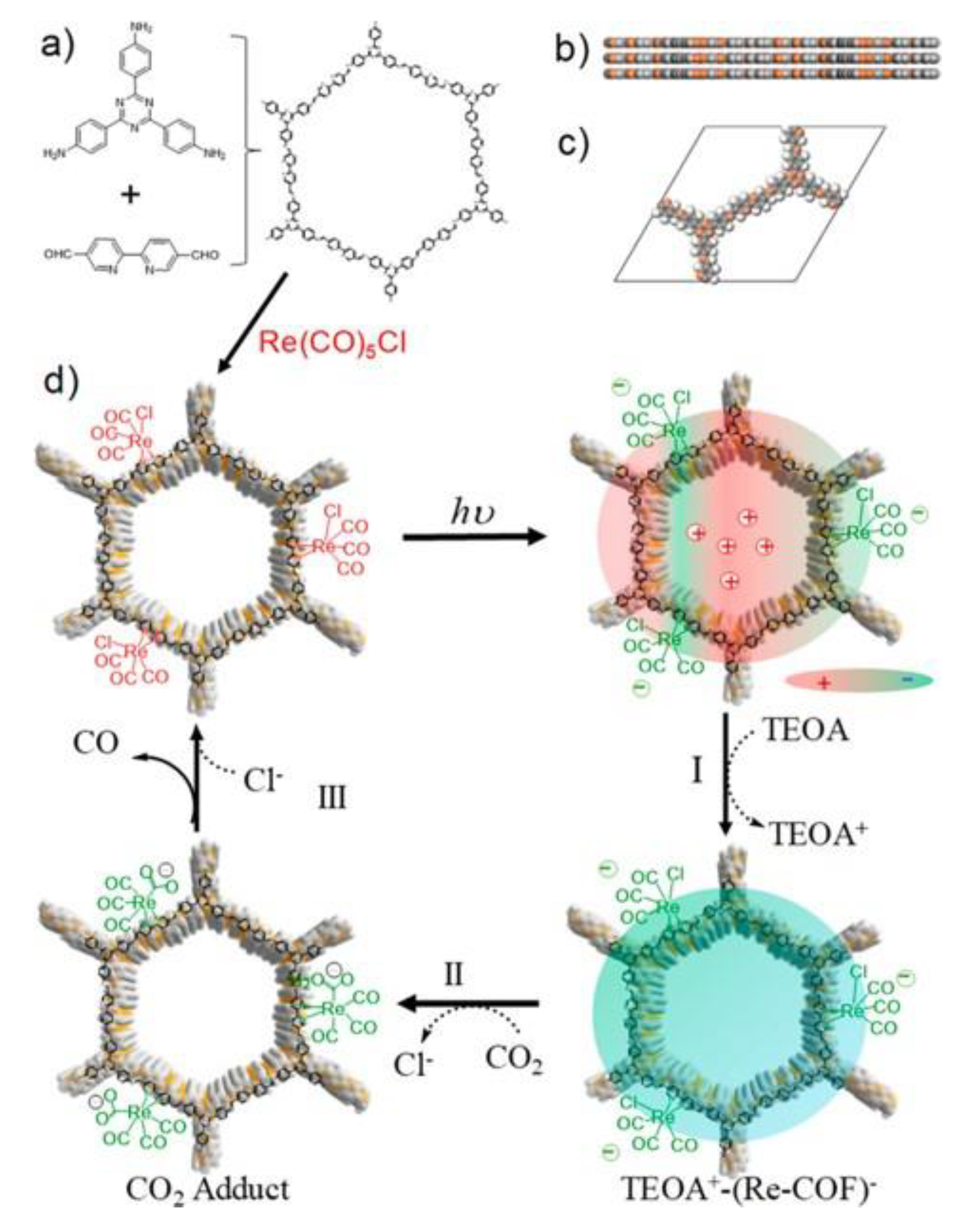
3.2.1. Crystalline Frameworks
3.2.2. Amorphous POPs
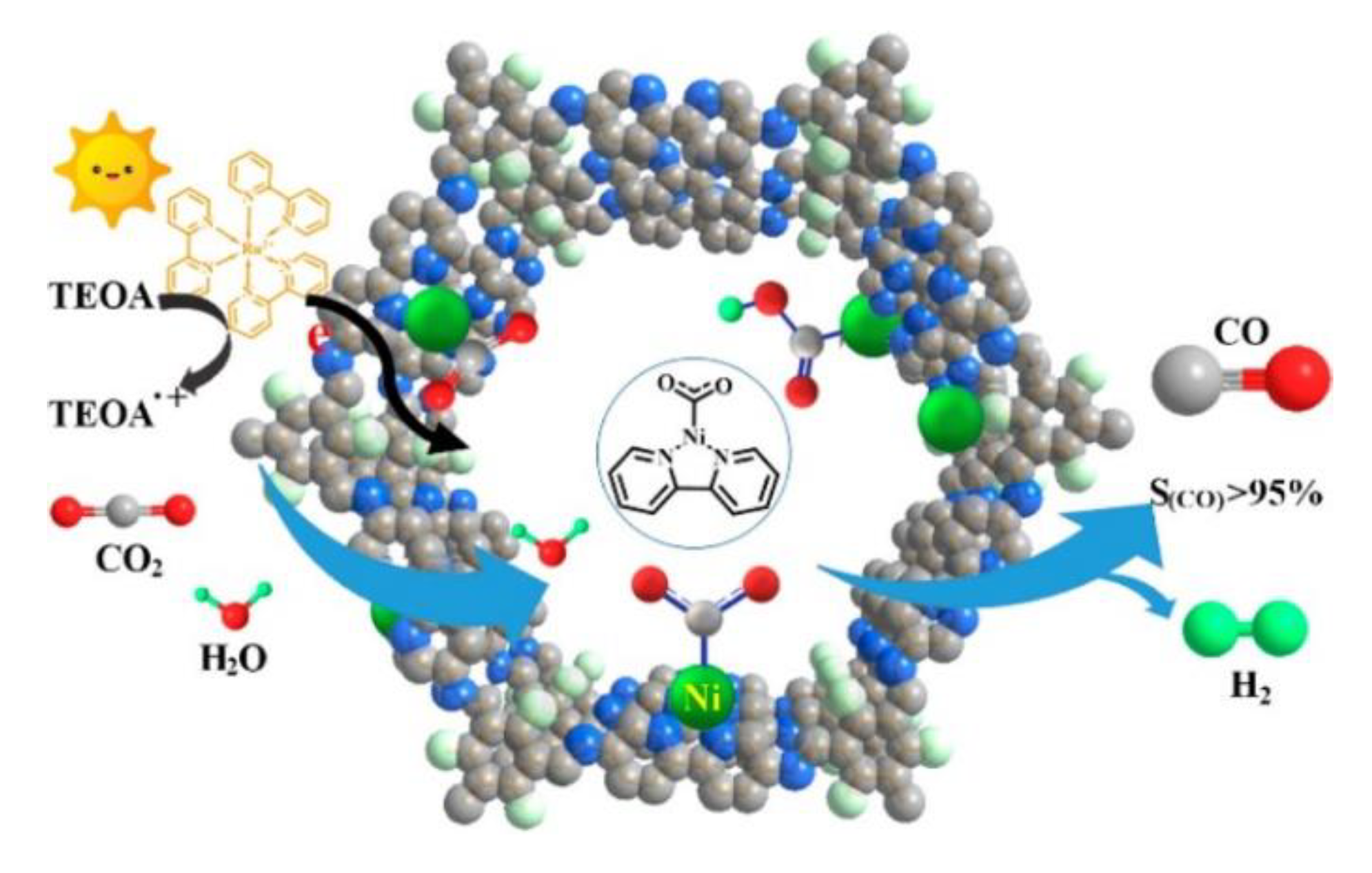
3.3. Metal-Organic Frameworks
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- UNDP. Sustainable Development Goals: Goal 13 Climate Action. Available online: https://www1.undp.org/content/oslo-governance-centre/en/home/sustainable-development-goals/goal-13-climate-action.html (accessed on 16 May 2022).
- Joos, F.; Spahni, R. Rates of change in natural and anthropogenic radiative forcing over the past 20,000 years. Proc. Natl. Acad. Sci. USA 2008, 105, 1425–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earth’s CO2 Home Page. Available online: https://www.co2.earth/ (accessed on 2 May 2022).
