
Predicting the Crystal Structure and Lattice Parameters of the Perovskite Materials via Different Machine Learning Models Based on Basic Atom Properties
Abstract
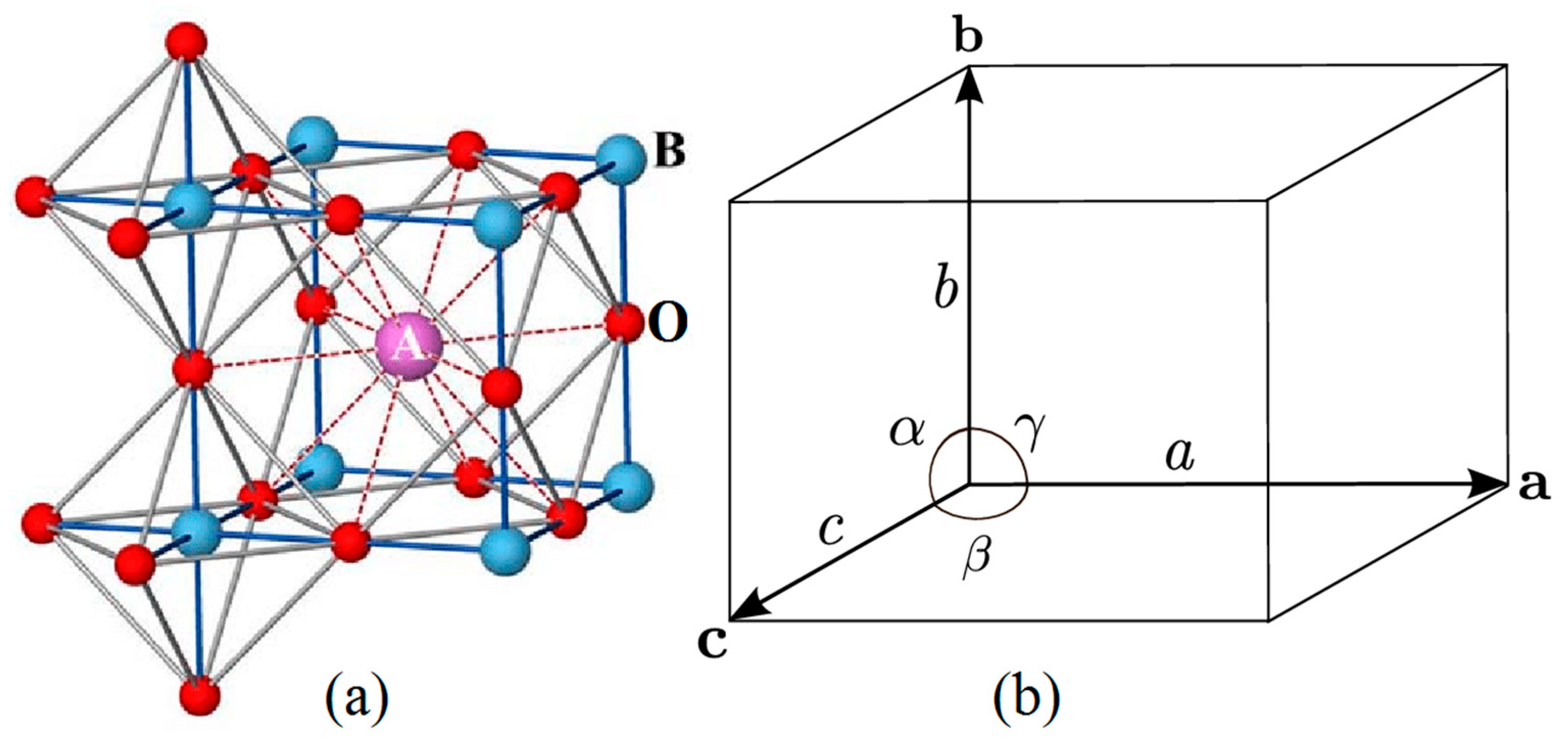
:1. Introduction
2. Modelling Strategy
- (i)
- Database construction—to develop a database system
- (ii)
- Feature selection.
- (iii)
- Data pre-processing.
- (iv)
- Hyper-parameter optimization—ML model tuning.
- (v)
- Testing model for accuracy.
2.1. Model Environment
2.2. Methods and ML Process
2.2.1. Database Construction
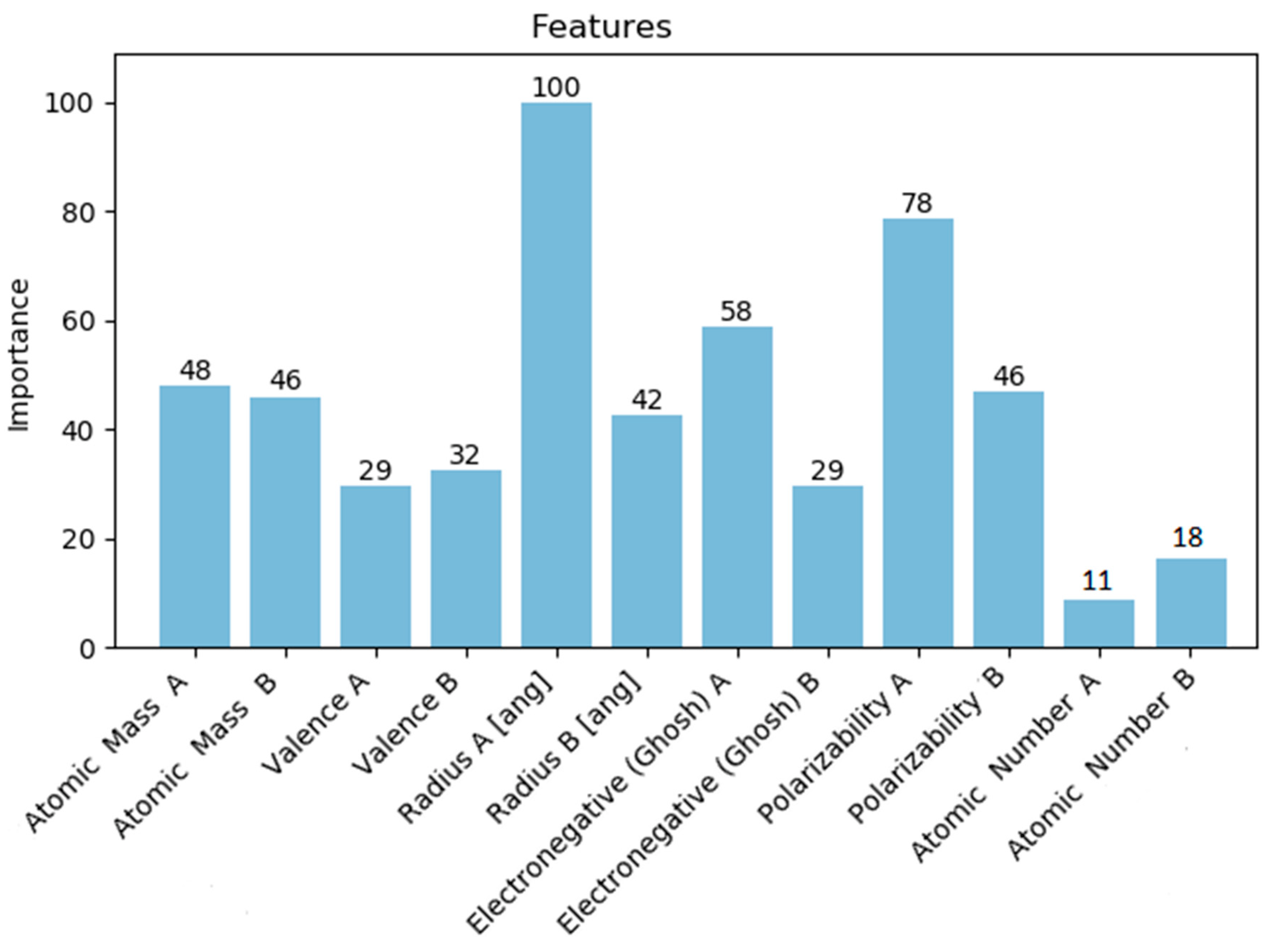
2.2.2. Feature Selection
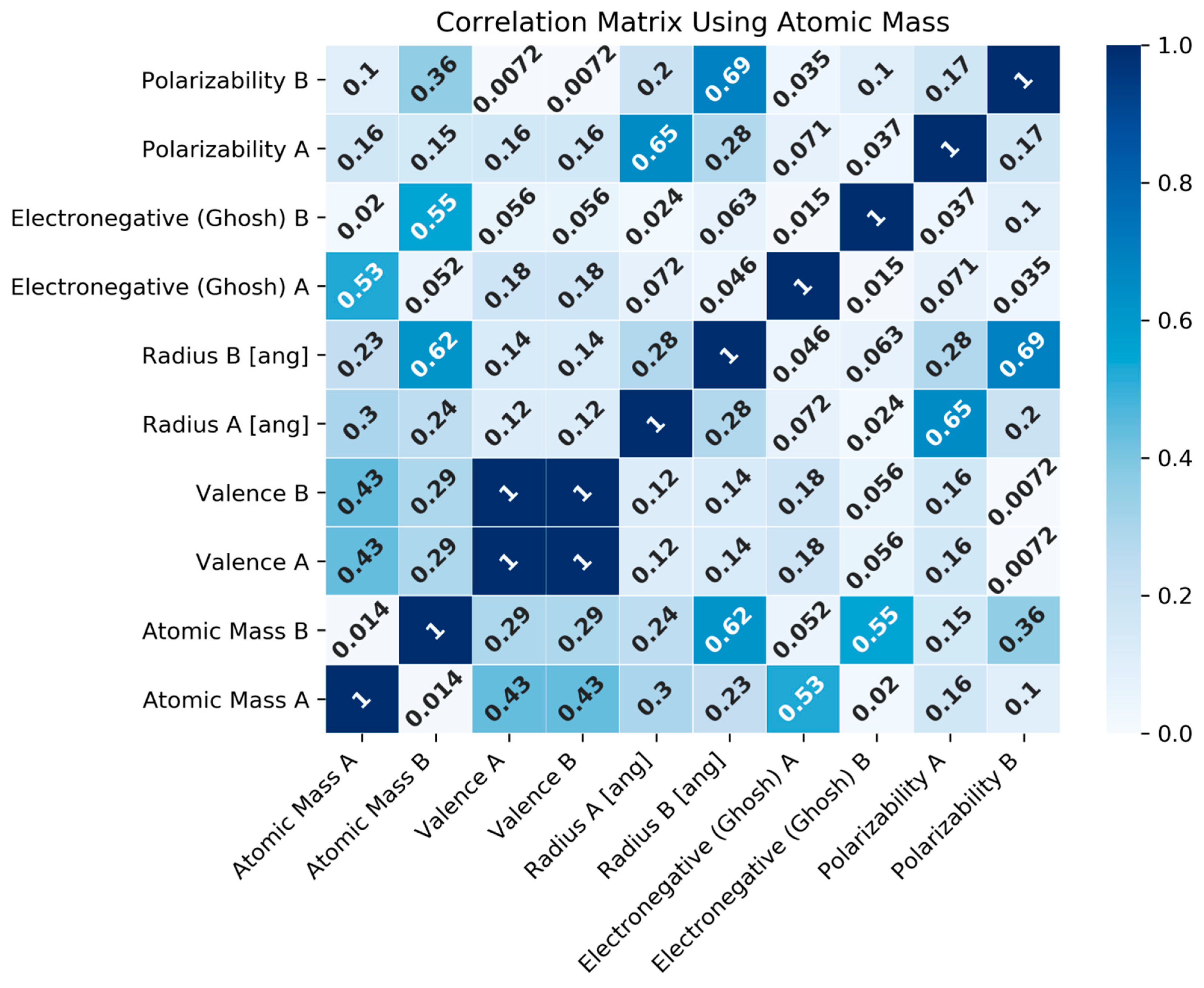
2.2.3. Feature Correlation
2.2.4. Feature Scaling
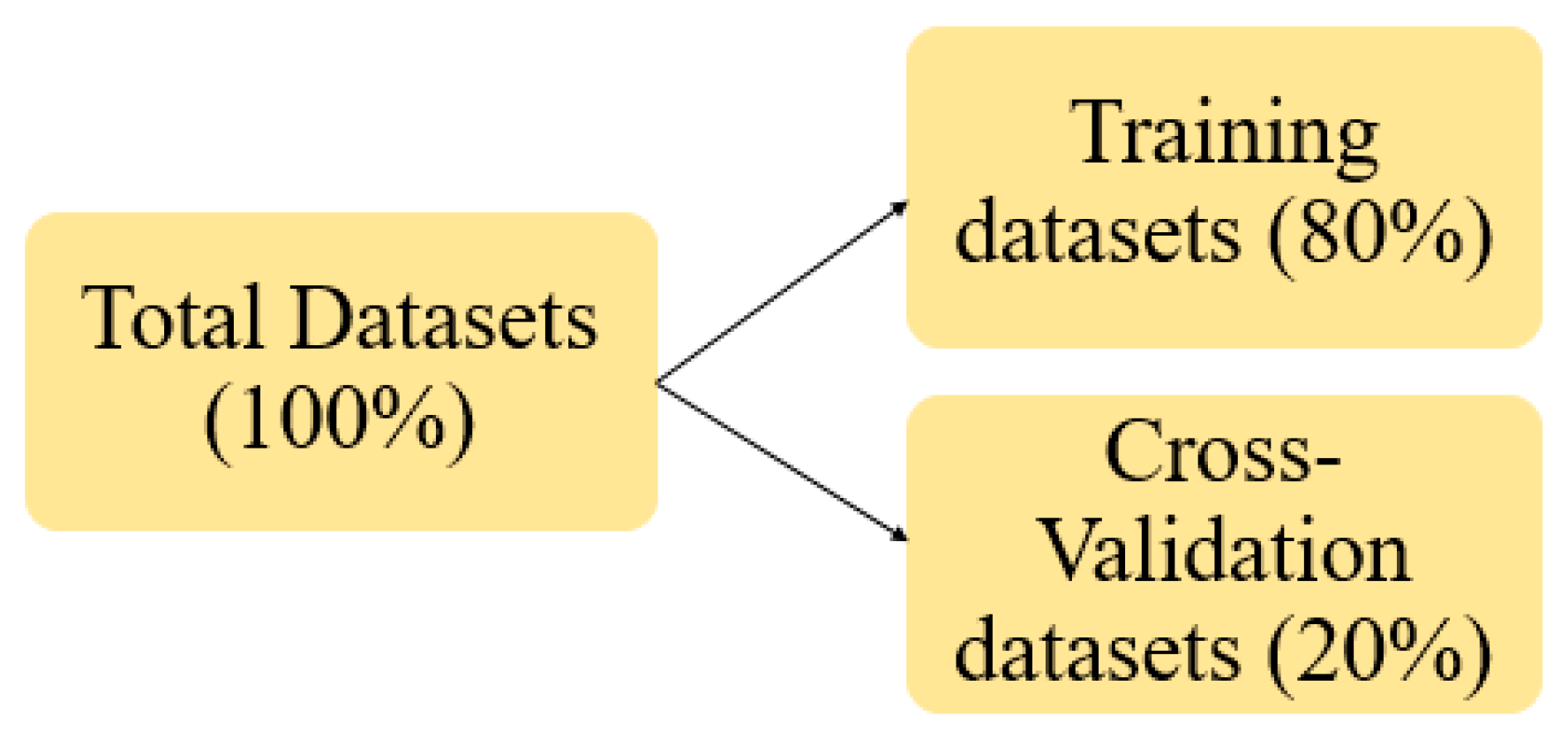
2.2.5. Datasets Pre-Processing (DP)
3. Machine Learning Models
3.1. Crystal Structure Prediction Models
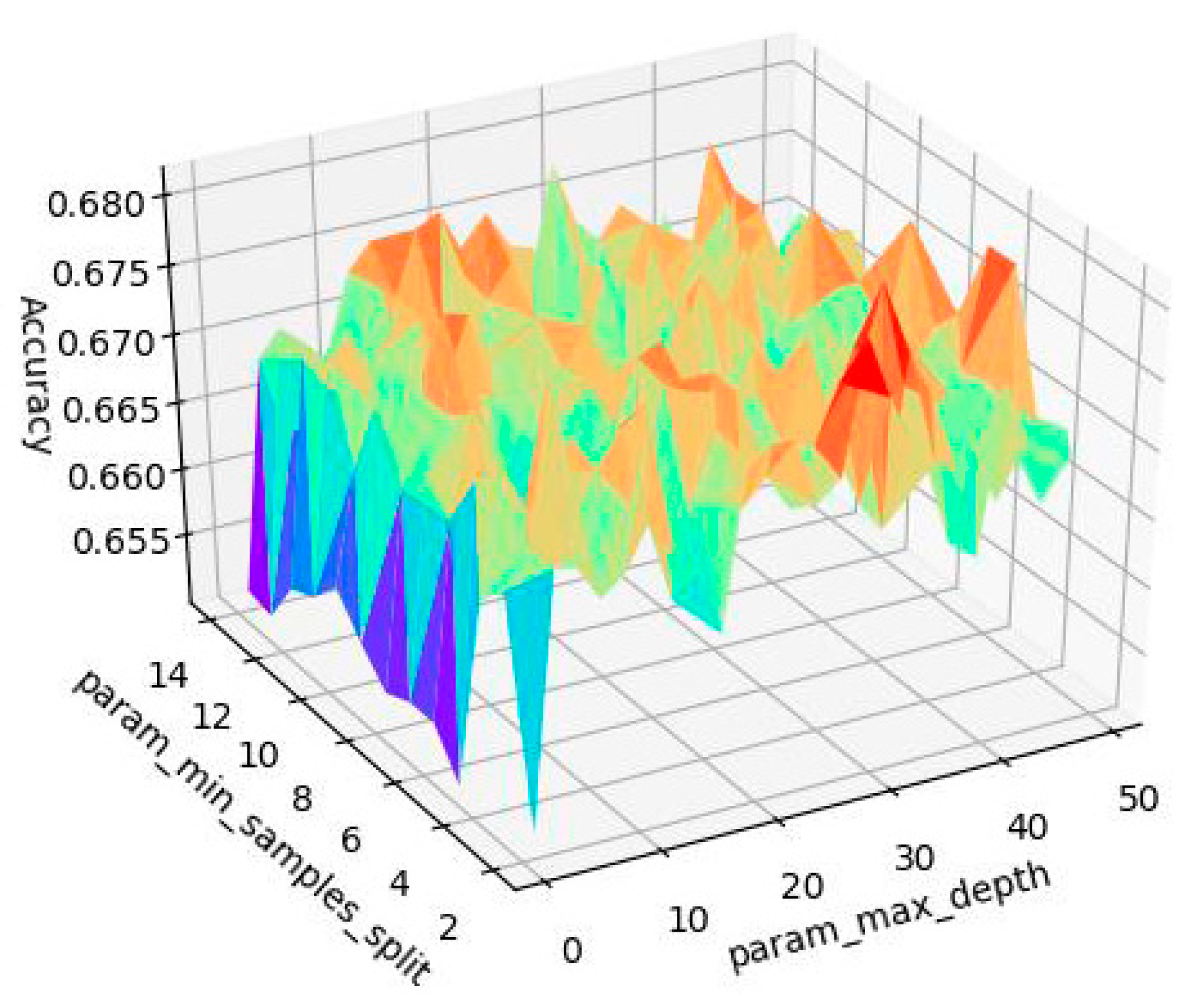
3.1.1. Random Forest—Modelling Process
Random Forest—Model Tuning
3.1.2. Support Vector Machine Modelling Process
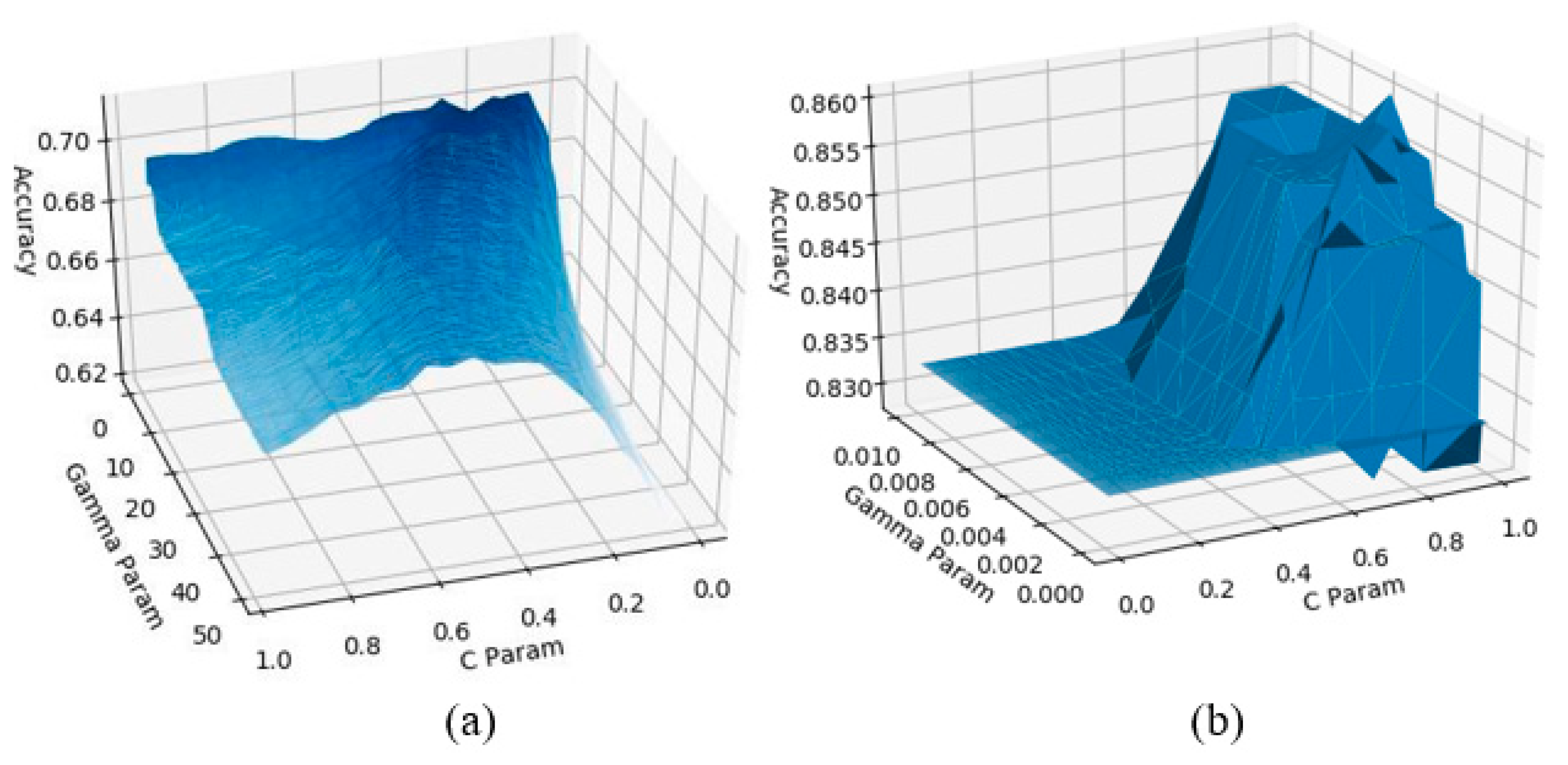
Support Vector Machine—Model Tuning
GA Supported SVM—Tuning Process
| Algorithm 1: GA Supported SVM Modelling Algorithm |
| Data: ABO3 perovskite materials datasets Crystal Structure Label, (for SVM classification) Lattice Parameters (a, b, c) Label, (for SVR regression) Output: Error function minimization Step 1: Initialize the SVM and GA parameters Step 2: Generate initial random population, Pi combining selected features and SVM parameters Step 3: Calculate the fitness evaluation function (accuracy score for 5 cross-validation of SVM) Step 4: Repeat Step 5: Performing random Selection, Crossover, and Mutation Step 6: Update population by evaluating the fitness function Step 7: Until satisfied the stopping criteria Step 8: Get the optimized C and γ for the SVM model and achieve the best accuracy for the model. |
