
Interfacial Characteristics of Graphene-Reinforced Iron Composites: A Molecular Dynamics Study
 , and
, and 
Abstract
:1. Introduction
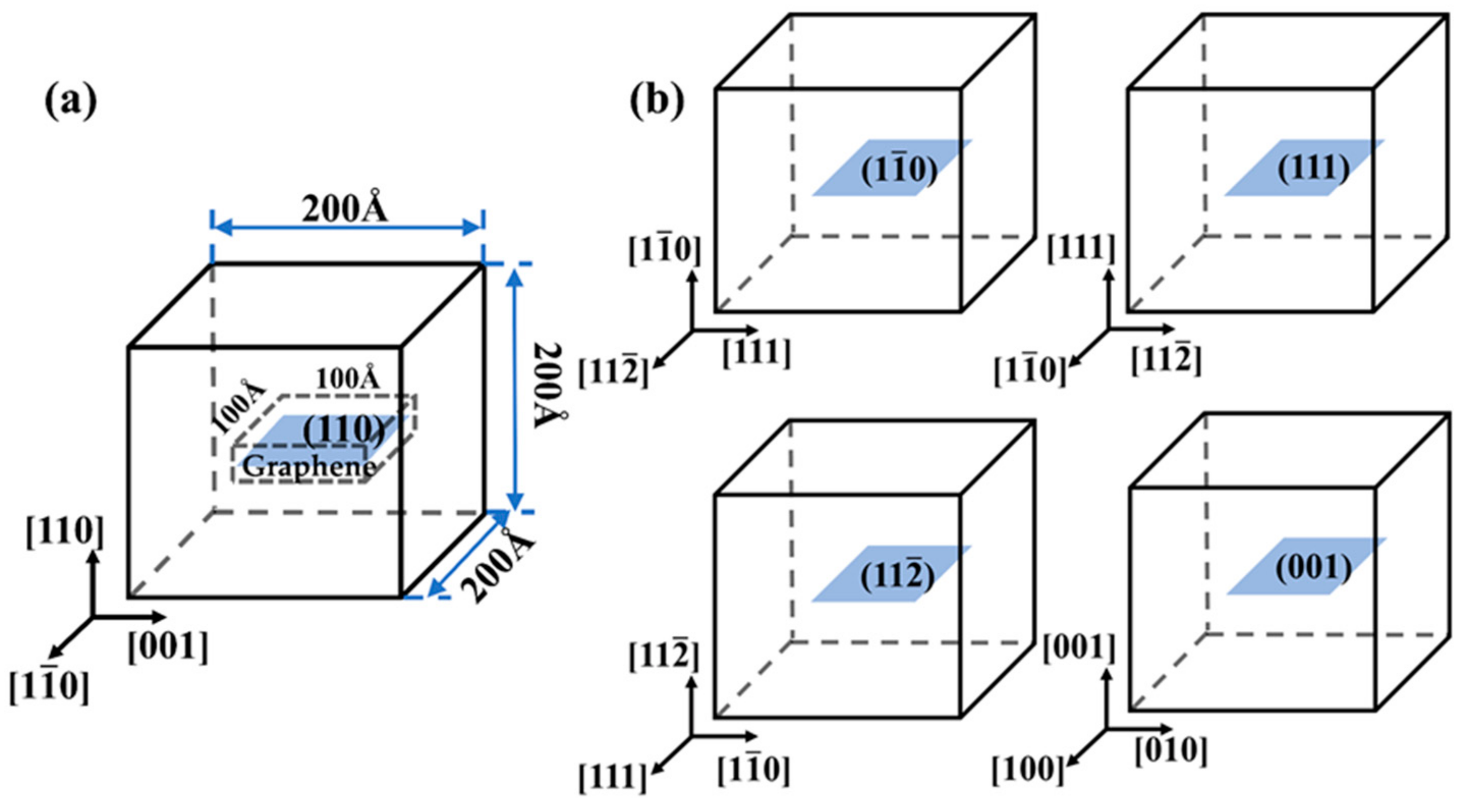
2. Molecular Dynamics (MD) Model and Setup
3. Results and Discussion
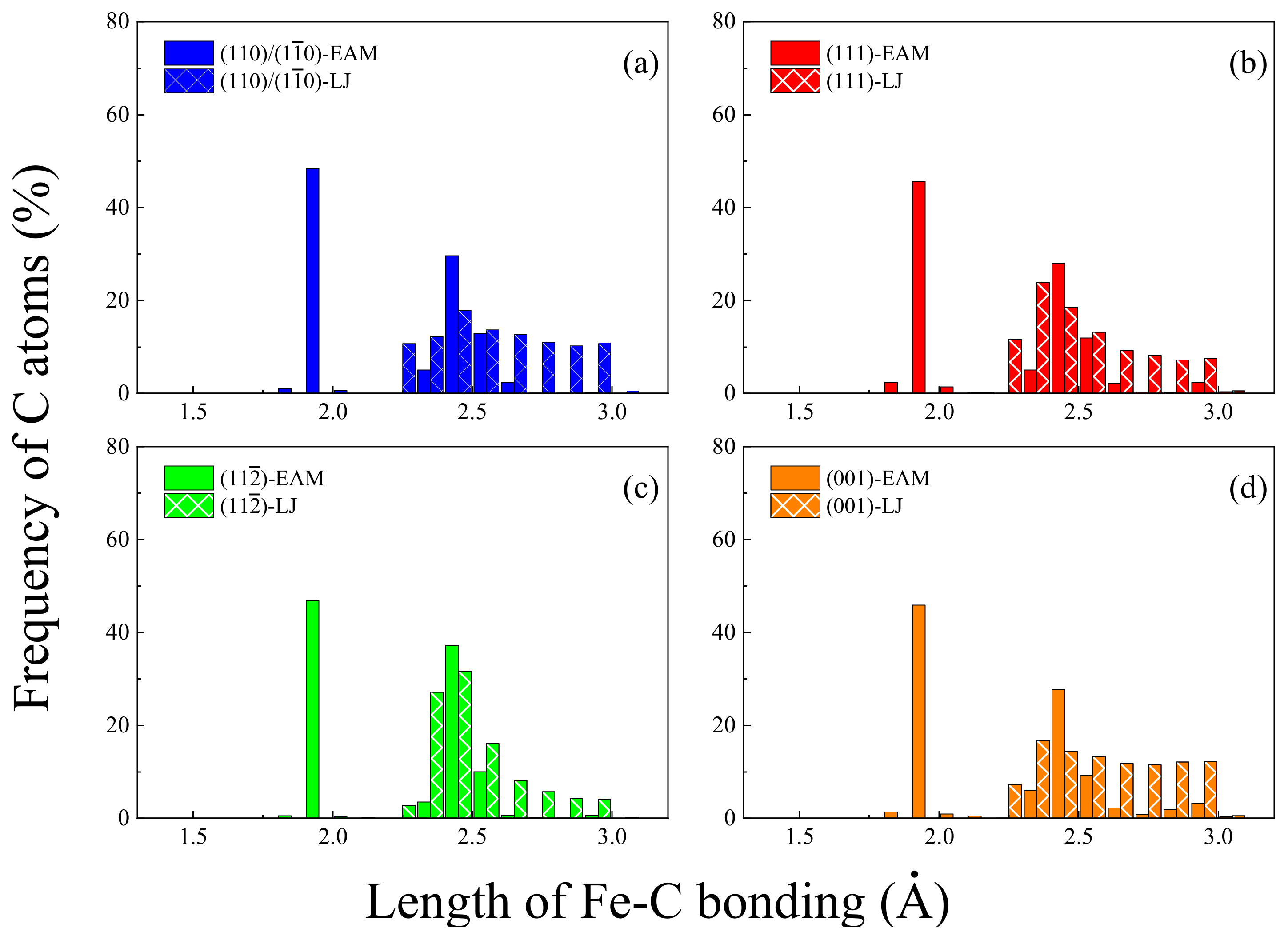
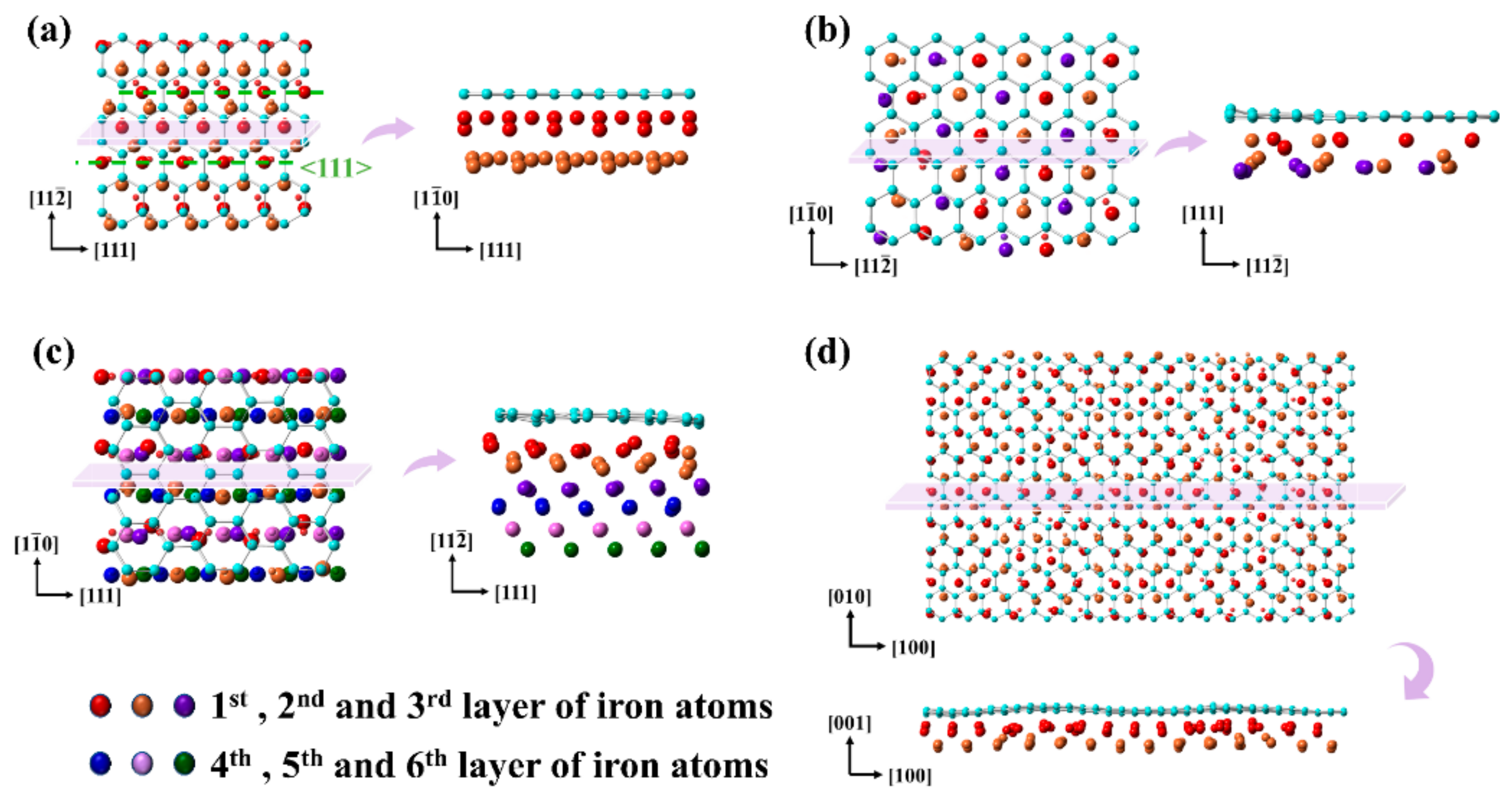
3.1. Interfacial Characteristics of Gr/Fe Interface
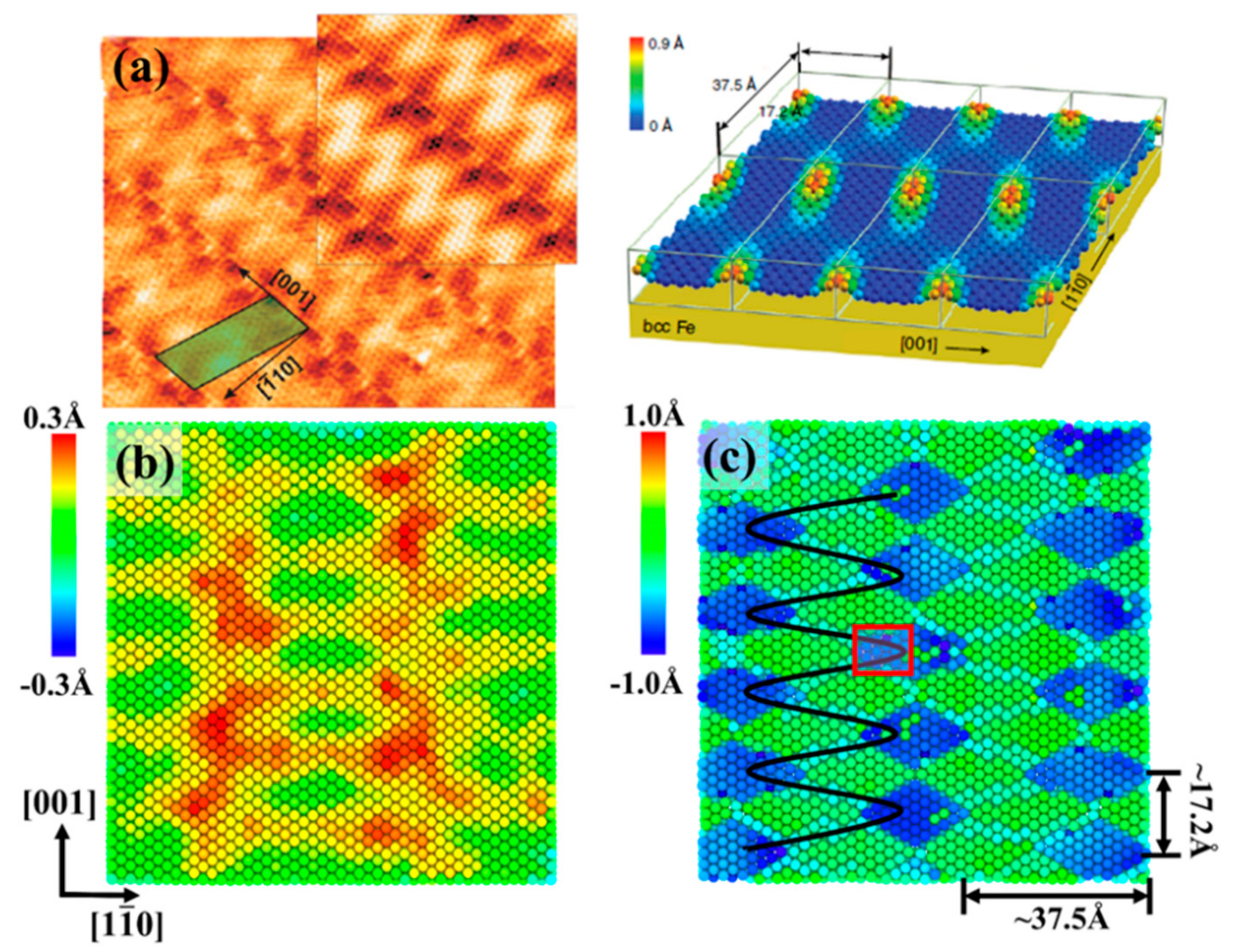
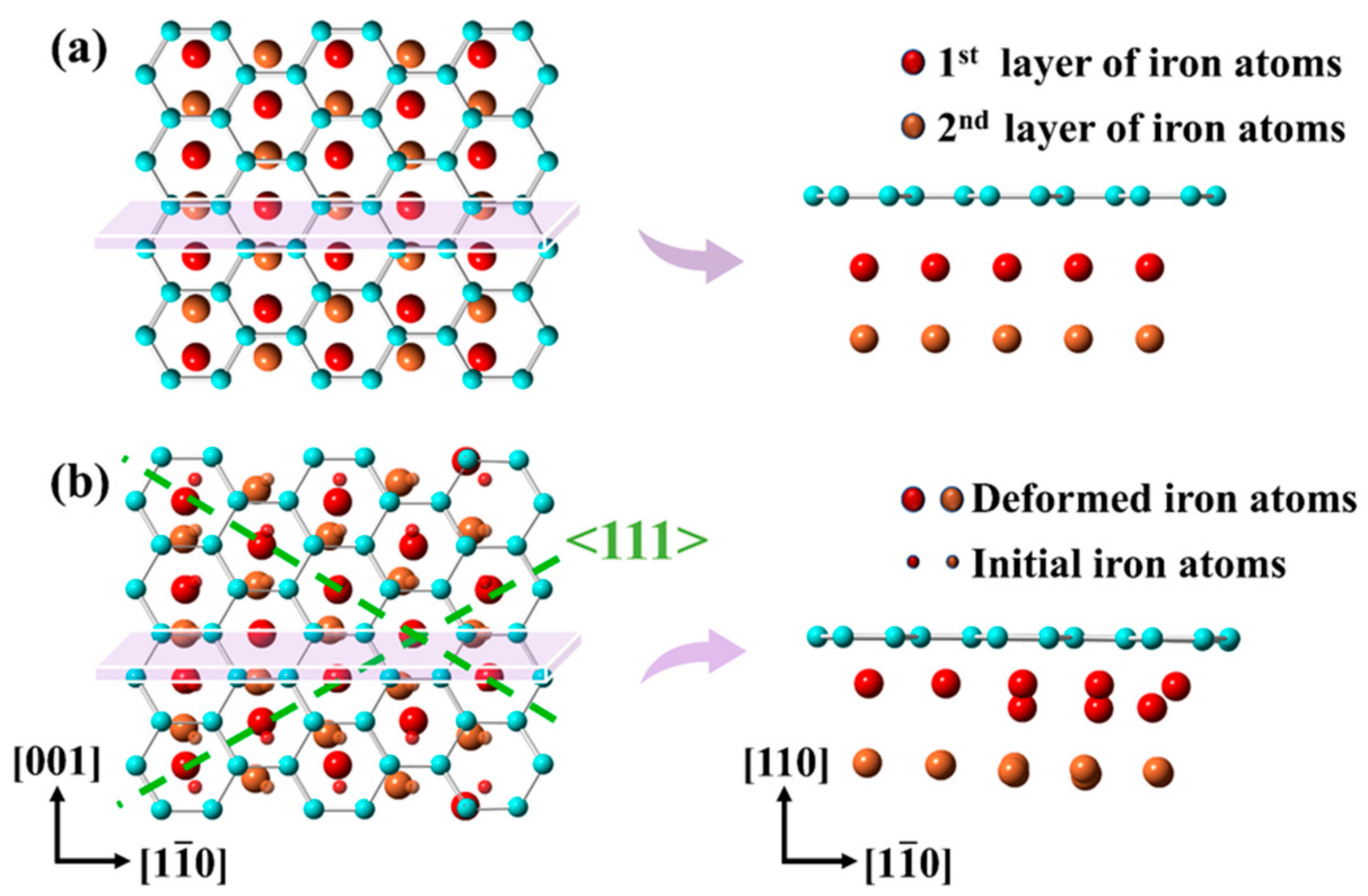
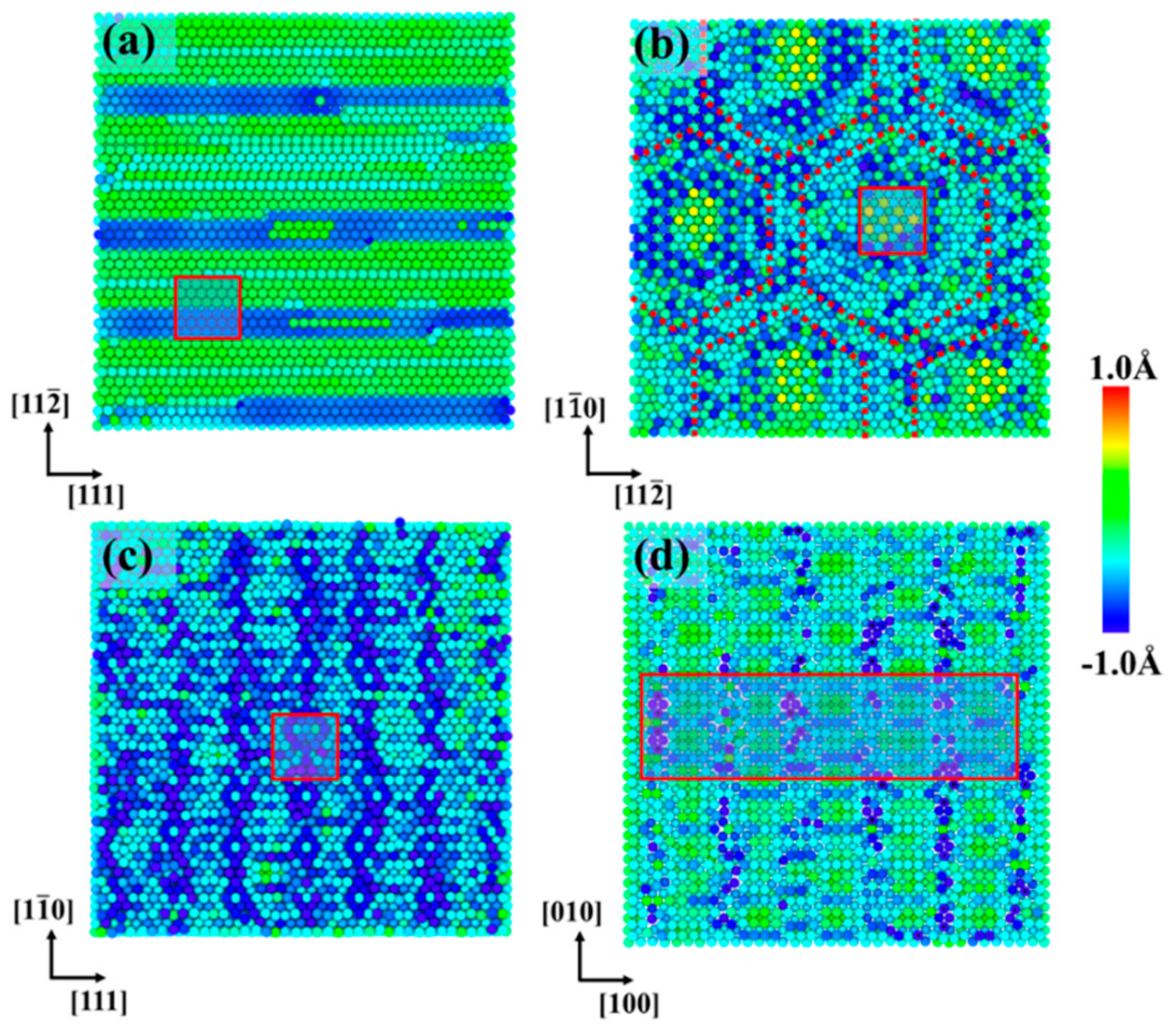
3.2. Moiré Pattern of Gr/Fe Typed-I {110} Interface
3.3. Moiré Pattern at Different Gr/Fe Interface
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Raccichini, R.; Varzi, A.; Passerini, S.; Scrosati, B. The role of graphene for electrochemical energy storage. Nat. Mater. 2015, 14, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Yoon, T.; Jin, S.H.; Lee, J.; Kim, T.-S.; Hong, S.H.; Jeon, S. Enhanced Mechanical Properties of Graphene/Copper Nanocomposites Using a Molecular-Level Mixing Process. Adv. Mater. 2013, 25, 6724–6729. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Galiotis, C.; Frank, O.; Koukaras, E.N.; Sfyris, D. Graphene Mechanics: Current Status and Perspectives. Annu. Rev. Chem. Biomol. Eng. 2015, 6, 121–140. [Google Scholar] [CrossRef]
