
Interplay of Ionic Species in Salts of Homoleptic Quaternary Phosphonium Cations Bearing Linear Biphenyl Moieties

 , and
, and 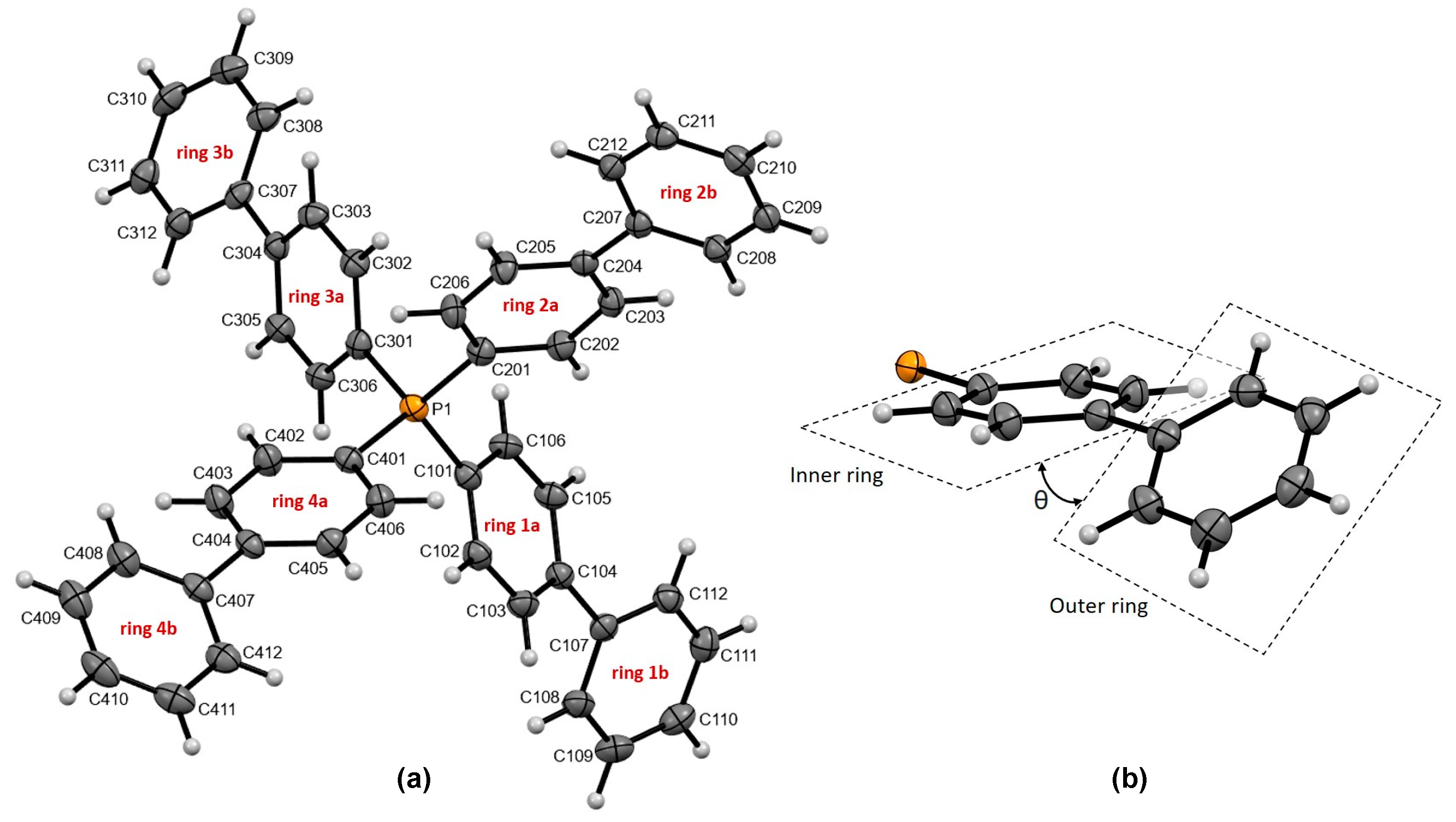{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
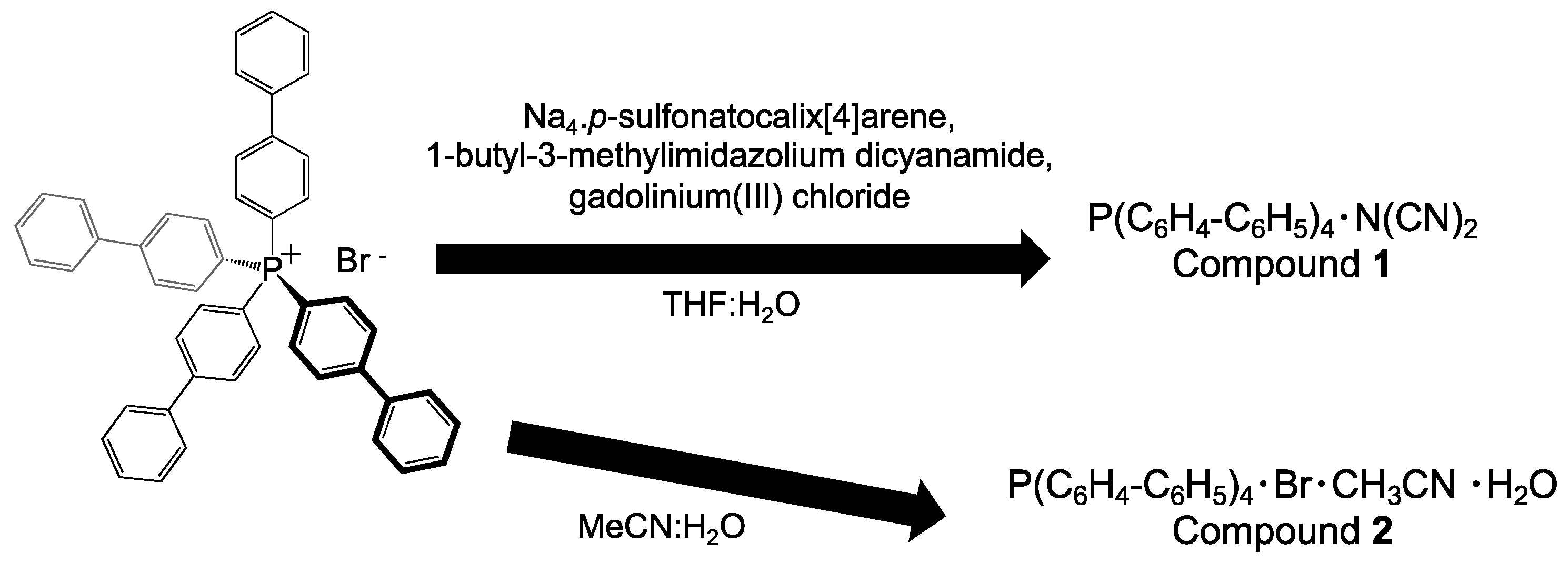
2.1. Synthesis
2.2. Single Crystal X-ray Crystallography
3. Results
3.1. Structural Description
3.1.1. Molecular Features of Tetra-Biphenyl Phosphonium Cation
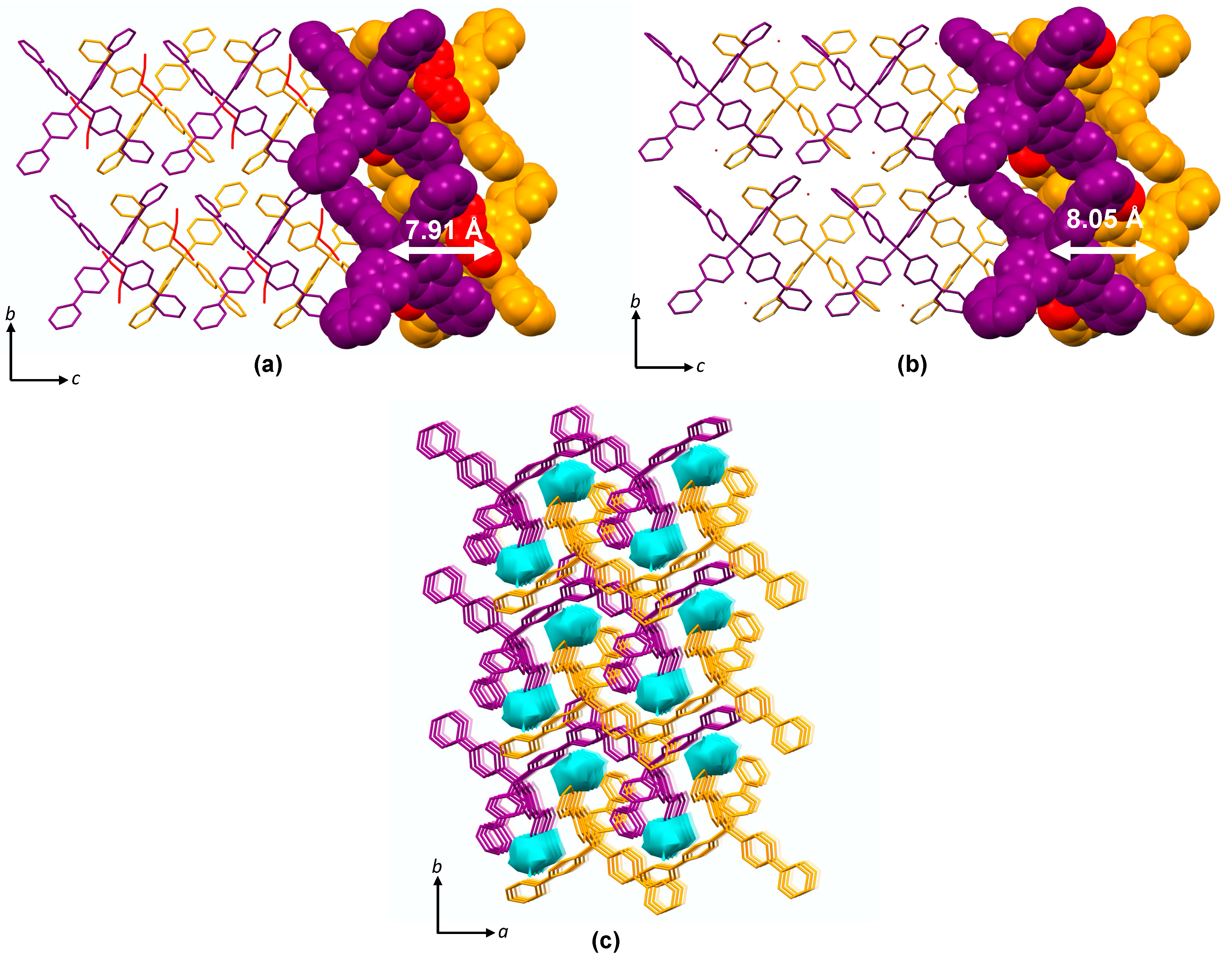
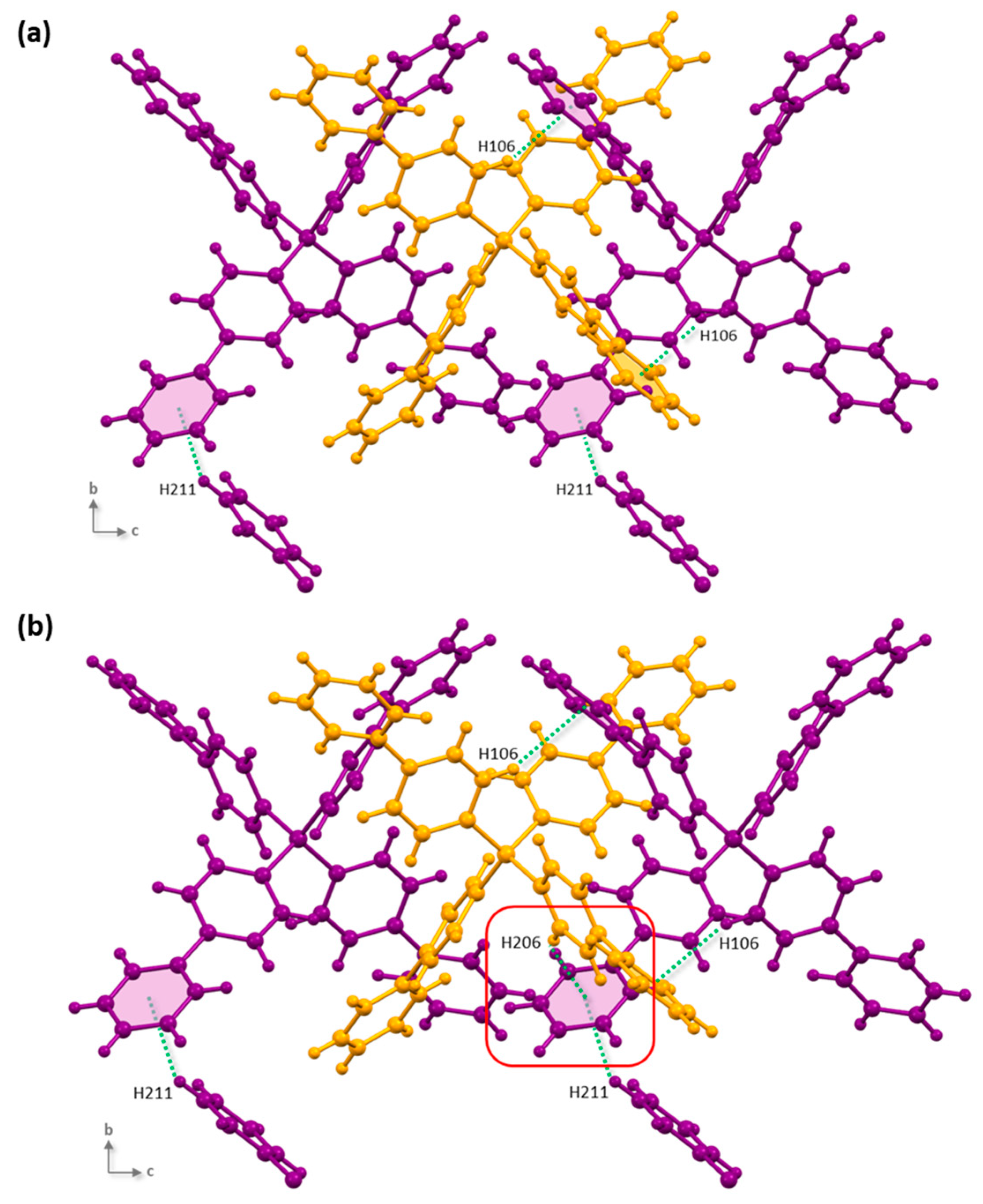
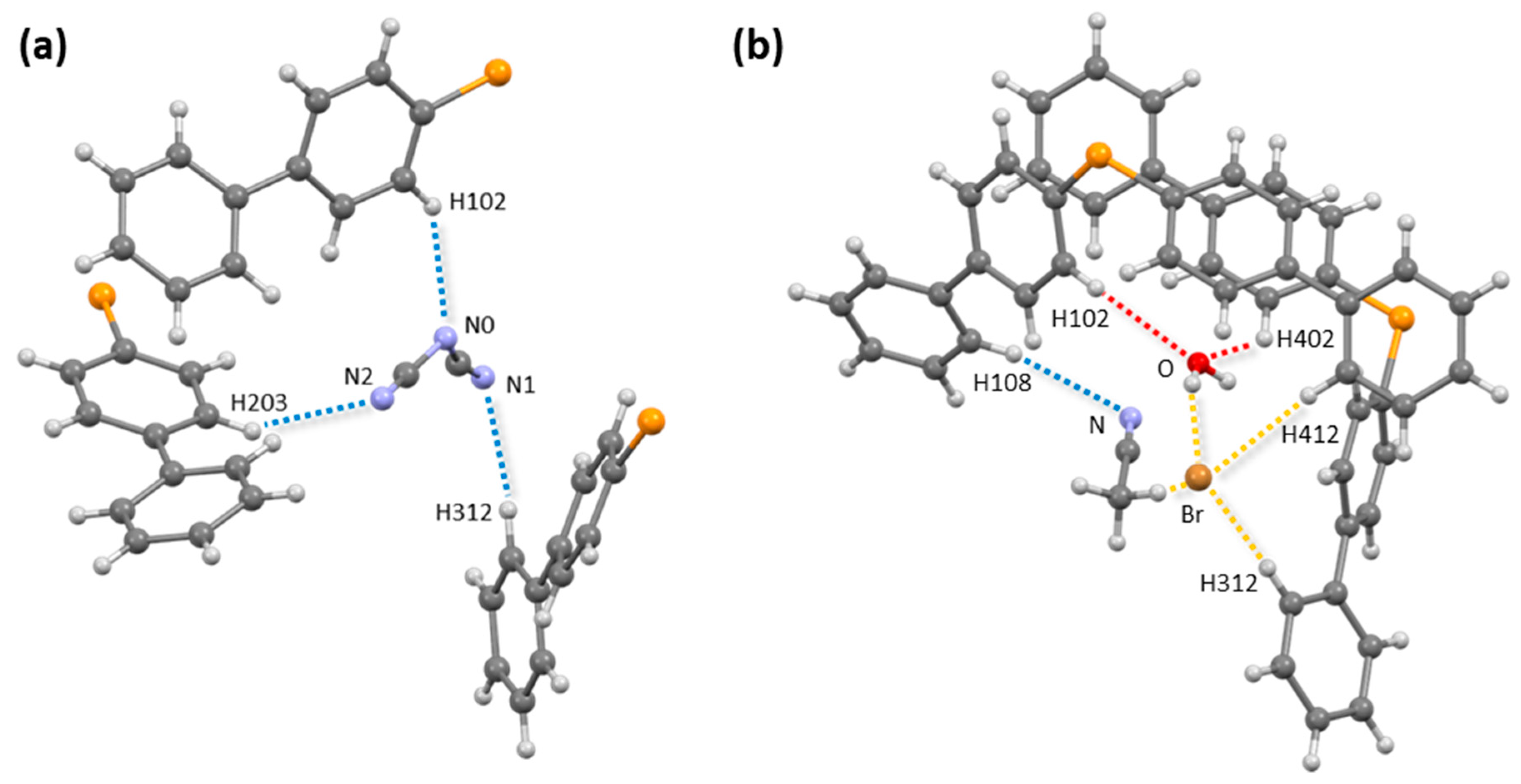
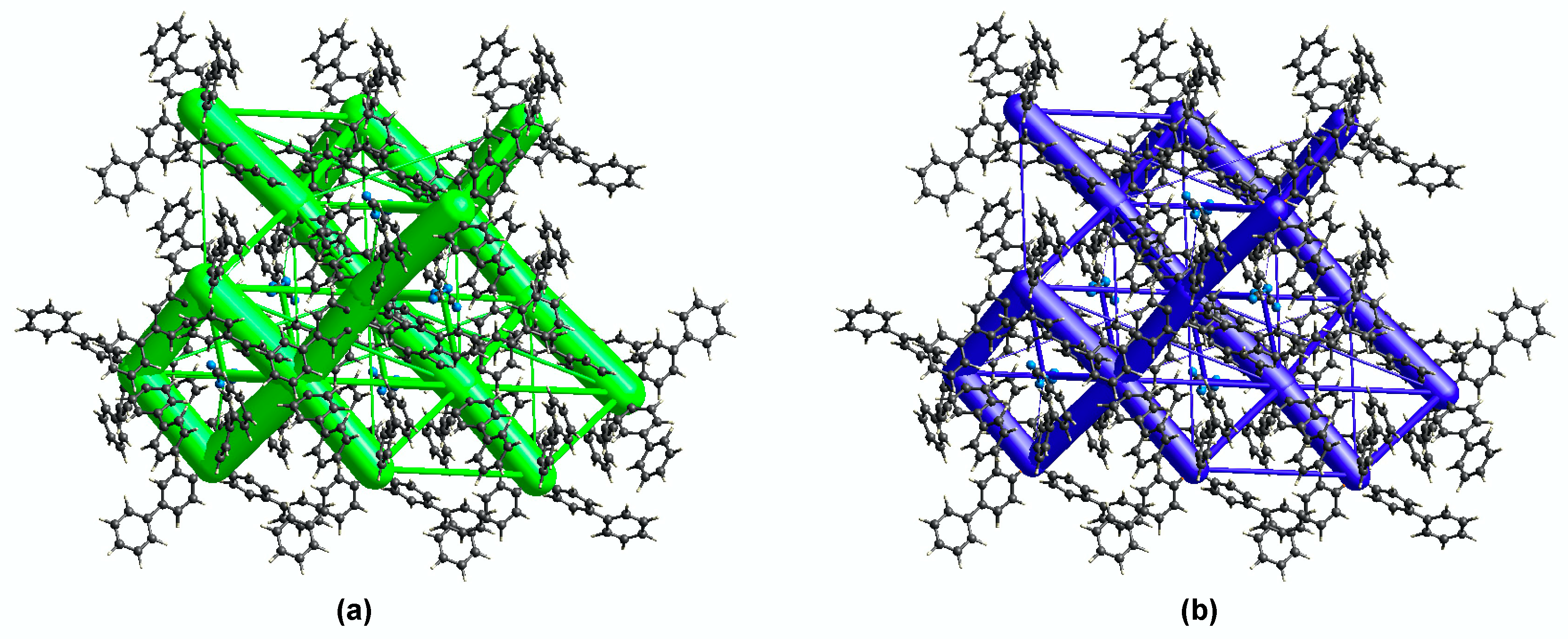
3.1.2. Self-Assembly
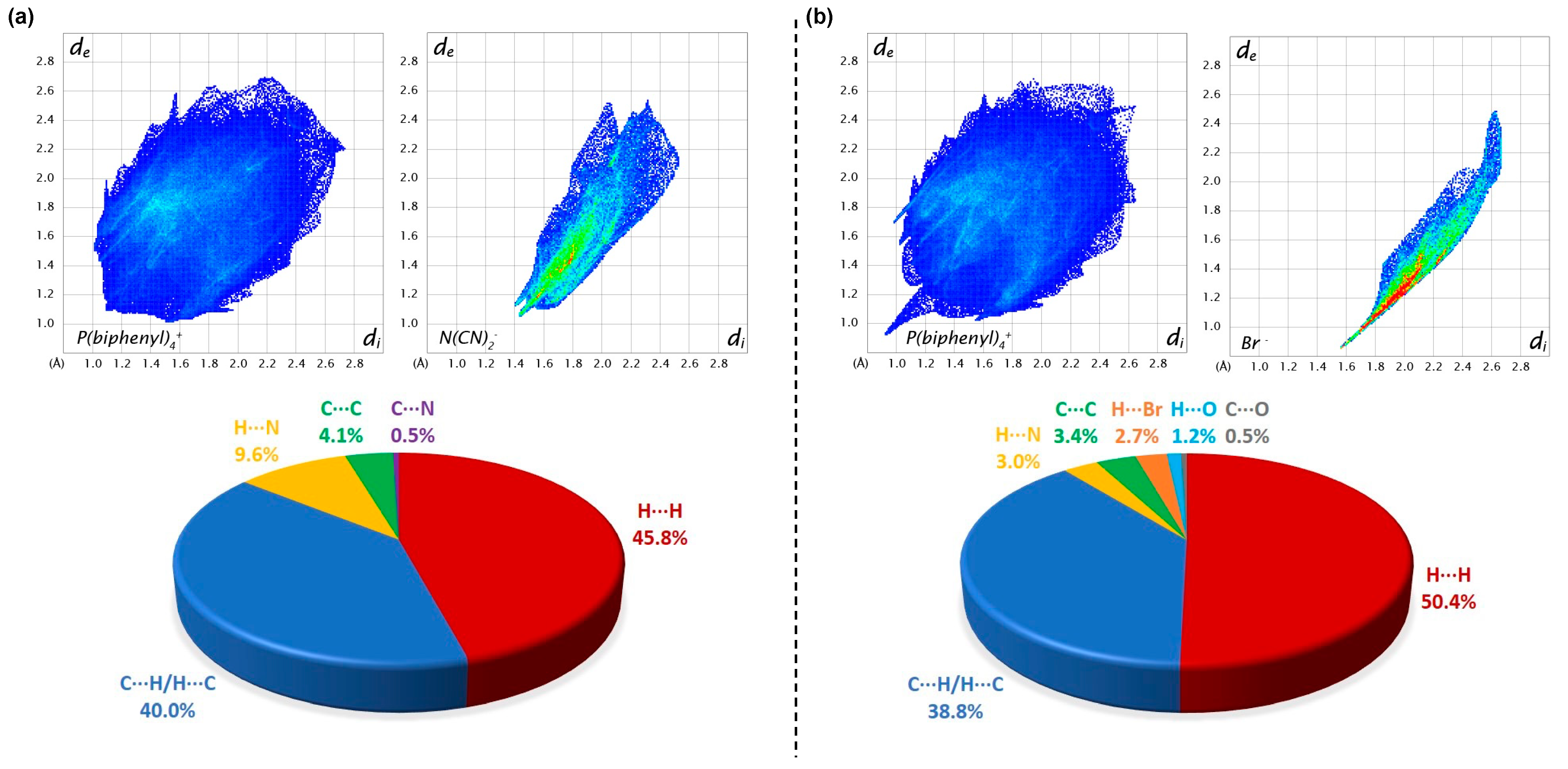
3.1.3. Hirshfeld Surface Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, M.-L.; Liu, B.-L.; Lin, S.-J. Synthesis of an active quaternary phosphonium salt and its application to the Wittig reaction: Kinetic study. J. Chin. Inst. Chem. Eng. 2007, 38, 451–459. [Google Scholar] [CrossRef]