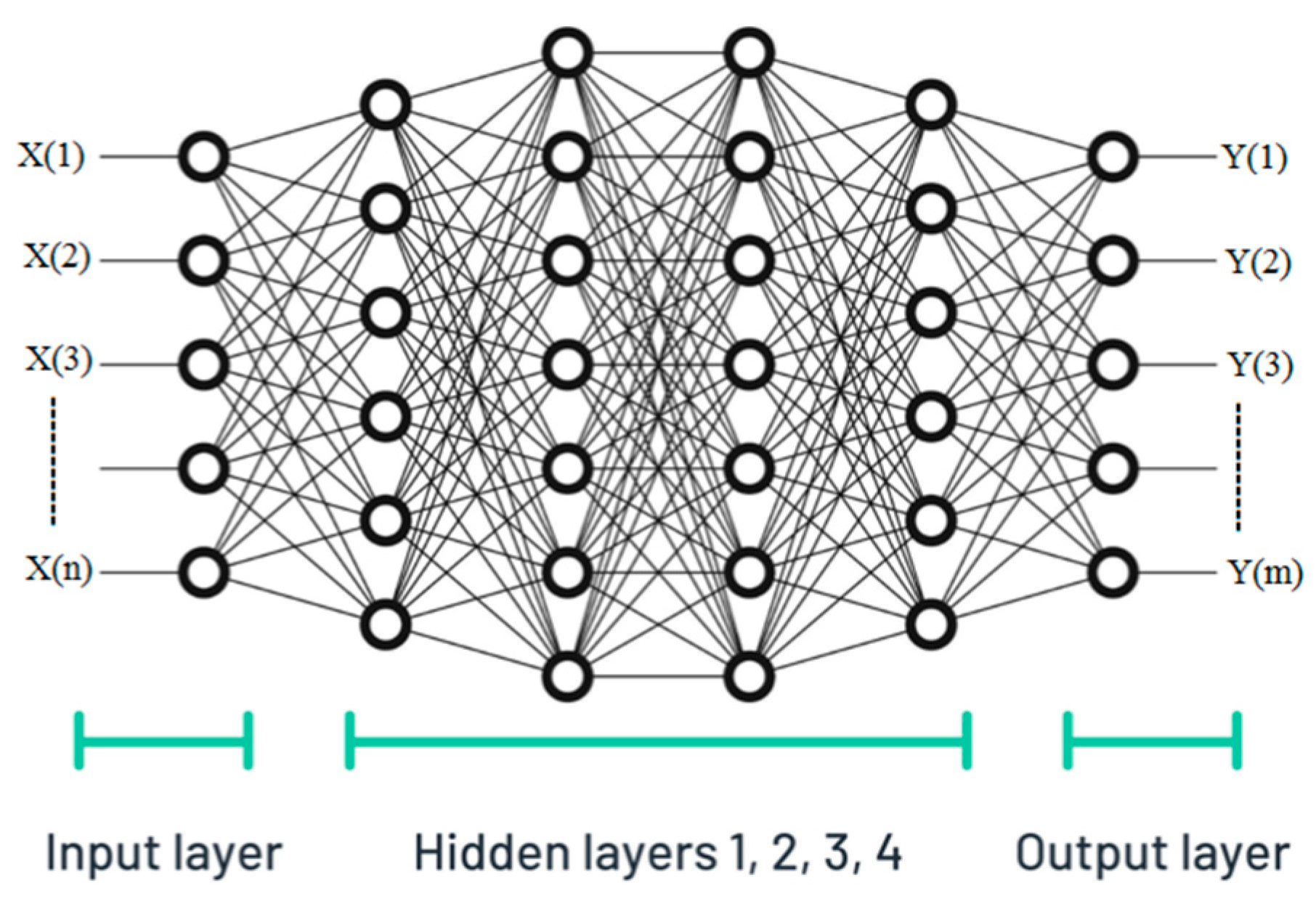
3.1.3. Neural Network—Modelling Process
NN—Model Tuning
3.1.4. GA Optimized Neural Network (GA-NN) Modelling Process
Tuning Methods for the GA-NN Model
| Algorithm 2: GA Optimized NN Modelling Algorithm |
| Data: ABO3 perovskite materials datasets Crystal Structure Label, Output: Loss function optimization Step 1: Initialize the NN model objective function as fitness function Initialize the number of chromosomes, x; mutation rate, mu rate; and crossover rate value Step 2: Generate random population, P Calculate the fitness function f(x) of each chromosome x in the population Step 3: Repeat Step 4: Update population by evaluating the objective function as fitness function Step 5: Update after performing cross over with a crossover probability Pc Step 6: Update with a mutation probability to mutate new offspring Step 7: Until satisfied the stopping criteria |
3.2. Lattice Parameters Prediction Models
3.2.1. SVR-Modeling Process
SVR-Model Tuning
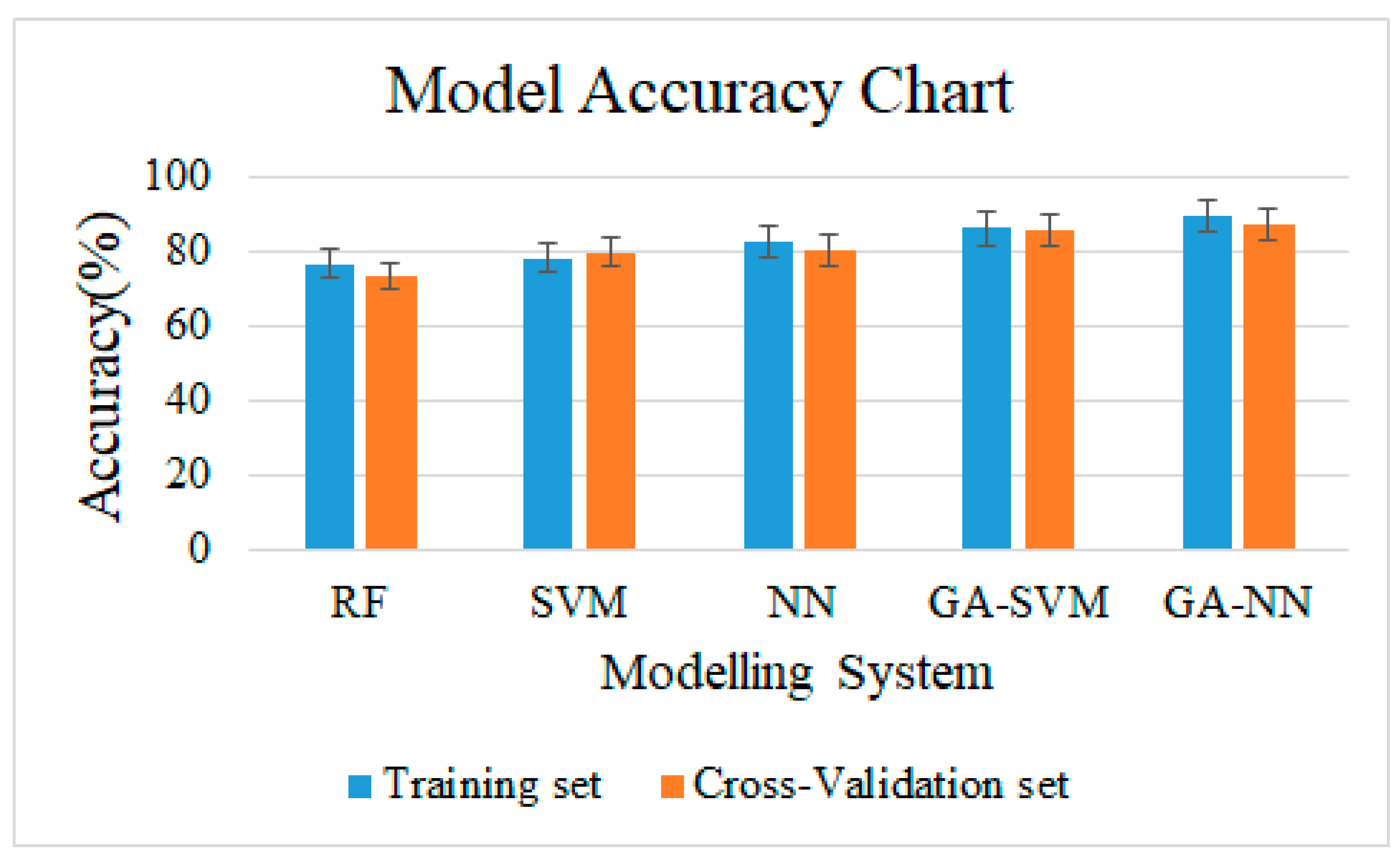
3.3. Model Accuracy
4. Results and Discussion
4.1. Crystal Structures Prediction Models: Accuracy
4.1.1. Performance Comparisons for NN and GA-NN Models
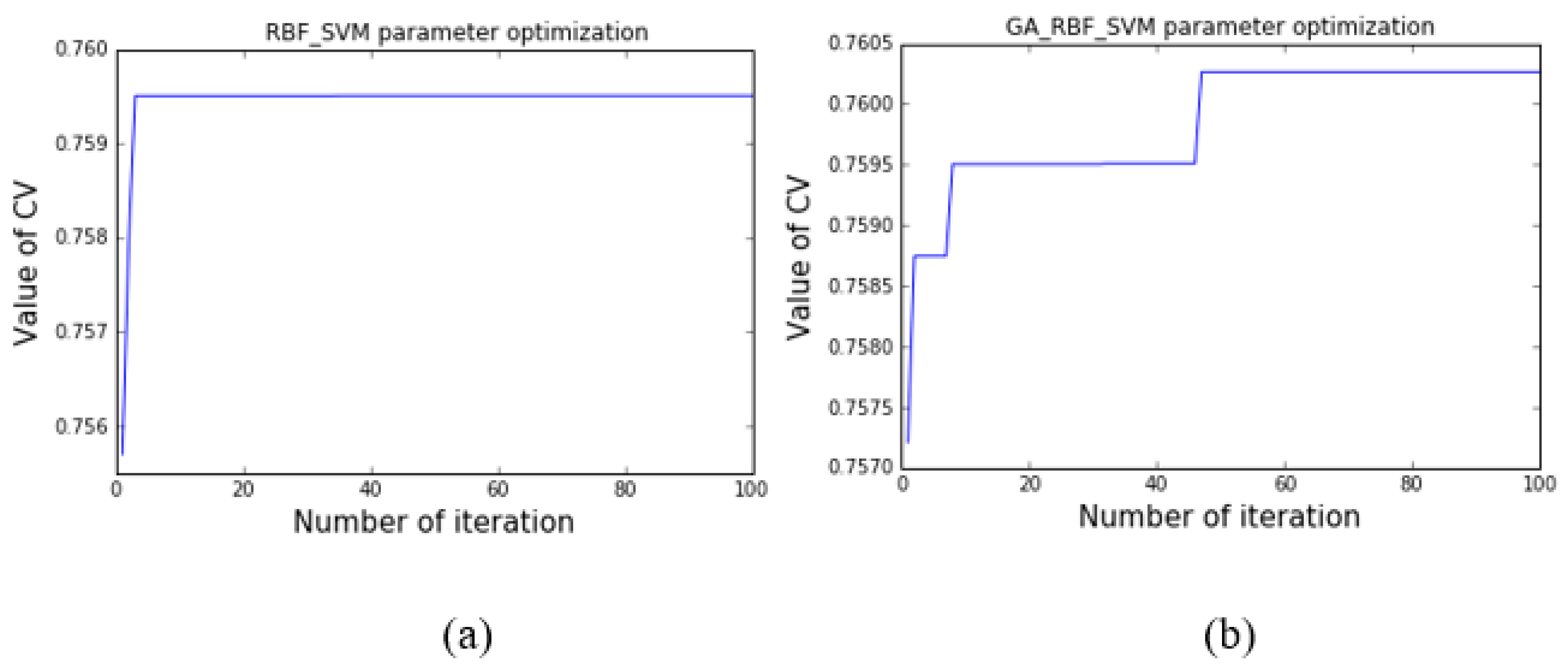
4.1.2. Performance Comparisons for SVM and GA-SVM Models
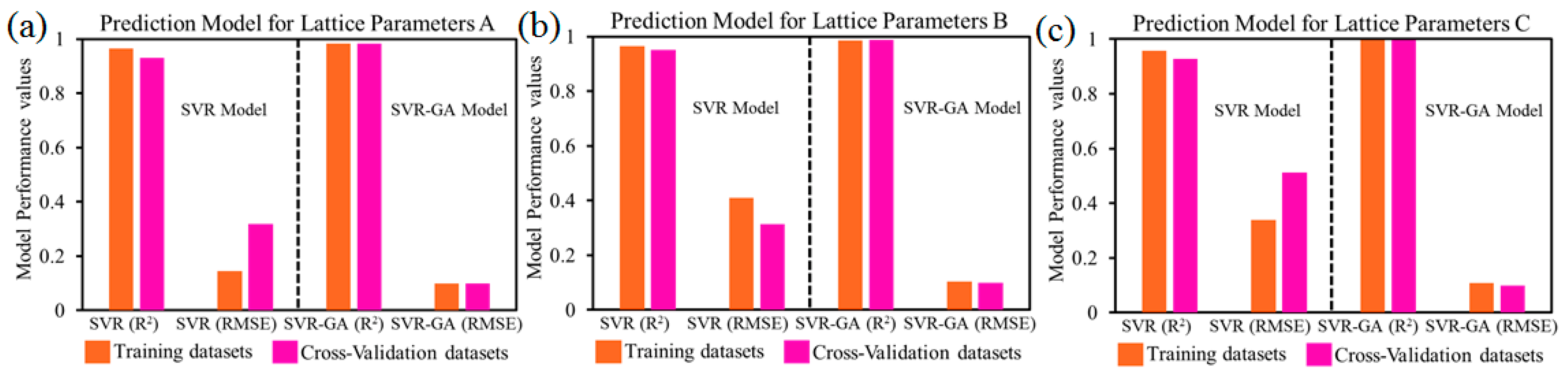
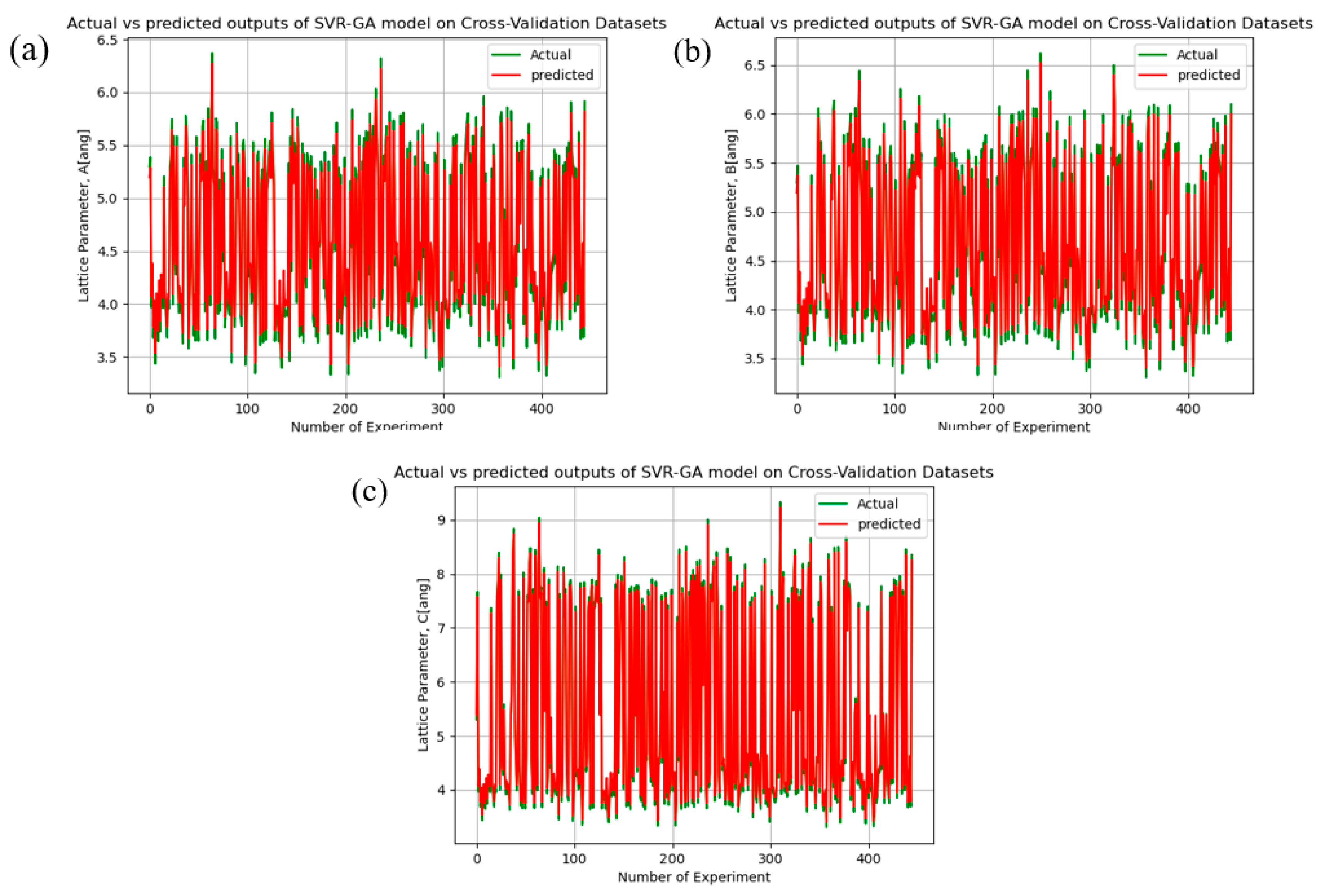
4.2. Lattice Parameters Prediction Models: Accuracy