- Agarwal, A.S.; Zhai, Y.; Hill, D.; Sridhar, N. The electrochemical reduction of carbon dioxide to formate/formic acid: Engineering and economic feasibility. ChemSusChem 2011, 4, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Meryem, S.S.; Nasreen, S.; Siddique, M.; Khan, R. An overview of the reaction conditions for an efficient photoconversion of CO2. Rev. Chem. Eng. 2018, 34, 409–425. [Google Scholar] [CrossRef]
- Verma, S.; Kim, B.; Jhong, H.-R.M.; Ma, S.; Kenis, P.J.A. A Gross-Margin Model for Defining Technoeconomic Benchmarks in the Electroreduction of CO2. ChemSusChem 2016, 9, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Cao, C.; et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Rosen Brian, A.; Salehi-Khojin, A.; Thorson Michael, R.; Zhu, W.; Whipple Devin, T.; Kenis Paul, J.A.; Masel Richard, I. Ionic Liquid–Mediated Selective Conversion of CO2 to CO at Low Overpotentials. Science 2011, 334, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M.K.; Song, N.; House, R.L.; Glass, J.T.; Meyer, T.J. Polyethylenimine-Enhanced Electrocatalytic Reduction of CO2 to Formate at Nitrogen-Doped Carbon Nanomaterials. J. Am. Chem. Soc. 2014, 136, 7845–7848. [Google Scholar] [CrossRef]
- Behera, A.; Kar, A.K.; Srivastava, R. Challenges and prospects in the selective photoreduction of CO2 to C1 and C2 products with nanostructured materials: A review. Mater. Horiz. 2022, 9, 607–639. [Google Scholar] [CrossRef]
- Gui, M.M.; Lee, W.P.C.; Putri, L.K.; Kong, X.Y.; Tan, L.-L.; Chai, S.-P. Photo-Driven Reduction of Carbon Dioxide: A Sustainable Approach Towards Achieving Carbon Neutrality Goal. Front. Chem. 2021, 3, 744911. [Google Scholar] [CrossRef]
- White, J.L.; Baruch, M.F.; Pander Iii, J.E.; Hu, Y.; Fortmeyer, I.C.; Park, J.E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; et al. Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes. Chem. Rev. 2015, 115, 12888–12935. [Google Scholar] [CrossRef]
- Vu, N.N.; Kaliaguine, S.; Do, T.O. Critical Aspects and Recent Advances in Structural Engineering of Photocatalysts for Sunlight-Driven Photocatalytic Reduction of CO2 into Fuels. Adv. Funct. Mater. 2019, 29, 1901825. [Google Scholar] [CrossRef]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 Reduction Using Water as an Electron Donor over Heterogeneous Photocatalysts Aiming at Artificial Photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Zhu, Q.; Murphy, C.J.; Baker, L.R. Opportunities for Electrocatalytic CO2 Reduction Enabled by Surface Ligands. J. Am. Chem. Soc. 2022, 144, 2829–2840. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, S.; Shi, G.; Zhang, L. Structure-Function Correlation and Dynamic Restructuring of Cu for Highly Efficient Electrochemical CO2 Conversion. ChemSusChem 2022, 15, e202200068. [Google Scholar] [CrossRef]
- Tang, T.; Wang, Z.; Guan, J. Optimizing the Electrocatalytic Selectivity of Carbon Dioxide Reduction Reaction by Regulating the Electronic Structure of Single-Atom M-N-C Materials. Adv. Funct. Mater. 2022, 32, 2111504. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Mills, J.P.; Du, C.; Kim, J.; Wen, J.; Wu, Y.A. Two-dimensional materials for electrochemical CO2 reduction: Materials, in situ/operando characterizations, and perspective. Nanoscale 2021, 13, 19712–19739. [Google Scholar] [CrossRef]
- Ochedi, F.O.; Liu, D.; Yu, J.; Hussain, A.; Liu, Y. Photocatalytic, electrocatalytic and photoelectrocatalytic conversion of carbon dioxide: A review. Environ. Chem. Lett. 2020, 19, 941–967. [Google Scholar] [CrossRef]
- Arai, T.; Sato, S.; Uemura, K.; Morikawa, T.; Kajino, T.; Motohiro, T. Photoelectrochemical reduction of CO2 in water under visible-light irradiation by a p-type InP photocathode modified with an electropolymerized ruthenium complex. Chem. Commun. 2010, 46, 6944–6946. [Google Scholar] [CrossRef]
