
Determination of Crystal Growth Rates in Multi-Component Solutions

Abstract
:1. Introduction
2. Materials and Method
2.1. Materials
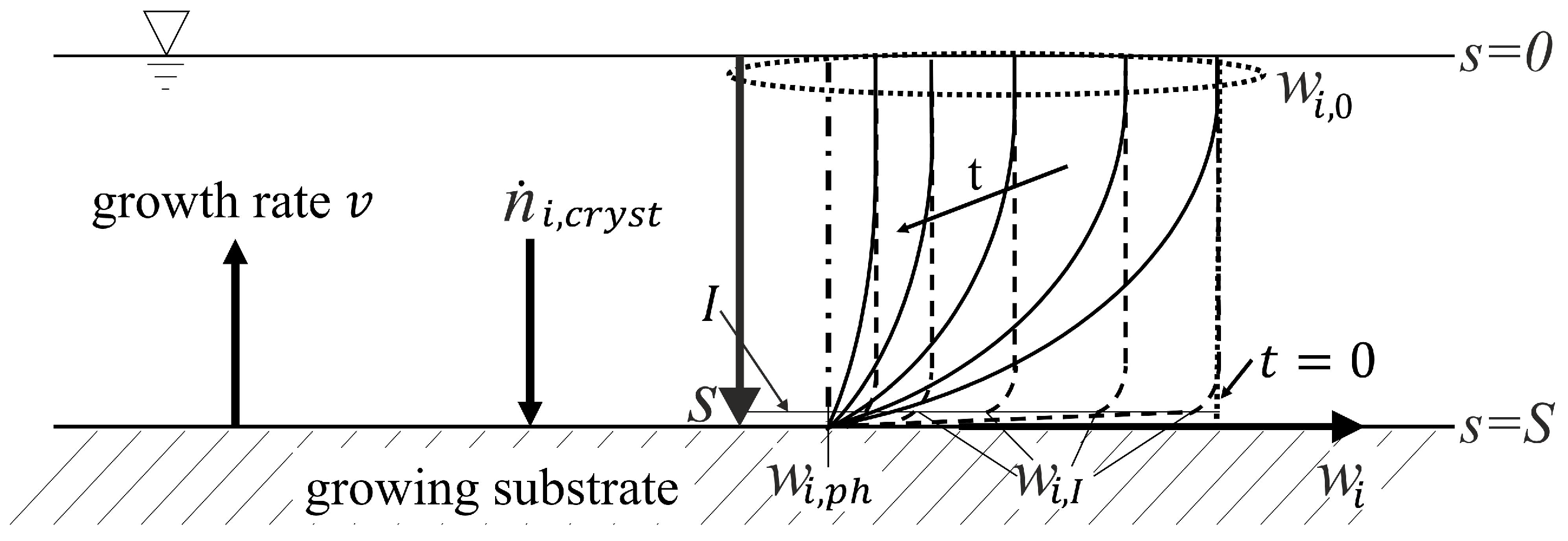
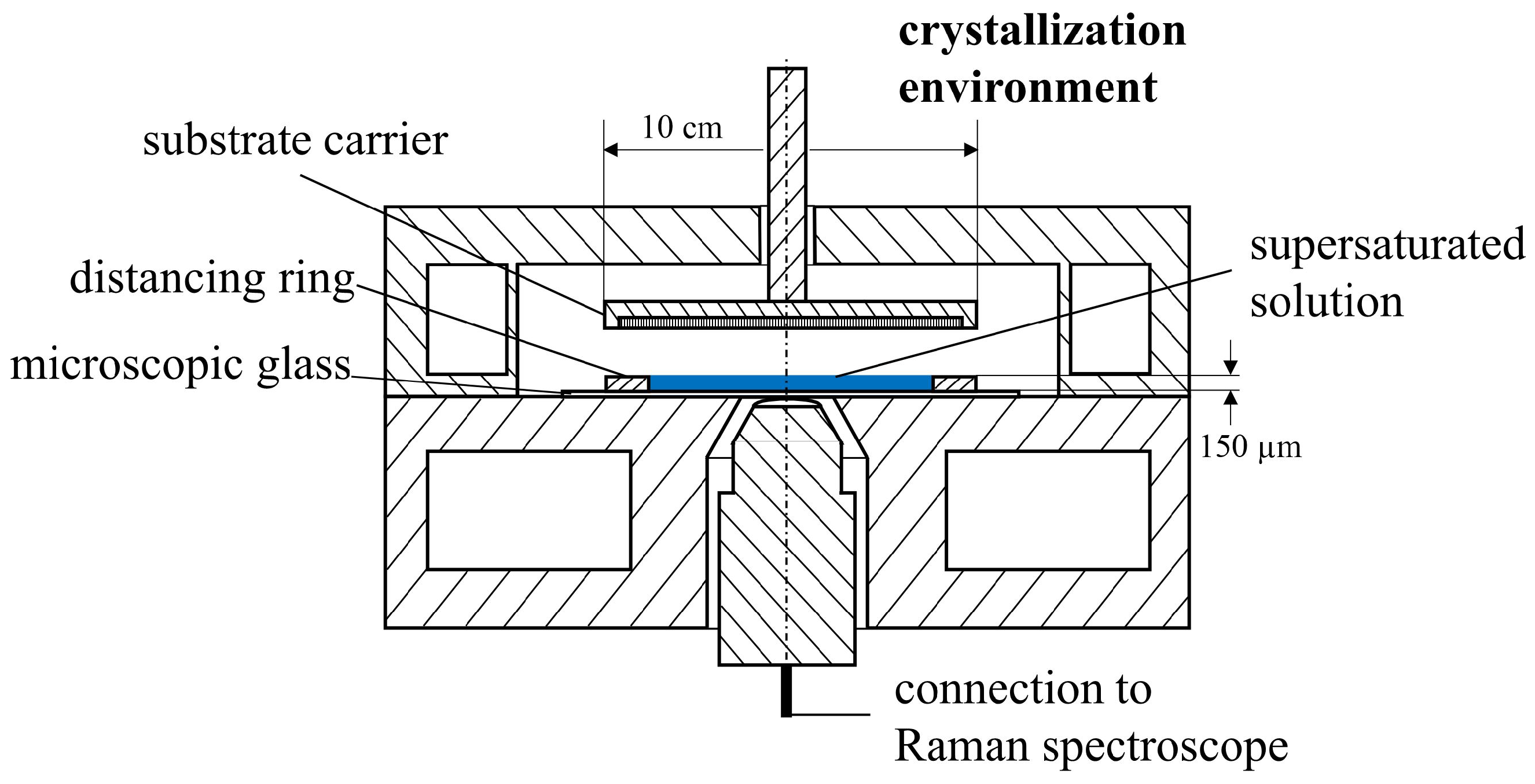
