
Supramolecular Structure of Tris(1,10-phenanthroline)zinc(II)-Cation and N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide-Anion: Synthesis, Crystal Structure, Vibrational Spectra, and Theoretical Investigations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of the Compound
2.2. Single-Crystal X-ray Crystallography
2.3. Powder X-ray Diffraction (PXRD)
2.4. Infrared and Raman Spectroscopy
2.5. Computional Chemistry
2.6. Thermal and Elemental Analysis
3. Results
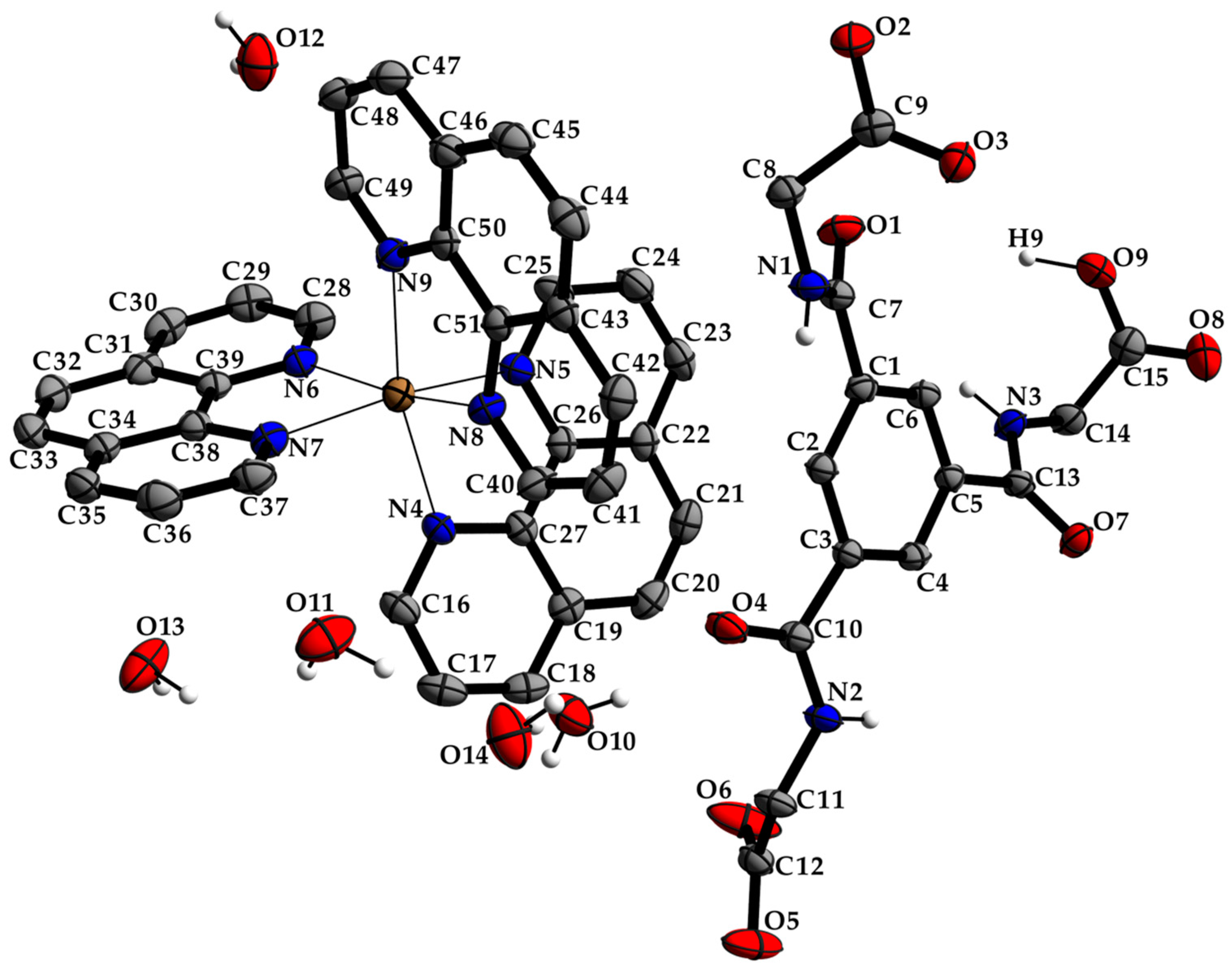
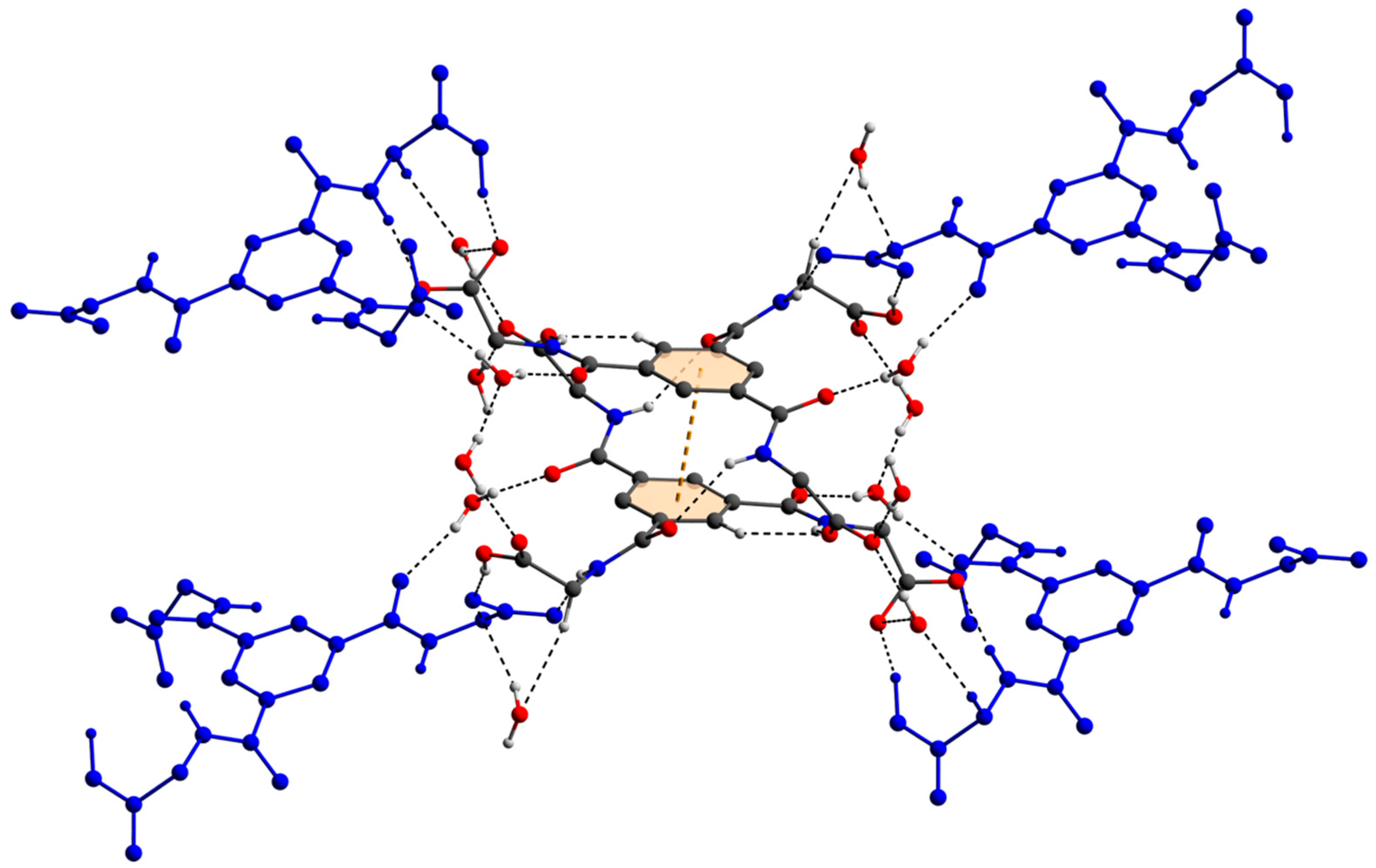
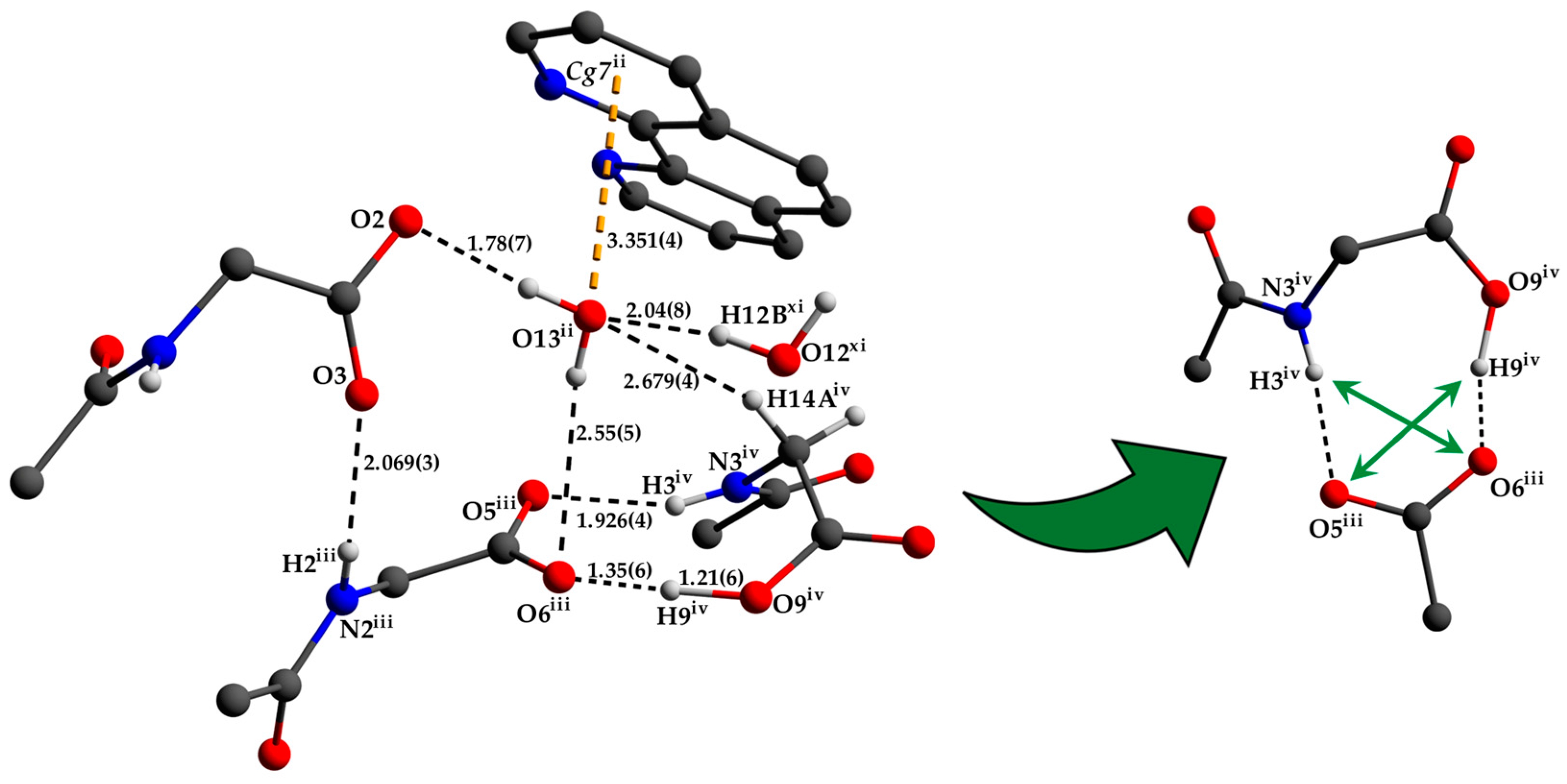
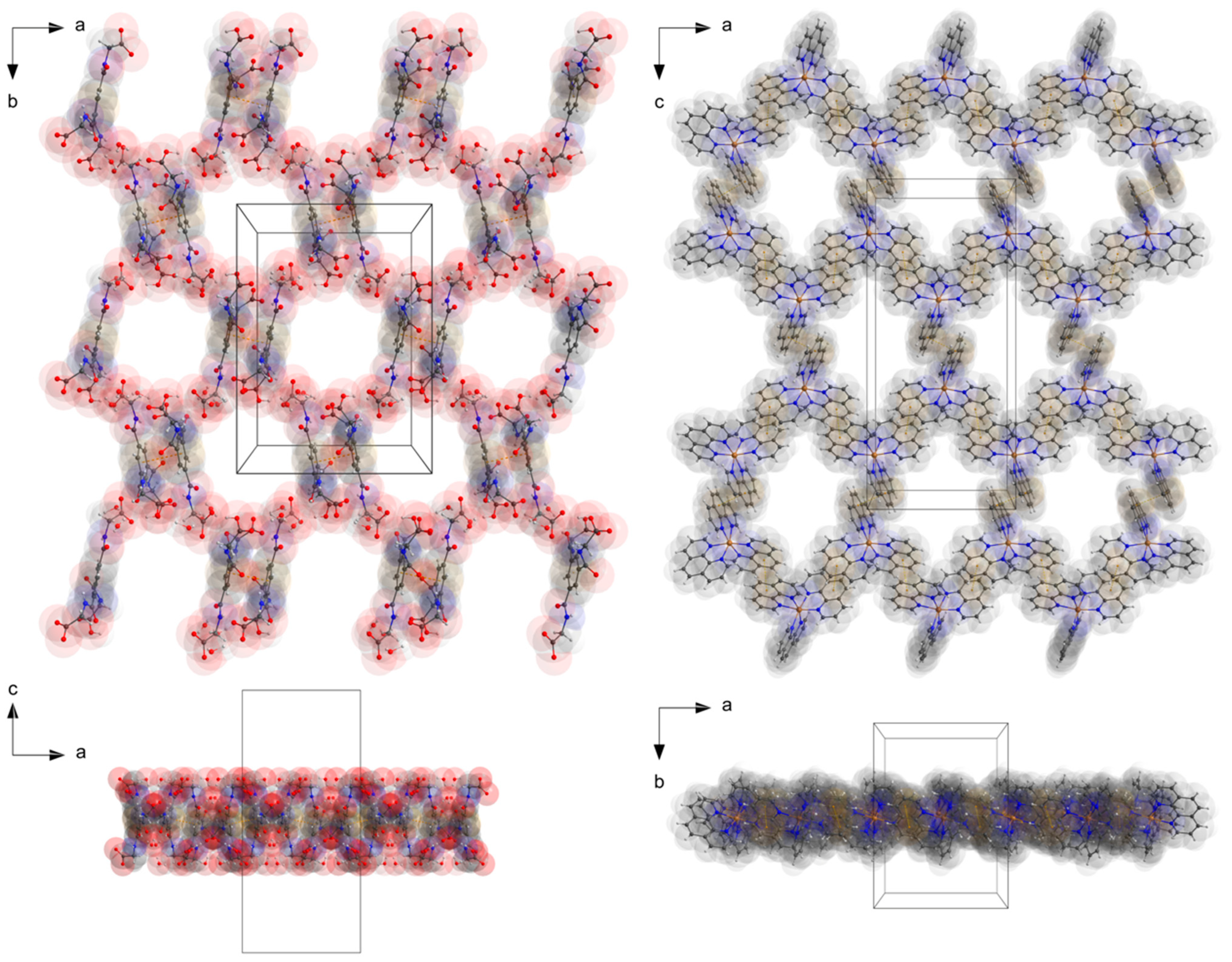
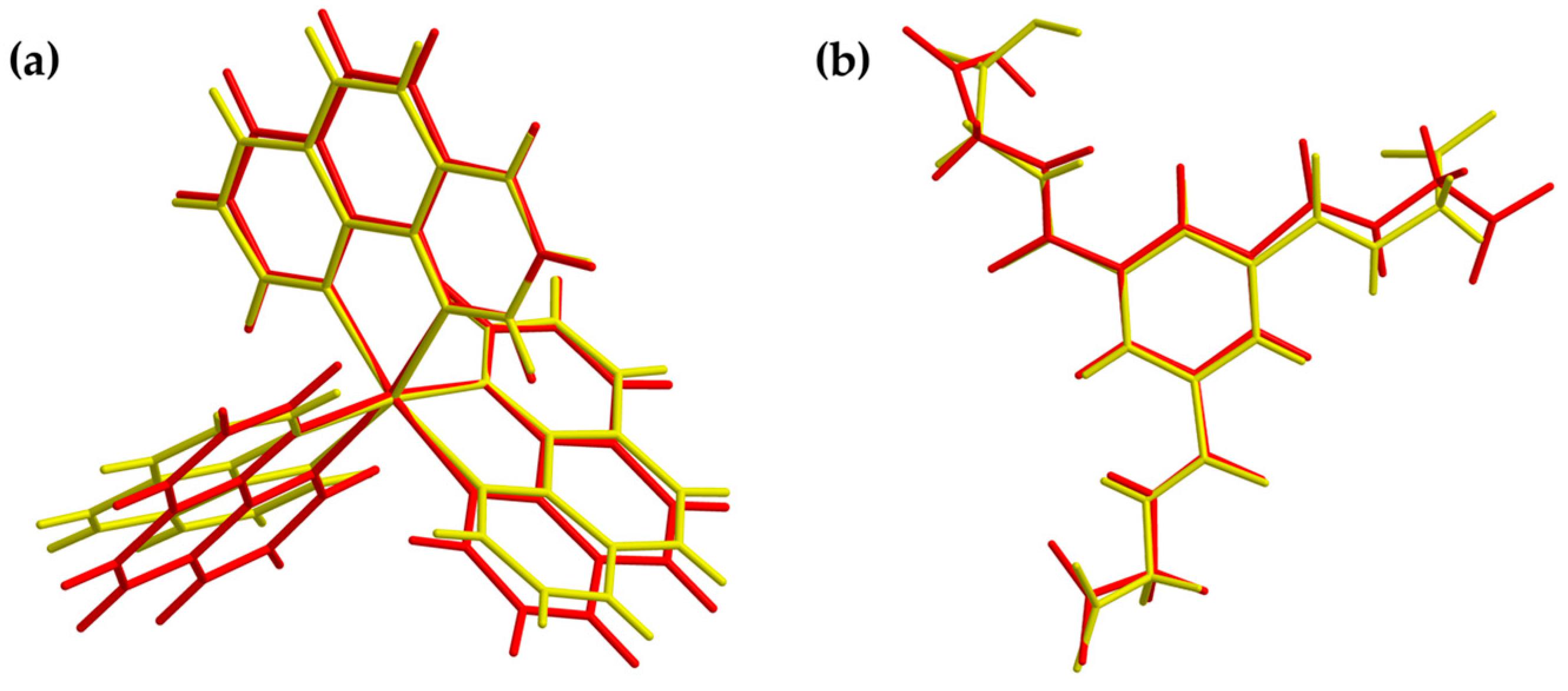
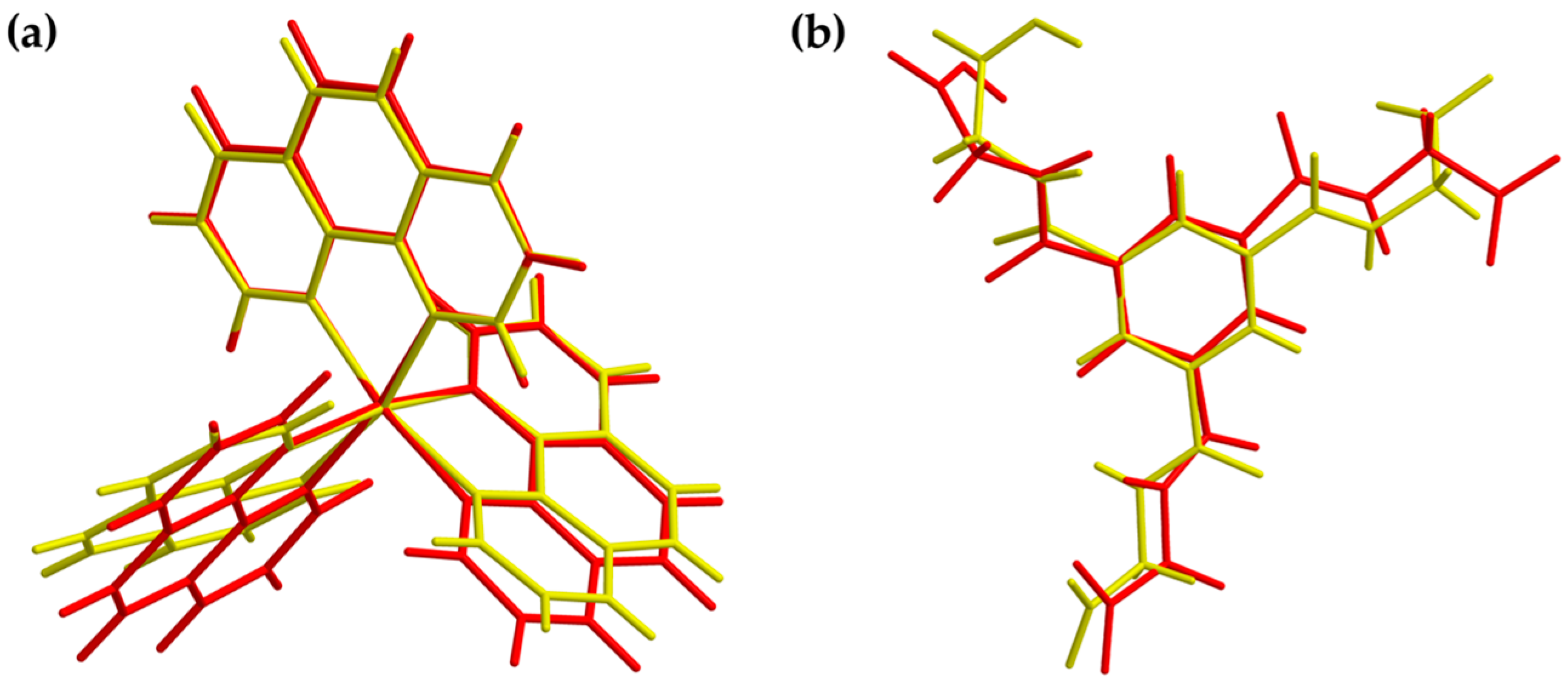
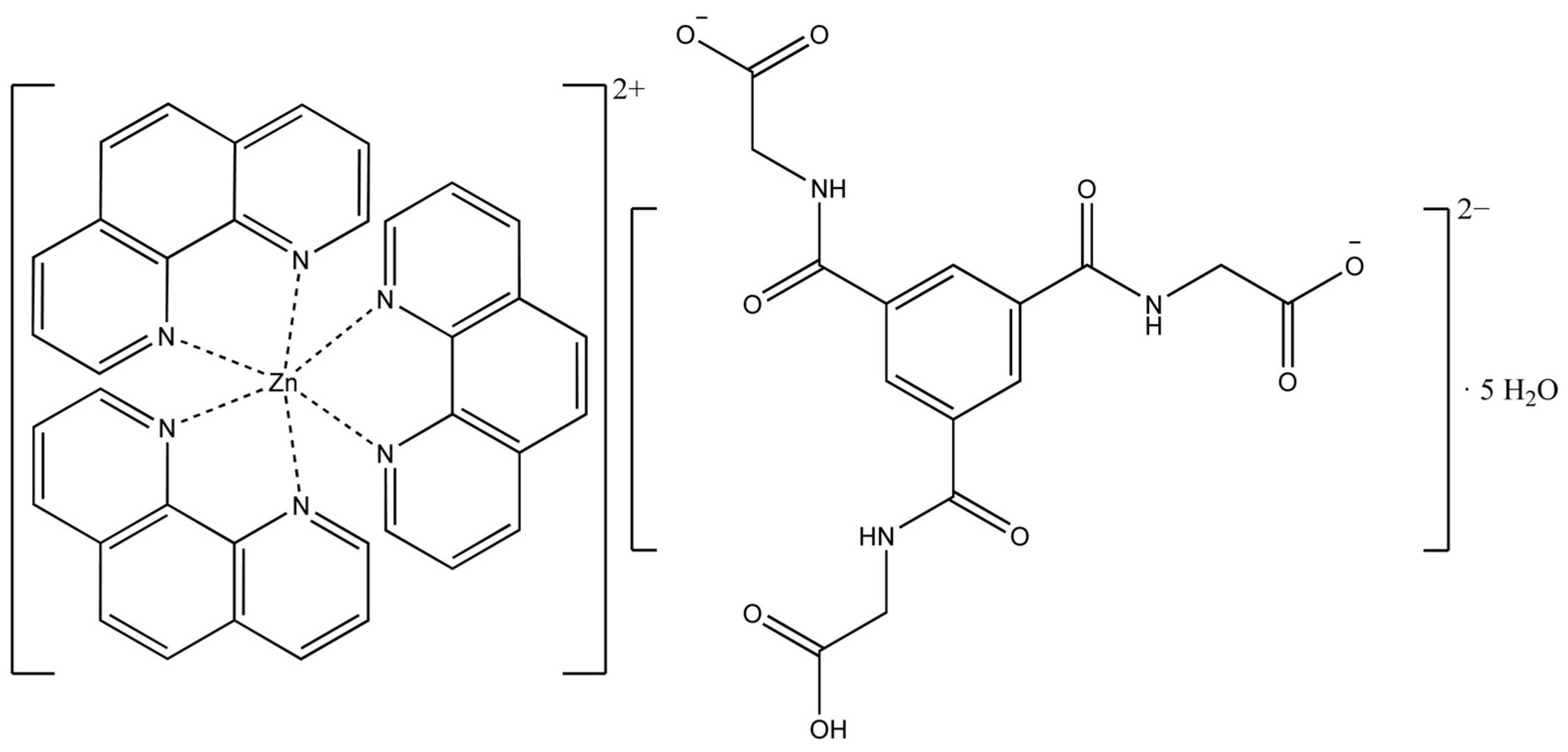
3.1. Description of the Crystal Structure
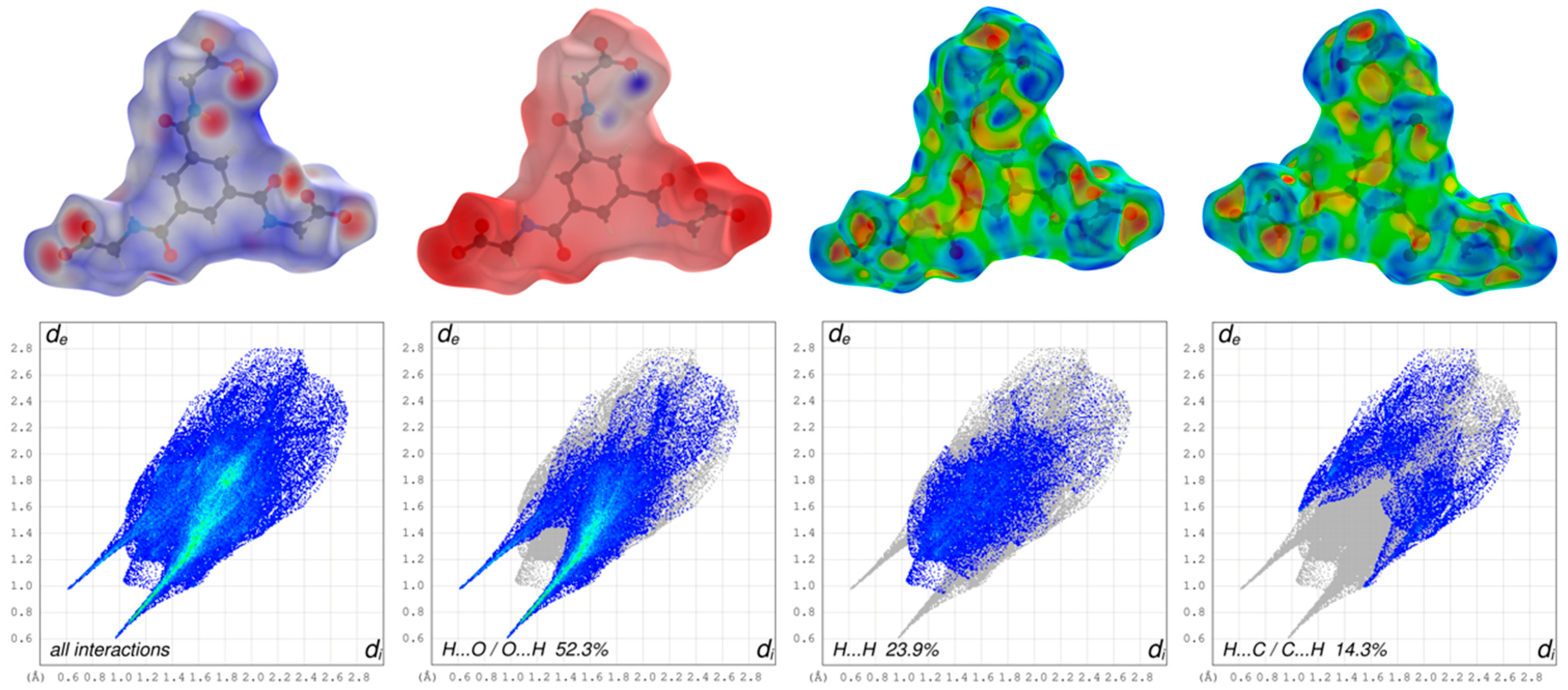
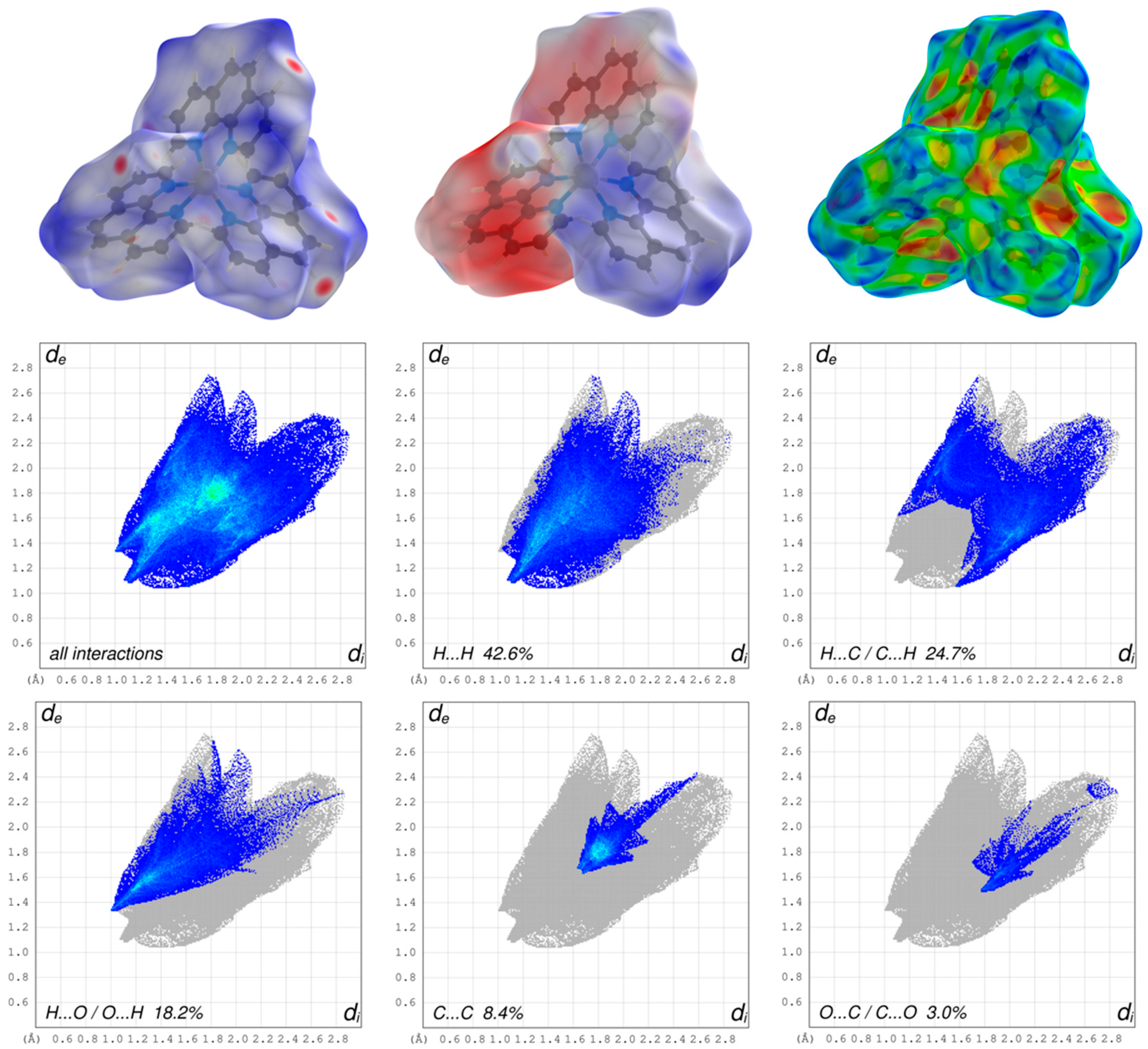
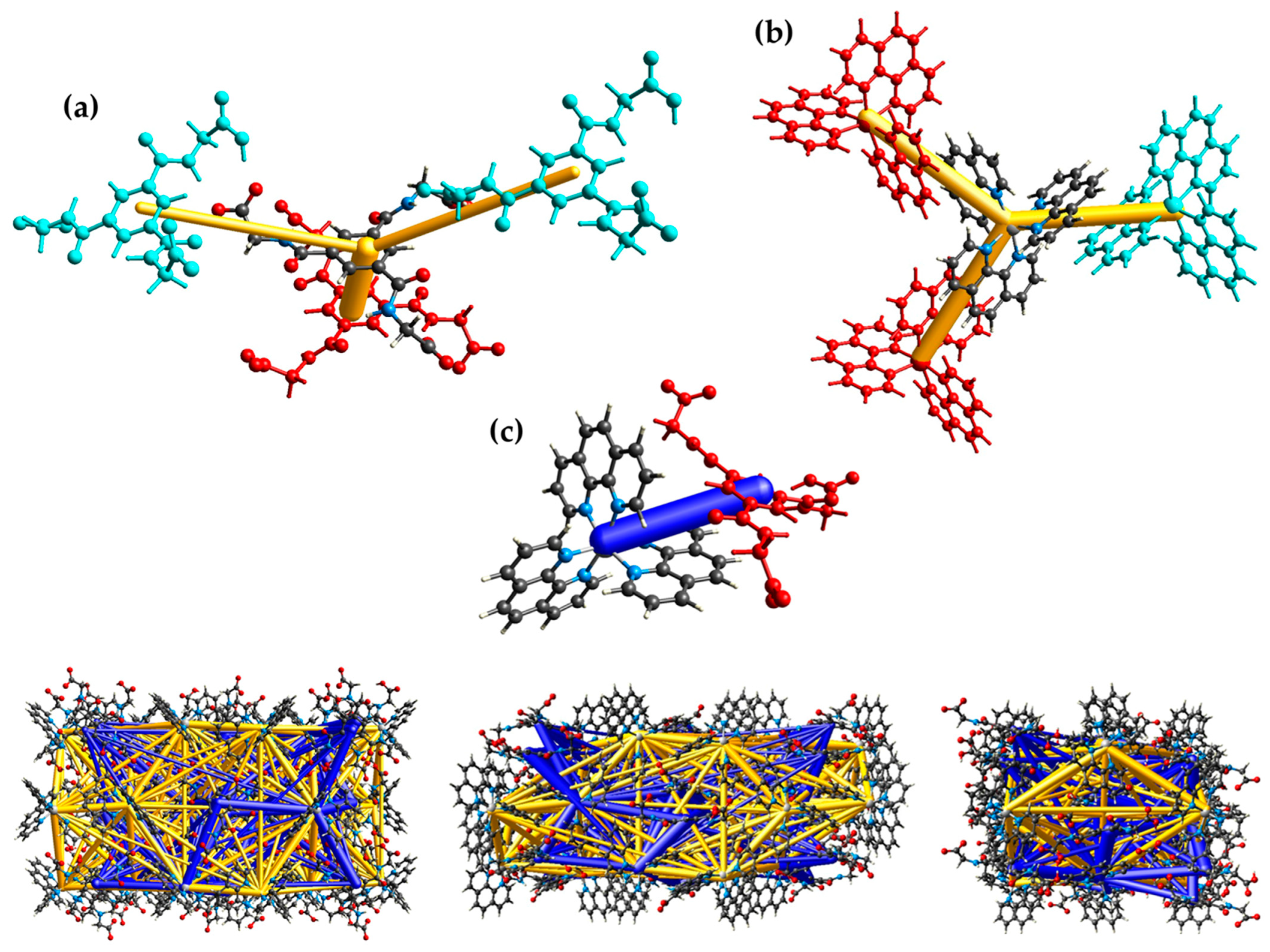
3.2. Hirshfeld Surface Analysis, Pairwise Interaction Energies and Energy Framework
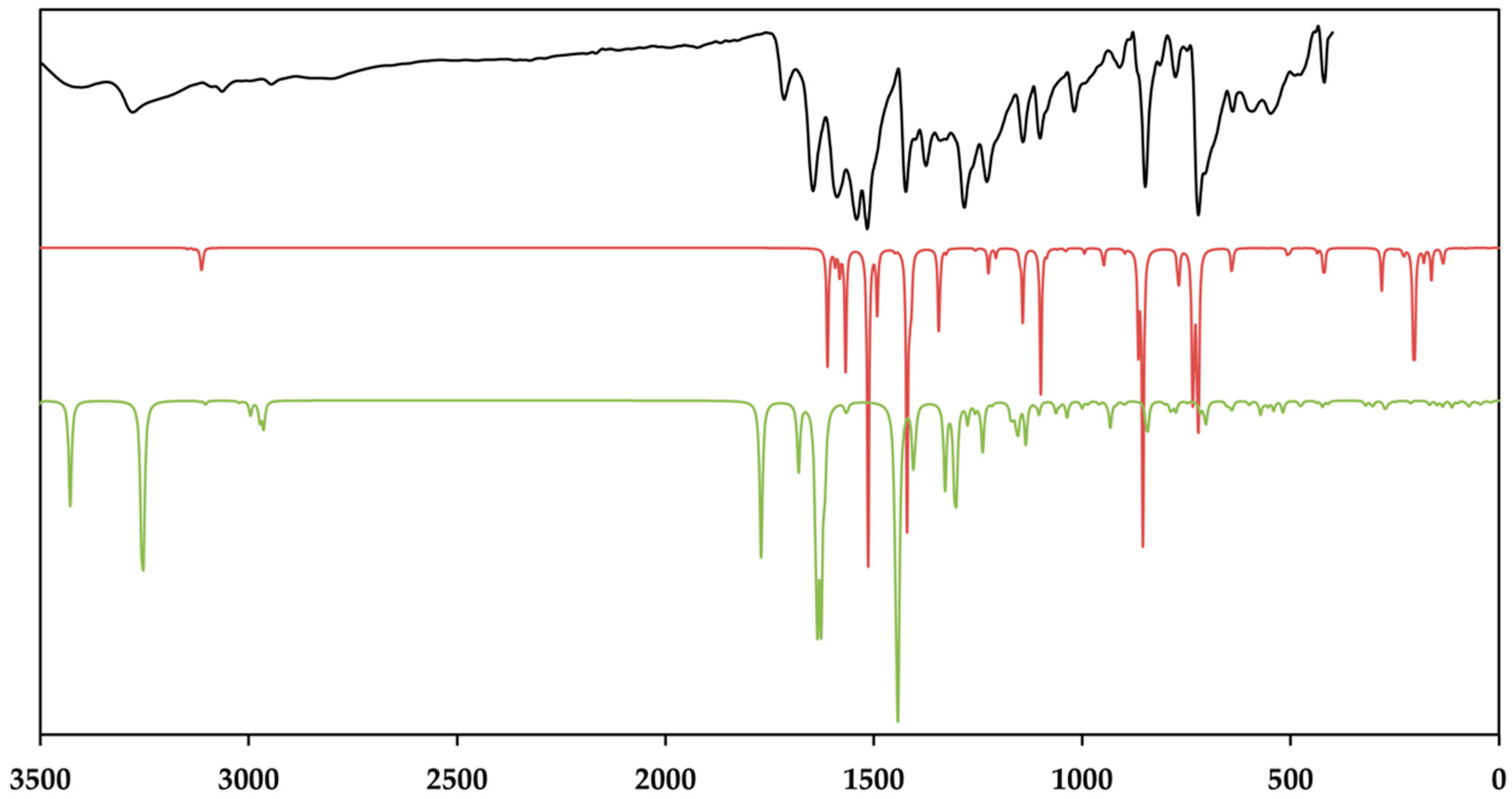
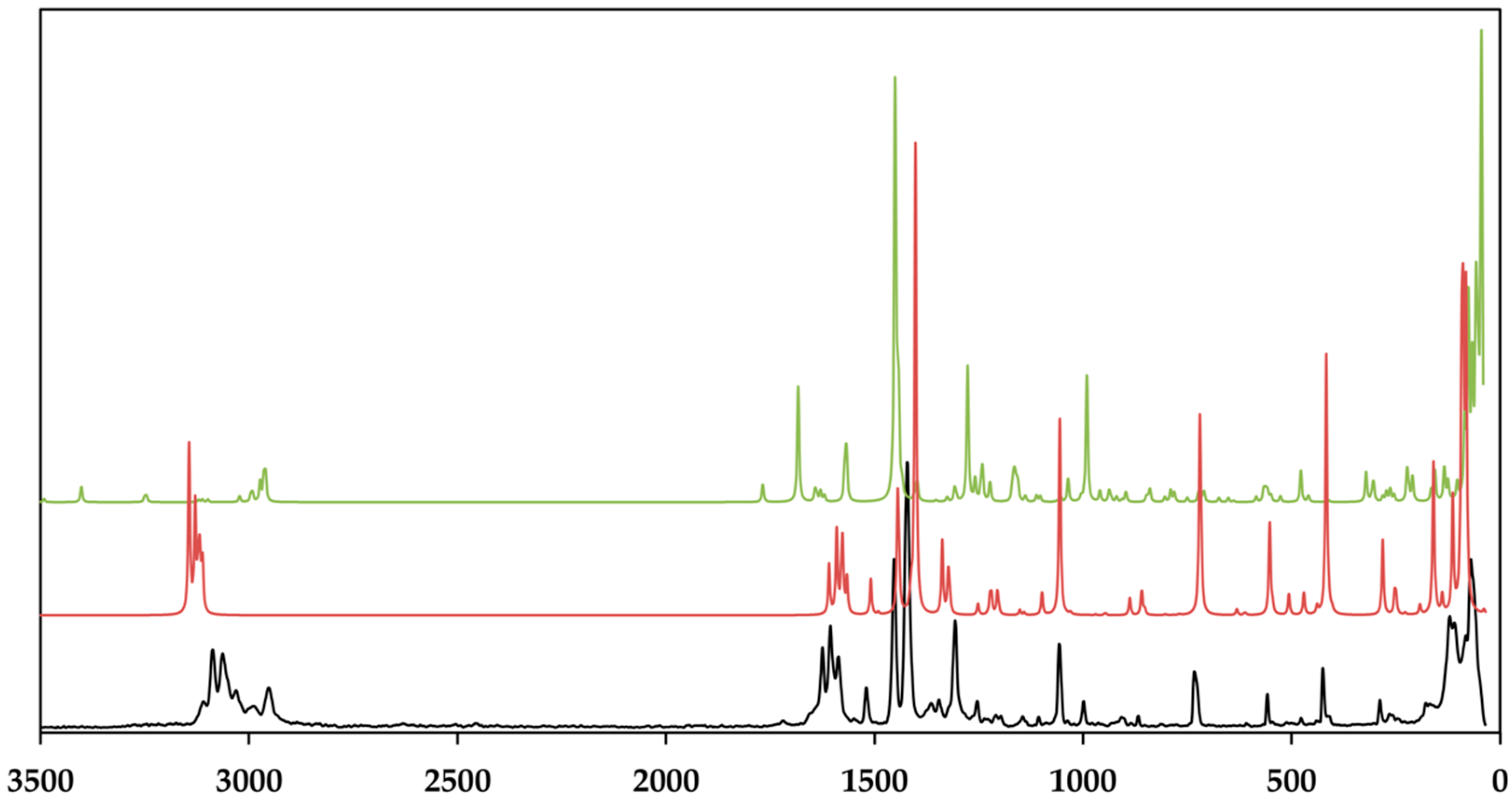
3.3. Vibrational and Theoretical Spektra
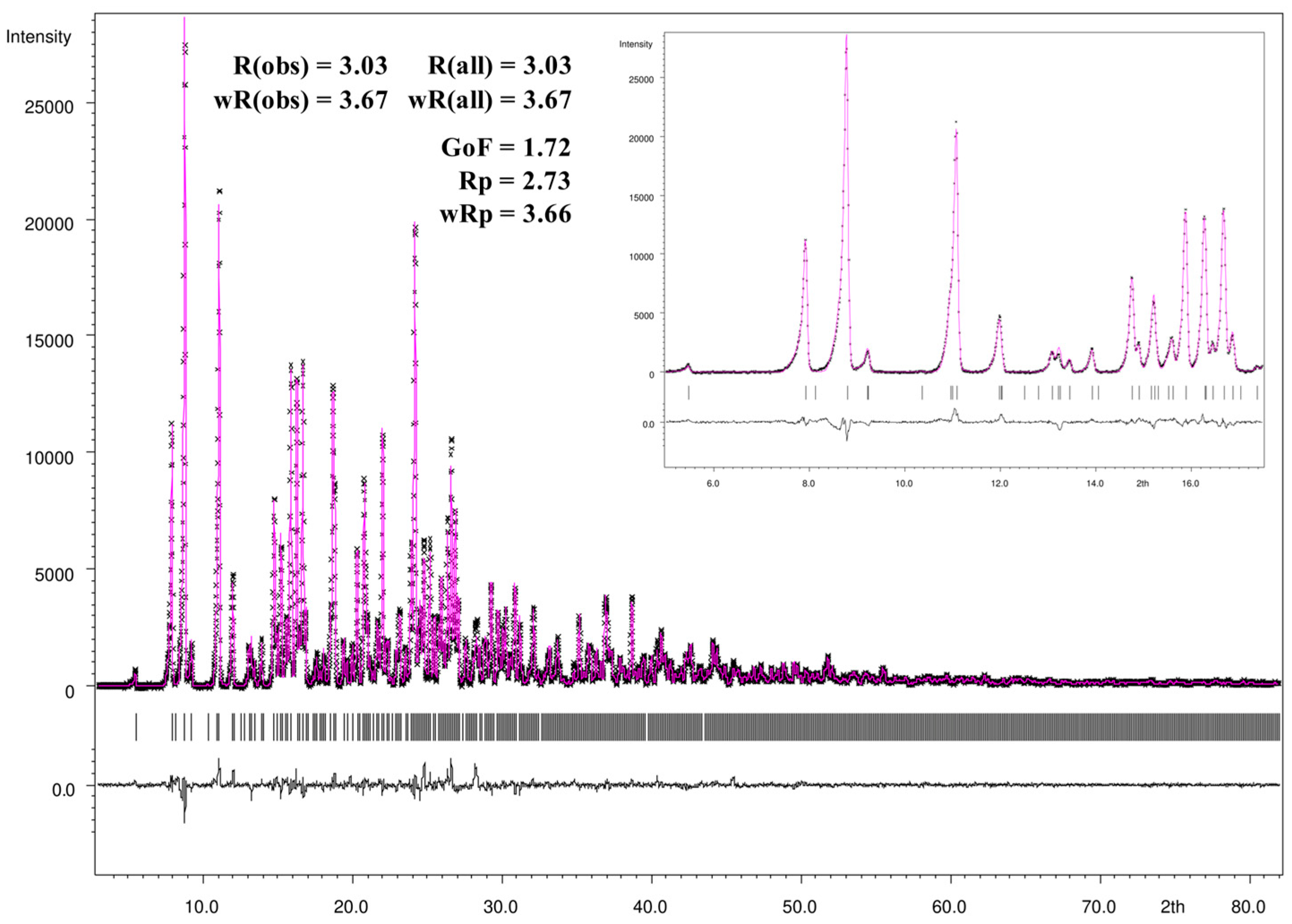
3.4. Rietveld Refinement
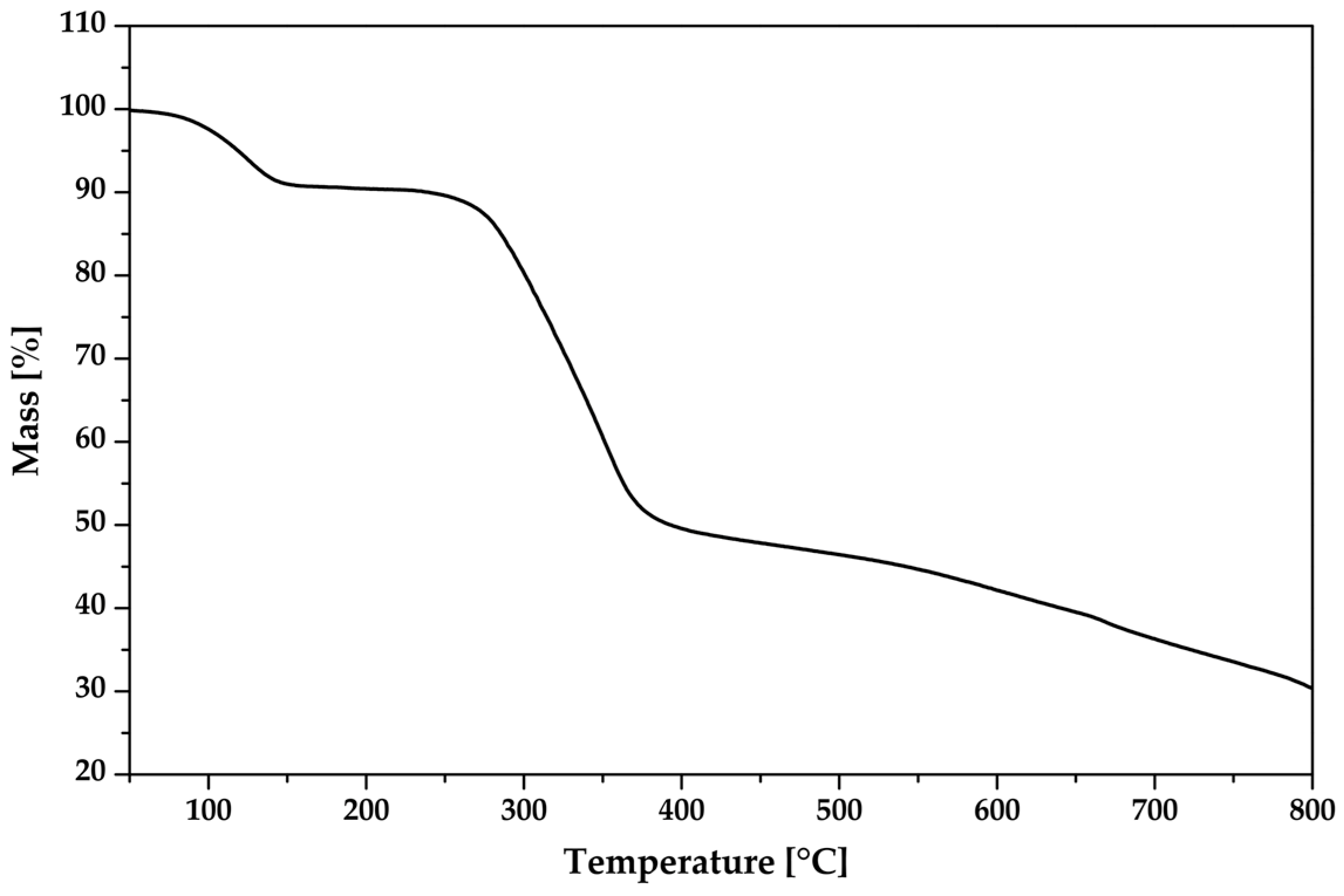
3.5. Thermal Analysis
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry, 2nd ed.; Wiley: Chichester, UK, 2009; ISBN 978-0-470-51234-0. [Google Scholar]
- Cerný, J.; Hobza, P. Non-covalent interactions in biomacromolecules. Phys. Chem. Chem. Phys. 2007, 9, 5291–5303. [Google Scholar] [CrossRef] [PubMed]