- Osterloh, F.E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kotyk, J.F.K.; Sheehan, S.W. Progress toward Commercial Application of Electrochemical Carbon Dioxide Reduction. Chem 2018, 4, 2571–2586. [Google Scholar] [CrossRef] [Green Version]
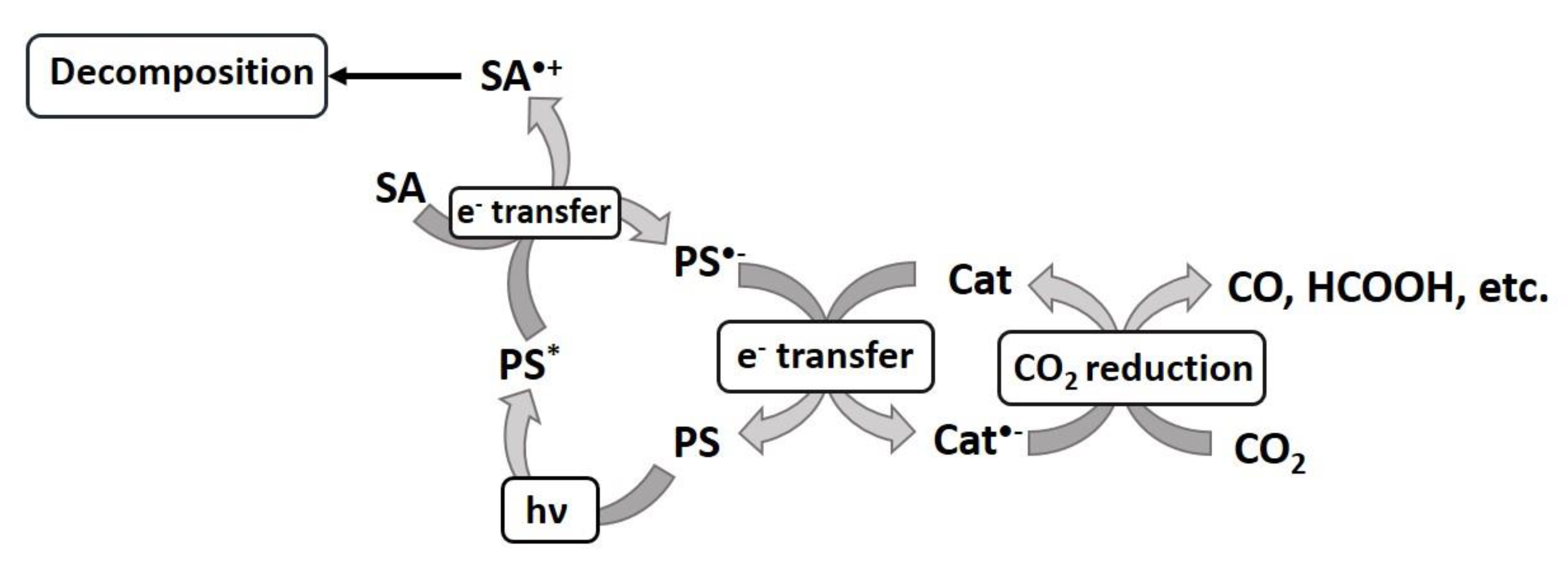
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kuhn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef]
- Samanta, S.; Srivastava, R. Catalytic conversion of CO2 to chemicals and fuels: The collective thermocatalytic/photocatalytic/electrocatalytic approach with graphitic carbon nitride. Adv. Mater. 2020, 1, 1506–1545. [Google Scholar] [CrossRef]
- Singh, R.; Dutta, S. Integrated photocatalytic hydrogen production and pollutants or wastes treatment: Prospects and challenges. In Sustainable Fuel Technologies Handbook; Dutta, S., Mustansar Hussain, C., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 541–549. [Google Scholar]
- Zhang, Y.; Lee, T.S.; Petersen, J.L.; Milsmann, C. A Zirconium Photosensitizer with a Long-Lived Excited State: Mechanistic Insight into Photoinduced Single-Electron Transfer. J. Am. Chem. Soc. 2018, 140, 5934–5947. [Google Scholar] [CrossRef]
- Arias-Rotondo, D.M.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef]
- Chang, X.X.; Wang, T.; Gong, J.L. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Gueymard, C.A. The sun’s total and spectral irradiance for solar energy applications and solar radiation models. Sol. Energy 2004, 76, 423–453. [Google Scholar] [CrossRef]
- Liu, Y.S.; Ji, G.B.; Dastageer, M.A.; Zhu, L.; Wang, J.Y.; Zhang, B.; Chang, X.F.; Gondal, M.A. Highly-active direct Z-scheme Si/TiO2 photocatalyst for boosted CO2 reduction into value-added methanol. RSC Adv. 2014, 4, 56961–56969. [Google Scholar] [CrossRef]
- Lehn, J.M.; Ziessel, R. Photochemical generation of carbon monoxide and hydrogen by reduction of carbon dioxide and water under visible light irradiation. Proc. Natl. Acad. Sci. USA 1982, 79, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Peng, B.S.; Peng, T.Y. Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Shen, H.D.; Peppel, T.; Stunk, J.; Sun, Z.Y. Photocatalytic Reduction of CO2 by Metal-Free-Based Materials: Recent Advances and Future Perspective. Sol. RRL 2020, 4, 1900546. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, Z.; Xu, X.; Wang, J. Review on porous nanomaterials for adsorption and photocatalytic conversion of CO2. Chin. J. Catal. 2017, 38, 1956–1969. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Coordination polymers, metal–organic frameworks and the need for terminology guidelines. CrystEngComm 2012, 14, 3001–3004. [Google Scholar] [CrossRef] [Green Version]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Paik Suh, M.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Adams, F.; Pschenitza, M.; Rieger, B. Yttrium-Catalyzed Synthesis of Bipyridine-Functionalized AB-Block Copolymers: Micellar Support for Photocatalytic Active Rhenium-Complexes. ChemCatChem 2018, 10, 4309–4316. [Google Scholar] [CrossRef]
- Ren, F.Y.; Chen, K.; Qiu, L.Q.; Chen, J.M.; Darensbourg, D.J.; He, L.N. Amphiphilic Polycarbonate Micellar Rhenium Catalysts for Efficient Photocatalytic CO2 Reduction in Aqueous Media. Angew. Chem. Int. Ed. 2022, 134, e202200751. [Google Scholar] [CrossRef]
- Deacy, A.C.; Moreby, E.; Phanopoulos, A.; Williams, C.K. Co(III)/Alkali-Metal(I) Heterodinuclear Catalysts for the Ring-Opening Copolymerization of CO2 and Propylene Oxide. J. Am. Chem. Soc. 2020, 142, 19150–19160. [Google Scholar] [CrossRef]
