
Multicomponent Solids of DL-2-Hydroxy-2-phenylacetic Acid and Pyridinecarboxamides

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Experimental Part
2.1. Materials and Physical Measurements
2.2. Cocrystal Screening
2.3. Cocrystal Synthesis
2.4. Solubility Determination
2.5. Theoretical Methods
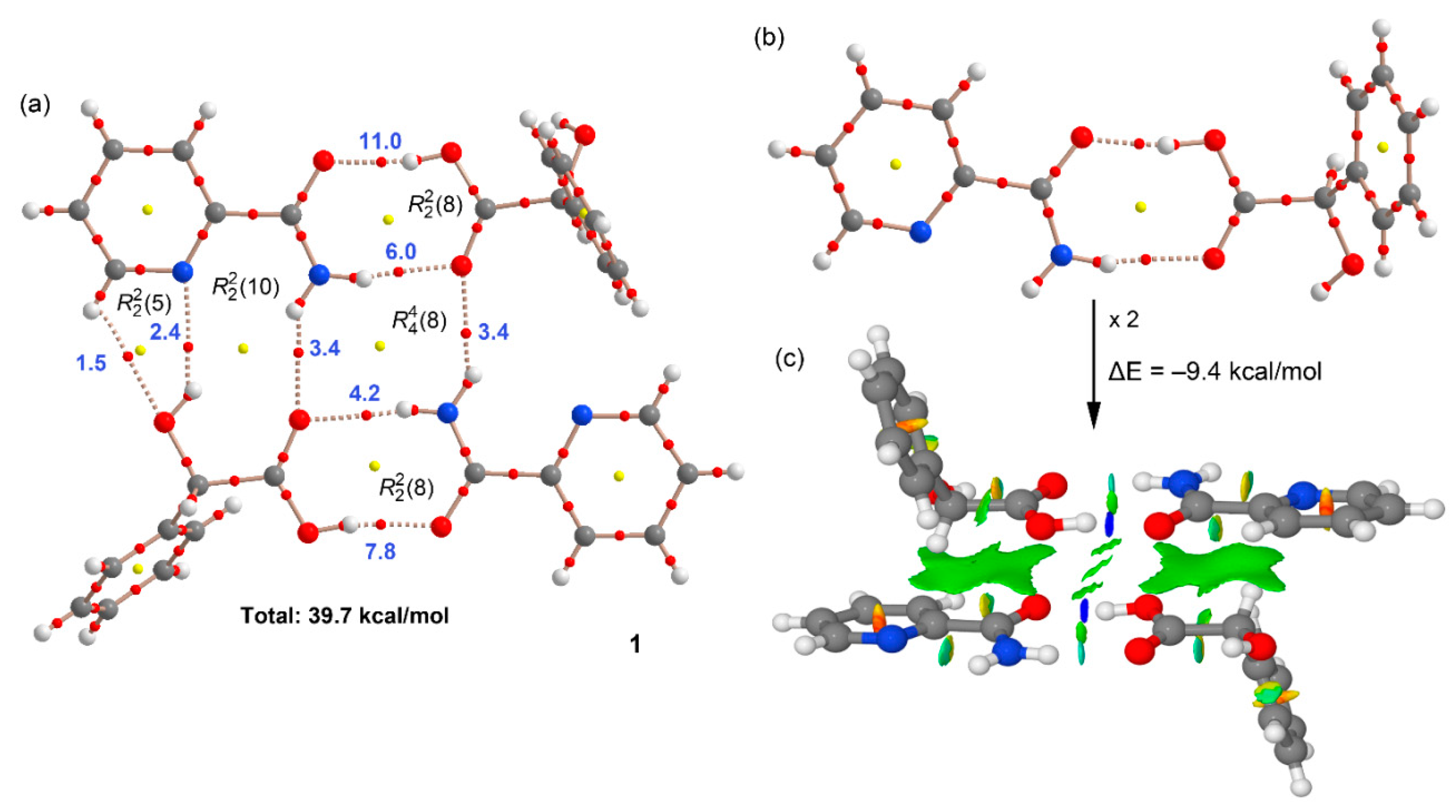
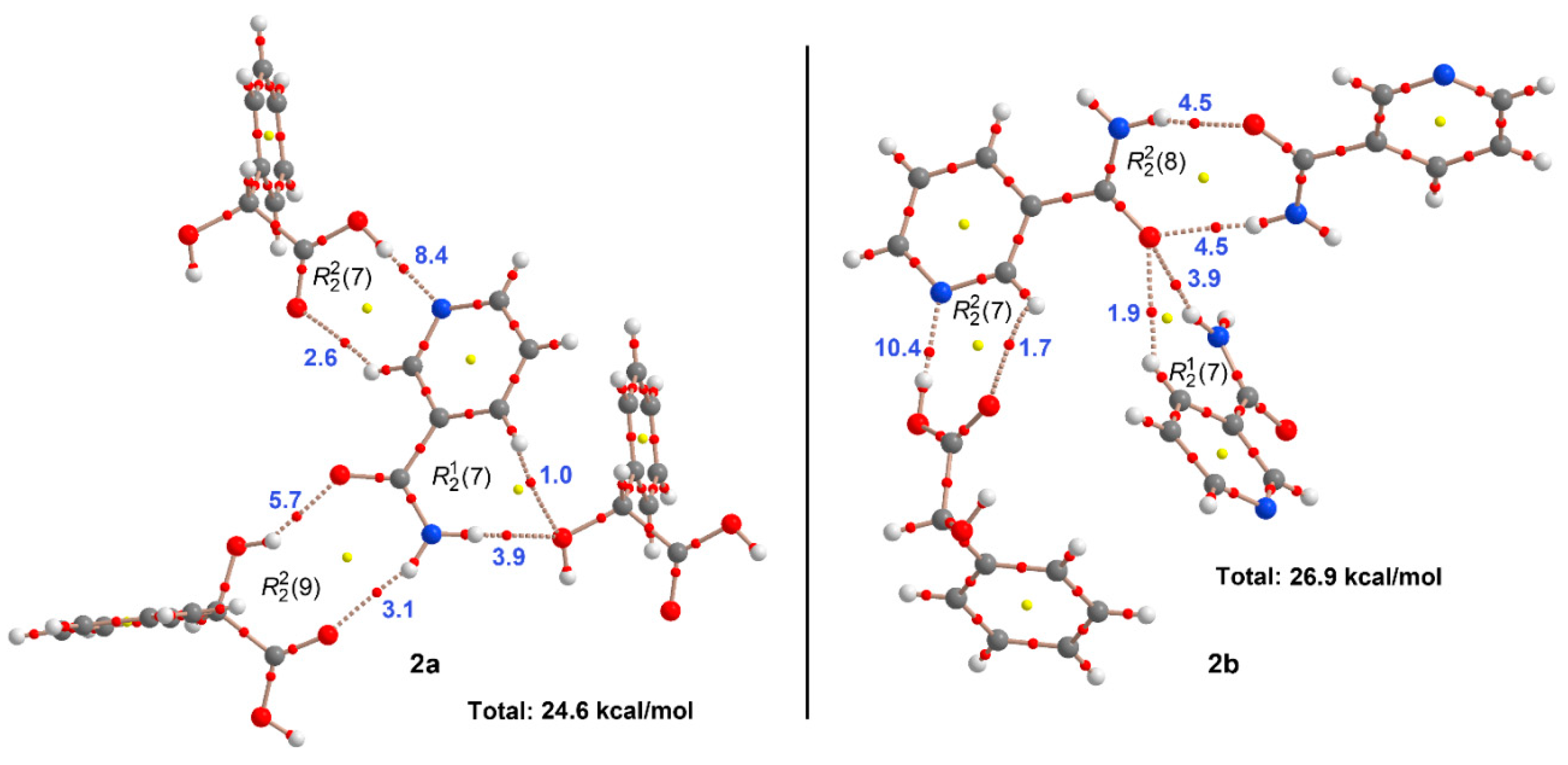
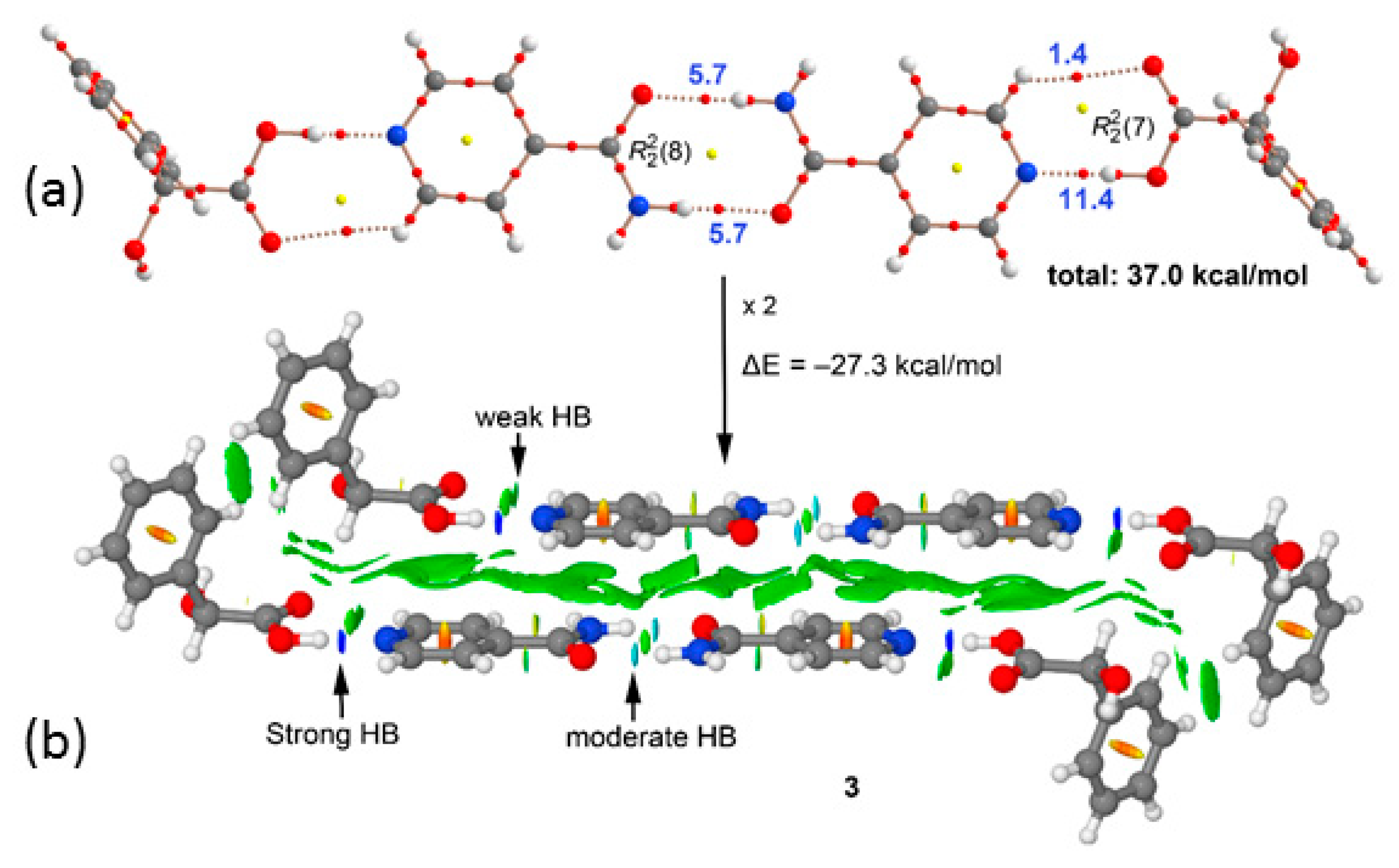
3. Results and Discussion
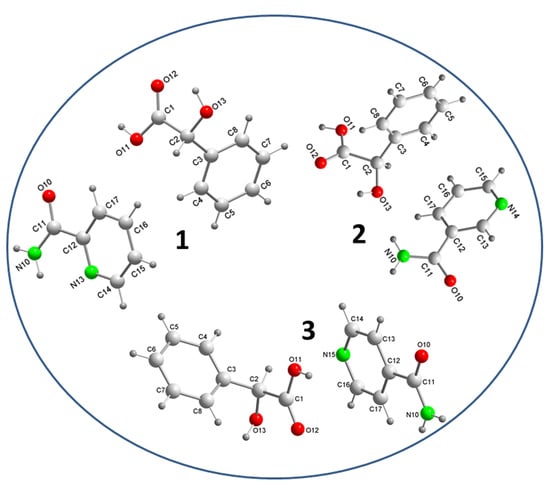
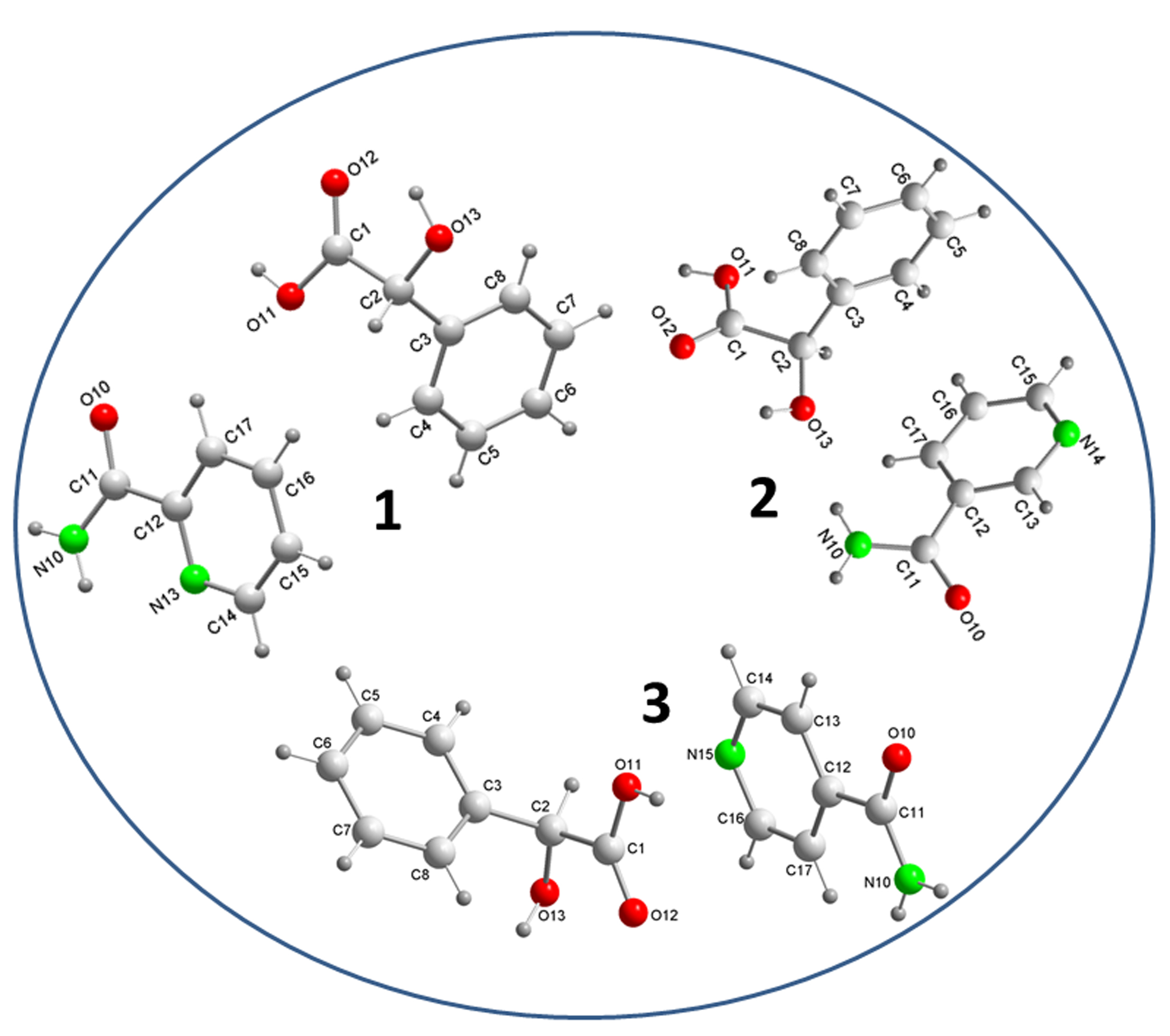
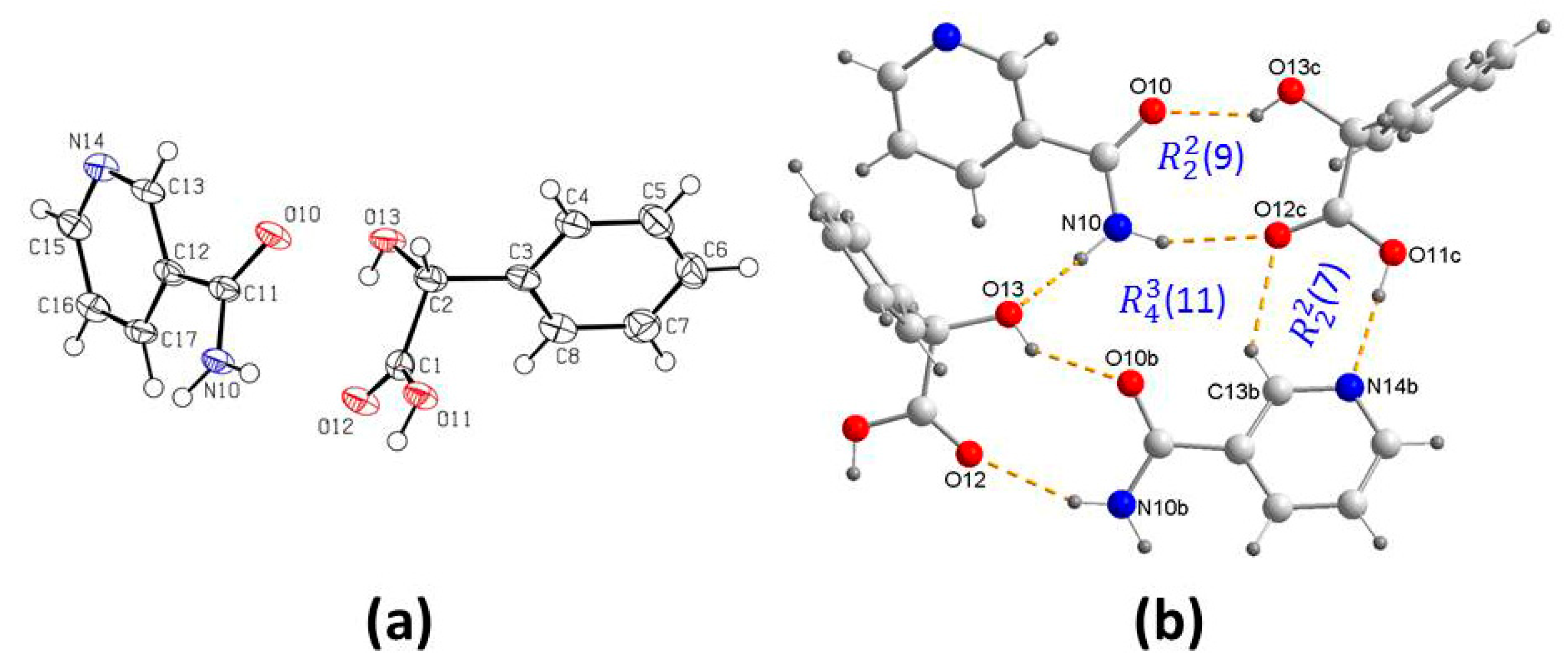
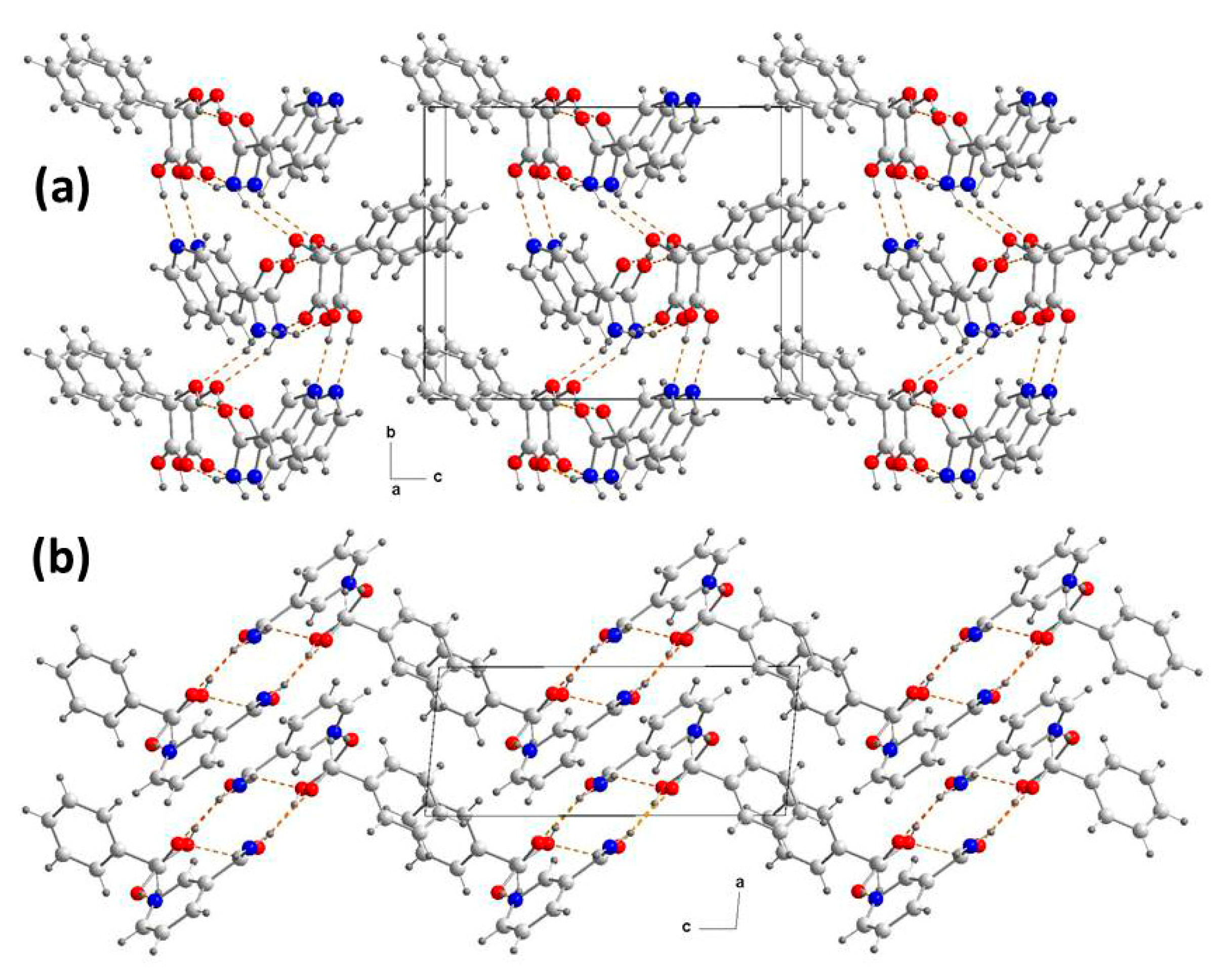
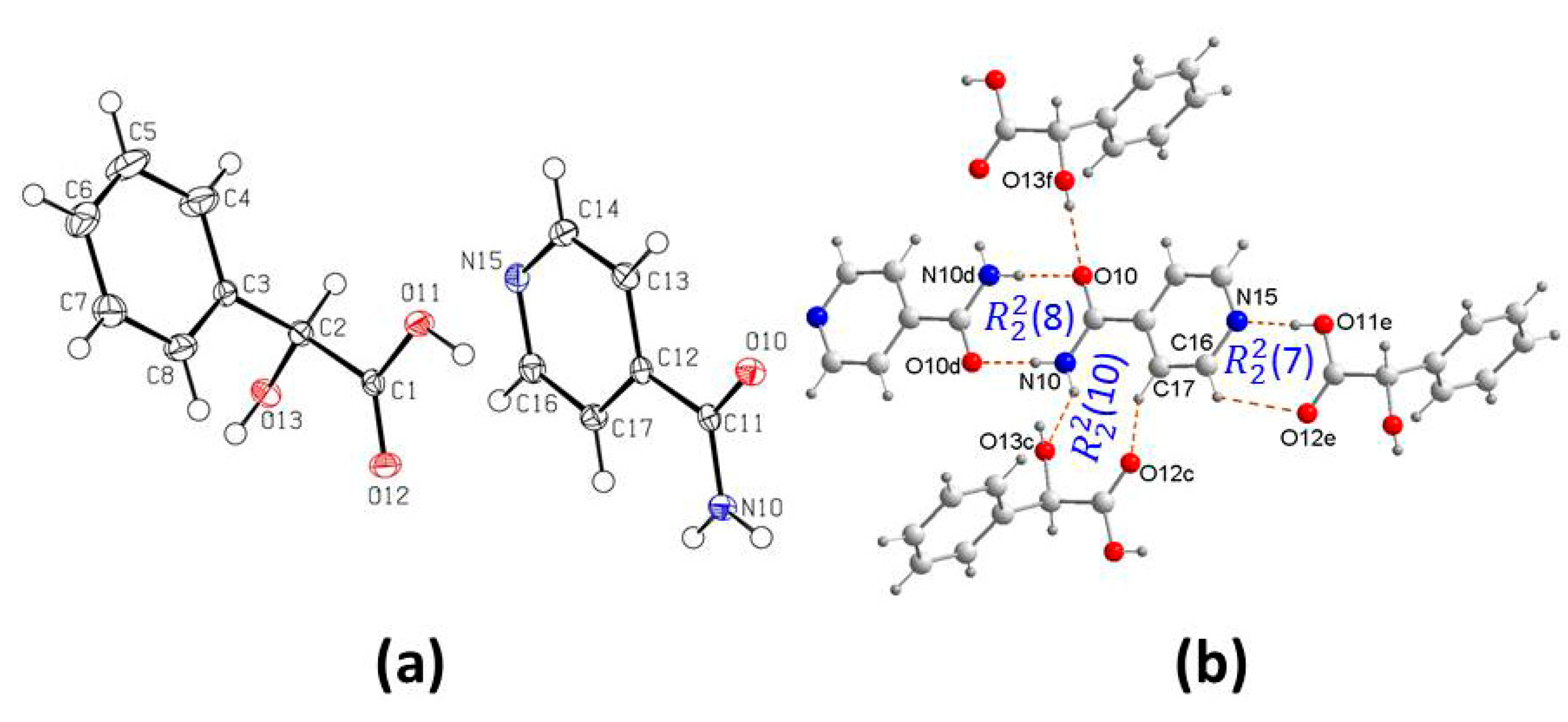
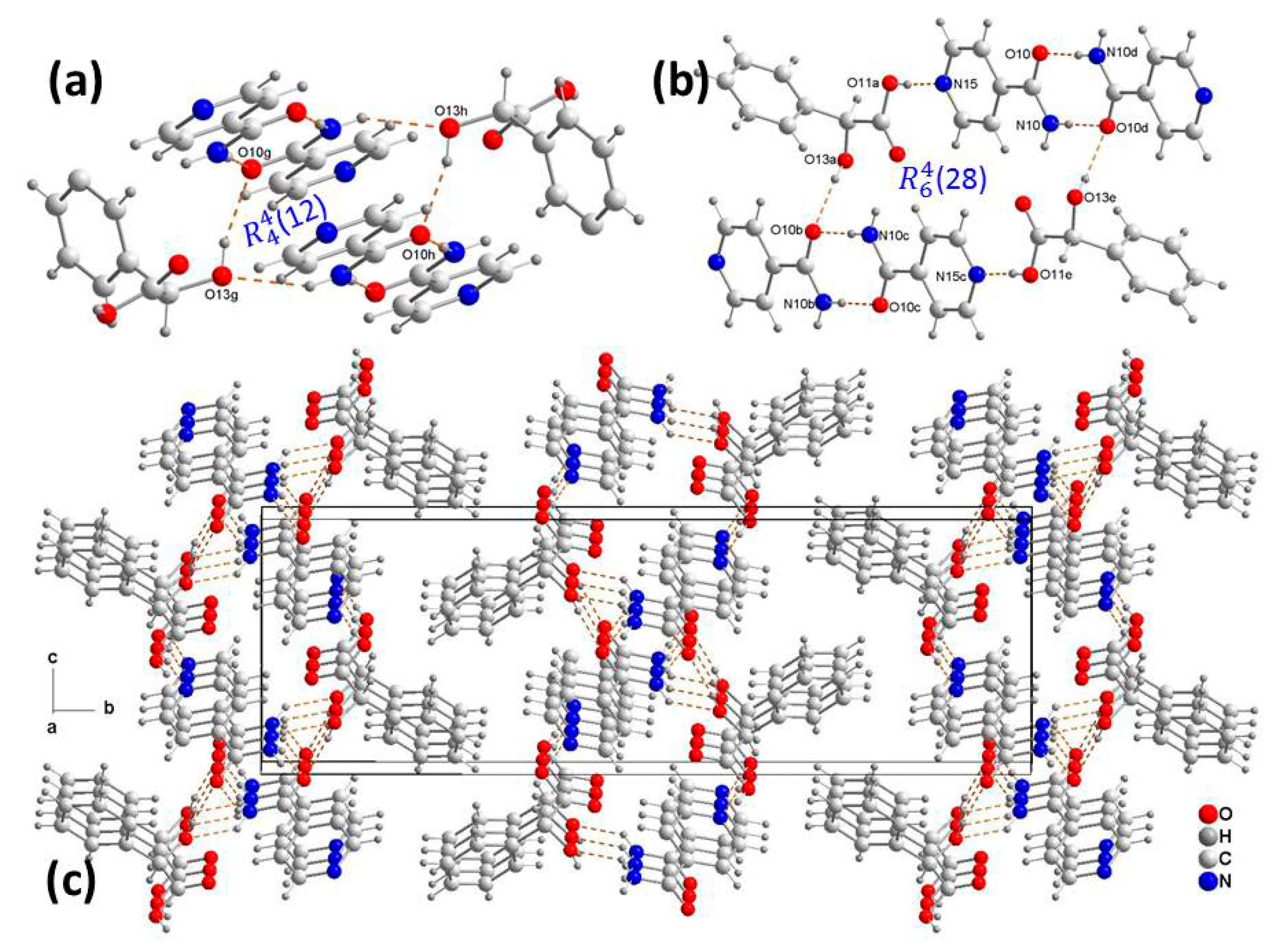
3.1. Crystal Structure Analysis
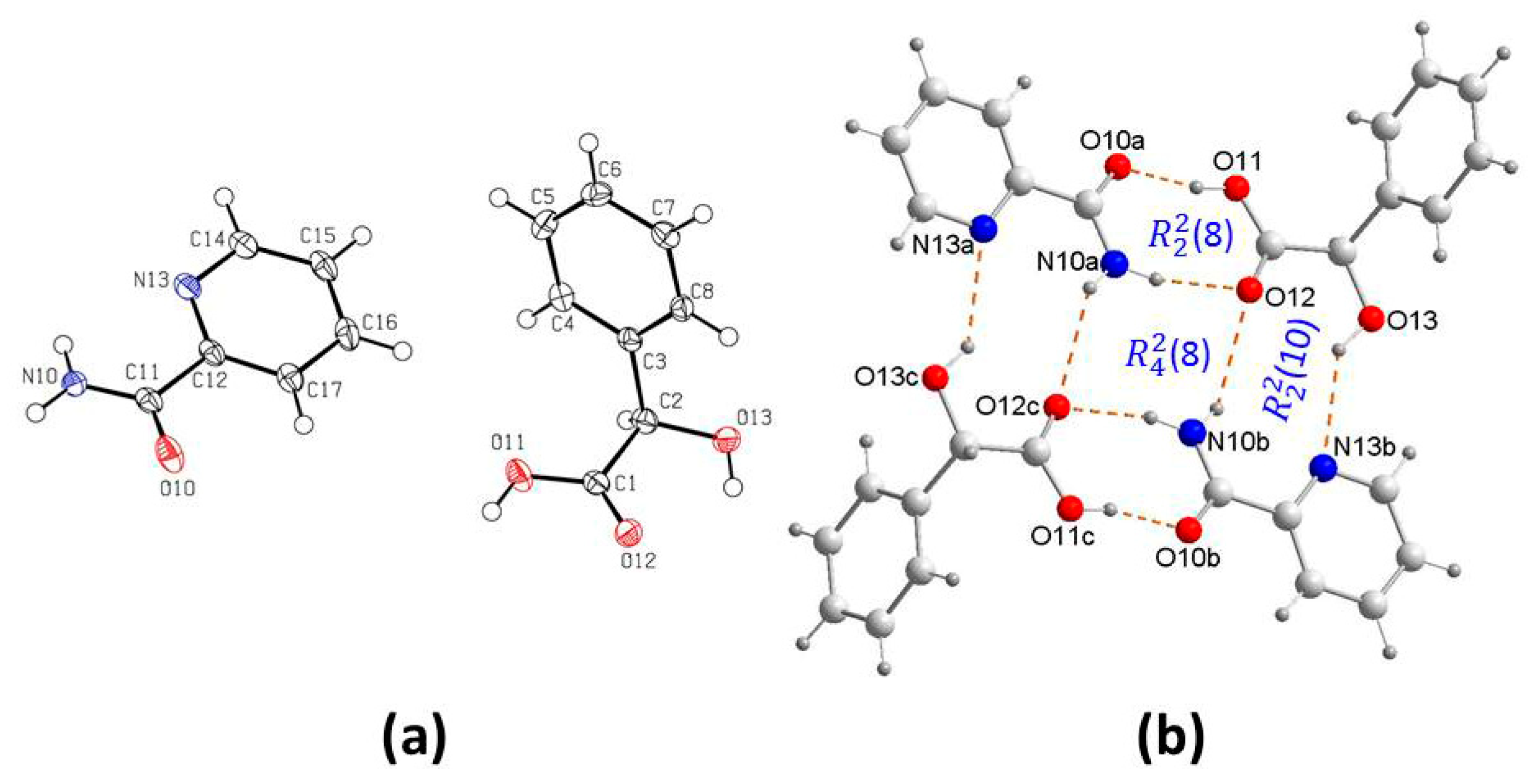
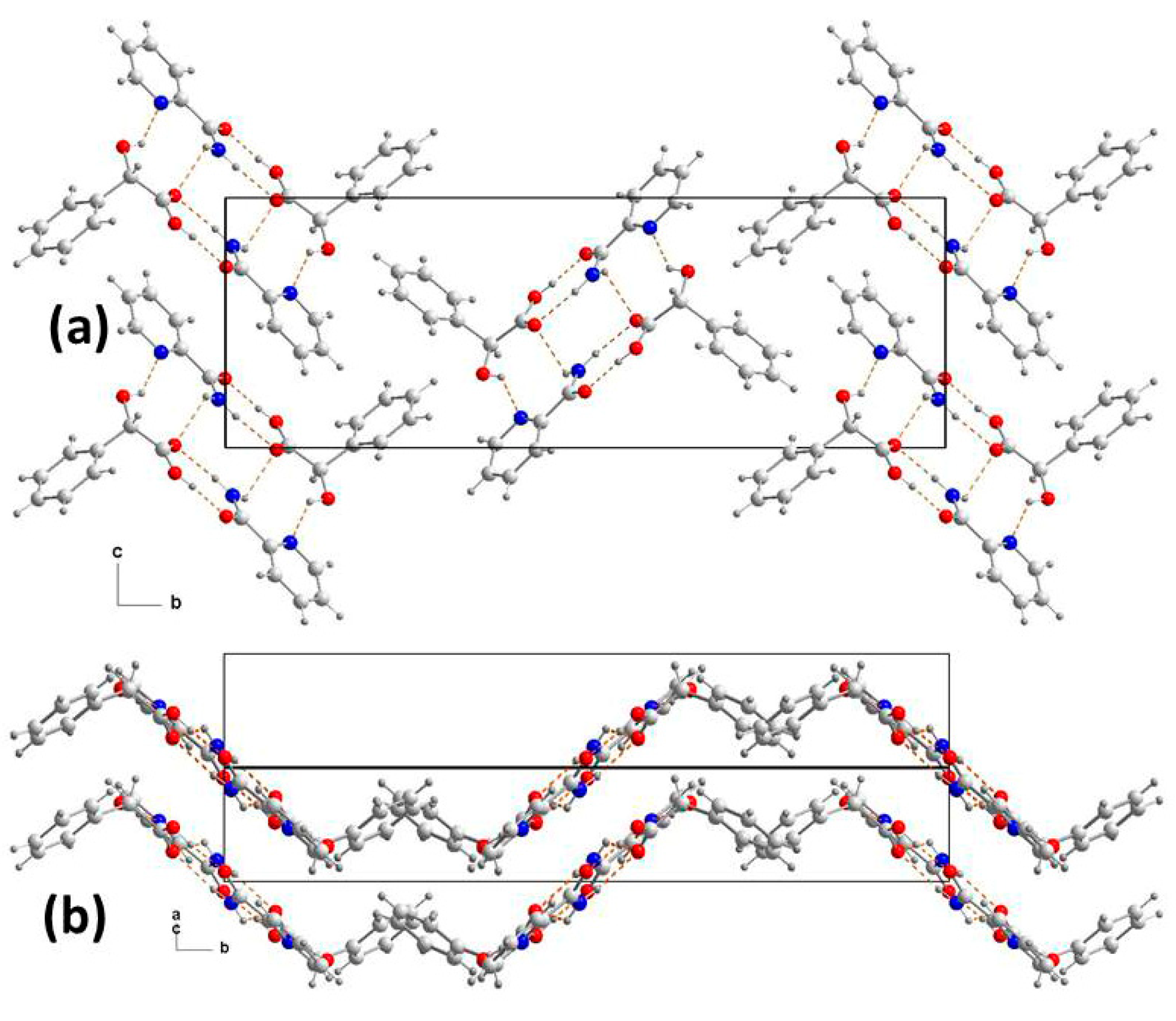
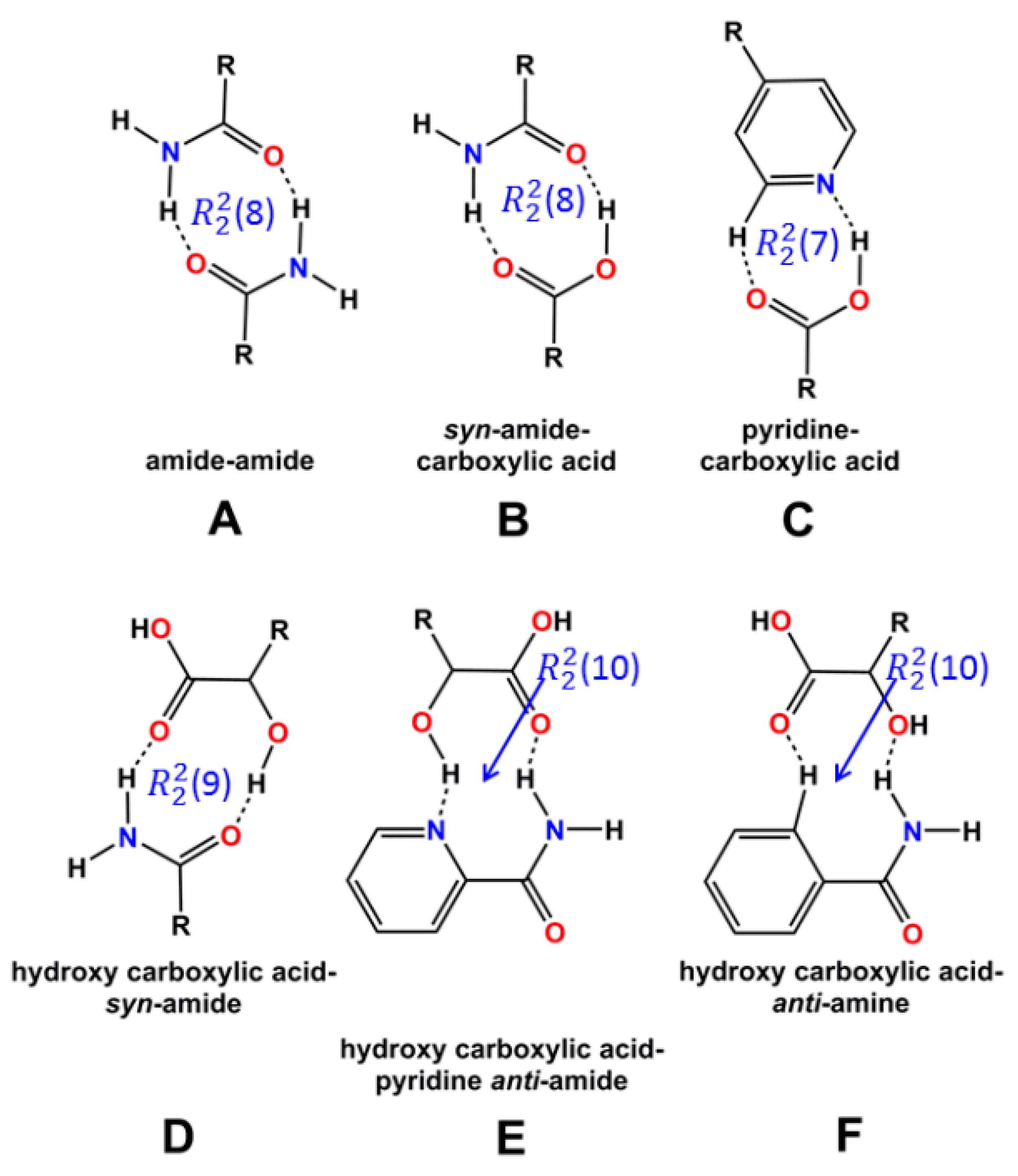
3.2. Supramolecular Synthons
3.3. Theoretical DFT Study
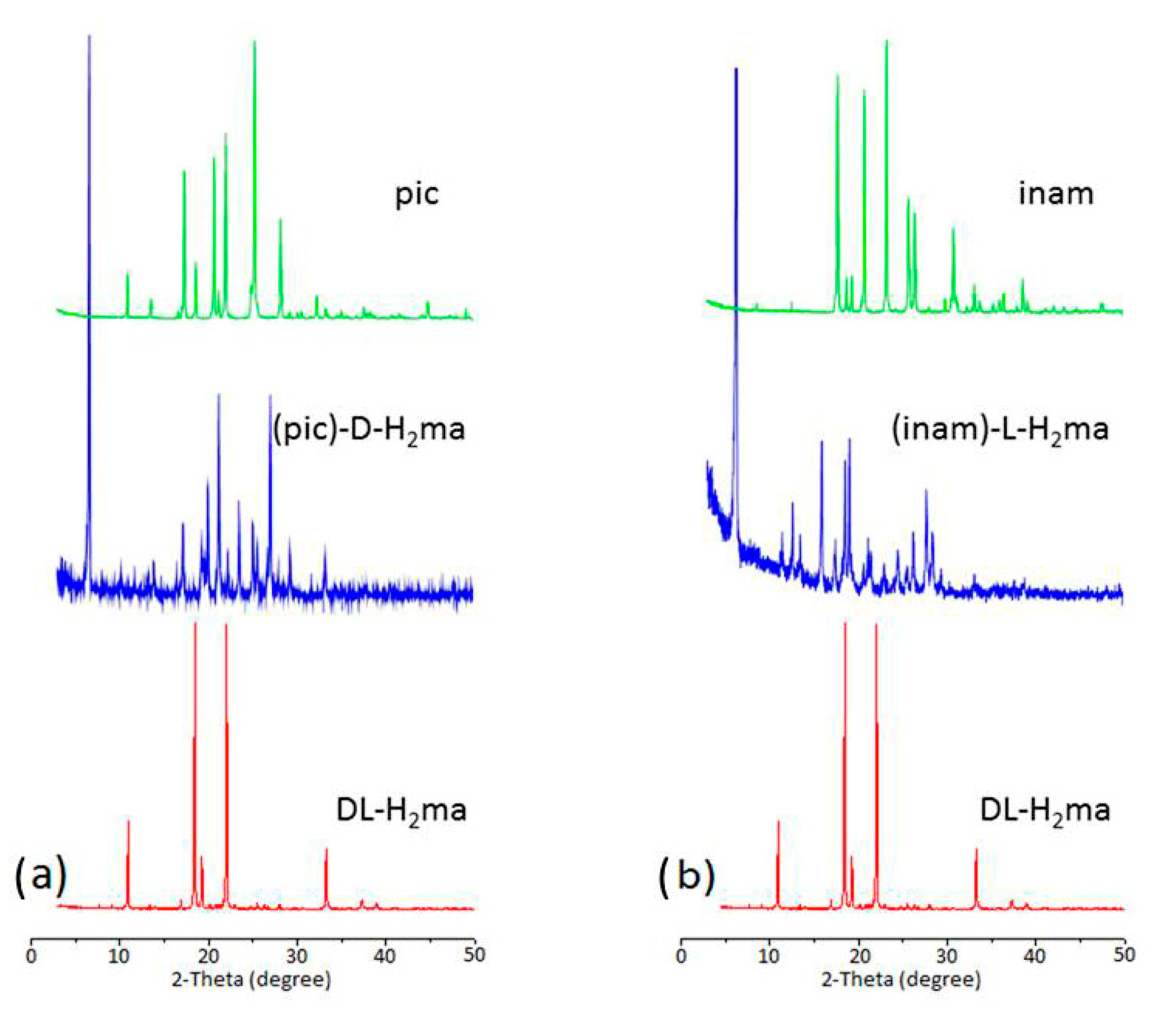
3.4. X-ray Powder Diffraction (XRPD) Analysis
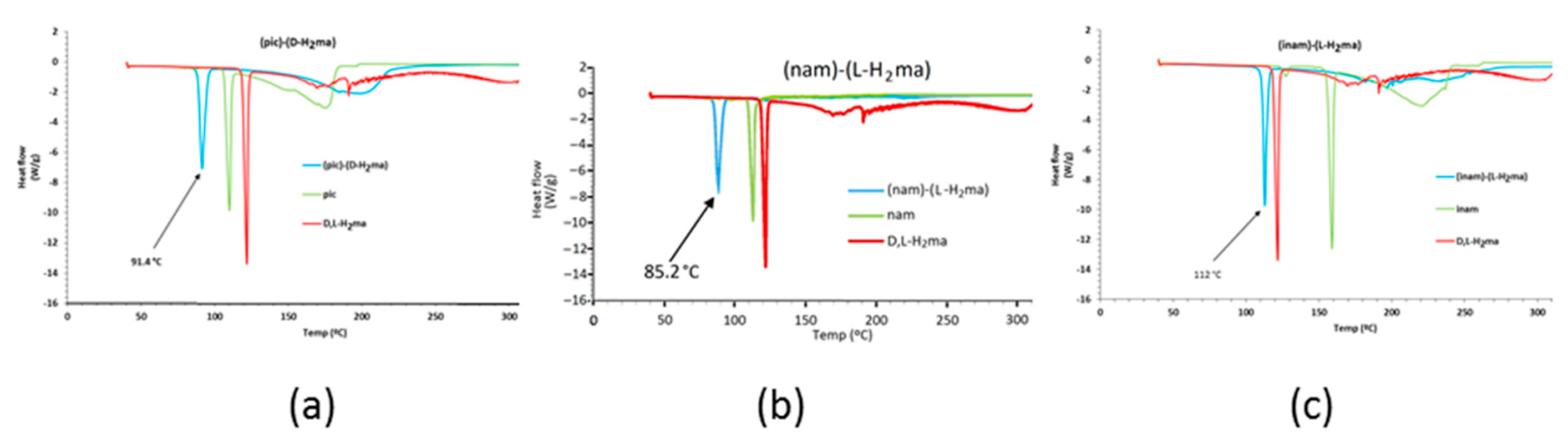
3.5. DSC Analysis
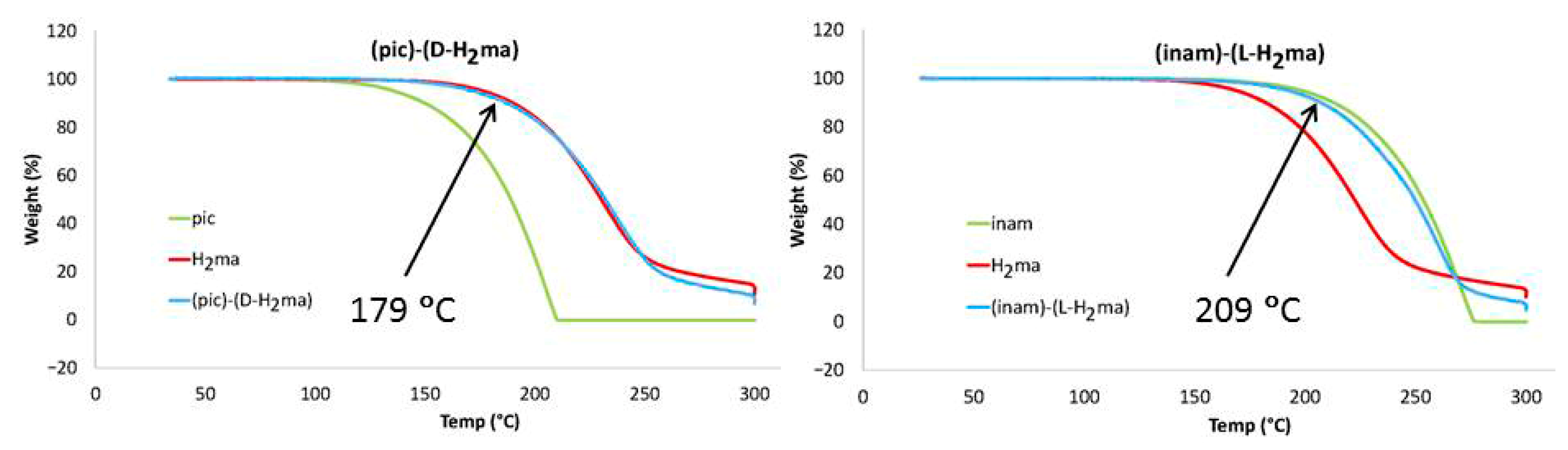
3.6. Thermal Analysis
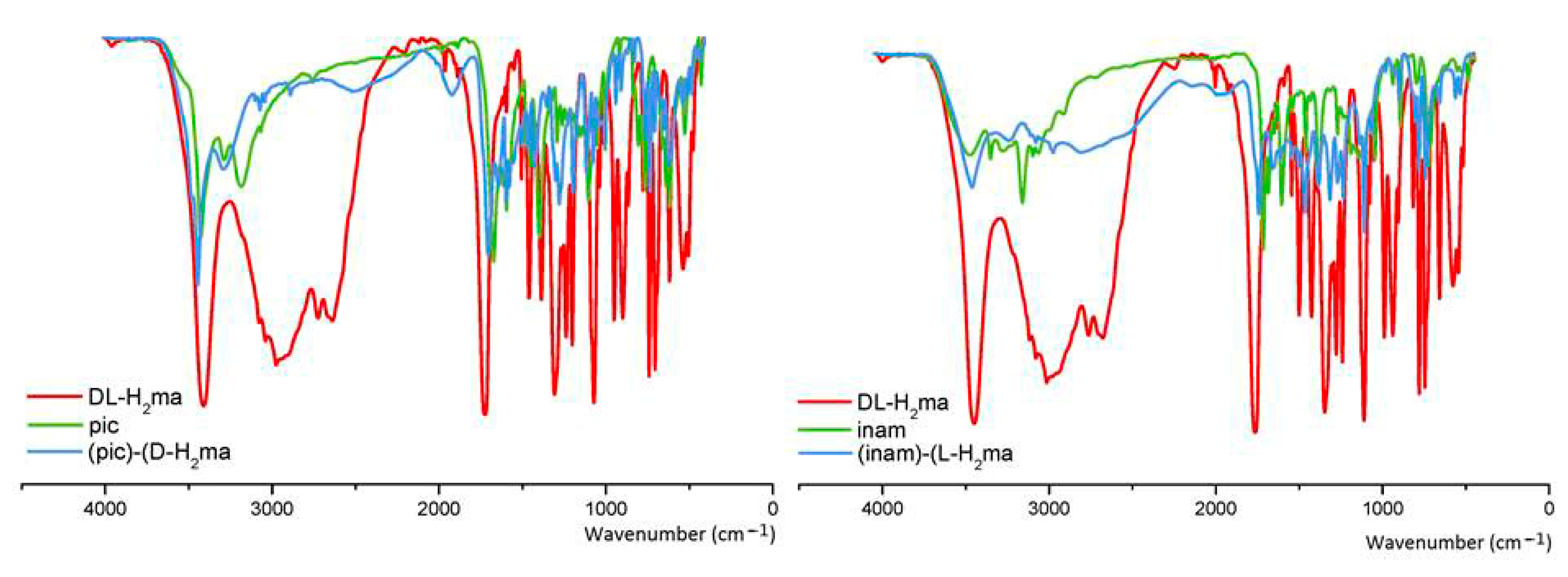
3.7. FT–IR Spectroscopy
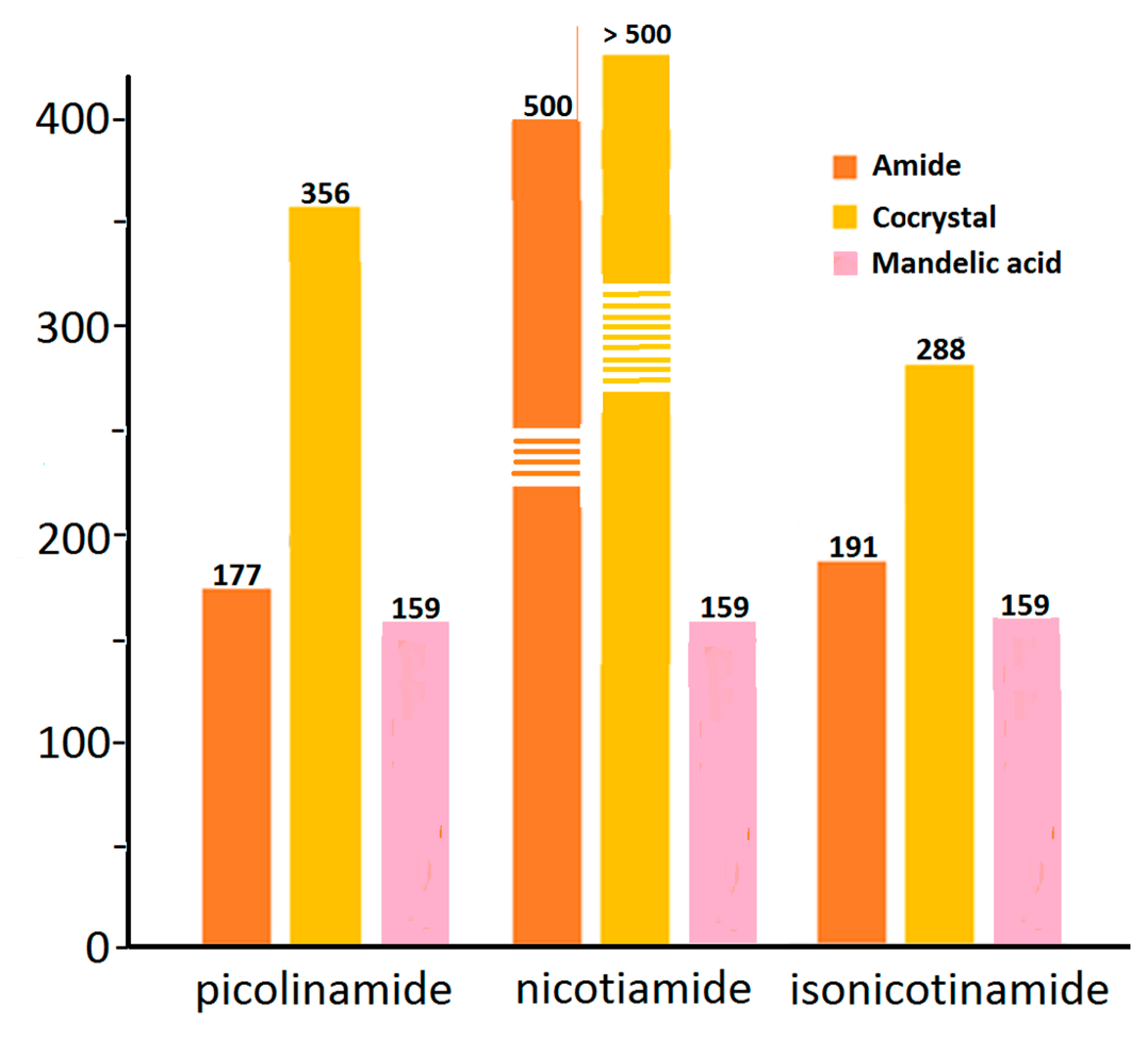
3.8. Solubility and Dissolution
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nangia, A.K.; Desiraju, G.R. Crystal Engineering: An Outlook for the Future. Angew. Chem. Int. Ed. 2019, 58, 4100–4107. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Vittal, J.J.; Ramanan, A. Crystal Engineering: A Text Book; World Scientific: Singapore, 2011. [Google Scholar]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Solomos, M.A.; Mohammadi, C.; Urbelis, J.H.; Koch, E.S.; Osborne, R.; Usala, C.C.; Swift, J.A. Predicting Cocrystallization Based on Heterodimer Energies: The Case of N,N′-Diphenylureas and Triphenylphosphine Oxide. Cryst. Growth Des. 2015, 15, 5068–5074. [Google Scholar] [CrossRef]