4.3. Models Performance Analysis: Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, J.; Zhang, Y.; Lu, C. Effects of wetting and misfit strain on the pattern formation of heteroepitaxially grown thin films. Comput. Mater. Sci. 2008, 44, 174–179. [Google Scholar] [CrossRef]
- Khranovskyy, V.; Minikayev, R.; Trushkin, S.; Lashkarev, G.; Lazorenko, V.; Grossner, U.; Paszkowicz, W.; Suchocki, A.; Svensson, B.G.; Yakimova, R. Improvement of ZnO thin film properties by application of ZnO buffer layers. J. Cryst. Growth 2007, 308, 93–98. [Google Scholar] [CrossRef]
- Bouville, M.; Ahluwalia, R. Effect of lattice-mismatch-induced strains on coupled diffusive and displacive phase transformations. Phys. Rev. B 2007, 75, 054110. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Ashcroft, N.W., Mermin, N.D., Eds.; Holt, Rinehart and Winston: New York, NY, USA, 1976. [Google Scholar]
- Terki, R.; Feraoun, H.; Bertrand, G.; Aourag, H. Full potential calculation of structural, elastic and electronic properties of BaZrO3 and SrZrO3. Phys. Status Solidi B 2005, 242, 1054–1062. [Google Scholar] [CrossRef]
- Li, C.; Soh, K.C.K.; Wu, P. Formability of ABO3 perovskites. J. Alloys Compd. 2004, 372, 40–48. [Google Scholar] [CrossRef]
- Cabuk, S.; Akkus, H.; Mamedov, A. Electronic and optical properties of KTaO3: Ab initio calculation. Phys. B Condens. Matter 2007, 394, 81–85. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Wang, R.; Li, Q. Ab initio study of structural and electronic properties of BiAlO3 and BiGaO3. Phys. B Condens. Matter 2007, 390, 96–100. [Google Scholar] [CrossRef]