- Maier, A.S.; Thomas, C.; Kränzlein, M.; Pehl, T.M.; Rieger, B. Macromolecular Rhenium–Ruthenium Complexes for Photocatalytic CO2 Conversion: From Catalytic Lewis Pair Polymerization to Well-Defined Poly(vinyl bipyridine)–Metal Complexes. Macromolecules 2022. [Google Scholar] [CrossRef]
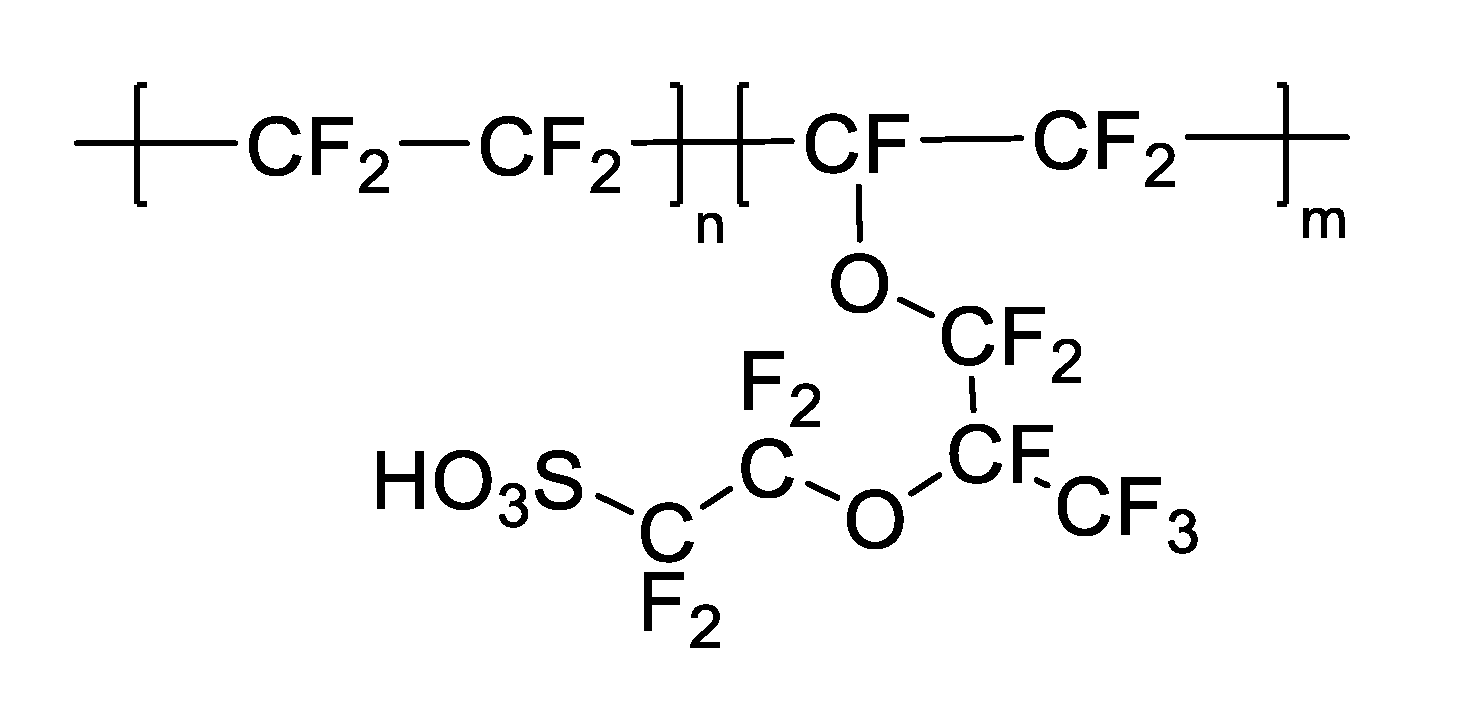
- Lee, S.; Kim, S.; Park, C.; Moon, G.H.; Son, H.J.; Baeg, J.O.; Kim, W.; Choi, W. Nafion-Assisted Noncovalent Assembly of Molecular Sensitizers and Catalysts for Sustained Photoreduction of CO2 to CO. ACS Sustain. Chem. Eng 2020, 8, 3709–3717. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Moore, R.B. State of understanding of nafion. Chem. Rev. 2004, 104, 4535–4585. [Google Scholar] [CrossRef] [PubMed]
- Orchanian, N.M.; Hong, L.E.; Skrainka, J.A.; Esterhuizen, J.A.; Popov, D.A.; Marinescu, S.C. Surface-Immobilized Conjugated Polymers Incorporating Rhenium Bipyridine Motifs for Electrocatalytic and Photocatalytic CO2 Reduction. ACS Appl. Energy Mater. 2018, 2, 110–123. [Google Scholar] [CrossRef]
- Wang, H.Y.; Xie, W.H.; Wei, D.D.; Hu, R.; Wang, N.; Chang, K.; Lei, S.L.; Wang, B.; Cao, R. A Hybrid Assembly with Nickel Poly-Pyridine Polymer on CdS Quantum Dots for Photo-Reducing CO2 into Syngas with Controlled H2/CO Ratios. ChemSusChem 2022, 15, e202200200. [Google Scholar] [CrossRef]
- Wang, J.J.; Li, Z.J.; Li, X.B.; Fan, X.B.; Meng, Q.Y.; Yu, S.; Li, C.B.; Li, J.X.; Tung, C.H.; Wu, L.Z. Photocatalytic hydrogen evolution from glycerol and water over nickel-hybrid cadmium sulfide quantum dots under visible-light irradiation. ChemSusChem 2014, 7, 1468–1475. [Google Scholar] [CrossRef]
- Otsuki, J. Supramolecular approach towards light-harvesting materials based on porphyrins and chlorophylls. J. Mater. Chem. A 2018, 6, 6710–6753. [Google Scholar] [CrossRef]
- Hestand, N.J.; Kazantsev, R.V.; Weingarten, A.S.; Palmer, L.C.; Stupp, S.I.; Spano, F.C. Extended-Charge-Transfer Excitons in Crystalline Supramolecular Photocatalytic Scaffolds. J. Am. Chem. Soc. 2016, 138, 11762–11774. [Google Scholar] [CrossRef] [Green Version]
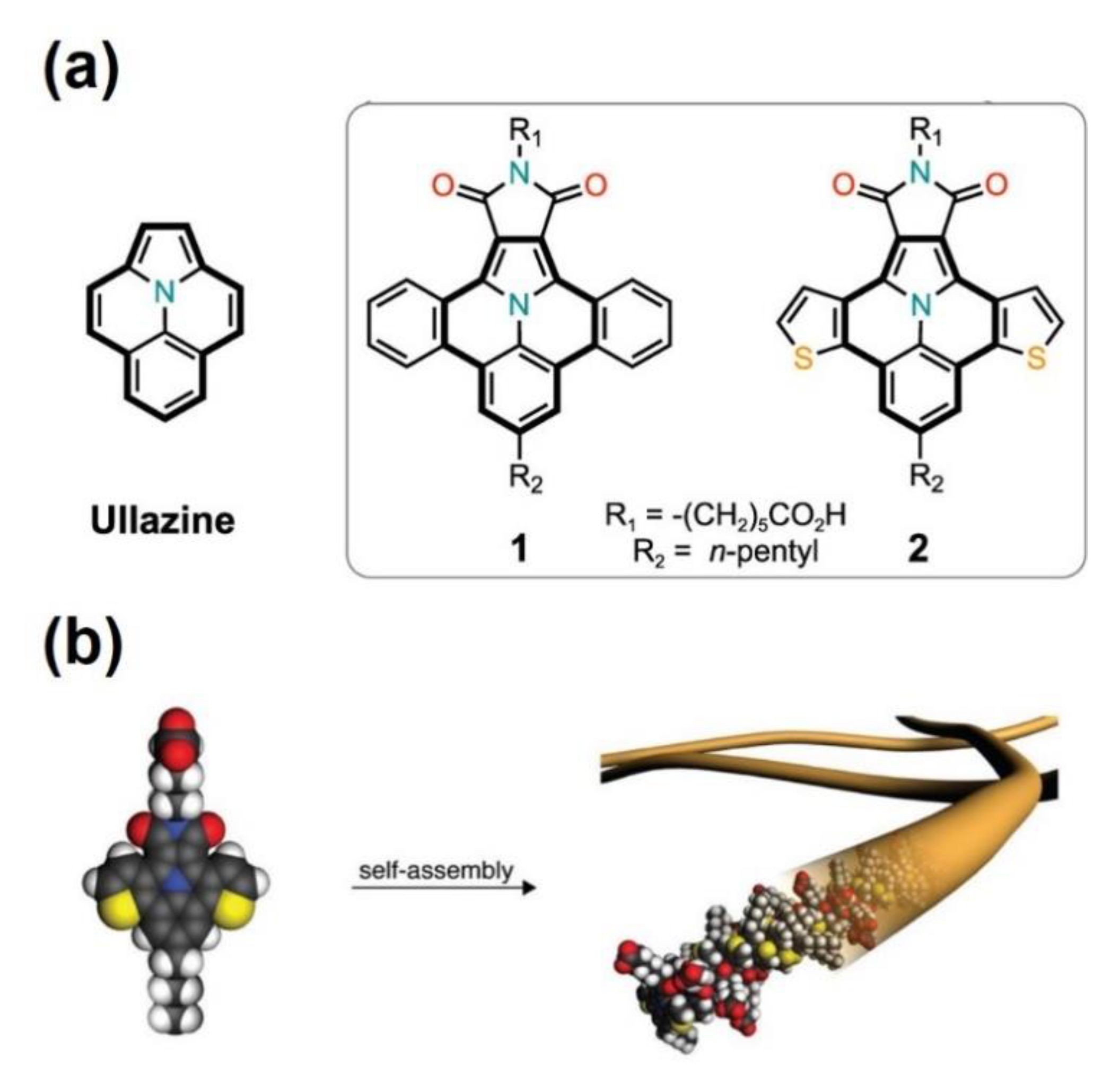
- Dumele, O.; Dordevic, L.; Sai, H.; Cotey, T.J.; Sangji, M.H.; Sato, K.; Dannenhoffer, A.J.; Stupp, S.I. Photocatalytic Aqueous CO2 Reduction to CO and CH4 Sensitized by Ullazine Supramolecular Polymers. J. Am. Chem. Soc. 2022, 144, 3127–3136. [Google Scholar] [CrossRef]
- Park, S.; Lim, J.H.; Chung, S.W.; Mirkin, C.A. Self-assembly of mesoscopic metal-polymer amphiphiles. Science 2004, 303, 348–351. [Google Scholar] [CrossRef] [Green Version]
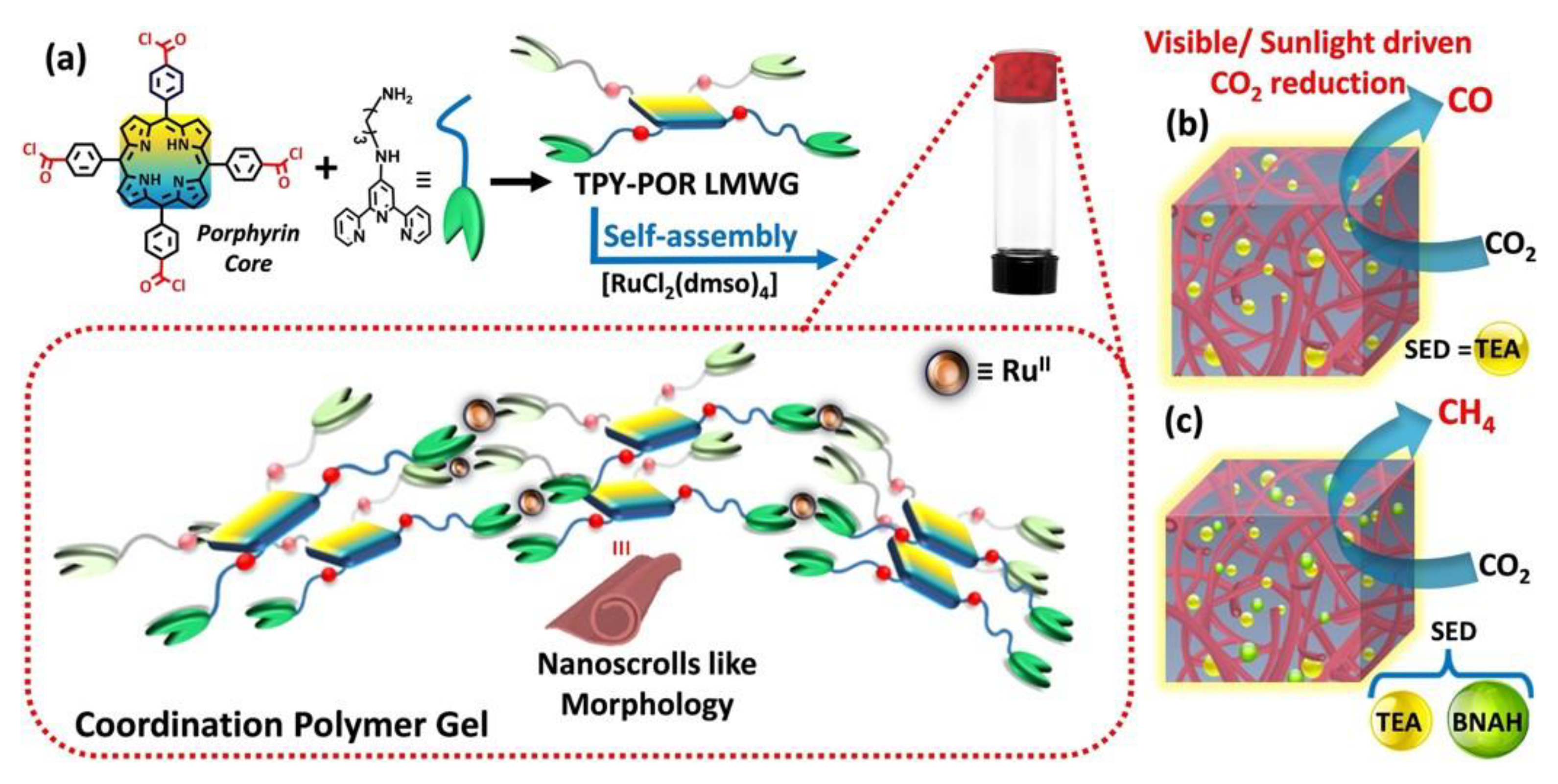
- Verma, P.; Rahimi, F.A.; Samanta, D.; Kundu, A.; Dasgupta, J.; Maji, T.K. Visible-Light-Driven Photocatalytic CO2 Reduction to CO/CH4 Using a Metal-Organic “Soft” Coordination Polymer Gel. Angew. Chem. Int. Ed. 2022, 61, e202116094. [Google Scholar] [CrossRef]
- Goze, C.; Sabatini, C.; Barbieri, A.; Barigelletti, F.; Ziessel, R. Ruthenium-terpyridine complexes with multiple ethynylpyrenyl or ethynyltoluyl subunits: X-ray structure, redox, and spectroscopic properties. Inorg. Chem. 2007, 46, 7341–7350. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.H.; Dao, X.Y.; Zhang, X.D.; Zhao, Y.; Sun, W.Y. Coordination polymers with a pyridyl-salen ligand for photocatalytic carbon dioxide reduction. Chem. Commun. 2020, 56, 4110–4113. [Google Scholar] [CrossRef]
- Costentin, C.; Sayeant, J.M.; Tard, C. Catalysis of CO2 Electrochemical Reduction by Protonated Pyridine and Similar Molecules. Useful Lessons from a Methodological Misadventure. ACS Energy Lett. 2018, 3, 695–703. [Google Scholar] [CrossRef]
- Cole, E.B.; Lakkaraju, P.S.; Rampulla, D.M.; Morris, A.J.; Abelev, E.; Bocarsly, A.B. Using a one-electron shuttle for the multielectron reduction of CO2 to methanol: Kinetic, mechanistic, and structural insights. J. Am. Chem. Soc. 2010, 132, 11539–11551. [Google Scholar] [CrossRef]
