
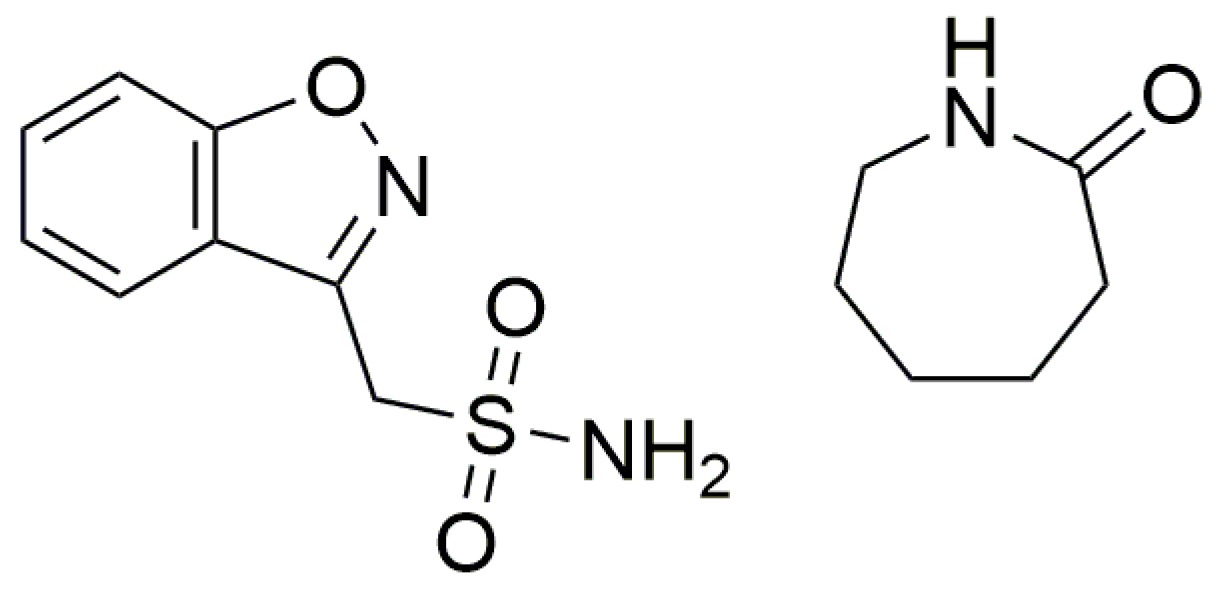
DFT–Assisted Structure Determination from Powder X-ray Diffraction Data of a New Zonisamide/ϵ-Caprolactam Cocrystal




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials, Experimental and Theoretical Methods
2.1. Materials
2.2. Sample Preparation
2.3. X-ray Powder Diffraction: Data Collection
2.4. DFT Calculations
3. Results and Discussion
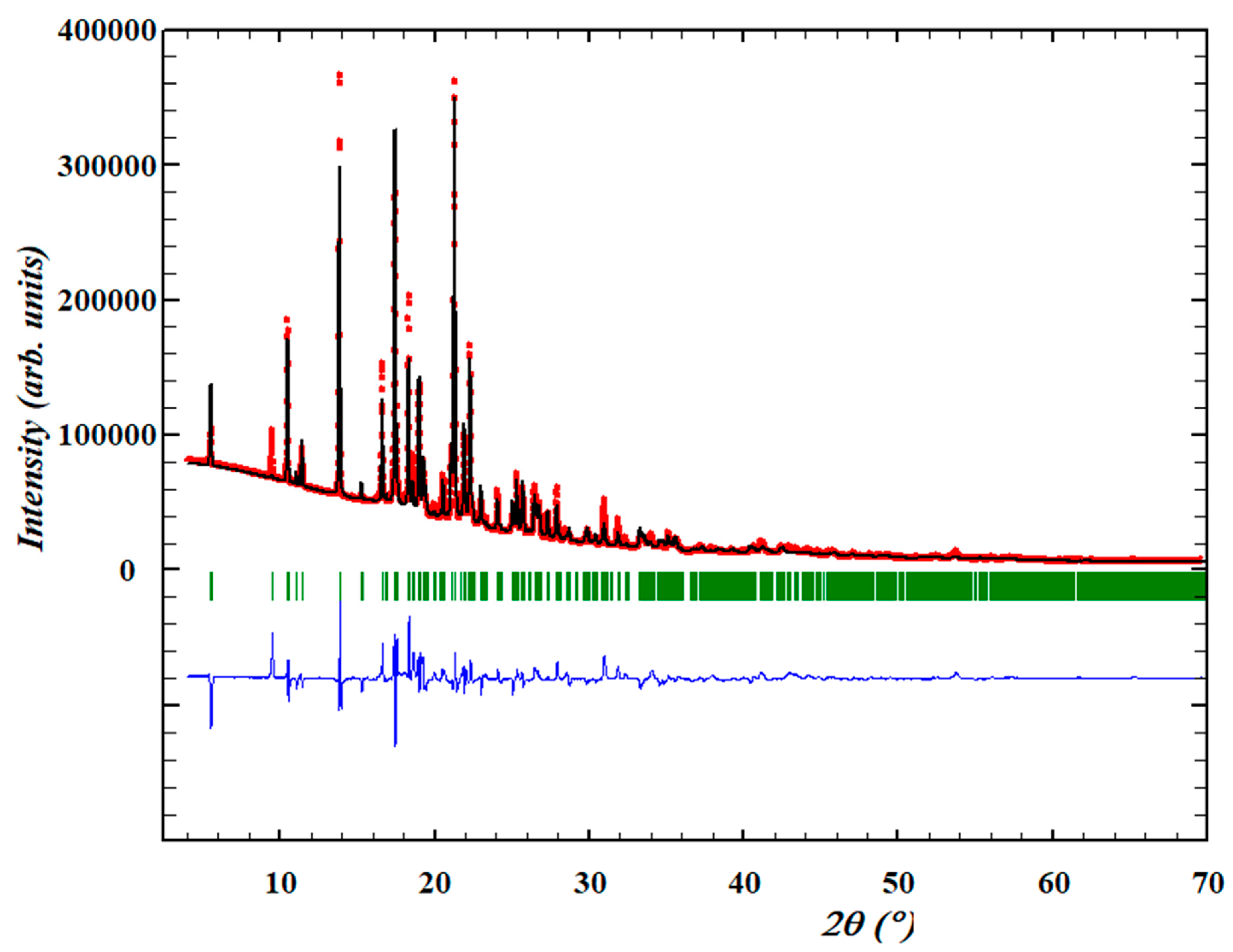
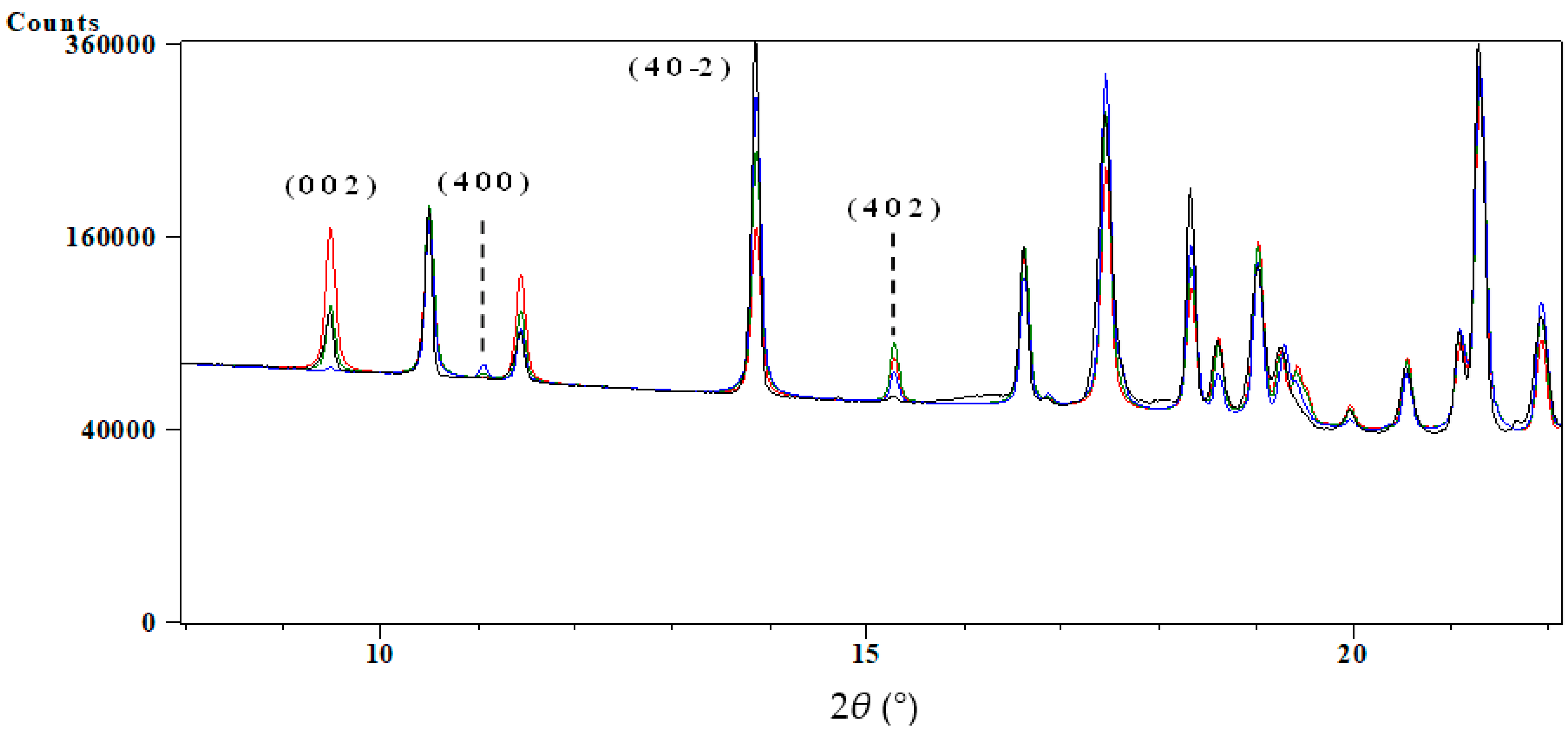
3.1. Structure Determination from Space Direct Methods
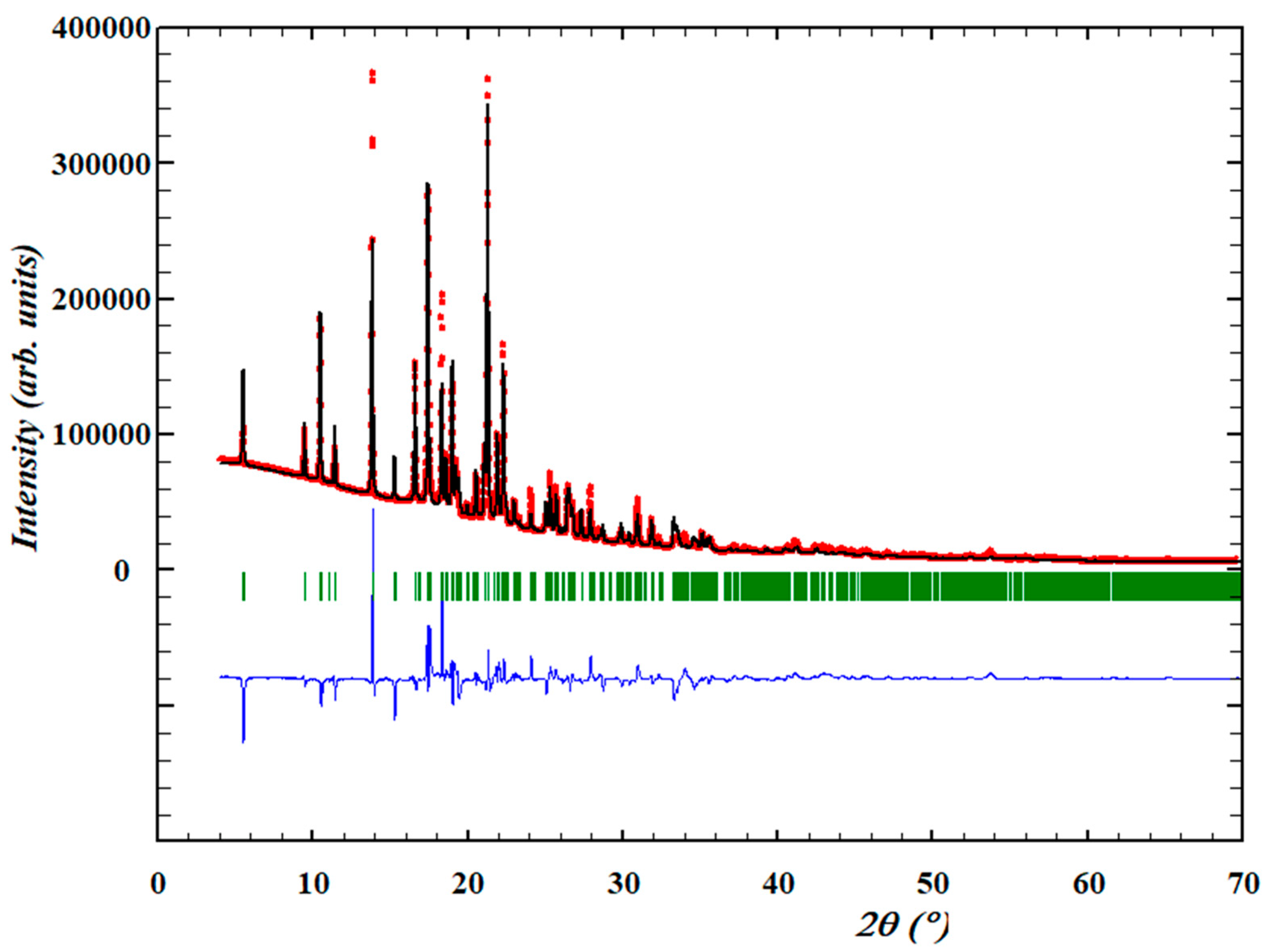
3.1.1. Structure Determination and Initial Rietveld Refinement
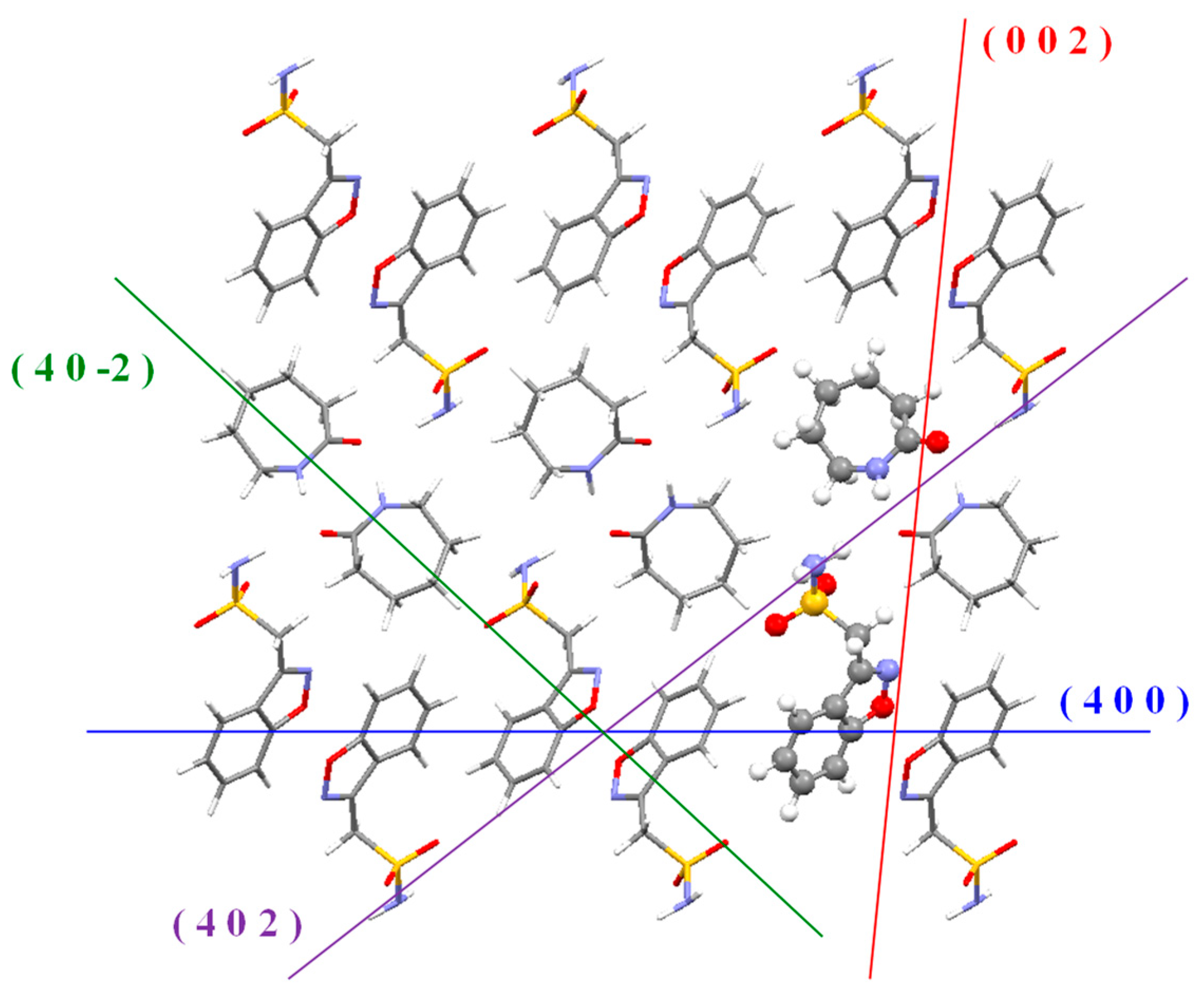
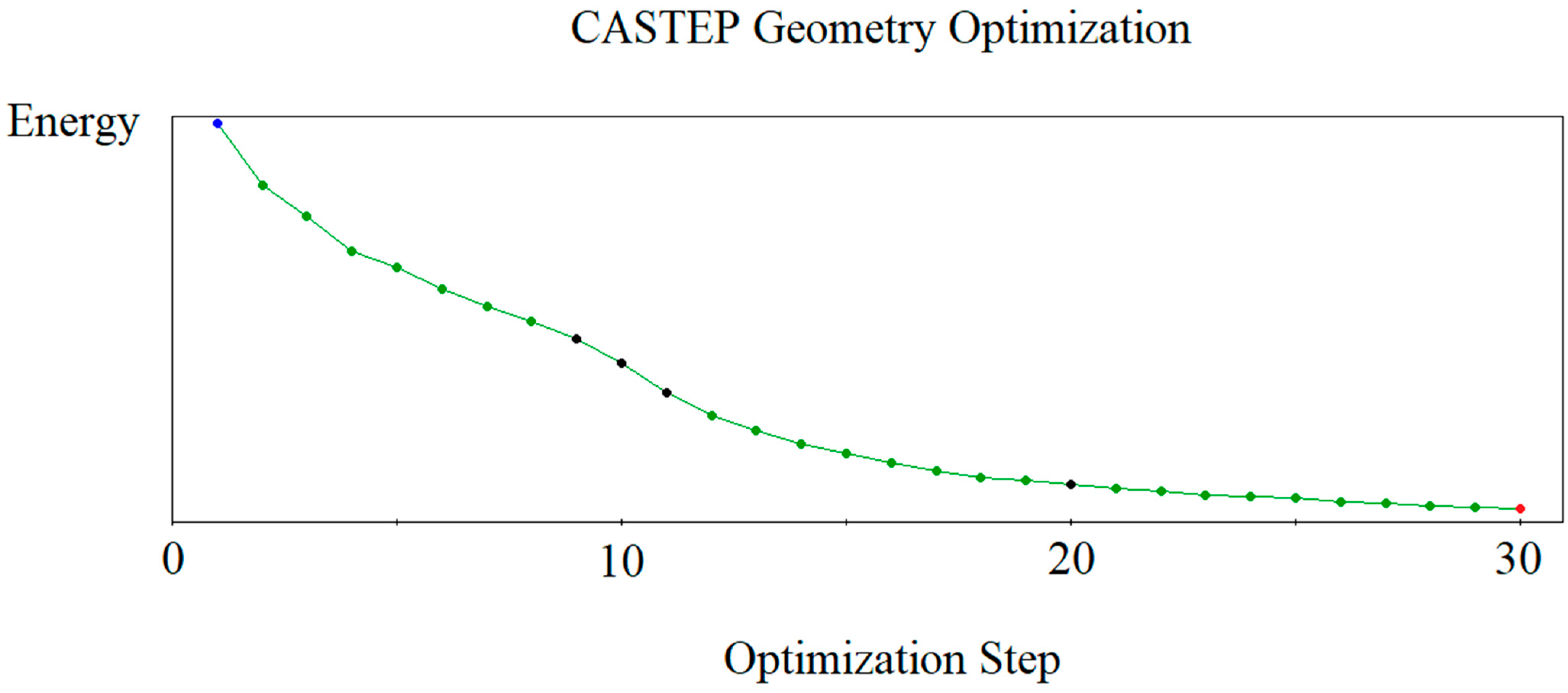
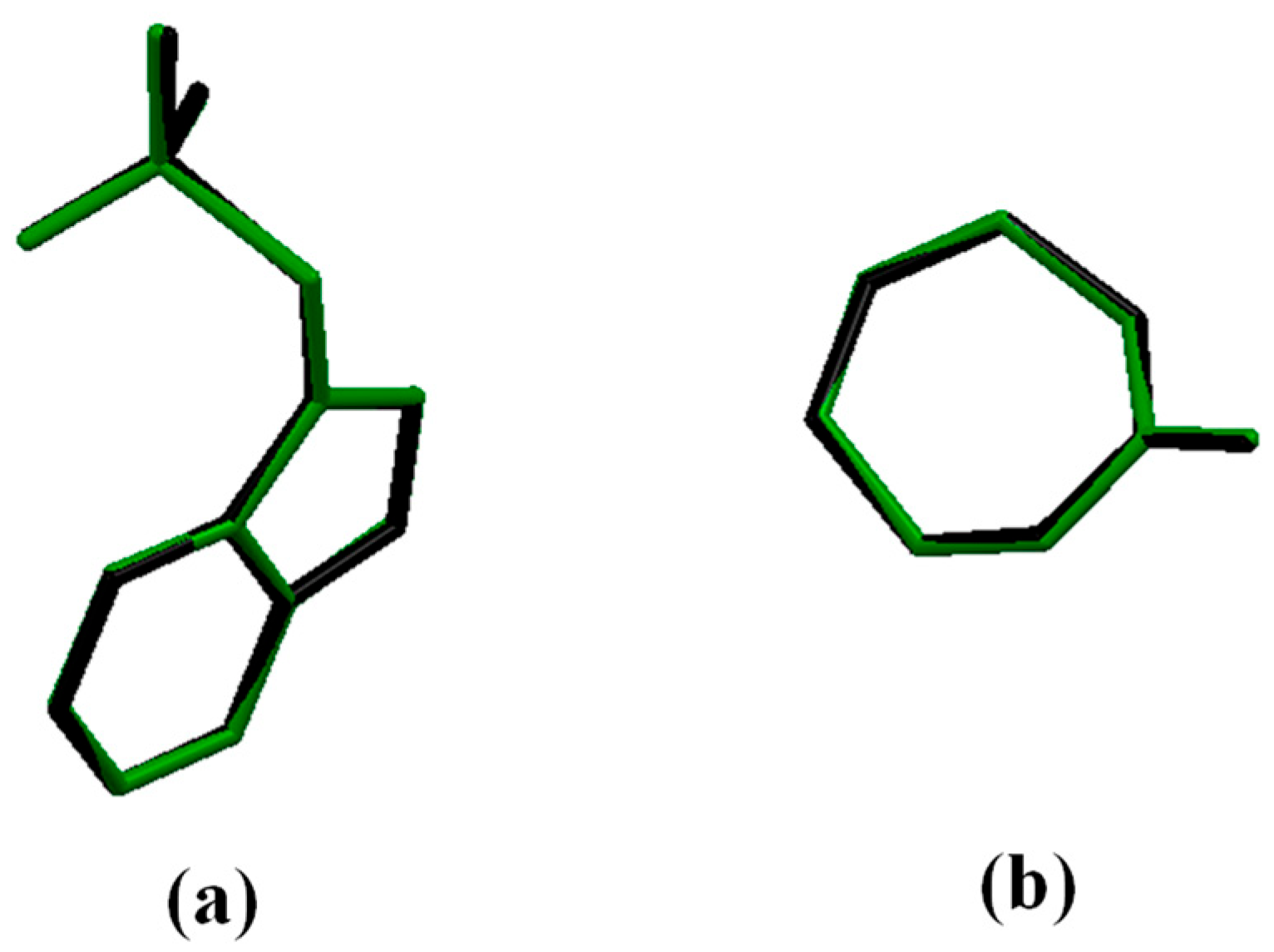
3.1.2. Crystal Structure Optimization