- Solomos, M.A.; Watts, T.A.; Swift, J.A. Predicting Cocrystallization Based on Heterodimer Energies: Part II. Cryst. Growth Des. 2017, 17, 5073–5079. [Google Scholar] [CrossRef]
- Berry, D.J.; Seaton, C.C.; Clegg, W.; Harrington, R.W.; Coles, S.J.; Horton, P.N.; Hursthouse, M.B.; Storey, R.; Jones, W.; Friščić, T.; et al. Applying Hot-Stage Microscopy to Co-Crystal Screening: A Study of Nicotinamide with Seven Active Pharmaceutical Ingredients. Cryst. Growth Des. 2008, 8, 1697–1712. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Beatty, A.M.; Helfrich, B.A. A High-Yielding Supramolecular Reaction. J. Am. Chem. Soc. 2002, 124, 14425–14432. [Google Scholar] [CrossRef] [PubMed]
- Sarma, B.; Reddy, L.S.; Nangia, A. The Role of π-Stacking in the Composition of Phloroglucinol and Phenazine Cocrystals. Cryst. Growth Des. 2008, 8, 4546–4552. [Google Scholar] [CrossRef]
- Shattock, T.R.; Arora, K.K.; Vishweshwar, P.; Zaworotko, M.J. Hierarchy of Supramolecular Synthons: Persistent Carboxylic Acid · · · Pyridine Hydrogen Bonds in Cocrystals That also Contain a Hydroxyl Moiety. Cryst. Growth Des. 2008, 8, 4533–4545. [Google Scholar] [CrossRef]
- Borba, A.; Gómez-Zavaglia, A.; Fausto, R. Molecular Structure, Vibrational Spectra, Quantum Chemical Calculations and Photochemistry of Picolinamide and Isonicotinamide Isolated in Cryogenic Inert Matrixes and in the Neat Low-Temperature Solid Phases. J. Phys. Chem. A 2008, 112, 45–57. [Google Scholar] [CrossRef]