- Chawla, M.; Chermak, E.; Zhang, Q.; Bujnicki, J.M.; Oliva, R.; Cavallo, L. Occurrence and stability of lone pair-π stacking interactions between ribose and nucleobases in functional RNAs. Nucleic Acids Res. 2017, 45, 11019–11032. [Google Scholar] [CrossRef] [Green Version]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191. [Google Scholar] [CrossRef] [Green Version]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives; VCH: Weinheim, UK, 1995; ISBN 978-3-527-29311-7. [Google Scholar]
- Culver, J.N. Tobacco mosaic virus assembly and disassembly: Determinants in pathogenicity and resistance. Annu. Rev. Phytopathol. 2002, 40, 287–308. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, S.; Uchimaru, T. Secondary Interaction Contribution in Hydrogen-Bonded Complex: Theoretical Model Study in Hydrogen Fluoride Trimer. J. Comput. Chem. Jpn. 2004, 3, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, S.C.; Murray, T.J. Hydrogen bonded complexes with the AA·DD, AA·DDD, and AAA·DD motifs: The role of three centered (bifurcated) hydrogen bonding. Tetrahedron Lett. 1994, 35, 4077–4080. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Pranata, J. Importance of secondary interactions in triply hydrogen bonded complexes: Guanine-cytosine vs uracil-2,6-diaminopyridine. J. Am. Chem. Soc. 1990, 112, 2008–2010. [Google Scholar] [CrossRef]