- Wolfram, T.; Ellialtioglu, S. Electronic and Optical Properties of D-Band Perovskites; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Galasso, F.S. Perovskites and High-T Sub c Superconductors; U.S. Department of Energy: Washington, DC, USA, 1990.
- Mizusaki, J. Nonstoichiometry, diffusion, and electrical properties of perovskite-type oxide electrode materials. Solid State Ion. 1992, 52, 79–91. [Google Scholar] [CrossRef]
- Glazer, A. Simple ways of determining perovskite structures. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1975, 31, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, V.M. Die gesetze der krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Pradhan, S.; Moschitti, A.; Xue, N.; Ng, H.T.; Björkelund, A.; Uryupina, O.; Zhang, Y.; Zhong, Z. Towards robust linguistic analysis using ontonotes. In Proceedings of the Seventeenth Conference on Computational Natural Language Learning, Sofia, Bulgaria, 8–9 August 2013. [Google Scholar]
- Mitchell, R.H.; Welch, M.D.; Chakhmouradian, A.R. Nomenclature of the perovskite supergroup: A hierarchical system of classification based on crystal structure and composition. Mineral. Mag. 2017, 81, 411–461. [Google Scholar] [CrossRef] [Green Version]
- Hudspeth, J. Short-Range Order in Ferroelectric Triglycine Sulphate. Ph.D. Thesis, The Australian National University, Canberra, Australia, 2012. [Google Scholar] [CrossRef]
- Behara, S.; Poonawala, T.; Thomas, T. Crystal structure classification in ABO3 perovskites via machine learning. Comput. Mater. Sci. 2021, 188, 110191. [Google Scholar] [CrossRef]