- Cao, Q.; Geng, X.; Wang, H.; Wang, P.; Liu, A.; Lan, Y.; Peng, Q. A Review of Current Development of Graphene Mechanics. Crystals 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Liang, C.; Ji, W.; De, S. A theoretical analysis of the effect of the hydrogenation of graphene to graphane on its mechanical properties. Phys. Chem. Chem. Phys. 2013, 15, 2003–2011. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Colombo, L.; Yu, G.; Stoller, M.; Tozzini, V.; Ferrari, A.C.; Ruoff, R.S.; Pellegrini, V. Graphene, related two-dimensional crystals, and hybrid systems for energy conversion and storage. Science 2015, 347, 1246501. [Google Scholar] [CrossRef]
- Hou, J.; Deng, B.; Zhu, H.; Lan, Y.; Shi, Y.; De, S.; Liu, L.; Chakraborty, P.; Gao, F.; Peng, Q. Magic auxeticity angle of graphene. Carbon 2019, 149, 350–354. [Google Scholar] [CrossRef]
- Deng, B.; Hou, J.; Zhu, H.; Liu, S.; Liu, E.; Shi, Y.; Peng, Q. The normal-auxeticity mechanical phase transition in graphene. 2D Mater. 2017, 4, 021020. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Yeom, M.S.; Shin, J.W.; Kim, H.; Cui, Y.; Kysar, J.W.; Hone, J.; Jung, Y.; Jeon, S.; et al. Strengthening effect of single-atomic-layer graphene in metal-graphene nanolayered composites. Nat. Commun. 2013, 4, 2114. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Sheng, J.; Wang, L.-D.; Yang, Z.-Y.; Shi, Z.-D.; Wang, X.