- Hisamatsu, Y.; Shirai, N.; Ikeda, S.; Odashima, K. A new quadruple hydrogen-bonding module with a DDAA array: Formation of a stable homodimer without competition from undesired hydrogen-bonded dimers. Org. Lett. 2009, 11, 4342–4345. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Zaccaria, F.; Sun, X.; Guerra, C.F. Secondary Electrostatic Interaction Model Revised: Prediction Comes Mainly from Measuring Charge Accumulation in Hydrogen-Bonded Monomers. J. Am. Chem. Soc. 2019, 141, 4878–4885. [Google Scholar] [CrossRef] [Green Version]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions: Theory and Experiment; Royal Society of Chemistry: Cambridge, UK, 2010; ISBN 978-1847558534. [Google Scholar]
- Schneider, H.-J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. Engl. 2009, 48, 3924–3977. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed Engl. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Jain, A.; Ramanathan, V.; Sankararamakrishnan, R. Lone pair … pi interactions between water oxygens and aromatic residues: Quantum chemical studies based on high-resolution protein structures and model compounds. Protein Sci. 2009, 18, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novotný, J.; Bazzi, S.; Marek, R.; Kozelka, J. Lone-pair-π interactions: Analysis of the physical origin and biological implications. Phys. Chem. Chem. Phys. 2016, 18, 19472–19481. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.-Q.; Chen, X.-D.; Mak, T.C.W. Supramolecular frameworks assembled via intermolecular lone pair-aromatic interaction between carbonyl and pyridyl groups. CrystEngComm 2008, 10, 475. [Google Scholar] [CrossRef]