- Podryabinkin, E.V.; Tikhonov, E.V.; Shapeev, A.V.; Oganov, A.R. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 2019, 99, 064114. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Cui, Y.; Xiong, Z.; Jin, J.; Liu, Z.; Dong, R.; Hu, J. Machine Learning-Based Prediction of Crystal Systems and Space Groups from Inorganic Materials Compositions. ACS Omega 2020, 5, 3596–3606. [Google Scholar] [CrossRef]
- Shandiz, M.A.; Gauvin, R. Application of machine learning methods for the prediction of crystal system of cathode materials in lithium-ion batteries. Comput. Mater. Sci. 2016, 117, 270–278. [Google Scholar] [CrossRef]
- Alade, I.O.; Olumegbon, I.A.; Bagudu, A. Lattice constant prediction of A2XY6 cubic crystals (A = K, Cs, Rb, TI.; X = tetravalent cation; Y = F, Cl, Br, I) using computational intelligence approach. J. Appl. Phys. 2020, 127, 015303. [Google Scholar] [CrossRef]
- Li, Y.; Yang, W.; Dong, R.; Hu, J. MLatticeABC: Generic lattice constant prediction of crystal materials using machine learning. ACS Omega 2021, 6, 11585–11594. [Google Scholar] [CrossRef]
- Javed, S.G.; Khan, A.; Majid, A.; Mirza, A.M.; Bashir, J. Lattice constant prediction of orthorhombic ABO3 perovskites using support vector machines. Comput. Mater. Sci. 2007, 39, 627–634. [Google Scholar] [CrossRef]
- Majid, A.; Khan, A.; Javed, G.; Mirza, A.M. Lattice constant prediction of cubic and monoclinic perovskites using neural networks and support vector regression. Comput. Mater. Sci. 2010, 50, 363–372. [Google Scholar] [CrossRef]
- Schütt, K.; Glawe, H.; Brockherde, F.; Sanna, A.; Müller, K.R.; Gross, E. How to represent crystal structures for machine learning: Towards fast prediction of electronic properties. Phys. Rev. B 2014, 89, 205118. [Google Scholar] [CrossRef] [Green Version]
