
Crystal Engineering of Cation-Radical Salts with Weakly Coordinating Carbadodecaborate Anions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization and X-ray Structural Analysis
2.3. Computations
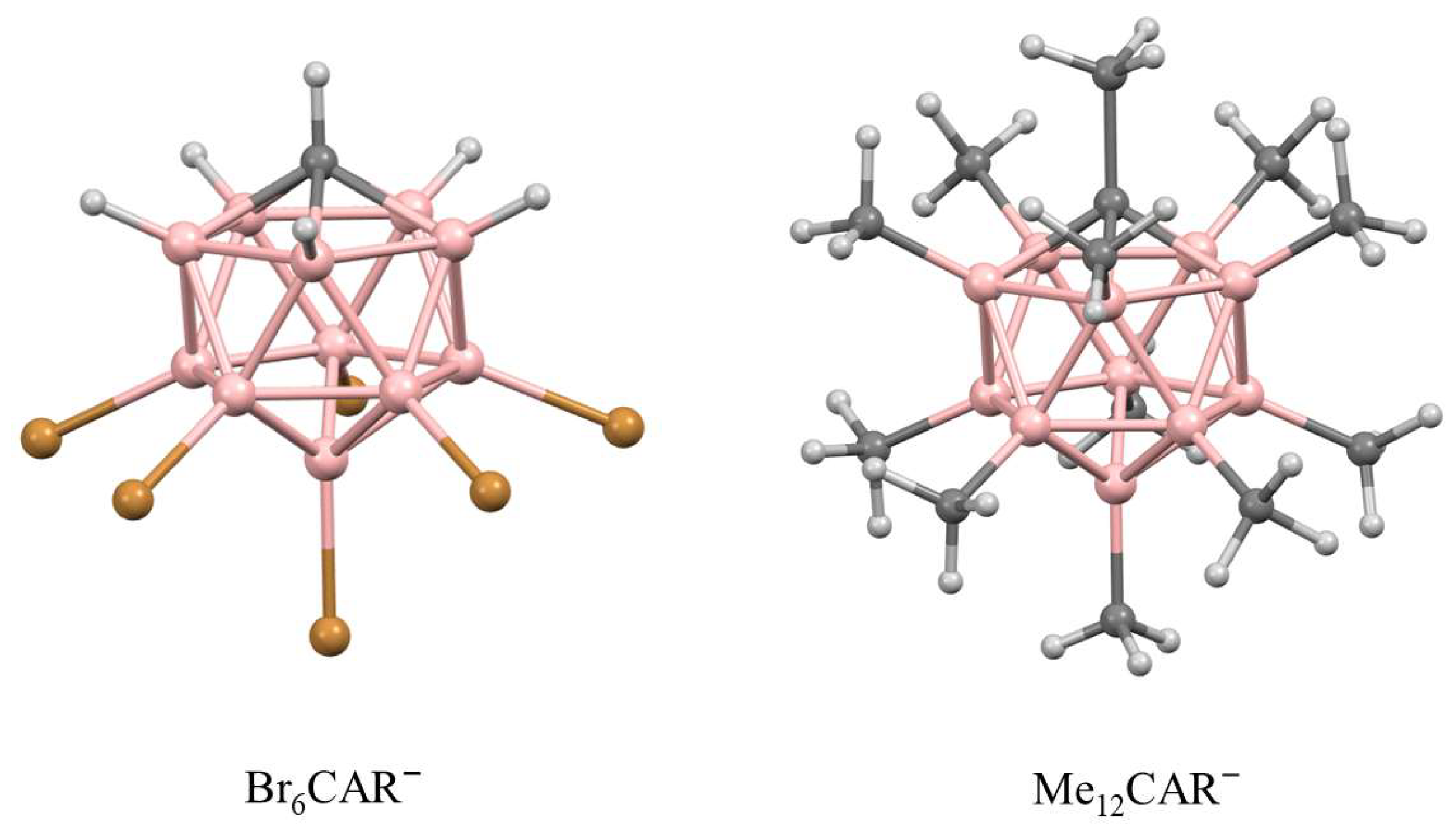
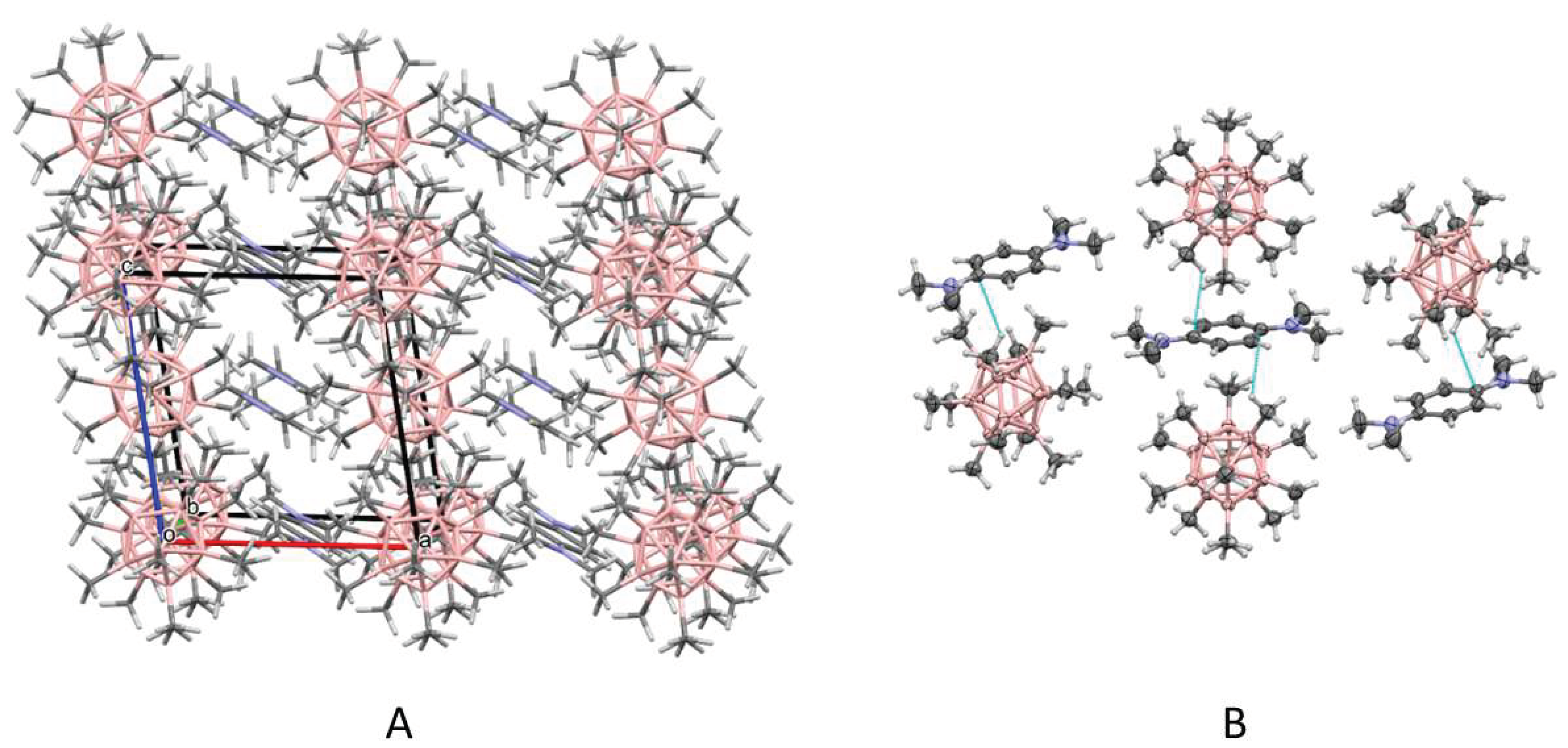
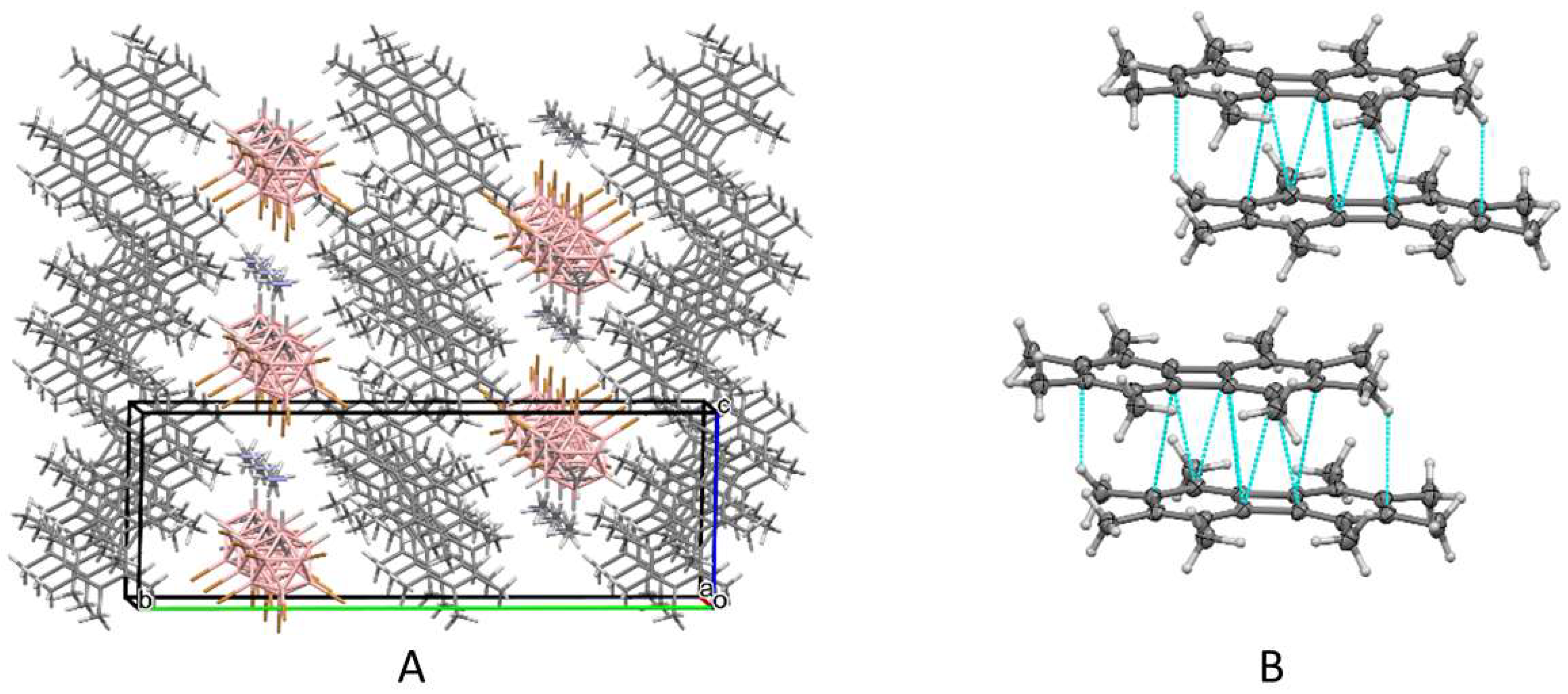
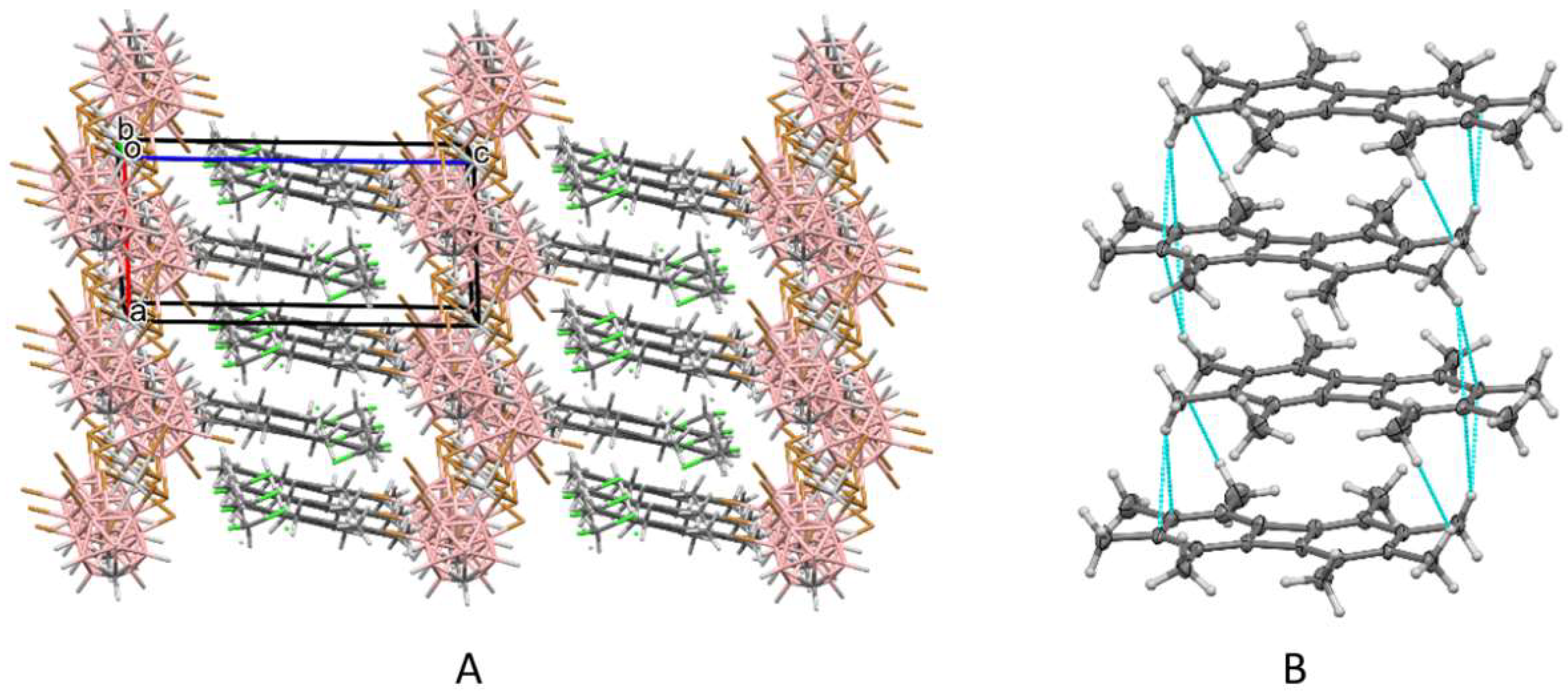
3. Results and Discussion
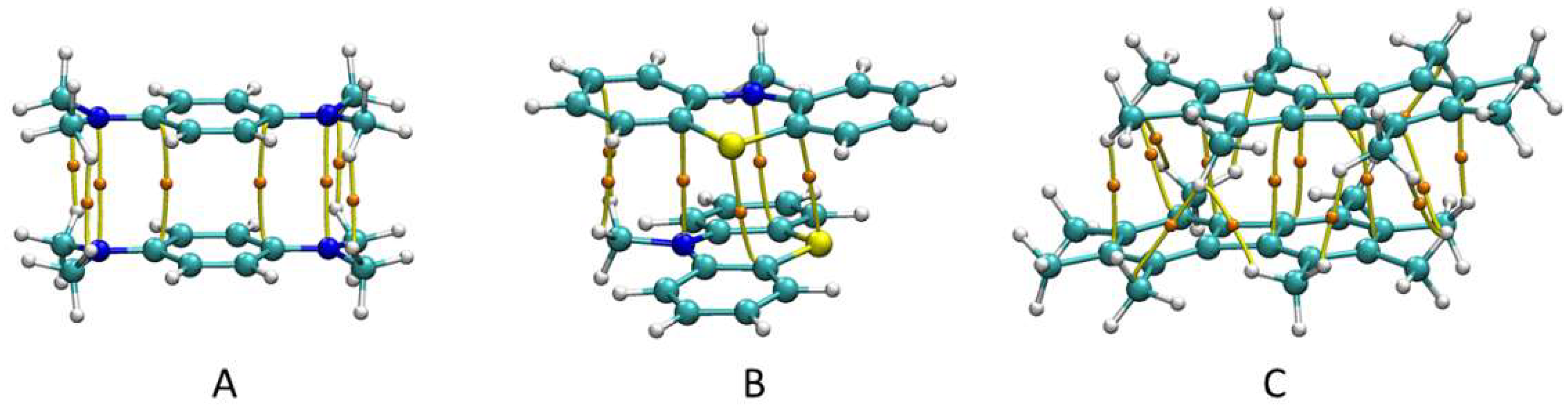
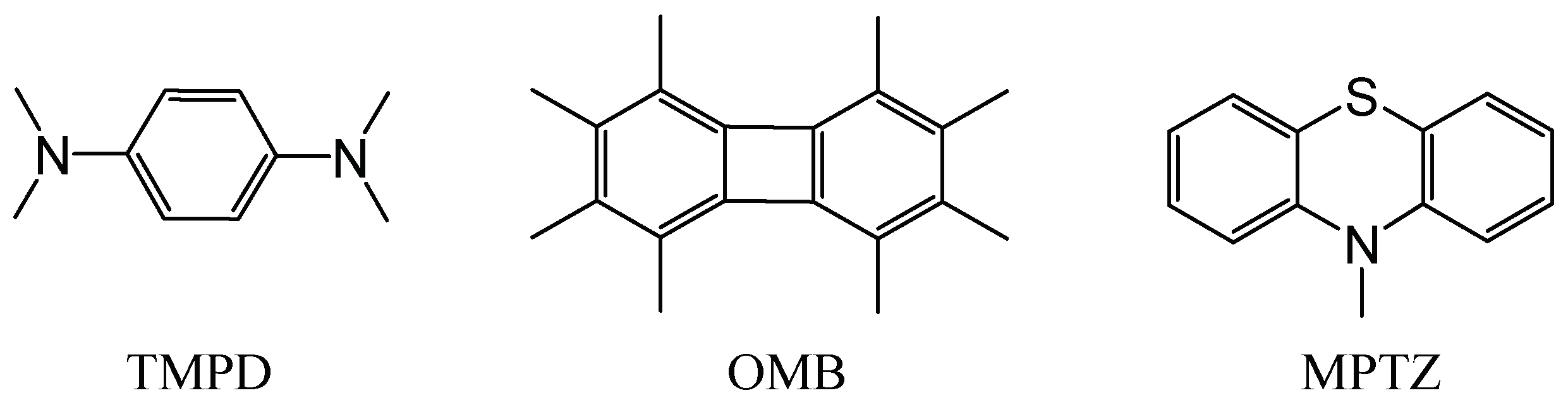
3.1. X-ray Structural Characterization of the Cation-Radical Salts
3.2. Quantum-Mechanical Computations of the Dimers of Cation Radicals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wudl, F.; Wobschall, D.; Hufnagel, E.J. Electrical conductivity by the bis(1,3-dithiole)-bis(1,3-dithiolium) system. J. Am. Chem. Soc. 1972, 94, 670–672. [Google Scholar] [CrossRef]
- Coleman, L.B.; Cohen, M.J.; Sandman, D.J.; Yamagishi, F.G.; Garito, A.F.; Heeger, A.J. Superconducting fluctuations and the Peierls instability in an organic solid. Solid State Commun. 1973, 12, 1125–1132. [Google Scholar] [CrossRef]