- Egli, M.; Sarkhel, S. Lone pair-aromatic interactions: To stabilize or not to stabilize. Acc. Chem. Res. 2007, 40, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Miao, H.; Hay, B.P. Crystal Structure Evidence for the Directionality of Lone Pair−π interactions: Fact or Fiction? Cryst. Growth Des. 2019, 19, 6806–6821. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]
- Brenig, C.; Prieto, L.; Oetterli, R.; Zelder, F. A Nickel(II)-Containing Vitamin B12 Derivative with a Cofactor-F430-type π-System. Angew. Chem. Int. Ed. Engl. 2018, 57, 16308–16312. [Google Scholar] [CrossRef] [Green Version]
- Eschenmoser, A. Vitamin B12: Experiments Concerning the Origin of Its Molecular Structure. Angew. Chem. Int. Ed. Engl. 1988, 27, 5–39. [Google Scholar] [CrossRef]
- Franck, B.; Nonn, A. Novel Porphyrinoids for Chemistry and Medicine by Biomimetic Syntheses. Angew. Chem. Int. Ed. Engl. 1995, 34, 1795–1811. [Google Scholar] [CrossRef]
- Doty, F.P.; Bauer, C.A.; Skulan, A.J.; Grant, P.G.; Allendorf, M.D. Scintillating Metal-Organic Frameworks: A New Class of Radiation Detection Materials. Adv. Mater. 2009, 21, 95–101. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Volotskova, O.; Lu, K.; Ahmad, M.; Sun, C.; Xing, L.; Lin, W. Synergistic assembly of heavy metal clusters and luminescent organic bridging ligands in metal-organic frameworks for highly efficient X-ray scintillation. J. Am. Chem. Soc. 2014, 136, 6171–6174. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wang, Y.; Liu, H.; Zhang, Y.; Liu, W.; Wang, X.; Wang, S. Color-tunable X-ray scintillation based on a series of isotypic lanthanide-organic frameworks. Chem. Commun. 2019, 56, 233–236. [Google Scholar] [CrossRef]