- Iqtidar, A.; Khan, N.B.; Kashif-Ur-Rehman, S.; Javed, M.F.; Aslam, F.; Alyousef, R.; Alabduljabbar, H.; Mosavi, A. Prediction of Compressive Strength of Rice Husk Ash Concrete through Different Machine Learning Processes. Crystals 2021, 11, 352. [Google Scholar] [CrossRef]
- Aslam, F.; Elkotb, M.A.; Iqtidar, A.; Khan, M.A.; Javed, M.F.; Usanova, K.I.; Alamri, S.; Musarat, M.A. Compressive strength prediction of rice husk ash using multiphysics genetic expression programming. Ain Shams Eng. J. 2022, 13, 101593. [Google Scholar] [CrossRef]
- Kumar, A.; Verma, A.; Bhardwaj, S. Prediction of formability in perovskite-type oxides. Predict. Formability Perovskite-Type Oxides 2008, 1, 11–19. [Google Scholar]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90. [Google Scholar] [CrossRef]
- Emery, A.A.; Wolverton, C. High-throughput dft calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci. Data 2017, 4, 170153. [Google Scholar] [CrossRef] [Green Version]
- Kantardzic, M. Data Mining: Concepts, Models, Methods, and Algorithms; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Sola, J.; Sevilla, J. Importance of input data normalization for the application of neural networks to complex industrial problems. IEEE Trans. Nucl. Sci. 1997, 44, 1464–1468. [Google Scholar] [CrossRef]
- Kotsiantis, S.; Kanellopoulos, D.; Pintelas, P.E. Data preprocessing for supervised leaning. Int. J. Comput. Sci. 2006, 1, 111–117. [Google Scholar]
- Smola, A.J.; Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 2005, 14, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Burges, C.J.C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Üstün, B.; Melssen, W.; Oudenhuijzen, M.; Buydens, L. Determination of optimal support vector regression parameters by genetic algorithms and simplex optimization. Anal. Chim. Acta 2005, 544, 292–305. [Google Scholar] [CrossRef]