- Olsen, R.A.; Liu, L.; Ghaderi, N.; Johns, A.; Hatcher, M.E.; Mueller, L.J. The Amide Rotational Barriers in Picolinamide and Nicotinamide: NMR and ab Initio Studies. J. Am. Chem. Soc. 2003, 125, 10125–10132. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Castiñeiras, A.; Frontera, A.; García-Santos, I.; González-Pérez, J.M.; Niclós-Gutiérrez, J.; Rodríguez-González, I.; Vílchez-Rodríguez, E.; Zaręba, J.K. Recurrent motifs in pharmaceutical cocrystals involving glycolic acid: X-ray characterization, Hirshfeld surface analysis and DFT calculations. Cryst. Eng. Comm. 2020, 22, 6674–6689. [Google Scholar] [CrossRef]
- Évora, A.O.L.; Castro, R.A.E.; Maria, T.M.R.; Rosado, M.T.S.; Ramos Silva, M.; Canotilho, J.; Eusébio, M.E.S. Resolved structures of two picolinamide polymorphs. Investigation of the dimorphic system behaviour under conditions relevant to co-crystal synthesis. Cryst. Eng. Comm. 2012, 14, 8649–8657. [Google Scholar] [CrossRef]
- Li, X.; Ou, X.; Wang, B.; Rong, H.; Wang, B.; Chang, C.; Shi, B.; Yu, L.; Lu, M. Rich polymorphism in nicotinamide revealed by melt crystallization and crystal structure prediction. Commun. Chem. 2020, 3, 152–160. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Beatty, A.M.; Helfrich, B.A.; Nieuwenhuyzen, M. Do Polymorphic Compounds Make Good Cocrystallizing Agents? A Structural Case Study that Demonstrates the Importance of Synthon Flexibility. Cryst. Growth Des. 2003, 3, 159–165. [Google Scholar] [CrossRef]
- Li, J.; Bourne, S.A.; Caira, M.R. New polymorphs of isonicotinamide and nicotinamide. Chem. Commun. 2011, 47, 1530–1532. [Google Scholar] [CrossRef]
- Eccles, K.S.; Deasy, R.E.; Fábián, L.; Braun, D.E.; Maguired, A.R.; Lawrence, S.E. Expanding the crystal landscape of isonicotinamide: Concomitant polymorphism and co-crystallisation. Cryst. Eng. Comm. 2011, 13, 6923–6925. [Google Scholar] [CrossRef]
- Vicatos, A.I.; Caira, M.R. A new polymorph of the common coformer isonicotinamide. Cryst. Eng. Comm. 2019, 21, 843–849. [Google Scholar] [CrossRef]