3.1.3. Crystal Structure Validation by DFT-D
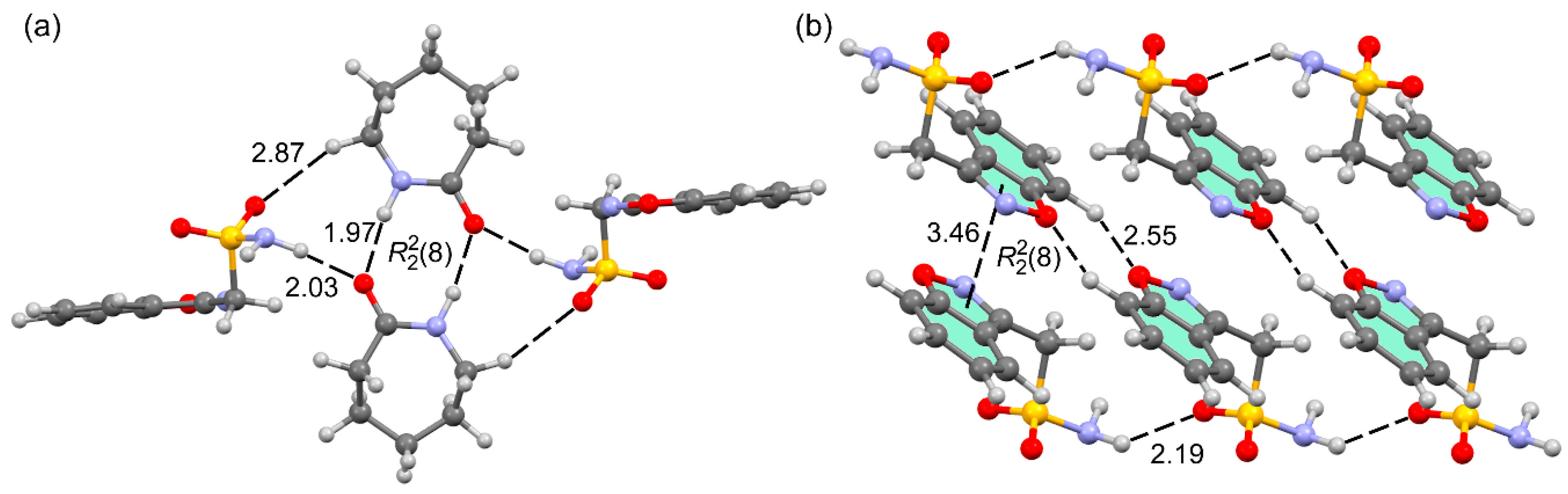
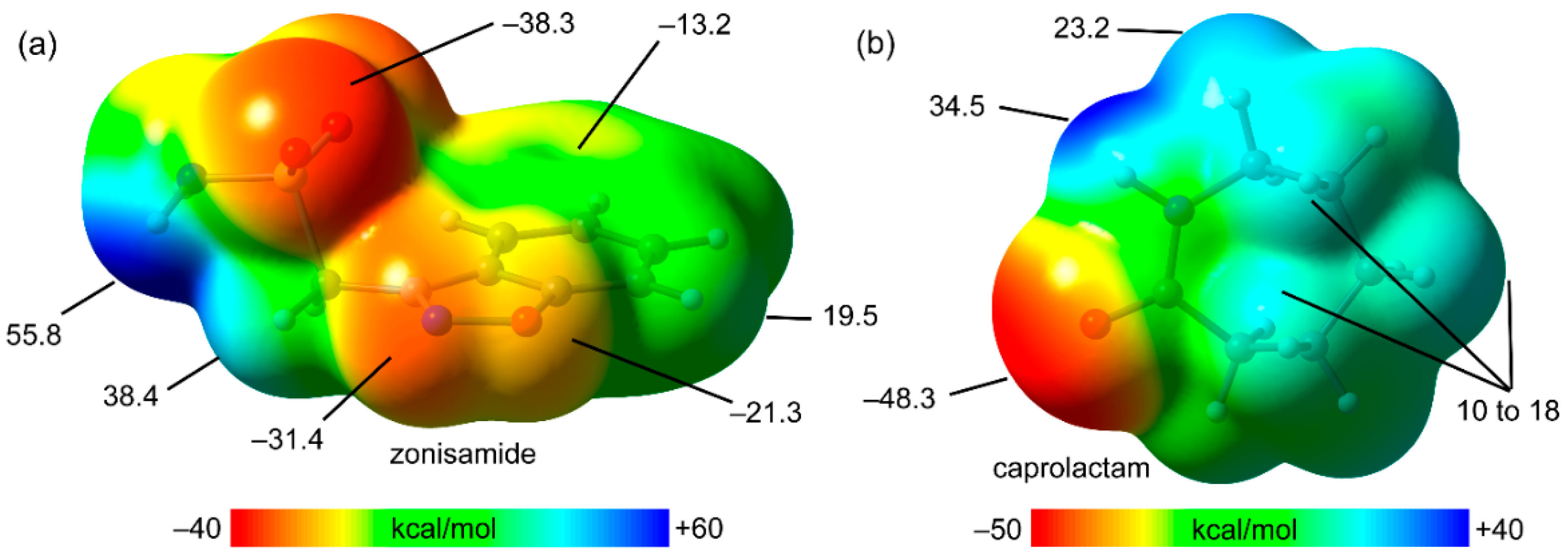
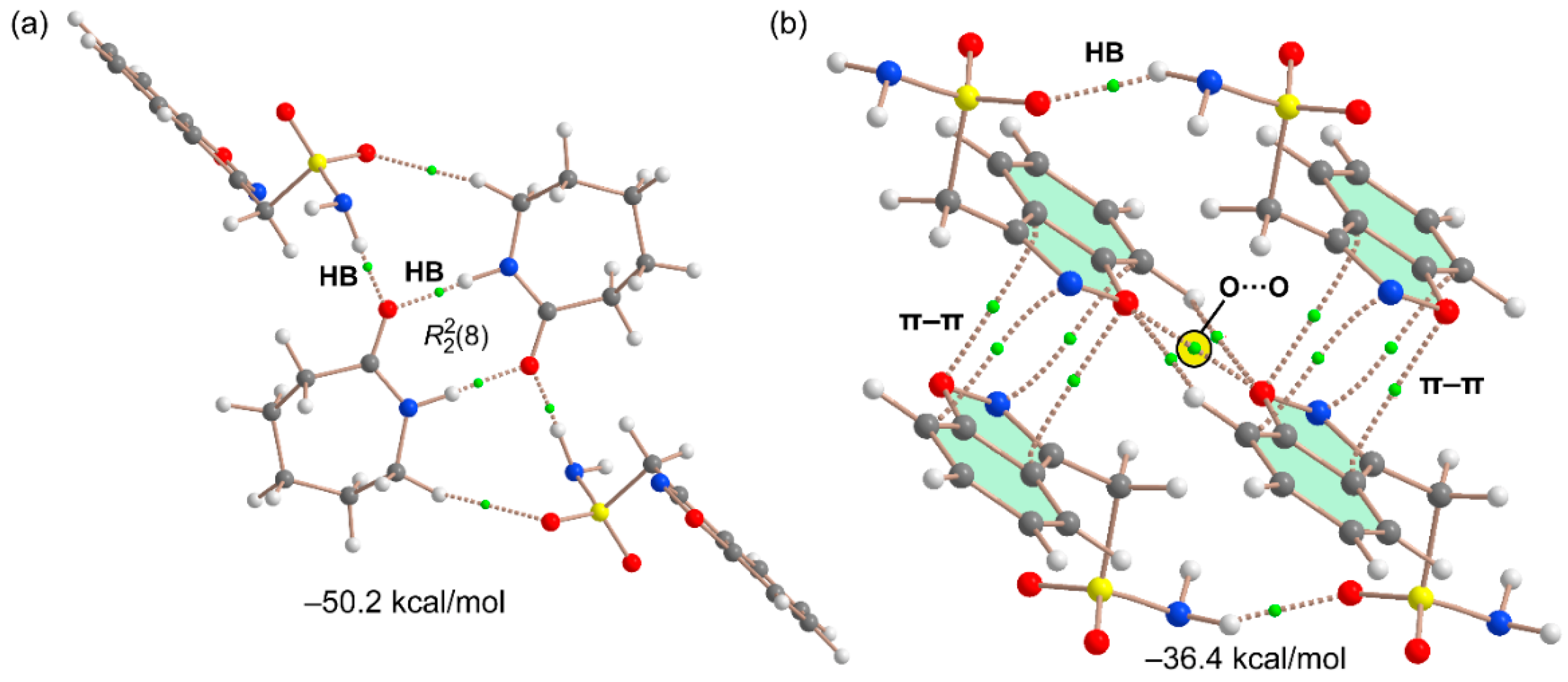
3.2. DFT Calculations of the Supramolecular Assemblies
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Bofill, L.; Barbas, R.; de Sande, D.; Font-Bardia, M.; Ràfols, C.; Albertí, J.; Prohens, R. A Novel, Extremely Bioavailable Cocrystal of Pterostilbene. Cryst. Growth Des. 2021, 21, 2315–2323. [Google Scholar] [CrossRef]
- Bofill, L.; de Sande, D.; Barbas, R.; Prohens, R. New Cocrystal of Ubiquinol with High Stability to Oxidation. Cryst. Growth Des. 2020, 20, 5583–5588. [Google Scholar] [CrossRef]