- Jarin, S.; Saleh, T. Artificial neural network modelling and analysis of carbon nanopowder mixed micro wire electro discharge machining of gold coated doped silicon. Int. J. Mater. Eng. Innov. 2019, 10, 346–363. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. IJCAI 1995, 14, 1137–1145. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature List (Basic Atom Characteristics) | Label for Classification Models (Different Crystal Structures) | Label for Regression Models (Lattice Parameters) | Experimental Datasets | DFT Calculated Datasets |
|---|---|---|---|---|
| Atomic number of A site | Cubic | Lattice parameter A | 222 | 2003 |
| Atomic number of B site | Rhombohedral | Lattice parameter B | ||
| Atomic mass of A site | Tetragonal | Lattice parameter C | ||
| Atomic mass of B site | Orthorhombic | |||
| Valance of A site | ||||
| Valance of B site | ||||
| Ionic radii of A site | ||||
| Ionic radii of B site | ||||
| Electronegativity of A site | ||||
| Electronegativity of B site | ||||
| Polarizability of A site | ||||
| Polarizability of B site | ||||
| Total number of Datasets | 2225 | 2225 |
| Model Output Parameters (Different Crystal Structure) | Model Output Values for Crystal Structures | Lattice Parameters Mapping | Number of Collected Datasets |
|---|---|---|---|
| Cubic | 0 | α = β = γ = 90° | 1379 |
| Rhombohedral | 1 | α = β = γ90° | 131 |
| Tetragonal | 2 | α = β = γ = 90° | 64 |
| Orthorhombic | 3 | α = β = γ = 90° | 651 |