- Van Putten, P.L. Mandelic acid and urinary tract infections. Ant. Van Leeuwenhoek 1979, 45, 622–623. [Google Scholar] [CrossRef]
- Cai, W.; Marciniak, J.; Andrzejewski, M.; Katrusiak, A. Pressure Effect on D,L-Mandelic Acid Racemate Crystallization. J. Phys. Chem. C 2013, 117, 7279–7285. [Google Scholar] [CrossRef]
- Marciniak, J.; Andrzejewski, M.; Cai, W.; Katrusiak, A. Wallach’s Rule Enforced by Pressure in Mandelic Acid. J. Phys. Chem. C 2014, 118, 4309–4313. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Harasimowicz, M.T.; de Villiers, M.M.; Yu, L. Cocrystals of Nicotinamide and (R)-Mandelic Acid in Many Ratios with Anomalous Formation Properties. J. Am. Chem. Soc. 2013, 135, 18981–18989. [Google Scholar] [CrossRef]
- Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; et al. Gaussian 16, Revision A.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll (Version 13.05.06); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Chan, H.C.S.; Woollam, G.R.; Wagner, T.; Schmidtc, M.U.; Lewis, R.A. Can picolinamide be a promising cocrystal former? Cryst. Eng. Comm. 2014, 16, 4365–4368. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-W.; Guzei, I.A.; de Villiers, M.M.; Yu, L.; Krzyzaniak, J.F. Formation Enthalpies and Polymorphs of Nicotinamide−R-Mandelic Acid Co-Crystals. Cryst. Growth Des. 2012, 12, 4090–4097. [Google Scholar] [CrossRef]
- Friščić, T.; Jones, W. Cocrystal architecture and properties: Design and building of chiral and racemic structures by solid–solid reactions. Faraday Discuss 2007, 136, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Gamidi, R.K.; Rasmuson, Å.C. Estimation of Melting Temperature of Molecular Cocrystals Using Artificial Neural Network Model. Cryst. Growth Des. 2017, 17, 175–182. [Google Scholar] [CrossRef]