- Dridi, H.; Comminges, C.; Morais, C.; Meledje, J.C.; Kokoh, K.B.; Costentin, C.; Saveant, J.M. Catalysis and Inhibition in the Electrochemical Reduction of CO2 on Platinum in the Presence of Protonated Pyridine. New Insights into Mechanisms and Products. J. Am. Chem. Soc. 2017, 139, 13922–13928. [Google Scholar] [CrossRef]
- Benseghir, Y.; Solé-Daura, A.; Mialane, P.; Marrot, J.; Dalecky, L.; Béchu, S.; Frégnaux, M.; Gomez-Mingot, M.; Fontecave, M.; Mellot-Draznieks, C.; et al. Understanding the Photocatalytic Reduction of CO2 with Heterometallic Molybdenum(V) Phosphate Polyoxometalates in Aqueous Media. ACS Catal. 2021, 12, 453–464. [Google Scholar] [CrossRef]
- Cheng, Y.Z.; Ding, X.S.; Han, B.H. Porous Organic Polymers for Photocatalytic Carbon Dioxide Reduction. ChemPhotoChem 2021, 5, 406–417. [Google Scholar] [CrossRef]
- Liu, M.Y.; Guo, L.P.; Jin, S.B.; Tan, B.E. Covalent triazine frameworks: Synthesis and applications. J. Mater. Chem. A 2019, 7, 5153–5172. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, M.; Liu, J.; Chen, Y.; Liao, J.P.; Yang, M.Y.; Cai, Y.P.; Li, S.L.; Lan, Y.Q. Covalent Organic Framework Based Functional Materials: Important Catalysts for Efficient CO2 Utilization. Angew. Chem. Int. Ed. 2022, 61, e202200003. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.W.; Winkler, D.A.; Liu, M.S.; Haranczyk, M.; Kennedy, D.F. Towards computational design of zeolite catalysts for CO2 reduction. RSC Adv. 2015, 5, 44361–44370. [Google Scholar] [CrossRef]
- Auerbach, S.M.; Carrado, K.A.; Dutta, P.K. Handbook of Zeolite Science and Technology; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Geng, K.; He, T.; Liu, R.; Dalapati, S.; Tan, K.T.; Li, Z.; Tao, S.; Gong, Y.; Jiang, Q.; Jiang, D. Covalent Organic Frameworks: Design, Synthesis, and Functions. Chem. Rev. 2020, 120, 8814–8933. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.P.; Acharjya, A.; Anito, D.A.; Vogl, S.; Wang, T.X.; Thomas, A.; Han, B.H. Rhenium-Metalated Polypyridine-Based Porous Polycarbazoles for Visible-Light CO2 Photoreduction. ACS Catal. 2019, 9, 3959–3968. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, X.; Gardner, A.M.; Wang, X.; Chong, S.Y.; Neri, G.; Cowan, A.J.; Liu, L.; Li, X.; Vogel, A.; et al. A stable covalent organic framework for photocatalytic carbon dioxide reduction. Chem. Sci. 2020, 11, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Hu, W.; Zhang, X.; He, P.; Pattengale, B.; Liu, C.; Cendejas, M.; Hermans, I.; Zhang, X.; Zhang, J.; et al. 2D Covalent Organic Frameworks as Intrinsic Photocatalysts for Visible Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2018, 140, 14614–14618. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Sekine, M.; Kitamura, K.; Maegawa, Y.; Goto, Y.; Shirai, S.; Inagaki, S.; Ishida, H. Photocatalytic CO2 Reduction by Periodic Mesoporous Organosilica (PMO) Containing Two Different Ruthenium Complexes as Photosensitizing and Catalytic Sites. Chem. Eur. J. 2017, 23, 10301–10309. [Google Scholar] [CrossRef]
- Lu, M.; Liu, J.; Li, Q.; Zhang, M.; Liu, M.; Wang, J.L.; Yuan, D.Q.; Lan, Y.Q. Rational Design of Crystalline Covalent Organic Frameworks for Efficient CO2 Photoreduction with H2O. Angew. Chem. Int. Ed. 2019, 58, 12392–12397. [Google Scholar] [CrossRef]
- Lv, H.; Sa, R.; Li, P.; Yuan, D.; Wang, X.; Wang, R. Metalloporphyrin-based covalent organic frameworks composed of the electron donor-acceptor dyads for visible-light-driven selective CO2 reduction. Sci. China Chem. 2020, 63, 1289–1294. [Google Scholar] [CrossRef]