-J.; Fei, W.-D. Hot rolling behavior of graphene/Cu composites. J. Alloys Compd. 2020, 816, 153204. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; He, Y.; Huang, H.; Wu, Y.; Hou, L.; Liu, X.; Yang, T.; Zou, J.; Huang, B. Synthesis, growth mechanism and thermal stability of copper nanoparticles encapsulated by multi-layer graphene. Carbon 2012, 50, 2119–2125. [Google Scholar] [CrossRef]
- Dixit, S.; Mahata, A.; Mahapatra, D.R.; Kailas, S.V.; Chattopadhyay, K. Multi-layer graphene reinforced aluminum-Manufacturing of high strength composite by friction stir alloying. Compos. Part B Eng. 2018, 136, 63–71. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, W.; Wang, L.; Shi, J.; Sun, L.; Cui, J.; Wang, J. Investigation on mechanical behavior of multilayer graphene reinforced aluminum composites. Phys. E Low-dimens. Syst. Nanostruct. 2020, 123, 114172. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, R.; Tan, Z.; Ji, G.; Fan, G.; Li, Z.; Xiong, D.-B.; Guo, Q.; Li, Z.; Zhang, D. Interface-induced strain hardening of graphene nanosheet/aluminum composites. Carbon 2019, 146, 17–27. [Google Scholar] [CrossRef]
- Kuang, D.; Xu, L.Y.; Liu, L.; Hu, W.B.; Wu, Y.T. Graphene-nickel composites. Appl. Surf. Sci. 2013, 273, 484–490. [Google Scholar] [CrossRef]