- Kerr, H.E.; Softley, L.K.; Suresh, K.; Hodgkinsona, P.; Evans, I.R. Structure and physicochemical characterization of a naproxen–picolinamide cocrystal. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2017, 73, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhogala, B.R.; Basavoju, S.; Nangia, A. Tape and layer structures in cocrystals of some di- and tricarboxylic acids with 4,49-bipyridines and isonicotinamide. From binary to ternary cocrystals. Cryst. Eng. Comm. 2005, 7, 551–562. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. Cryst. Eng. Comm. 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Lopes, L.C.; Orlando, T.; Simões, P.H.B.; Farias, F.F.S.; Bonacorso, H.G.; Zanatta, N.; Salbego, P.R.S.; Martins, M.A.P. Persistence of N-H···O=C Interactions in the Crystallization Mechanisms of Trisubstituted Bis-Ureas with Bulky Substituents. Cryst. Growth Des. 2021, 21, 5740–5751. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Guo, D. Sijbesma, R.P.; Zuilhof, H. π-Stacked Quadruply Hydrogen-Bonded Dimers: π-Stacking Influences H-Bonding. Org. Lett. 2004, 6, 3667–3670. [Google Scholar] [CrossRef]
- Bhattacharyya, M.K.; Saha, U.; Dutta, D.; Frontera, A.; Verma, A.K.; Sharma, P.; Das, A. Unconventional DNA-relevant π-stacked hydrogen bonded arrays involving supramolecular guest benzoate dimers and cooperative anion–π/π–π/π–anion contacts in coordination compounds of Co(ii) and Zn(ii) phenanthroline: Experimental and theoretical studies. New J. Chem. 2020, 44, 4504–4518. [Google Scholar] [CrossRef]
- Badawi, H.M.; Förner, W. Analysis of the infrared and Raman spectra of phenylacetic acid and mandelic (2-hydroxy-2-phenylacetic) acid. Spectrochim. Acta Part A 2011, 78, 1162–1167. [Google Scholar] [CrossRef]