- Zhong, W.; Sa, R.; Li, L.; He, Y.; Li, L.; Bi, J.; Zhuang, Z.; Yu, Y.; Zou, Z. A Covalent Organic Framework Bearing Single Ni Sites as a Synergistic Photocatalyst for Selective Photoreduction of CO2 to CO. J. Am. Chem. Soc. 2019, 141, 7615–7621. [Google Scholar] [CrossRef]
- Wang, W.Y.; Xi, Y.; Yang, C.; Byun, J.; Cheng, J.J.; Wang, S.B.; Wang, X.C. Incorporation of Metal Active Sites on Porous Polycarbazoles for Photocatalytic CO2 Reduction. ChemCatChem 2022, 14, e202101872. [Google Scholar] [CrossRef]
- Xu, R.; Wang, X.S.; Zhao, H.; Lin, H.; Huang, Y.B.; Cao, R. Rhenium-modified porous covalent triazine framework for highly efficient photocatalytic carbon dioxide reduction in a solid-gas system. Catal. Sci. Technol. 2018, 8, 2224–2230. [Google Scholar] [CrossRef]
- Thomas, A. Functional materials: From hard to soft porous frameworks. Angew. Chem. Int. Ed. 2010, 49, 8328–8344. [Google Scholar] [CrossRef]
- Kuhn, P.; Thomas, A.; Antonietti, M. Toward Tailorable Porous Organic Polymer Networks: A High-Temperature Dynamic Polymerization Scheme Based on Aromatic Nitriles. Macromolecules 2009, 42, 319–326. [Google Scholar] [CrossRef]
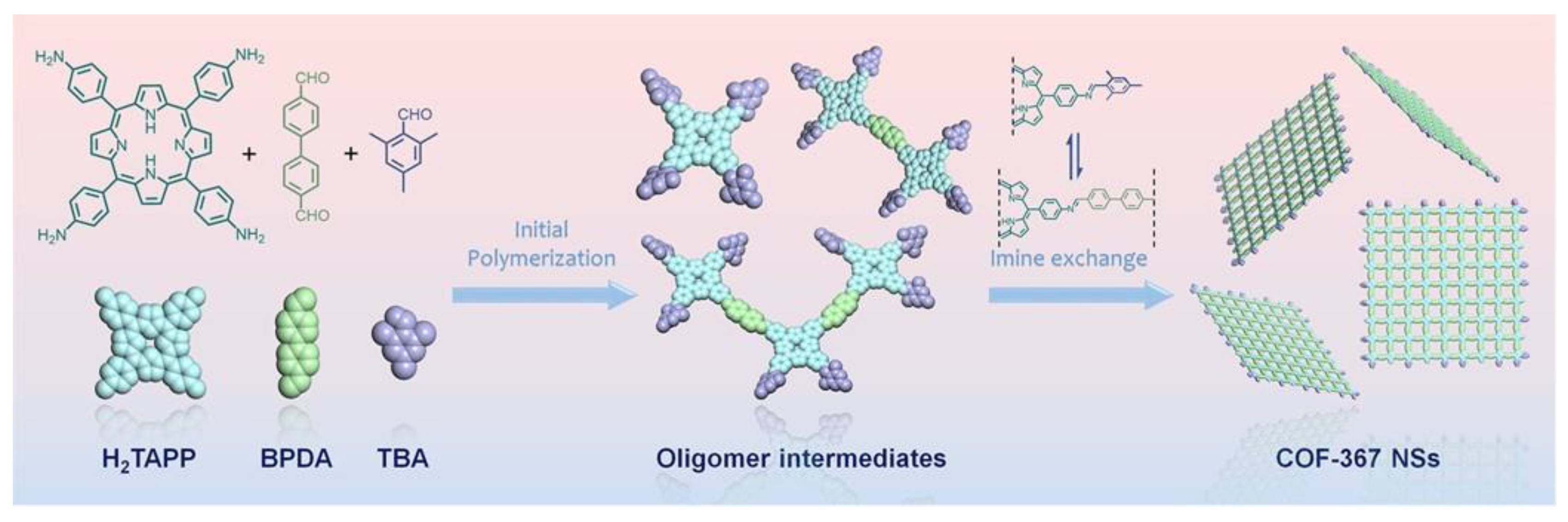
- Liu, W.; Li, X.; Wang, C.; Pan, H.; Liu, W.; Wang, K.; Zeng, Q.; Wang, R.; Jiang, J. A Scalable General Synthetic Approach toward Ultrathin Imine-Linked Two-Dimensional Covalent Organic Framework Nanosheets for Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 17431–17440. [Google Scholar] [CrossRef]
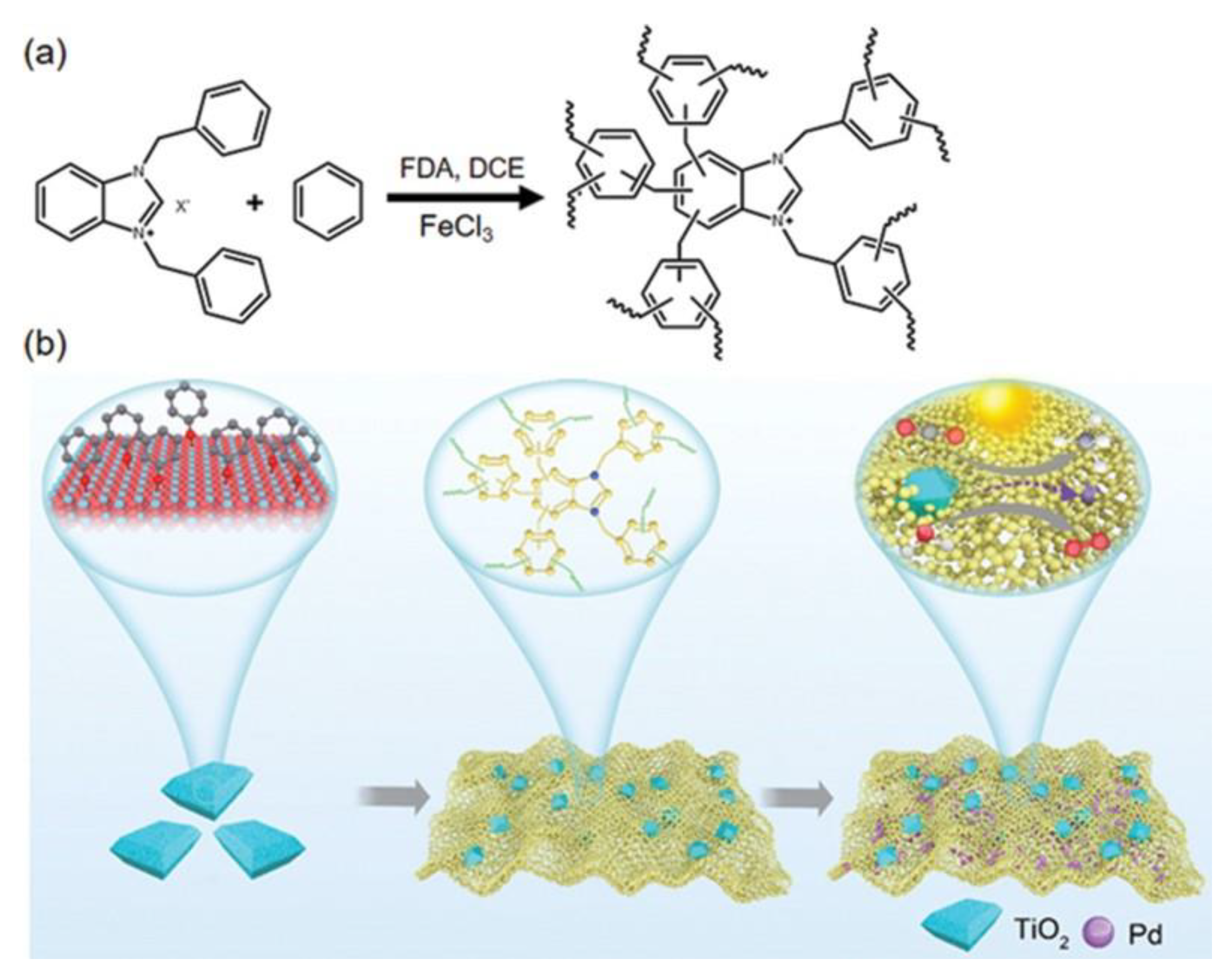
- Wang, S.; Xu, M.; Peng, T.; Zhang, C.; Li, T.; Hussain, I.; Wang, J.; Tan, B. Porous hypercrosslinked polymer-TiO2-graphene composite photocatalysts for visible-light-driven CO2 conversion. Nat. Commun. 2019, 10, 676. [Google Scholar] [CrossRef]
- Gu, L.; Wang, J.; Cheng, H.; Zhao, Y.; Liu, L.; Han, X. One-step preparation of graphene-supported anatase TiO2 with exposed {001} facets and mechanism of enhanced photocatalytic properties. ACS Appl. Mater. Interfaces 2013, 5, 3085–3093. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Shu, Y.; Jiang, S.; Xia, Q.; Chen, L.; Jin, S.; Hussain, I.; Cooper, A.I.; Tan, B. Layered microporous polymers by solvent knitting method. Sci. Adv. 2017, 3, e1602610. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Z.; Wang, H.; Huang, Q.; Li, S.; Yi, X.; Tang, Q.; Wang, J.; Tan, B. Grafting Hypercrosslinked Polymers on TiO2 Surface for Anchoring Ultrafine Pd Nanoparticles: Dramatically Enhanced Efficiency and Selectivity toward Photocatalytic Reduction of CO2 to CH4. Small 2022, 18, e2105083. [Google Scholar] [CrossRef]
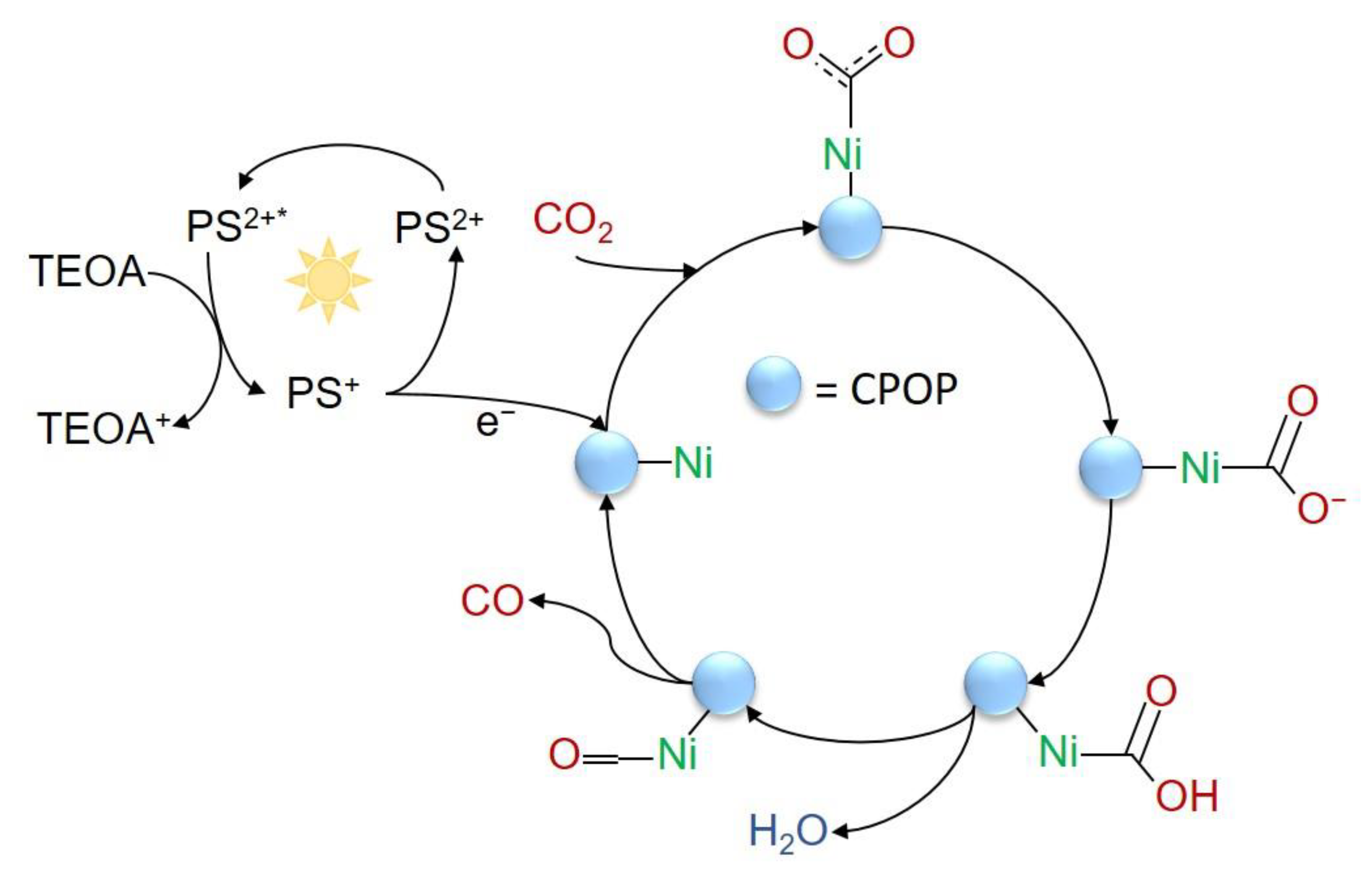
- Dong, X.Y.; Si, Y.N.; Wang, Q.Y.; Wang, S.; Zang, S.Q. Integrating Single Atoms with Different Microenvironments into One Porous Organic Polymer for Efficient Photocatalytic CO2 Reduction. Adv. Mater. 2021, 33, e2101568. [Google Scholar] [CrossRef]
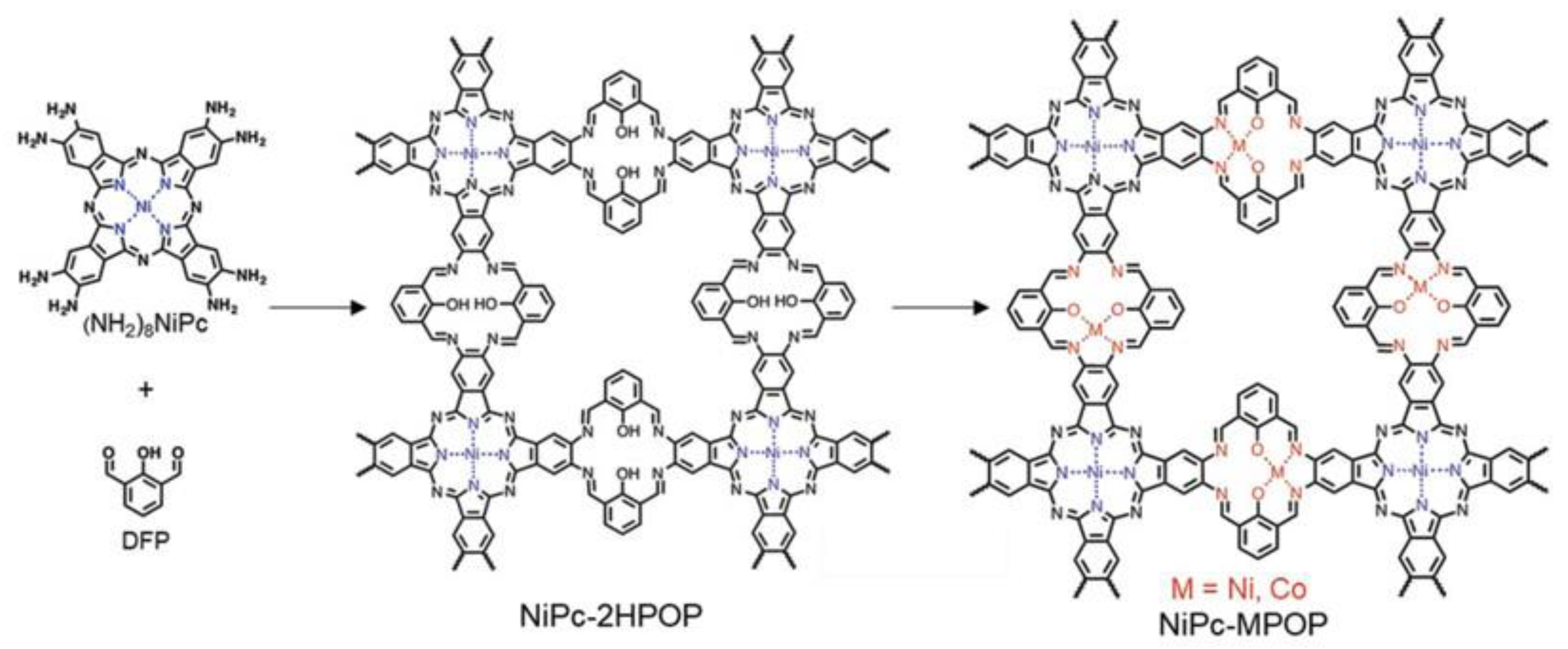
- Huang, N.; Lee, K.H.; Yue, Y.; Xu, X.; Irle, S.; Jiang, Q.; Jiang, D. A Stable and Conductive Metallophthalocyanine Framework for Electrocatalytic Carbon Dioxide Reduction in Water. Angew. Chem. Int. Ed. 2020, 59, 16587–16593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.D.; Si, D.H.; Yi, J.D.; Yin, Q.; Huang, Y.B.; Cao, R. Conductive phthalocyanine-based metal-organic framework as a highly efficient electrocatalyst for carbon dioxide reduction reaction. Sci. China Chem. 2021, 64, 1332–1339. [Google Scholar] [CrossRef]
- Kuila, A.; Surib, N.A.; Mishra, N.S.; Nawaz, A.; Leong, K.H.; Sim, L.C.; Saravanan, P.; Ibrahim, S. Metal Organic Frameworks: A New Generation Coordination Polymers for Visible Light Photocatalysis. ChemistrySelect 2017, 2, 6163–6177. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, S.M.; Nandy, A.; Jablonka, K.M.; Ongari, D.; Janet, J.P.; Boyd, P.G.; Lee, Y.; Smit, B.; Kulik, H.J. Understanding the diversity of the metal-organic framework ecosystem. Nat. Commun. 2020, 11, 4068. [Google Scholar] [CrossRef]