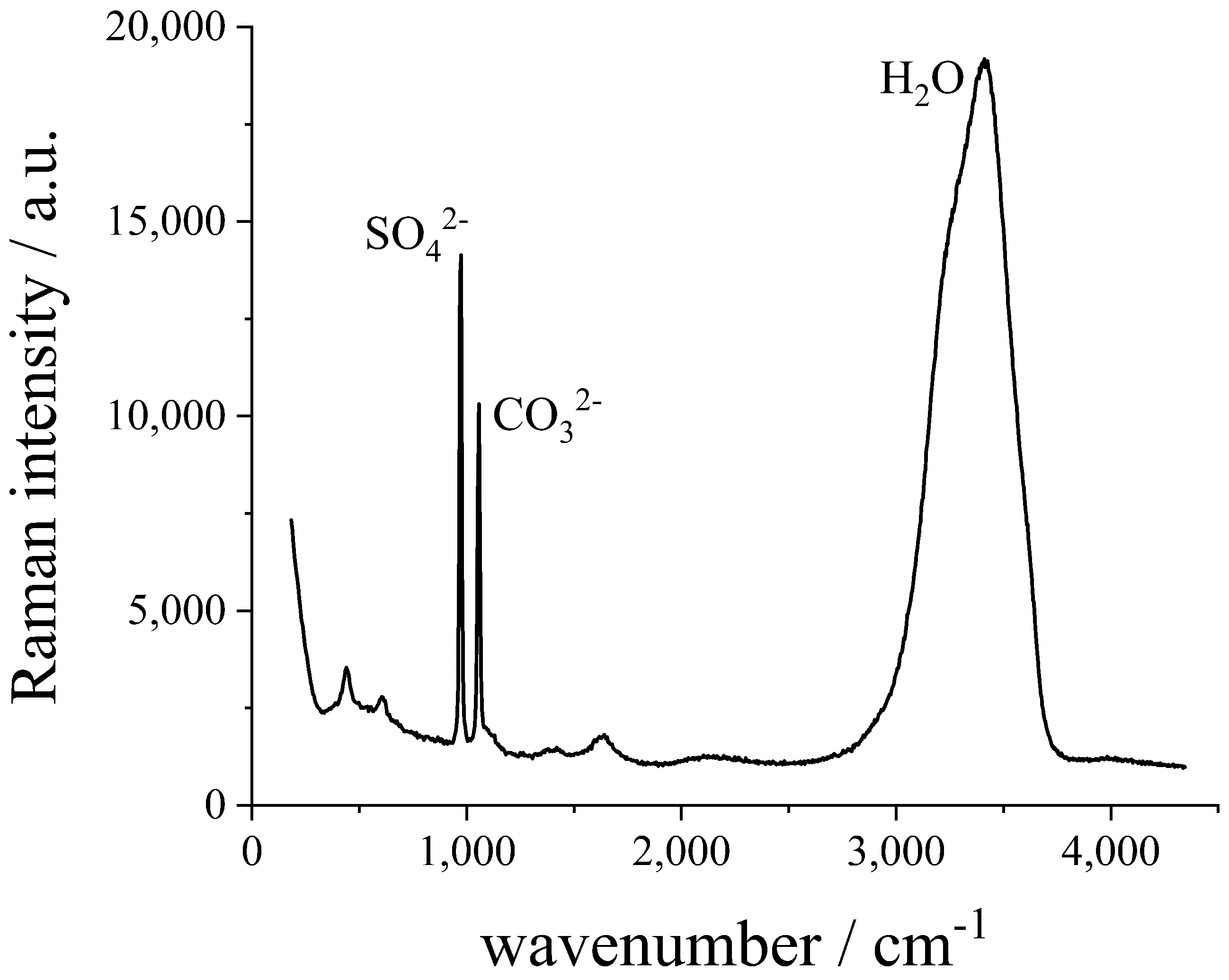
2.2. Method
3. Results
3.1. Binary Experiments