- Bakiler, M.; Bolukbasi, O.; Yilmaz, A. An experimental and theoretical study of vibrational spectra of picolinamide, nicotinamide, and isonicotinamide. J. Mol. Struct. 2007, 826, 6–16. [Google Scholar] [CrossRef]
- Castro, R.A.E.; Ribeiro, J.D.B.; Maria, T.M.R.; Ramos Silva, M.; Yuste-Vivas, C.; Canotilho, J.; Eusébio, M.E.S. Naproxen Cocrystals with Pyridinecarboxamide Isomers. Cryst. Growth Des. 2011, 11, 5396–5404. [Google Scholar] [CrossRef]
- Suresh, K.; Minkov, V.S.; Namila, K.K.; Derevyannikova, E.; Losev, E.; Nangia, A.; Boldyreva, E.V. Novel Synthons in Sulfamethizole Cocrystals: Structure−Property Relations and Solubility. Cryst. Growth Des. 2015, 15, 3498–3510. [Google Scholar] [CrossRef]
- Mannava, M.K.C.; Gunnam, A.; Lodagekar, A.; Shastri, N.R.; Nangia, A.K.; Solomon, K.A. Enhanced solubility, permeability, and tabletability of nicorandil by salt and cocrystal formation. CrystEngComm 2021, 23, 227–237. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (pic)-(D-H2ma) (1) | HOGGOB [31] | JILZOU01 (2a) [32] | JILZOU (2b) [33] | (inam)-(L-H2ma) (3) |
|---|---|---|---|---|---|
| Empirical formula | C14H14N2O4 | C14H14N2O4 | C14H14N2O4 | C14H14N2O4 | C14H14N2O4 |
| Formula weight | 274.27 | 274.27 | 274.27 | 274.27 | 274.27 |
| Temperature/K | 100 (2) | 100 (2) | 100 (2) | 150 (2) | 100 (2) |
| Wavelength/Å | 0.71073 | 1.54178 | 1.54178 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21 | P21 | C2 | P21/c |
| a/Å | 5.4240 (3) | 5.390 (2) | 5.2406 (3) | 32.6557 (9) | 5.2201 (8) |
| b/Å | 26.1177 (14) | 9.897 (3) | 10.0477 (6) | 5.4751 (10) | 27.662 (4) |
| c/Å | 9.3622 (5) | 24.214 (6) | 12.6006 (7) | 14.9264 (5) | 9.1862 (15) |
| α/º | |||||
| β/º | 104.715 (2) | 90.699 (13) | 95.678 (4) | 99.400 (1) | 99.935 (10) |
| γ/º | |||||
| Volume/Å–3 | 1282.77 (12) | 1291.6 (7) | 660.24 (7) | 2632.9 (5) | 1306.6 (4) |
| Z | 4 | 4 | 2 | 8 | 4 |