- Lecoq, P. Development of new scintillators for medical applications. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2016, 809, 130–139. [Google Scholar] [CrossRef]
- Perry IV, J.J.; Feng, P.L.; Meek, S.T.; Leong, K.; Doty, F.P.; Allendorf, M.D. Connecting structure with function in metal–organic frameworks to design novel photo- and radioluminescent materials. J. Mater. Chem. 2012, 22, 10235. [Google Scholar] [CrossRef]
- Pook, N.-P. A Scintillating One-Dimensional Coordination Polymer Based on Cadmium(II), N,N′-(1,4-Phenylenedicarbonyl)diglycinate, and 2,2′-Bipyridine: Crystal Structure, Hirshfeld Surface Analysis, and Luminescence Lifetime Properties †. Solids 2021, 2, 371–384. [Google Scholar] [CrossRef]
- Pook, N.-P.; Fruhner, C.-J.; Franzl, T.; Denzer, U.; Adam, A. Instrumentation for X-ray Excited and Laser Induced Fluorescence Lifetime Spectroscopy Using Two-Dimensional Photon Counting. IEEE Trans. Nucl. Sci. 2012, 59, 2319–2323. [Google Scholar] [CrossRef]
- Pook, N.-P.; Fruhner, C.-J.; Franzl, T.; Denzer, U.; Adam, A. Further performance tests of a picosecond X-ray and laser induced streak camera system with fast scintillation materials. Radiat. Meas. 2013, 56, 281–284. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands †. J. Chem. Soc. Dalton Trans. 2000, 21, 3885–3896. [Google Scholar] [CrossRef]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. Engl. 2011, 50, 4808–4842. [Google Scholar] [CrossRef]