- Kurapova, O.Y.; Lomakin, I.V.; Sergeev, S.N.; Solovyeva, E.N.; Zhilyaev, A.P.; Archakov, I.Y.; Konakov, V.G. Fabrication of nickel-graphene composites with superior hardness. J. Alloys Compd. 2020, 835, 155463. [Google Scholar] [CrossRef]
- Xu, J.; Yao, Y.; Jiang, X. Research on preparation process and performance of graphene reinforced iron matrix composites. Mach. Bldg. Auto. 2018, 47, 53–57. [Google Scholar]
- Lin, D.; Motlag, M.; Saei, M.; Jin, S.; Rahimi, R.M.; Bahr, D.; Cheng, G.J. Shock engineering the additive manufactured graphene-metal nanocomposite with high density nanotwins and dislocations for ultra-stable mechanical properties. Acta Mater. 2018, 150, 360–372. [Google Scholar] [CrossRef]
- Wang, L.; Jin, J.; Cao, J.; Yang, P.; Peng, Q. Interaction of Edge Dislocations with Graphene Nanosheets in Graphene/Fe Composites. Crystals 2018, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Guerrero, A.; Sánchez, S.; Narciso, J.; Louis, E.; Rodríguez-Reinoso, F. Pressure infiltration of Al-12wt.% Si-X (X=Cu, Ti, Mg) alloys into graphite particle preforms. Acta Mater. 2006, 54, 1821–1831. [Google Scholar] [CrossRef]
- You, Y.; Yoshimura, M.; Cholake, S.; Lee, G.-H.; Sahajwalla, V.; Joshi, R. A Controlled Carburization Process to Obtain Graphene-Fe3C-Fe Composites. Adv. Mater. Interfaces 2018, 5, 1800599. [Google Scholar] [CrossRef]