- Noroozi-Shad, N.; Gholizadeh, M.; Sabet-Sarvestani, H. Quaternary phosphonium salts in the synthetic chemistry: Recent progress, development, and future perspectives. J. Mol. Struct. 2022, 1257, 132628. [Google Scholar] [CrossRef]
- Liu, S.; Kumatabara, Y.; Shirakawa, S. Chiral quaternary phosphonium salts as phase-transfer catalysts for environmentally benign asymmetric transformations. Green Chem. 2016, 18, 331–341. [Google Scholar] [CrossRef]
- Enders, D.; Nguyen, T. Chiral quaternary phosphonium salts, a new class of organocatalysts. Org. Biomol. Chem. 2012, 10, 5327–5331. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Wang, X.; Hashimoto, T.; Maruoka, K. Binaphthyl-Modified Quaternary Phosphonium Salts as Chiral Phase-Transfer Catalysts, Asymmetric Amination of β-Keto Esters. Angew. Chem. Int. Ed. 2008, 47, 9466–9468. [Google Scholar] [CrossRef]
- Manabe, K. Asymmetric phase-transfer alkylation catalyzed by a chiral quaternary phosphonium salt with a multiple hydrogen-bonding site. Tetrahedron Lett. 1998, 39, 5807–5810. [Google Scholar] [CrossRef]
- Castillo, J.; Teresa, M.; Fortuny, A.; Navarro, P.; Sepúlveda, R.; María, A. Cu(II) extraction using quaternary ammonium and quaternary phosphonium based ionic liquid. Hydrometallurgy 2014, 141, 89–96. [Google Scholar] [CrossRef]
- Fraser, K.J.; MacFarlane, D. Phosphonium-Based Ionic Liquids, An Overview. Aust. J. Chem. 2009, 62, 309–321. [Google Scholar] [CrossRef]
- Del Sesto, R.E.; Corley, C.; Robertson, A.; Wilkes, J.S. Tetraalkylphosphonium-based ionic liquids. J. Organomet. Chem. 2005, 690, 2536–2542. [Google Scholar] [CrossRef]