- Riwar, L.-J.; Trapp, N.; Kuhn, B.; Diederich, F. Substituent Effects in Parallel-Displaced π-π Stacking Interactions: Distance Matters. Angew. Chem. Int. Ed. Engl. 2017, 56, 11252–11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Gamez, P.; Mutikainen, I.; Turpeinen, U.; Reedijk, J. Supramolecular Assemblies Generated from Both Lone-Pair···π and C−H···π Binding Interactions. Cryst. Growth Des. 2007, 7, 1669–1671. [Google Scholar] [CrossRef]
- Li, W.; Usabiaga, I.; Calabrese, C.; Evangelisti, L.; Maris, A.; Favero, L.B.; Melandri, S. Characterizing the lone pairπ-hole interaction in complexes of ammonia with perfluorinated arenes. Phys. Chem. Chem. Phys. 2021, 23, 9121–9129. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.P.; Drew, M.G.B.; Das, A.K.; Banerjee, A. Formation of triple helical nanofibers using self-assembling chiral benzene-1,3,5-tricarboxamides and reversal of the nanostructure’s handedness using mirror image building blocks. Chem. Commun. 2006, 30, 3196–3198. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Zheng, C.; Yan, Y. Structure of N,N′,N′′-tris(carboxymethyl)-1,3,5benzenetricarboxamide trihydrate. J. Chem. Crystallogr. 1999, 29, 649–652. [Google Scholar] [CrossRef]
- Dokorou, V.N.; Powell, A.K.; Kostakis, G.E. Two pseudopolymorphs derived from alkaline earth metals and the pseudopeptidic ligand trimesoyl-tris-glycine. Polyhedron 2013, 52, 538–544. [Google Scholar] [CrossRef]