- Wintterlin, J.; Bocquet, M.-L. Graphene on metal surfaces. Surf. Sci. 2009, 603, 1841–1852. [Google Scholar] [CrossRef]
- Politano, A.; Slotman, G.J.; Roldán, R.; Chiarello, G.; Campi, D.; Katsnelson, M.I.; Yuan, S. Effect of moiré superlattice reconstruction in the electronic excitation spectrum of graphene-metal heterostructures. 2D Mater. 2017, 4, 021001. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Han, Y.; Evans, J.W.; Engstfeld, A.K.; Behm, R.J.; Tringides, M.C.; Hupalo, M.; Lin, H.-Q.; Huang, L.; Ho, K.-M.; et al. Growth morphology and properties of metals on graphene. Prog. Surf. Sci. 2015, 90, 397–443. [Google Scholar] [CrossRef] [Green Version]
- Jacobberger, R.M.; Arnold, M.S. Graphene Growth Dynamics on Epitaxial Copper Thin Films. Chem. Mater. 2013, 25, 871–877. [Google Scholar] [CrossRef]
- Rasool, H.I.; Song, E.B.; Mecklenburg, M.; Regan, B.C.; Wang, K.L.; Weiller, B.H.; Gimzewski, J.K. Atomic-Scale Characterization of Graphene Grown on Copper (100) Single Crystals. J. Am. Chem. Soc. 2011, 133, 12536–12543. [Google Scholar] [CrossRef]
- Vinogradov, N.; Zakharov, A.; Kocevski, V.; Rusz, J.; Simonov, K.A.; Eriksson, O.; Mikkelsen, A.; Lundgren, E.; Vinogradov, A.; Mårtensson, N.; et al. Formation and Structure of Graphene Waves on Fe (110). Phys. Rev. Lett. 2012, 109, 026101. [Google Scholar] [CrossRef]