- Nahlé, A.H.; Harvey, T.; Walsh, F. Quaternary aryl phosphonium salts as corrosion inhibitors for iron in HCl. J. Alloys Compd. 2018, 765, 812–825. [Google Scholar] [CrossRef]
- Wang, C.; Tao, Z.; Zhao, X.; Li, J.; Ren, Q. Poly(aryl ether nitrile)s containing flexible side-chain-type quaternary phosphonium cations as anion exchange membranes. Sci. China Mater. 2020, 63, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Cai, R.; Luo, T.; Jensen, K.; Contreras, C.; Yan, Y. Quaternary Phosphonium-Based Polymers as Hydroxide Exchange Membranes. ChemSusChem 2010, 3, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-J.; Biswal, S.; Deroose, C.; Gambhir, S.S. Tetraphenylphosphonium as a Novel Molecular Probe for Imaging Tumors. J. Nucl. Med. 2004, 45, 636–643. [Google Scholar] [PubMed]
- Dance, I.; Scudder, M. Molecules embracing in crystals. CrystEngComm 2009, 11, 2233–2247. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Supramolecular Motifs: Concerted Multiple Phenyl Embraces between Ph4P+Cations Are Attractive and Ubiquitous. Chem.-Eur. J. 1996, 2, 481–486. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Concerted supramolecular motifs: Linear columns and zigzag chains of multiple phenyl embraces involving Ph4P+cations in crystals. J. Chem. Soc. Dalton Trans. 1996, 19, 3755–3769. [Google Scholar] [CrossRef]
- Scudder, M.; Dance, I. Crystal supramolecular motifs. Two- and three-dimensional networks of Ph4P+ cations engaged in sixfold phenyl embraces. J. Chem. Soc. Dalton Trans. 1998, 19, 3167–3176. [Google Scholar] [CrossRef]
- Scudder, M.; Dance, I. Crystal supramolecular motifs. Ladders, layers and labyrinths of Ph4P+ cations engaged in fourfold phenyl embraces. J. Chem. Soc. Dalton Trans. 1998, 19, 3155–3166. [Google Scholar] [CrossRef]