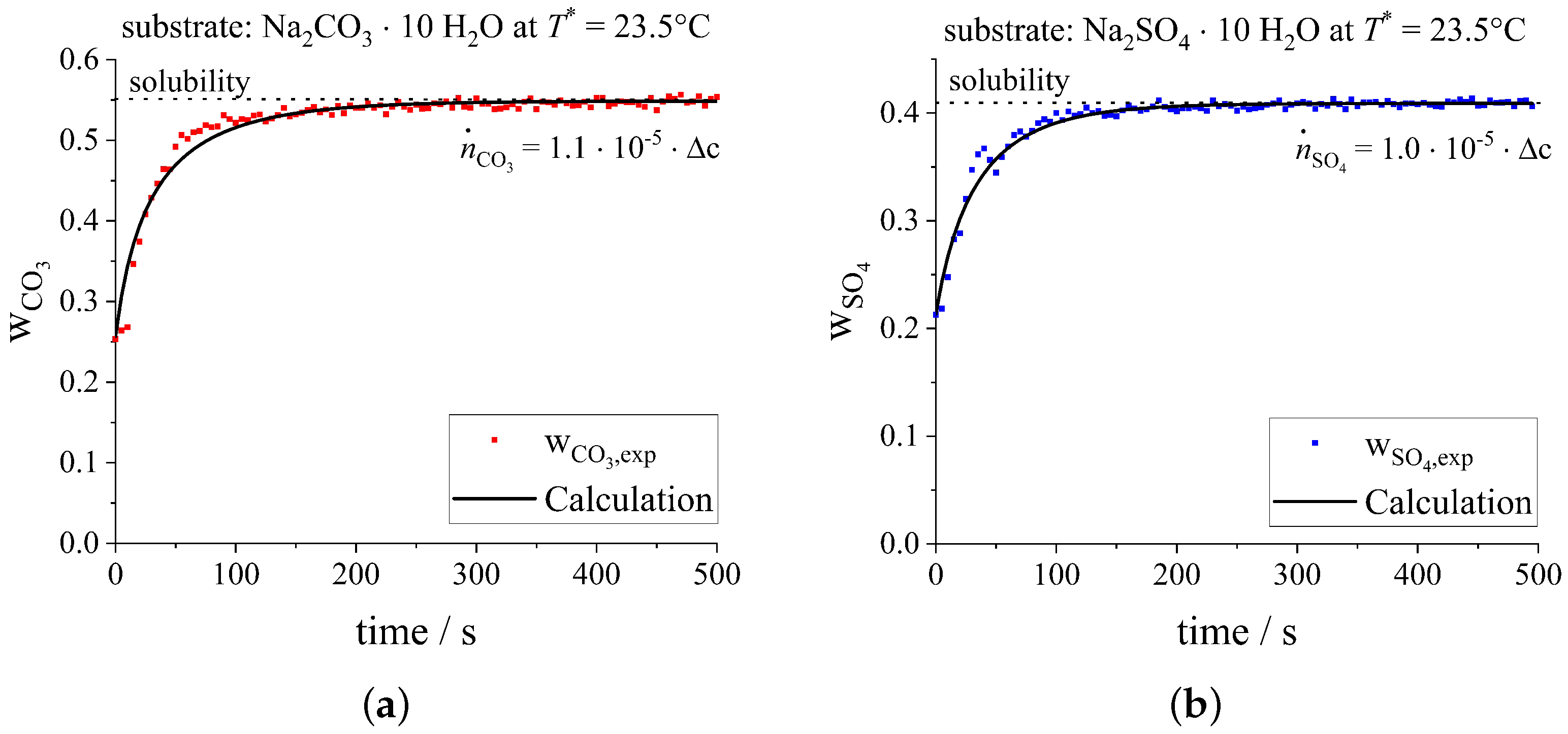
3.1.1. Binary Dissolution
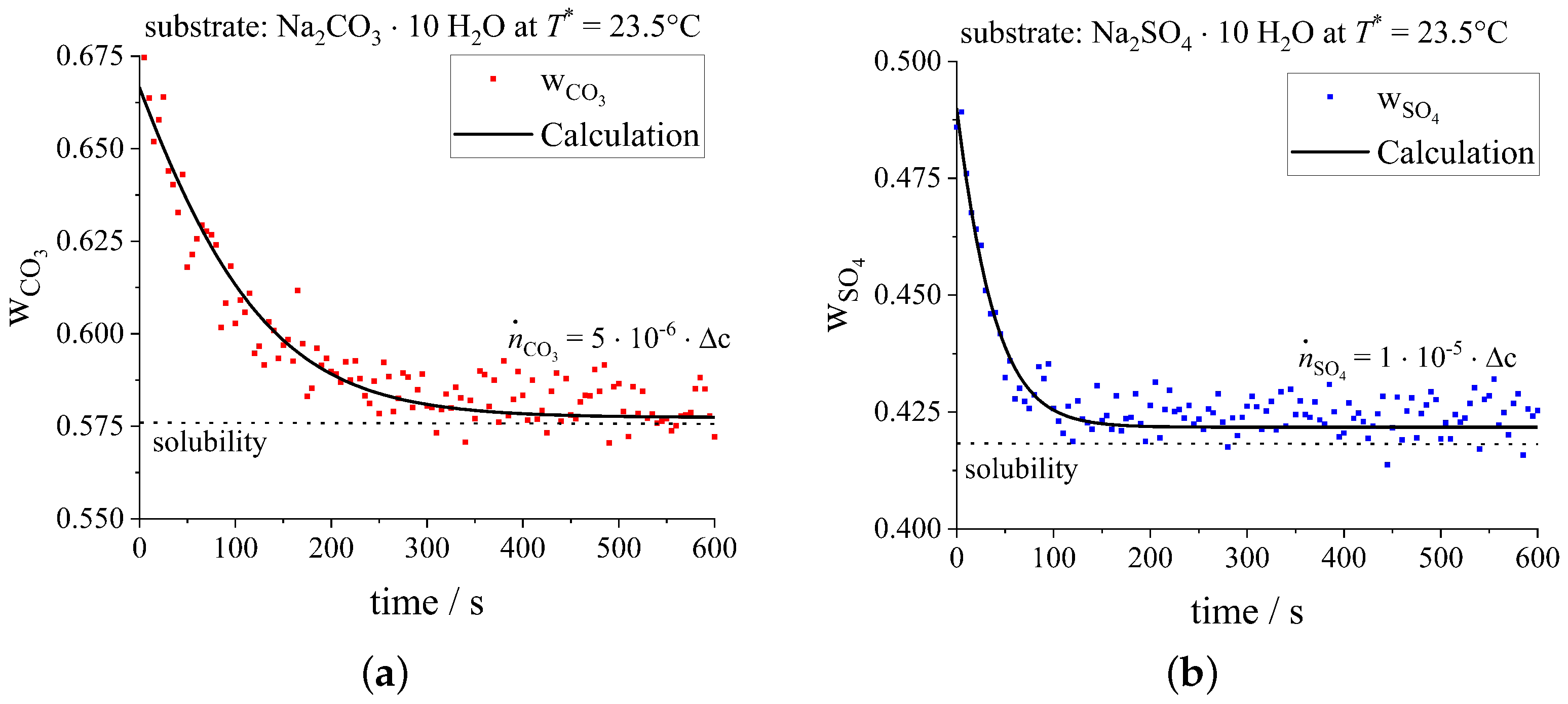