- Preobrajenski, A.B.; Ng, M.L.; Vinogradov, A.S.; Mårtensson, N. Controlling graphene corrugation on lattice-mismatched substrates. Phys. Rev. B 2008, 78, 073401. [Google Scholar] [CrossRef]
- Pang, Z.; Deng, B.; Liu, Z.; Peng, H.; Wei, Y. Defects guided wrinkling in graphene on copper substrate. Carbon 2019, 143, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jin, J.; Yang, P.; Li, S.; Tang, S.; Zong, Y.; Peng, Q. Effect of interfacial bonding on dislocation strengthening in graphene nanosheet reinforced iron composite: A molecular dynamics study. Comput. Mater. Sci. 2021, 191, 110309. [Google Scholar] [CrossRef]
- Shuang, F.; Aifantis, K.E. Relating the strength of graphene/metal composites to the graphene orientation and position. Scr. Mater. 2020, 181, 70–75. [Google Scholar] [CrossRef]
- Shuang, F.; Aifantis, K.E. Modelling dislocation-graphene interactions in a BCC Fe matrix by molecular dynamics simulations and gradient plasticity theory. Appl. Surf. Sci. 2021, 535, 147602. [Google Scholar] [CrossRef]
- Shuang, F.; Aifantis, K.E. Dislocation-graphene interactions in Cu/graphene composites and the effect of boundary conditions: A molecular dynamics study. Carbon 2021, 172, 50–70. [Google Scholar] [CrossRef]