- Miller, J.S. Organometallic and Organic-based magnets: New chemistry and new materials for the new millennium. Inorg. Chem. 2000, 39, 4392–4408. [Google Scholar] [CrossRef]
- Williams, J.M. Organic Superconductors: Synthesis, Structure, Properties and Theory; Prentice Hall: Englewood Cliffs, NJ, USA, 1992. [Google Scholar]
- Miller, J.S.; Drillon, M. (Eds.) Magnetism: Molecules to Materials; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Deumal, M.; Vela, S.; Fumanal, M.; Ribas-Arino, J.; Novoa, J.J. Insights into the magnetism and phase transitions of organic radical-based materials. J. Mater. Chem. C 2021, 9, 10624–10646. [Google Scholar] [CrossRef]
- Bredas, J.L.; Calbert, J.P.; da Silva Filho, D.A.; Cornil, J. Organic semiconductors: A theoretical characterization of the basic parameters governing charge transport. Proc. Nat. Acad. Sci. USA 2002, 99, 5804. [Google Scholar] [CrossRef] [Green Version]
- Giamarchi, T. Theoretical framework for quasi-one dimensional systems. Chem. Rev. 2004, 104, 5037–5056. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.-I.; Sugimoto, T. TTF-Foreword. TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene; Springer: New York, NY, USA, 2004. [Google Scholar]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, oligoacenenes, and their Buckminsterfullerene derivatives: The brick and mortar of organic electronics. Chem. Rev. 2004, 104, 4891. [Google Scholar] [CrossRef]
- Chi, X.; Itkis, M.E.; Kirschbaum, K.; Pinkerton, A.A.; Oakley, R.T.; Cordes, A.W.; Haddon, R.C. Dimeric phenalenyl-based neutral radical molecular conductors. J. Am. Chem. Soc. 2001, 123, 4041–4048. [Google Scholar] [CrossRef]
- Enkelmann, V. Radical-cation salts of arenes: A new family of organic metals. Adv. Chem. Ser. 1988, 217, 177–200. [Google Scholar]
- Nishinaga, T.; Komatsu, K. Persistent π radical cations: Self-association and its steric control in the condensed phase. Org. Biomol. Chem. 2005, 3, 5610569. [Google Scholar] [CrossRef]
- Krossing, I.; Raabe, I. Noncoordinating anions—fact or fiction? A survey of likely candidates. Angew. Chem. Int. Ed. 2004, 43, 2066–2090. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.A. Carboranes: A new class of weakly coordinating anions for strong electrophiles, oxidants, and superacids. Acc. Chem. Res. 1998, 31, 133–139. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Sivaev, I.; Bregadze, V. Borane, Carborane and Metallacarborane Anions for Stabilization of Transient and Highly Reactive Intermediates: With Applications in Organometallics, Catalysis, Materials and Medicine. In Handbook of Boron Science. V.1. Boron in Organometallic Chemistry; Hosmane, N.S., Eagling, R., Eds.; World Scientific Books: London, UK, 2018; pp. 147–203. [Google Scholar]
- Fisher, S.P.; Tomich, A.W.; Guo, J.; Lavallo, V. Teaching an Old Dog New Tricks: New Directions in Fundamental and Applied Closo-Carborane Anion Chemistry. Chem. Commun. 2019, 55, 1684–1701. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.A.; Kim, K.S.; Stoyanov, E.S.; Stasko, D.; Tham, F.S.; Mueller, L.J.; Boyd, P.D.W. Isolating benzenium ion salts. J. Am. Chem. Soc. 2003, 125, 1796–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Juhasz, M.; Reed, C.A. The X-ray structure of a vinyl cation. Angew. Chem. Int. Ed. 2004, 43, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Küppers, T.; Bernhardt, E.; Eujen, R.; Willner, H.; Lehmann, C.W. [Me 3 Si][R-CB 11 F 11 ]-Synthesis and Properties. Angew. Chem. Int. Ed. 2007, 46, 6346–6349. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.M.; Molinos, E.; Brayshaw, S.K.; Weller, A.S. Rhodium phosphine olefin complexes of the weakly coordinating anions [BArF4]− and [1-closo-CB11H6Br6]−. Kinetic versus thermodynamic factors in anion coordination and complex reactivity. Organometallics 2007, 26, 463–465. [Google Scholar] [CrossRef]
- Priego, J.L.; Doerrer, L.H.; Rees, L.H.; Green, M.L.H. Weakly-coordinating anions stabilize the unprecedented monovalent and divalent η-benzene nickel cations [(η-C5H5)-Ni(η-C6H6)Ni(η-C5H5)]2+ and [Ni(η-C6H6)2]2+. Chem. Commun. 2000, 779–780. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Kochi, J.K. Molecular and electronic structures of the long-bonded π-dimers of tetrathiafulvalene cation radical in intermolecular electron transfer and in (solid-state) conductivity. J. Am. Chem. Soc. 2007, 129, 828–838. [Google Scholar] [CrossRef]