| Calc. density/Mg/m3 | 1.420 | 1.410 | 1.380 | 1.384 | 1.394 |
| Absorp. coefc./mm−1 | 0.106 | 0.876 | 0.857 | 0.103 | 0.104 |
| F(000) | 576 | 576 | 288 | 1152 | 576 |
| Crystal size | 0.34 × 0.18 × 0.12 | 0.25 × 0.10 × 0.03 | 0.28 × 0.18 × 0.16 | 0.46 × 0.07 × 0.07 | 0.54 × 0.26 × 0.16 |
| θ range/º | 2.381–27.484 | 1.82–66.57 | 3.52–67.60 | 3.77–27.43 | 2.368–29.571 |
| Limiting indices/h,k,l | −7/6, −33/33, −12/12 | −6/6, −11/11, −28/28 | −6/6, −9/11, −15/14 | −42/41, −6/7, −15/19 | −7/7, −38/38, −12/12 |
| Absorp. correct. | Multiscan | Multiscan | Multiscan | Multiscan | Multiscan |
| Max. /min. transm. | 1.000–0.946 | 1.000–0.832 | 1.000–0.909 | 1.000–0.569 | 1.000–0.913 |
| Data/parameters | 2926/196 | 4517/365 | 1235/183 | 5773/385 | 3655/196 |
| Goodness-of-fit on F2 | 1.026 | 1.066 | 1.011 | 1.122 | 1.097 |
| Final R indices | R1 = 0.0378, wR2 = 0.0871 | R1 = 0.0271, wR2 = 0.0695 | R1 = 0.0430, wR2 = 0.1138 | R1 = 0.0550, wR2 = 0.1278 | R1 = 0.0468, wR2 = 0.1149 |
| R indices (all data) | R1 = 0.0492, wR2 = 0.0936 | R1 = 0.0287, wR2 = 0.0708 | R1 = 0.0472, wR2 = 0.1174 | R1 = 0.0703, wR2 = 0.1374 | R1 = 0.0633, wR2 = 0.1234 |
| Largest dif. peak/hole | 0.305/−0.248 | 0.159/−0.189 | 0.306/−0.248 | 0.291/−0.298 | 0.444/−0.276 |
| CCDC number | 2072590 | 977791 | 904263 | 626647 | 2072591 |
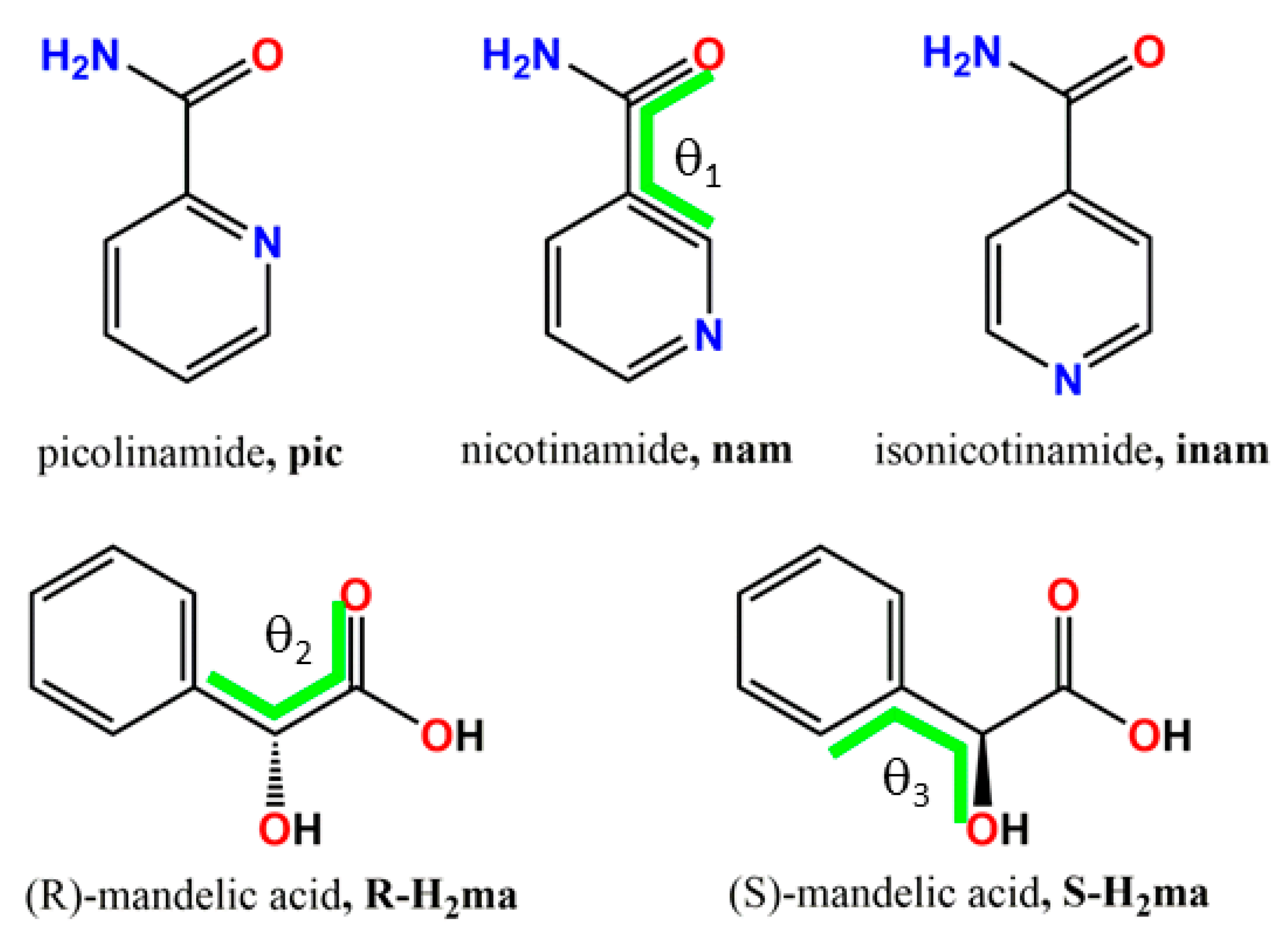
| Compound | Polymorph | θ1 * | θ2 * | θ3 * | Molecule |
|---|---|---|---|---|---|
| pic | I | −161.3 | |||
| DL-H2ma | I | −105.0 | −65.8 | ||
| DL-H2ma | II | −122.1 | −91.5 | 1 | |
| II | −125.7 | −42.5 | 2 | ||
| 1 | −168.5 | −111.0 | −25.8 | ||
| HOGGOB [31] | −173.3 | 125.5 | 90.9 | 1 | |
| 175.5 | −79.2 | 54.3 | 2 | ||
| nam | α | 157.6 | |||
| ε | 26.8 | ||||
| ι | 151.5 | ||||
| D-H2ma | −124.5 | −80.5 | 1 | ||
| −122.0 | −30.5 | 2 | |||
| JILZOU01 (2a) [32] | −28.7 | −106.1 | −72.8 | ||