- Yang, Y.; Song, G.; Fu, Q.; Pan, C. Preparation of Fe-Gr composite layer via DC electro-plating for high performances. J. Alloys Compd. 2018, 768, 859–865. [Google Scholar] [CrossRef]
- Wang, L.; Jin, J.; Yang, P.; Zong, Y.; Peng, Q. Graphene Adhesion Mechanics on Iron Substrates: Insight from Molecular Dynamic Simulations. Crystals 2019, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Hepburn, D.J.; Ackland, G.J. Metallic-covalent interatomic potential for carbon in iron. Phys. Rev. B 2008, 78, 165115. [Google Scholar] [CrossRef]
- He, X.; Bai, Q.; Shen, R. Atomistic perspective of how graphene protects metal substrate from surface damage in rough contacts. Carbon 2018, 130, 672–679. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Modelling. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Bai, Q.-S.; Dou, Y.-H.; He, X.; Zhang, A.-M.; Guo, Y.-B. Deposition and growth mechanism of graphene on copper crystal surface based on molecular dynamics simulation. Acta Phys. Sin. 2020, 69, 226102. [Google Scholar] [CrossRef]
- Ding, J.; Qiao, Z.H.; Feng, W.X.; Yao, Y.; Niu, Q. Engineering quantum anomalous/valley Hall states in graphene via metal-atom adsorption: An ab-initio study. Phys. Rev. B 2011, 84, 195444. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Hu, X.; Wu, X.; Du, C.; Dai, Y.; Deng, J. Density functional calculation of transition metal adatom adsorption on graphene. Phys. B Condens. Matter 2010, 405, 3337–3341. [Google Scholar] [CrossRef]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface Properties | (110) | () | (111) | () | (001) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| LJ | EAM | LJ | EAM | LJ | EAM | LJ | EAM | LJ | EAM | |
| E (J/m2) | −1.00 | −5.63 | −1.00 | −5.63 | −0.73 | −4.32 | −0.82 | −4.39 | −0.81 | −4.52 |
| (Å) | 2.02 | 2.02 | 2.06 | 2.32 | 2.15 | |||||
| (Å) | 2.15 | 1.73 | 2.15 | 1.73 | 2.05 | 1.66 | 2.17 | 1.75 | 2.15 | 1.76 |
| (Å) | 4.16 | 3.78 | 4.16 | 3.78 | 3.61 | 2.36 | 3.27 | 2.76 | 4.16 | 3.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.; Jin, J.; Wang, L.; Li, S.; Wang, H.; Tang, S.; Peng, Q. Interfacial Characteristics of Graphene-Reinforced Iron Composites: A Molecular Dynamics Study. Crystals 2023, 13, 27. https://doi.org/10.3390/cryst13010027
Song M, Jin J, Wang L, Li S, Wang H, Tang S, Peng Q. Interfacial Characteristics of Graphene-Reinforced Iron Composites: A Molecular Dynamics Study. Crystals. 2023; 13(1):27. https://doi.org/10.3390/cryst13010027
Chicago/Turabian StyleSong, Miaomiao, Jianfeng Jin, Lu Wang, Shaojie Li, Huiming Wang, Shuai Tang, and Qing Peng. 2023. "Interfacial Characteristics of Graphene-Reinforced Iron Composites: A Molecular Dynamics Study" Crystals 13, no. 1: 27. https://doi.org/10.3390/cryst13010027