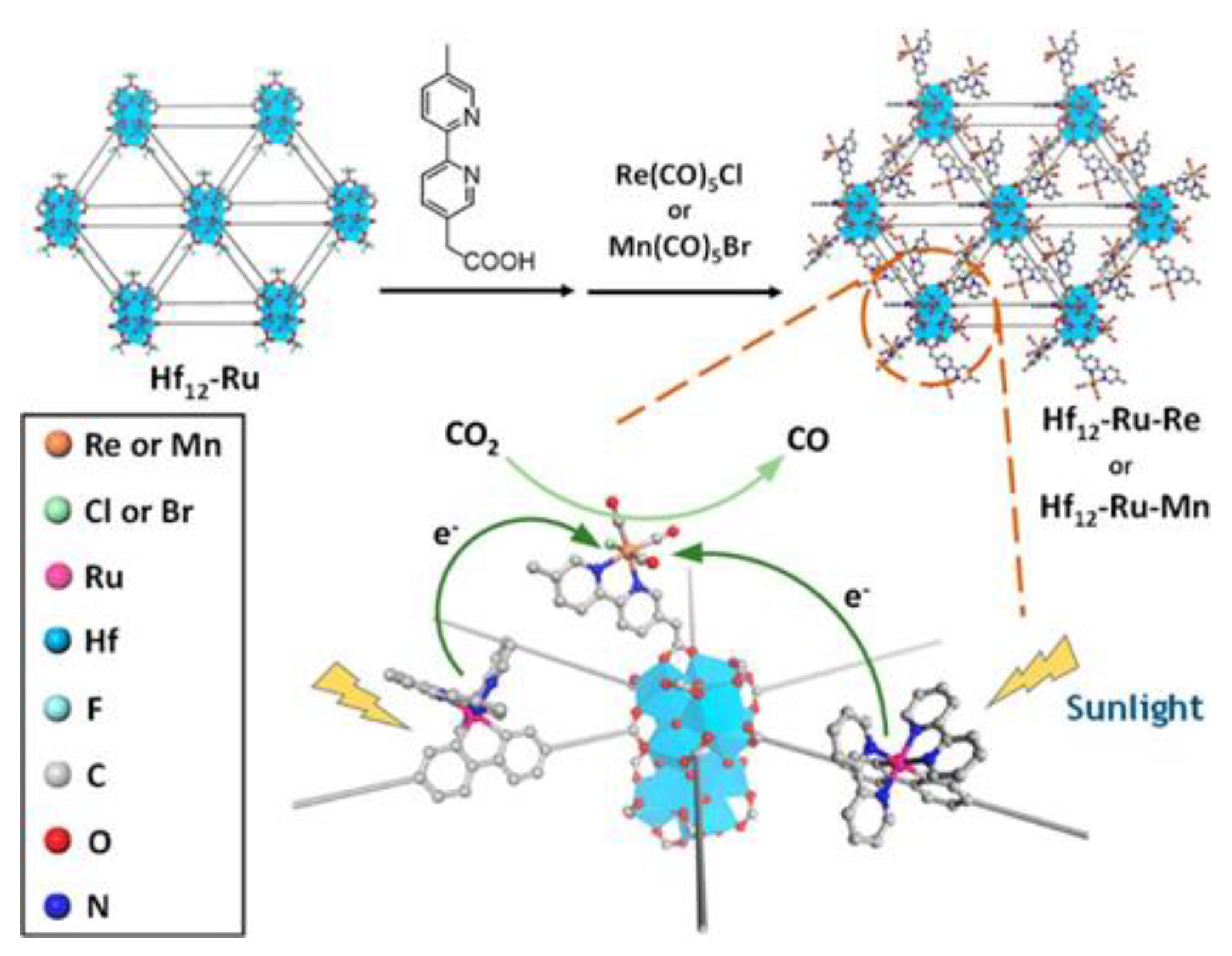
- Dao, X.Y.; Sun, W.Y. Single- and mixed-metal-organic framework photocatalysts for carbon dioxide reduction. Inorg. Chem. Front. 2021, 8, 3178–3204. [Google Scholar] [CrossRef]
- Kidanemariam, A.; Lee, J.; Park, J. Recent Innovation of Metal-Organic Frameworks for Carbon Dioxide Photocatalytic Reduction. Polymers 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Redfern, L.R.; Farha, O.K. Mechanical properties of metal-organic frameworks. Chem. Sci. 2019, 10, 10666–10679. [Google Scholar] [CrossRef] [Green Version]
- Santaclara, J.G.; Nasalevich, M.A.; Castellanos, S.; Evers, W.H.; Spoor, F.C.; Rock, K.; Siebbeles, L.D.; Kapteijn, F.; Grozema, F.; Houtepen, A.; et al. Organic Linker Defines the Excited-State Decay of Photocatalytic MIL-125(Ti)-Type Materials. ChemSusChem 2016, 9, 388–395. [Google Scholar] [CrossRef]
- Lu, G.; Li, S.; Guo, Z.; Farha, O.K.; Hauser, B.G.; Qi, X.; Wang, Y.; Wang, X.; Han, S.; Liu, X.; et al. Imparting functionality to a metal-organic framework material by controlled nanoparticle encapsulation. Nat. Chem. 2012, 4, 310–316. [Google Scholar] [CrossRef]
- Maina, J.W.; Pozo-Gonzalo, C.; Kong, L.X.; Schutz, J.; Hill, M.; Dumee, L.F. Metal organic framework based catalysts for CO2 conversion. Mater. Horiz. 2017, 4, 345–361. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Metal-organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Li, L.; Zhang, S.; Xu, L.; Wang, J.; Shi, L.-X.; Chen, Z.-N.; Hong, M.; Luo, J. Effective visible-light driven CO2photoreduction via a promising bifunctional iridium coordination polymer. Chem. Sci. 2014, 5, 3808–3813. [Google Scholar] [CrossRef]
- Li, R.; Hu, J.; Deng, M.; Wang, H.; Wang, X.; Hu, Y.; Jiang, H.L.; Jiang, J.; Zhang, Q.; Xie, Y.; et al. Integration of an inorganic semiconductor with a metal-organic framework: A platform for enhanced gaseous photocatalytic reactions. Adv. Mater. 2014, 26, 4783–4788. [Google Scholar] [CrossRef]
- Lin, Z.; Thacker, N.C.; Sawano, T.; Drake, T.; Ji, P.; Lan, G.; Cao, L.; Liu, S.; Wang, C.; Lin, W. Metal-organic layers stabilize earth-abundant metal-terpyridine diradical complexes for catalytic C-H activation. Chem. Sci. 2018, 9, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Wang, Y.; Ma, Q.; Huang, Y.; Zhang, X.; Ping, J.; Zhang, Z.; Lu, Q.; Yu, Y.; Xu, H.; et al. Ultrathin 2D Metal-Organic Framework Nanosheets. Adv. Mater. 2015, 27, 7372–7378. [Google Scholar] [CrossRef]
- Lan, G.; Ni, K.; Xu, R.; Lu, K.; Lin, Z.; Chan, C.; Lin, W. Nanoscale Metal-Organic Layers for Deeply Penetrating X-ray-Induced Photodynamic Therapy. Angew. Chem. Int. Ed. 2017, 56, 12102–12106. [Google Scholar] [CrossRef]
- Cao, L.; Lin, Z.; Peng, F.; Wang, W.; Huang, R.; Wang, C.; Yan, J.; Liang, J.; Zhang, Z.; Zhang, T.; et al. Self-Supporting Metal-Organic Layers as Single-Site Solid Catalysts. Angew. Chem. Int. Ed. 2016, 55, 4962–4966. [Google Scholar] [CrossRef]
- Lan, G.; Li, Z.; Veroneau, S.S.; Zhu, Y.Y.; Xu, Z.; Wang, C.; Lin, W. Photosensitizing Metal-Organic Layers for Efficient Sunlight-Driven Carbon Dioxide Reduction. J. Am. Chem. Soc. 2018, 140, 12369–12373. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, C.F.; Li, Q.; Xiong, L.K.; Chen, R.X.; Wan, X.B.; Wang, Z.; Chen, W.; Deng, Z.; Peng, Y. Selective reduction of CO2 by conductive MOF nanosheets as an efficient co-catalyst under visible light illumination. Appl. Catal. B 2018, 238, 339–345. [Google Scholar] [CrossRef]