- Bofill, L.; de Sande, D.; Barbas, R.; Prohens, R. A New and Highly Stable Cocrystal of Vitamin D3 for Use in Enhanced Food Supplements. Cryst. Growth Des. 2021, 21, 1418–1423. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, O.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef]
- Thipparaboina, R.; Kumar, D.; Chavan, R.B.; Shastri, N.R. Multidrug co-crystals: Towards the development of effective therapeutic hybrids. Drug Discov. Today 2016, 21, 481–490. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Delivery Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef]
- Wilfong, A.A.; Willmore, L.J. Zonisamide–A review of experience and use in partial seizures. Neuropsychiatr. Dis. Treat. 2006, 2, 269–280. [Google Scholar] [CrossRef]
- Masuda, Y.; Karasawa, T.; Shiraishi, Y.; Hori, M.; Yoshida, K.; Shimizu, M. 3-Sulfamoylmethyl-1, 2-benzisoxazole, a new type of anticonvulsant drug. Pharmacological profile. Arzneim. Forsch. 1980, 30, 477–483. [Google Scholar] [CrossRef]
- Ojemann, L.M.; Shastri, R.A.; Wilensky, A.J.; Friel, P.N.; Levy, R.H.; McLean, J.R.; Buchanan, R.A. Comparative pharmacokinetics of zonisamide (CI-912) in epileptic patients on carbamazepine or phenytoin monotherapy. Ther. Drug Monit. 1986, 8, 293–296. [Google Scholar] [CrossRef]
- Shinoda, M.; Akita, M.; Hasegawa, M.; Hasegawa, T.; Nabeshima, T. The necessity of adjusting the dosage of zonisamide when coadministered with other anti-epileptic drugs. Biol. Pharm. Bull. 1996, 19, 1090–1092. [Google Scholar] [CrossRef]
- Aitipamula, S.; Cadden, J.; Chow, P.S. Cocrystals of zonisamide: Physicochemical characterization and sustained release solid forms. CrystEngComm 2018, 20, 2923–2931. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Materials Studio, version 7.0; Accelrys Software, Inc.: San Diego, CA, USA, 2013; Available online: https://www.scientific-computing.com/press-releases/accelrys-materials-studio-70 (accessed on 12 July 2022).
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter Mater. Phys. 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- van de Streek, J.; Neumann, M.A. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Cryst. 2014, B70, 1020–1032. [Google Scholar] [CrossRef]
- van de Streek, J.; Neumann, M.A. Validation of experimental molecular crystal structures with dispersion-corrected density functional theory calculations. Acta Cryst. 2010, B66, 544–558. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154118. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. J. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Todd, A.K. AIMAll (Version 13.05.06); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Boultif, A.; Louër, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- Boultif, A.; Louër, D. Powder pattern indexing with the dichotomy method. J. Appl. Cryst. 2004, 37, 724. [Google Scholar] [CrossRef]
- Vallcorba, O.; Rius, J.; Frontera, C.; Peral, I.; Miravitlles, C. DAJUST: A suite of computer programs for pattern matching, space-group determination and intensity extraction from powder diffraction data. J. Appl. Cryst. 2012, 45, 844–848. [Google Scholar] [CrossRef]
- Spartan’10, Irvin, CA, USA, 2014.
- Favre-Nicolin, V.; Černý, R. FOX, ‘free objects for crystallography’: A modular approach to ab initio structure determination from powder diffraction. J. Appl. Cryst. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- Rodriguez–Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter. 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Sykes, R.A.; McCabe, P.; Allen, F.H.; Battle, G.M.; Bruno, I.J.; Wood, P.A. New software for statistical analysis of Cambridge Structural Database data. J. Appl. Crystallogr. 2011, 44, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Jabłonski, M. Counterintuitive bond paths: An intriguing case of the C(NO2)3– ion. Chem. Phys. Lett. 2020, 759, 137946. [Google Scholar] [CrossRef]
- Jabłonski, M. Bond Paths Between Distant Atoms Do Not Necessarily Indicate Dominant Interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbas, R.; Portell, A.; Prohens, R.; Frontera, A. DFT–Assisted Structure Determination from Powder X-ray Diffraction Data of a New Zonisamide/ϵ-Caprolactam Cocrystal. Crystals 2022, 12, 1020. https://doi.org/10.3390/cryst12081020
Barbas R, Portell A, Prohens R, Frontera A. DFT–Assisted Structure Determination from Powder X-ray Diffraction Data of a New Zonisamide/ϵ-Caprolactam Cocrystal. Crystals. 2022; 12(8):1020. https://doi.org/10.3390/cryst12081020
Chicago/Turabian StyleBarbas, Rafael, Anna Portell, Rafel Prohens, and Antonio Frontera. 2022. "DFT–Assisted Structure Determination from Powder X-ray Diffraction Data of a New Zonisamide/ϵ-Caprolactam Cocrystal" Crystals 12, no. 8: 1020. https://doi.org/10.3390/cryst12081020