- Scudder, M.; Dance, I. Sixfold phenyl embraces with substituted phenyl in PPh3. J. Chem. Soc. Dalton Trans. 2000, 17, 2909–2915. [Google Scholar] [CrossRef]
- Ling, I.; Raston, C.L. Sulfonato and Phosphonatocalix[n]arenes in Self-Assembly. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Gokel, G.W., Barbour, L.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 6, pp. 19–73. [Google Scholar]
- Ling, I.; Skelton, B.W.; Sobolev, A.N.; Alias, Y.; Khor, Z.C.; Raston, C.L. Effect of anions on the solid-state interplay of symmetric and unsymmetric phosphonium cations. New J. Chem. 2020, 44, 10220–10228. [Google Scholar] [CrossRef]
- Ling, I.; Alias, Y.; Sobolev, A.N.; Raston, C.L. Hirshfeld surface analysis of phosphonium salts. CrystEngComm 2010, 12, 4321–4327. [Google Scholar] [CrossRef]
- Schwarzer, A.; Schilling, I.C.; Seichter, W.; Weber, E. Synthesis and X-ray Crystal Structures of New Tetrahedral Arylethynyl Substituted Silanes. Silicon 2009, 1, 3–12. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M.; Van Doorn, J.A.; Meijboom, N. Reductive cleavage of the carbon-phosphorus bond with alkali metals. I. Cleavage of functionalised triphenylphosphines; formation of secondary and primary phosphines. Recl. Trav. Chim. Pays-Bas 1991, 110, 420–432. [Google Scholar] [CrossRef]
- Worrall, D.E. Studies in The Diphenyl Series. III. Some Phosphorus Derivatives of Diphenyl. J. Am. Chem. Soc. 1930, 52, 2933–2937. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Casalone, G.; Mariani, C.; Mugnoli, A.; Simonetta, M. The molecular structure of biphenyl in the gas and solid phases. Mol. Phys. 1968, 15, 339–348. [Google Scholar] [CrossRef]
- Johnson, C.K. ORTEPII. Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Nishio, M. CH/π hydrogen bonds in crystals. CrystEngComm 2004, 6, 130–158. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, M.B.; Sobolev, A.N.; Raston, C.L.; Dalgarno, S.J.; Ling, I. Interplay of Ionic Species in Salts of Homoleptic Quaternary Phosphonium Cations Bearing Linear Biphenyl Moieties. Crystals 2023, 13, 59. https://doi.org/10.3390/cryst13010059
Tan MB, Sobolev AN, Raston CL, Dalgarno SJ, Ling I. Interplay of Ionic Species in Salts of Homoleptic Quaternary Phosphonium Cations Bearing Linear Biphenyl Moieties. Crystals. 2023; 13(1):59. https://doi.org/10.3390/cryst13010059
Chicago/Turabian StyleTan, Monica Bernard, Alexandre N. Sobolev, Colin L. Raston, Scott J. Dalgarno, and Irene Ling. 2023. "Interplay of Ionic Species in Salts of Homoleptic Quaternary Phosphonium Cations Bearing Linear Biphenyl Moieties" Crystals 13, no. 1: 59. https://doi.org/10.3390/cryst13010059