- Raynal, M.; Li, Y.; Troufflard, C.; Przybylski, C.; Gontard, G.; Maistriaux, T.; Idé, J.; Lazzaroni, R.; Bouteiller, L.; Brocorens, P. Experimental and computational diagnosis of the fluxional nature of a benzene-1,3,5-tricarboxamide-based hydrogen-bonded dimer. Phys. Chem. Chem. Phys. 2021, 23, 5207–5221. [Google Scholar] [CrossRef]
- Sun, R.; Li, Y.-Z.; Bai, J.; Pan, Y. Synthesis, Structure, Water-Induced Reversible Crystal-to-Amorphous Transformation, and Luminescence Properties of Novel Cationic Spacer-Filled 3D Transition Metal Supramolecular Frameworks from N, N′, N′′-Tris(carboxymethyl)-1,3,5-benzenetricarboxamide. Cryst. Growth Des. 2007, 7, 890–894. [Google Scholar] [CrossRef]
- Sun, R.; Wang, S.; Xing, H.; Bai, J.; Li, Y.; Pan, Y.; You, X. Unprecedented 4(2)6(4) topological 2-D rare-earth coordination polymers from a flexible tripodal acid with additional amide groups. Inorg. Chem. 2007, 46, 8451–8453. [Google Scholar] [CrossRef]
- Loukopoulos, E.; Michail, A.; Kostakis, G. A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand. Crystals 2018, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Checkcif validation ALERTS: What they mean and how to respond. Acta Crystallogr. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. What makes a crystal structure report valid? Inorg. Chim. Acta 2018, 470, 232–237. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. Diamond—Crystal and Molecular Structure Visualization. Available online: http://www.crystalimpact.com/diamond (accessed on 6 March 2023).