- Sheberla, D.; Sun, L.; Blood-Forsythe, M.A.; Er, S.; Wade, C.R.; Brozek, C.K.; Aspuru-Guzik, A.; Dinca, M. High electrical conductivity in Ni(3)(2,3,6,7,10,11-hexaiminotriphenylene)(2), a semiconducting metal-organic graphene analogue. J. Am. Chem. Soc. 2014, 136, 8859–8862. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.Q.; Zhu, W.; Li, X.; Zhang, C.F.; Su, Y.H.; Lian, Y.B.; Qi, P.W.; Deng, Z.; Zhang, D.; Wang, S.; et al. Electrostatic charge transfer for boosting the photocatalytic CO2 reduction on metal centers of 2D MOF/rGO heterostructure. Appl. Catal. B 2020, 262, 118144. [Google Scholar] [CrossRef]
- Ye, L.; Gao, Y.; Cao, S.Y.; Chen, H.; Yao, Y.; Hou, J.G.; Sun, L.C. Assembly of highly efficient photocatalytic CO2 conversion systems with ultrathin two-dimensional metal-organic framework nanosheets. Appl. Catal. B 2018, 227, 54–60. [Google Scholar] [CrossRef]
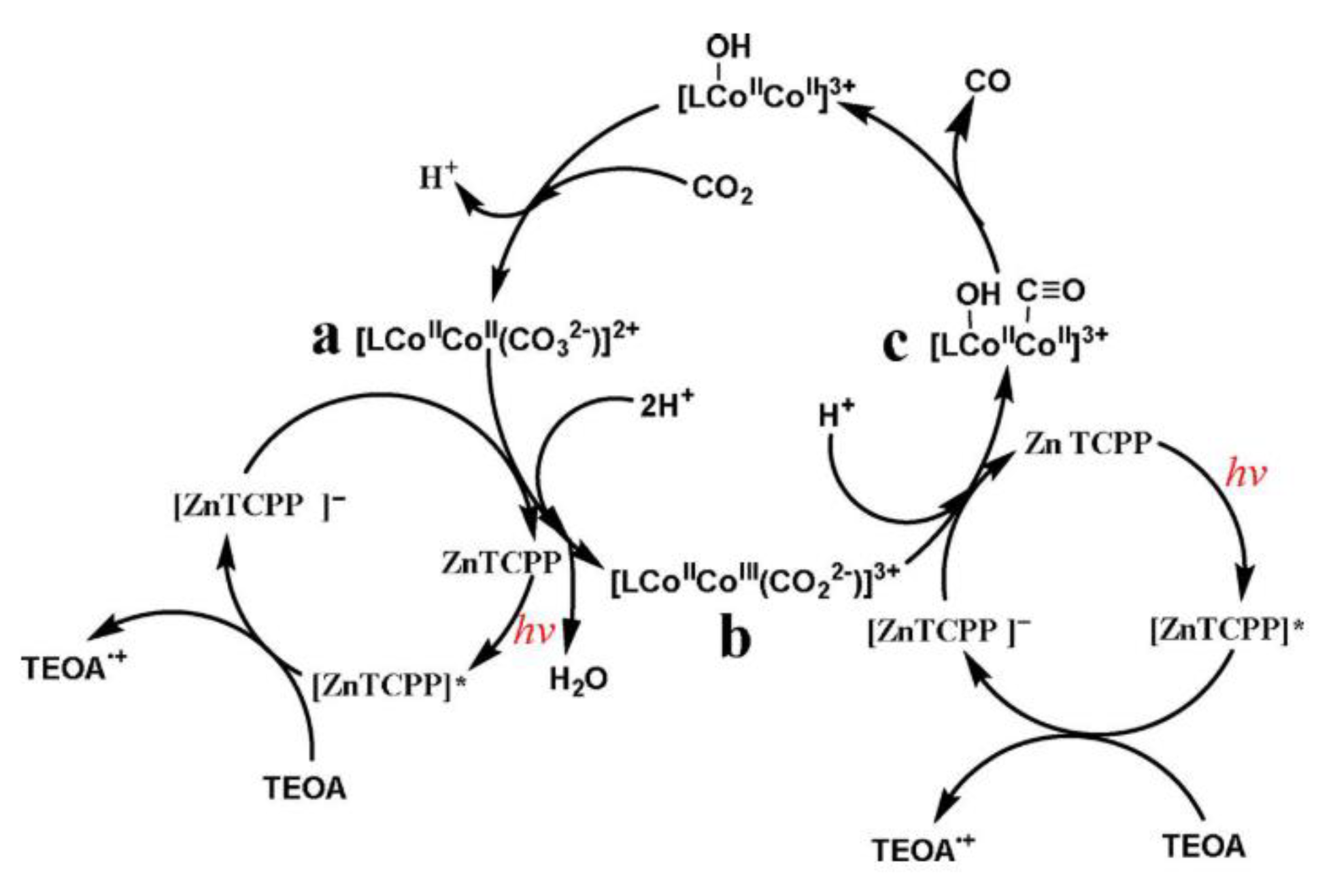
- Ouyang, T.; Huang, H.H.; Wang, J.W.; Zhong, D.C.; Lu, T.B. A Dinuclear Cobalt Cryptate as a Homogeneous Photocatalyst for Highly Selective and Efficient Visible-Light Driven CO2 Reduction to CO in CH3 CN/H2 O Solution. Angew. Chem. Int. Ed. 2017, 56, 738–743. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Half-Cell Reaction | E0’ in V |
|---|---|
| 2 H2O → O2 + 4 H+ + 4 e− | +0.82 |
| 2 H+ + 2 e− → H2 | −0.41 |
| CO2 + 2 H+ + 2 e− → CO + H2O | −0.52 |
| CO2 + 2 H+ + 2 e− → HCOOH | −0.61 |
| CO2 + 4 H+ + 4 e− → C + 2 H2O | −0.20 |
| CO2 + 4 H+ + 4 e− → HCHO + H2O | −0.48 |
| CO2 + 6 H+ + 6 e− → CH3OH + H2O | −0.38 |
| CO2 + 8 H+ + 8 e− → CH4 + 2 H2O | −0.24 |
| Catalyst Support | Photocatalytic System | Solvent | Light Source | Main Products | Ref. |
|---|---|---|---|---|---|
| Triblock amphiphilic micelles | Re (Cat, PS), TEOA (SA) | H2O | 500 W Xe (λ ≥ 400 nm) | CO, H2 | [42] |
| Poly(vinyl bipyridine) | Re (Cat), Ru (PS), TEOA+BIH (SA) | DMF | green LED (λ = 520 ± 30 nm) | CO | [44] |
| Polymers on quantum dots | CdS-Ni assembly (Cat, PS), TEOA (SA) | H2O | LED (λ = 420 nm) | CO, H2 | [48] |
| Ullazine supramolecular polymers | Co (Cat), Chromophores (PS), TEOA (SA) | MeCN/H2O | blue LED (λ = 450 nm) | CO, CH4 | [52] |
| Metal-organic coordination polymer gel | Ru (Cat), Porphyrin (PS), TEA/BNAH (SA) | MeCN/H2O | 300 W Xe lamp (λ > 400 nm) | CO, CH4 | [54] |
| Bipyridine COF | Re (Cat), Ir (PS), TEOA (SA) | MeCN | 300 W Xe (λ > 420 nm cut-off filter) | CO, H2 | [69] |
| PMO with bipyridyl ligands | Ru (Cat), Ru (PS), BNAH (SA) | DMA/H2O | 500 W Hg (λ > 430 nm) | CO, HCOO− | [71] |
| Porous CTF | Re (Cat, PS), TEOA | none | 300 W Xe (λ = 200–1100 nm) | CO | [76] |
| HCP-TiO2-Pd | TiO2 (Cat), Pd (co-Cat) | none | 300 W Xe, UV-vis light | CH4 | [83] |
| Metallophthalocyanine and salen POPs | Ni (Cat), Ru (PS), TEOA (SA) | MeCN/H2O | white LED (λ = 400–800 nm) | CO | [84] |
| Hybrid MOF-metal core–shell structure | Cu (Cat), TiO2 (PS) | none | 300 W Xe (λ < 400 nm) | CH4 | [98] |
| MOLs made from Ni3(HITP)2 | Ni (Cat), MOL (co-Cat), Ru (PS), TEOA (SA) | TEOA/H2O/MeCN | 100 W LED (λ = 420 nm) | CO | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hornberger, L.-S.; Adams, F. Photocatalytic CO2 Conversion Using Metal-Containing Coordination Polymers and Networks: Recent Developments in Material Design and Mechanistic Details. Polymers 2022, 14, 2778. https://doi.org/10.3390/polym14142778
Hornberger L-S, Adams F. Photocatalytic CO2 Conversion Using Metal-Containing Coordination Polymers and Networks: Recent Developments in Material Design and Mechanistic Details. Polymers. 2022; 14(14):2778. https://doi.org/10.3390/polym14142778
Chicago/Turabian StyleHornberger, Lea-Sophie, and Friederike Adams. 2022. "Photocatalytic CO2 Conversion Using Metal-Containing Coordination Polymers and Networks: Recent Developments in Material Design and Mechanistic Details" Polymers 14, no. 14: 2778. https://doi.org/10.3390/polym14142778