- Brown, J.; Zeller, M.; Rosokha, S.V. Effects of structural variations on-dimer formation: Long-distance multicenter bonding of cation-radicals of tetrathiafulvalene analogues Phys. Chem. Chem. Phys. 2020, 22, 25054–25065. [Google Scholar] [CrossRef]
- Brown, J.T.; Grounds, O.; Zeller, M.; Dilley, N.G.; Rosokha, S.V. Structures, multicenter π-bonding, and spin equilibria in the mixed-valence trimers of tetramethyltetrathiafulvalene cation-radicals. Cryst. Growth Des. 2021, 21, 7257–7268. [Google Scholar] [CrossRef]
- Nakayama, S.; Suzuki, K. The electronic absorption spectra of Würster’s cation radicals and their dimerization in solution. Bull. Chem. Soc. Jpn. 1973, 46, 3694–3698. [Google Scholar] [CrossRef]
- Sun, D.; Rosokha, S.V.; Kochi, J.K. Donor-acceptor (electronic) coupling in the precursor complex to organic electron transfer: Intermolecular and intramolecular self-exchange between phenothiazine redox centers. J. Am. Chem. Soc. 2004, 126, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.-M.; Rosokha, S.V.; Kochi, J.K. Stable (long-bonded) dimers via the quantitative self-association of different cationic, anionic, and uncharged π-radicals: Structures, energetics, and optical transitions. J. Am. Chem. Soc. 2003, 125, 12161–12171. [Google Scholar] [CrossRef]
- Hart, H.; Teuerstein, A. Octamethylnaphthalene (improved synthesis) and octamethylbiphenylene. Synthesis 1979, 693–695. [Google Scholar] [CrossRef]
- King, B.T.; Noll, B.C.; McKinley, A.J.; Michl, J. Dodecamethylcarba-closo-dodecarboranyl (CB11Me12∙), a stable free radical. J. Am. Chem. Soc. 1996, 118, 10902. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. π-Bonded molecular wires: Self-assembly of mixed-valence cation-radical stacks within the nanochannels formed by inert tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anions. CrystEngComm 2013, 15, 10638–10647. [Google Scholar] [CrossRef]
- Bruker (Apex3 v2019.1-0, SAINT V8.40A, Bruker AXS Inc.: Madison, WI, USA, 2019.
- SHELXTL Suite of Programs, Version 6.14, 2000–2003, Bruker Advanced X-ray Solutions, Bruker AXS Inc.: Madison, WI, USA, 2003.