| JILZOU (2b) [33] | 35.3 | −129.6 | −48.2 | 1 | |
| 12.0 | −103.5 | −41.6 | 2 | ||
| inam | I | 30.5 | |||
| III | 30.9 | ||||
| V | 19.6 | ||||
| L-H2ma | I | 119.4 | 22.9 | 1 | |
| 120.1 | −87.4 | 2 | |||
| II | 115.5 | 10.9 | 1 | ||
| 116.8 | 100.0 | 2 | |||
| 3 | −0.5 | 112.4 | 73.5 |
| Compound | EBSSE | ΣEHB |
|---|---|---|
| (pic)-(D-H2ma) (1) (Figure 8a) | −42.7 | 39.7 |
| (pic)-(D-H2ma) (1) (Figure 8b) | −17.0 | 16.9 |
| JILZOU01 (2a) (Figure 9a) | −22.5 | 24.6 |
| JILZOU (2b) (Figure 9b) | −32.3 | 26.9 |
| (inam)-(L-H2ma) (3) (Figure 10a) | −39.5 | 37.0 |
| Coformer | M. p. (°C) | Cocrystal | M. p. (°C) |
|---|---|---|---|
| DL-Mandelic acid (D,L-H2ma) | 118–121 | ||
| D/L-Mandelic acid (L-H2ma) | 131–135 | ||
| Picolinamide (pic) | 104–108 | (pic)-(D-H2ma) (1) | 91.4 |
| Nicotinamide (nam) | 128–131 | (nam)-(L-H2ma) (2) [25] | 85.2 |
| Isonicotinamide (inam) | 155–157 | (inam)-(L-H2ma) (3) | 112.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiñeiras, A.; Frontera, A.; García-Santos, I.; González-Pérez, J.M.; Niclós-Gutiérrez, J.; Torres-Iglesias, R. Multicomponent Solids of DL-2-Hydroxy-2-phenylacetic Acid and Pyridinecarboxamides. Crystals 2022, 12, 142. https://doi.org/10.3390/cryst12020142
Castiñeiras A, Frontera A, García-Santos I, González-Pérez JM, Niclós-Gutiérrez J, Torres-Iglesias R. Multicomponent Solids of DL-2-Hydroxy-2-phenylacetic Acid and Pyridinecarboxamides. Crystals. 2022; 12(2):142. https://doi.org/10.3390/cryst12020142
Chicago/Turabian StyleCastiñeiras, Alfonso, Antonio Frontera, Isabel García-Santos, Josefa M. González-Pérez, Juan Niclós-Gutiérrez, and Rocío Torres-Iglesias. 2022. "Multicomponent Solids of DL-2-Hydroxy-2-phenylacetic Acid and Pyridinecarboxamides" Crystals 12, no. 2: 142. https://doi.org/10.3390/cryst12020142