- Persistence of Vision Raytracer (Version 3.6)[Computer Software]. Persistence of Vision Pty. Ltd. 2004. Available online: http://www.povray.org/download/ (accessed on 6 March 2023).
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Für Krist. —Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Chem. Eur. J. 1998, 4, 2136–2141. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J.; Jayatilaka, D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377–388. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Parkin, A.; Barr, G.; Dong, W.; Gilmore, C.J.; Jayatilaka, D.; McKinnon, J.J.; Spackman, M.A.; Wilson, C.C. Comparing entire crystal structures: Structural genetic fingerprinting. CrystEngComm 2007, 9, 648. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. Psi4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Wei, D.-Y.; Zheng, Y.-Q.; Lin, J.-L. Self—Assembly of Zinc Ions with Suberic Acid and Phenanthroline—Crystal Structures of five New Zinc Phenanthroline Suberato Complexes. Z. Anorg. Allg. Chem. 2002, 628, 2005–2012. [Google Scholar] [CrossRef]
- Pook, N.-P.; Adam, A.; Gjikaj, M. Crystal structure and Hirshfeld surface analysis of (μ-2-{4-(carboxyl-atometh-yl)carbamo-ylbenz-amido}-acetato-κ2O:O′)bis-bis-(1,10-phenanthroline-κ2N,N′)copper(II) dinitrate N,N′-(1,4-phenyl-enedicarbon-yl)diglycine monosolvate octa-hydrate. Acta Crystallogr. E Crystallogr. Commun. 2019, 75, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, J. Very strong hydrogen bonding. Chem. Soc. Rev. 1980, 9, 91. [Google Scholar] [CrossRef]
- Feng, G.; Favero, L.B.; Maris, A.; Vigorito, A.; Caminati, W.; Meyer, R. Proton transfer in homodimers of carboxylic acids: The rotational spectrum of the dimer of acrylic acid. J. Am. Chem. Soc. 2012, 134, 19281–19286. [Google Scholar] [CrossRef]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Limbach, H.-H. Hydrogen bond geometries and proton tautomerism of homoconjugated anions of carboxylic acids studied via H/D isotope effects on 13C NMR chemical shifts. J. Phys. Chem. A 2012, 116, 11180–11188. [Google Scholar] [CrossRef]
- Lin, J.; Pozharski, E.; Wilson, M.A. Short Carboxylic Acid-Carboxylate Hydrogen Bonds Can Have Fully Localized Protons. Biochemistry 2017, 56, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Silvi, B.; Ratajczak, H. Hydrogen bonding and delocalization in the ELF analysis approach. Phys. Chem. Chem. Phys. 2016, 18, 27442–27449. [Google Scholar] [CrossRef] [Green Version]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. Engl. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Fonseca Guerra, C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem. Asian J. 2019, 14, 2760–2769. [Google Scholar] [CrossRef] [Green Version]
- Sarkhel, S.; Desiraju, G.R. N-H…O, O-H…O, and C-H…O hydrogen bonds in protein-ligand complexes: Strong and weak interactions in molecular recognition. Proteins 2004, 54, 247–259. [Google Scholar] [CrossRef]
- Lüning, U.; Kühl, C.; Uphoff, A. Four Hydrogen Bonds−DDAA, DADA, DAAD and ADDA Hydrogen Bond Motifs. Eur. J. Org. Chem. 2002, 2002, 4063–4070. [Google Scholar] [CrossRef]
- Kumar Seth, S.; Dey, B.; Kar, T.; Mukhopadhyay, S. Experimental observation of supramolecular carbonyl–π/π–π/π–carbonyl assemblies of CuII complex of iminodiacetate and dipyridylamine. J. Mol. Struct. 2010, 973, 81–88. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theoret. Chim. Acta 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Hesse, M.; Maier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie: 96 Tabellen, 4th ed.; Thieme: Stuttgart, Germany, 1991; ISBN 3-13-576104-5. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th ed.; Wiley-Blackwell: Oxford, UK, 2008; ISBN 978-0-471-74493-1. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; Wiley: Chichester, UK, 2001; ISBN 0-471-85298-8. [Google Scholar]
- Larkin, P.J. Infrared and Raman Spectroscopy: Principles and Spectral Interpretation; Elsevier: Amsterdam, The Netherlands, 2011; ISBN 978-0-12-386984-5. [Google Scholar]
- Krishnan, K.; Plane, R.A. Raman and infrared spectra of o-phenanthroline and its complexes with Zn(II) and Hg(II). Spectrochim. Acta Part A Mol. Spectrosc. 1969, 25, 831–837. [Google Scholar] [CrossRef]
- Thornton, D.A.; Watkins, G.M. The Infrared Spectra (4000–50 cm−1) of Complexes of 2,2′-Bipyridine, 1,10-Phenanthroline and their Perdeuterated Analogues with Metal(II) Perchlorates of the First Transition Series. J. Coord. Chem. 1992, 25, 299–315. [Google Scholar] [CrossRef]
- Altmann, V.W.; Perkampus, H.-H. Normalkoordinatenanalyse der phenanthroline teil. I. Die planaren grundschwingungen. Spectrochim. Acta Part A Mol. Spectrosc. 1979, 35, 253–257. [Google Scholar] [CrossRef]
- Perkampus, H.-H.; Rother, W. Die Infrarot- und Ramanspektren der Phenanthroline. Spectrochim. Acta Part A Mol. Spectrosc. 1974, 30, 597–610. [Google Scholar] [CrossRef]
- Reiher, M.; Brehm, G.; Schneider, S. Assignment of Vibrational Spectra of 1,10-Phenanthroline by Comparison with Frequencies and Raman Intensities from Density Functional Calculations. J. Phys. Chem. A 2004, 108, 734–742. [Google Scholar] [CrossRef]
- Thornton, D.A.; Watkins, G.M. A full vibrational assignment (4000-50 cm−1) of 1, 10-phenanthroline and its perdeuterated analogue. Spectrochim. Acta Part A Mol. Spectrosc. 1991, 47, 1085–1096. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | [Zn(C12H8N2)3]C15H13N3O9∙5H2O |
|---|---|
| Empirical formula | C51H47N9O14Zn |
| Formula weight | 1075.34 |
| Temperature (K) | 223 |
| Diffractometer | Stoe IPDS II |
| Wavelength (Å) | 0.71073 |
| Crystal system | orthorhombic |
| Color | colorless |
| Space group | Pbca (No. 61) |
| a, b, c (Å) | 14.643(1), 20.095(1), 32.345(2) |
| α, β, γ (°) | 90 |
| V (Å3) | 9517.0(10) |
| Z | 8 |
| Dcalc (g·cm3) | 1.501 |
| µ (mm−1) | 0.597 |
| F(000) | 4464.0 |
| Crystal size (mm) | 0.250, 0.290, 0.170 |
| θ Range (°) | 1.832–25.100 |
| Index ranges | –17 ≤ h ≤ 16 –23 ≤ k ≤ 18 –38 ≤ l ≤ 35 |
| Reflection collected/unique | 24712/8439 |
| Completeness to θ (%) | 0.995 |
| Absorption correction | Numerical; X-AREA, X-RED (2008) |
| Max. and min. transmission | 0.8040/0.9159 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/parameters/restrains | 8439/709/4 |
| Goodness-of-Fit on F2 | 0.908 |
| R1 [I ≥ 2σ(I)]/R1 (all data) | 0.0536/0.1162 |
| wR2 [I ≥ 2σ(I)]/wR2 (all data) | 0.0948/0.1277 |
| Largest diff. peak and hole (e·Å3) | 0.280/−0.579 |
| Deposition number | 2,234,251 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pook, N.-P. Supramolecular Structure of Tris(1,10-phenanthroline)zinc(II)-Cation and N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide-Anion: Synthesis, Crystal Structure, Vibrational Spectra, and Theoretical Investigations. Crystals 2023, 13, 569. https://doi.org/10.3390/cryst13040569
Pook N-P. Supramolecular Structure of Tris(1,10-phenanthroline)zinc(II)-Cation and N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide-Anion: Synthesis, Crystal Structure, Vibrational Spectra, and Theoretical Investigations. Crystals. 2023; 13(4):569. https://doi.org/10.3390/cryst13040569
Chicago/Turabian StylePook, Niels-Patrick. 2023. "Supramolecular Structure of Tris(1,10-phenanthroline)zinc(II)-Cation and N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide-Anion: Synthesis, Crystal Structure, Vibrational Spectra, and Theoretical Investigations" Crystals 13, no. 4: 569. https://doi.org/10.3390/cryst13040569