- Sheldrick, G. Crystal Structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Capdevila-Cortada, M.; Ribas-Arino, J.; Novoa, J.J. Assessing the performance of CASPT2 and DFT methods for the description of long, multicenter bonding in dimers between radical ions. J. Chem. Theory Comput. 2014, 10, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Lischka, H.; Beneberu, H.Z.; Kertesz, M. Rotational barrier in phenalenyl neutral radical dimer: Separating pancake and van der waals interactions. J. Am. Chem. Soc. 2014, 136, 5539–5542. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.P.; Ellern, A.; Winter, A.H. Spin delocalization, polarization, and London dispersion forces govern the formation of diradical pimers. J. Am. Chem. Soc. 2020, 142, 5304–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ikemoto, I.; Katagiri, G.; Nishimura, S.; Yakushi, K.; Kuroda, H. Structure of N,N,N′,N′-tetramethyl-p-phenylenediamine. Acta Cryst. B. 1979, B35, 2264–2265. [Google Scholar] [CrossRef]
- Chu, S.S.C.; van der Helm, D. The refinement of the crystal structure of N-methylphenothiazine Acta Cryst. B 1974, 30, 2489. [Google Scholar]
- Kertesz, M.K. Pancake bonding: An unusual pi-stacking interaction Chem. -Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Preuss, K.E. Pancake bonds: π-Stacked dimers of organic and light-atom radicals. Polyhedron 2014, 79, 1–15. [Google Scholar] [CrossRef]
- Molcanov, K.; Jelsch, C.; Wenger, E.; Mou, Z.; Kertesz, M.; Landeros-Rivera, B.; Hernandez-Trujillo, J.; Stilinovic, V.; Kojic-Prodic, B. Multicentric two-electron covalent bonding (pancake bonding) between semiquinone radicals determines bulk properties. Acta Crystallogr. Sect. A 2018, 74, E80. [Google Scholar] [CrossRef]
- Ganesan, V.; Rosokha, S.V.; Kochi, J.K. Isolation of the latent precursor complex in electron-transfer dynamics. intermolecular association and self-exchange with acceptor anion radicals. J. Am. Chem. Soc. 2003, 125, 2559–2571. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bau, R.; Reed, C.A. “Free” [Fe(tpp)]+ cation: A new concept in the search for the least coordinating anion. Angew. Chem. Int. Ed. 1995, 33, 2433–2434. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Lu, J.J.; Rosokha, T.Y.; Kochi, J.K. Tris(thianthrene)(2+)-bis(dodecamethylcarba-closo-dodeca bo rate) dichloromethane tetrasolvate: A crossed triple-decker π-trimer dication. Acta Crystallogr. C 2007, C63, o347–o349. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Salt i | a ii | b ii | c ii | d ii | e ii | f ii |
| (TMPD)Me12CAR | 1.357 | 1.421 | 1.342 | 1.453 | ||
| (TMPD)Br6CAR | 1.354 | 1.424 | 1.350 | 1.460 | ||
| (MPTZ)Br6CAR | 1.403 | 1.359 | 1.408 | 1.393 | 1.723 | 1.391 |
| (MPTZ)Me12CAR iii | 1.399 | 1.366 | 1.408 | 1.411 | 1.715 | 1.389 |
| (OMB)2Br6CAR | 1.421 | 1.422 | 1.376 | 1.434 | 1.501 | |
| (OMB)2[Ag(Br6CAR)2] | 1.418 | 1.422 | 1.378 | 1.426 | 1.4915 | |
| π-Dimer | ΔE a | EST b | Edisp c | EES d |
|---|---|---|---|---|
| TMPD22+ | −14.5 | −34.0 | −101.9 | 245.8 |
| MPTZ22+ | −30.7 | −37.2 | −92.8 | 239.0 |
| OMB22+ | −74.0 | −71.5 | −177.0 | 216.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeniyi, E.; Zeller, M.; Rosokha, S.V. Crystal Engineering of Cation-Radical Salts with Weakly Coordinating Carbadodecaborate Anions. Crystals 2023, 13, 99. https://doi.org/10.3390/cryst13010099
Adeniyi E, Zeller M, Rosokha SV. Crystal Engineering of Cation-Radical Salts with Weakly Coordinating Carbadodecaborate Anions. Crystals. 2023; 13(1):99. https://doi.org/10.3390/cryst13010099
Chicago/Turabian StyleAdeniyi, Emmanuel, Matthias Zeller, and Sergiy V. Rosokha. 2023. "Crystal Engineering of Cation-Radical Salts with Weakly Coordinating Carbadodecaborate Anions" Crystals 13, no. 1: 99. https://doi.org/10.3390/cryst13010099