| Parameter | Data Range | Type of Technique Used |
|---|---|---|
| No. of input neuron | 12 | |
| No. of hidden layers | 4 | |
| No. of neurons in different hidden layer | 6, 6, 6, 4 | |
| No. of output neuron | 4 | |
| Total no. of sample | 2225 | |
| Proportion of training and Cross-Validation datasets | 80% & 20% | |
| Data normalization | 0 to 1 | Min-max data normalization technique |
| Weight initialization | −0.5 to 0.5 | Random weight initialization technique |
| Transfer function | 0 to 1 | ReLu function for hidden layers and SoftMax for output layer. |
| Error function | Loss function | |
| Type of Learning rule | Supervised learning rule | |
| Stopping criteria | Early stopping |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarin, S.; Yuan, Y.; Zhang, M.; Hu, M.; Rana, M.; Wang, S.; Knibbe, R. Predicting the Crystal Structure and Lattice Parameters of the Perovskite Materials via Different Machine Learning Models Based on Basic Atom Properties. Crystals 2022, 12, 1570. https://doi.org/10.3390/cryst12111570
Jarin S, Yuan Y, Zhang M, Hu M, Rana M, Wang S, Knibbe R. Predicting the Crystal Structure and Lattice Parameters of the Perovskite Materials via Different Machine Learning Models Based on Basic Atom Properties. Crystals. 2022; 12(11):1570. https://doi.org/10.3390/cryst12111570
Chicago/Turabian StyleJarin, Sams, Yufan Yuan, Mingxing Zhang, Mingwei Hu, Masud Rana, Sen Wang, and Ruth Knibbe. 2022. "Predicting the Crystal Structure and Lattice Parameters of the Perovskite Materials via Different Machine Learning Models Based on Basic Atom Properties" Crystals 12, no. 11: 1570. https://doi.org/10.3390/cryst12111570