3.1.2. Binary Crystallization
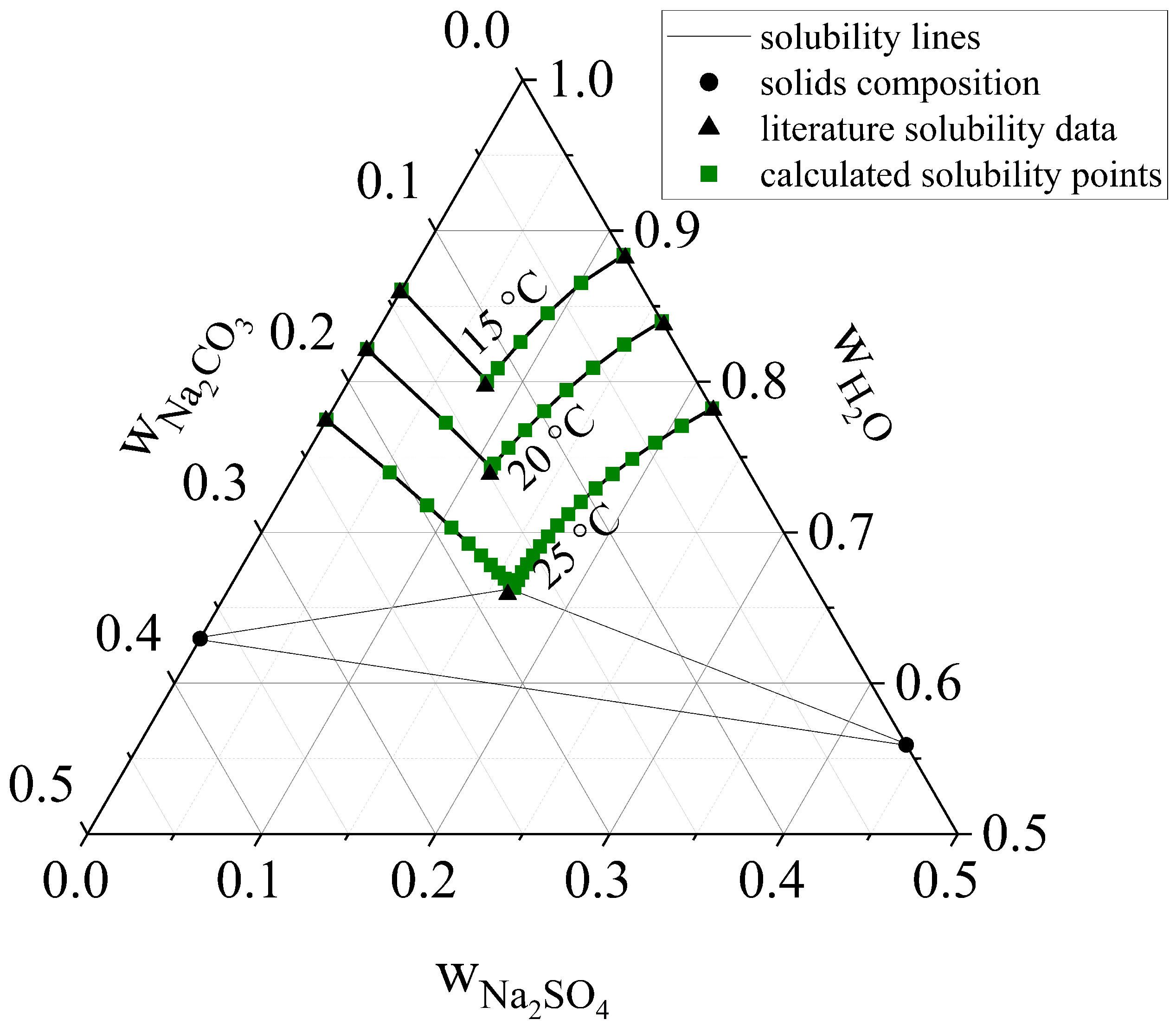
3.2. Ternary Experiments
Ternary Dissolution
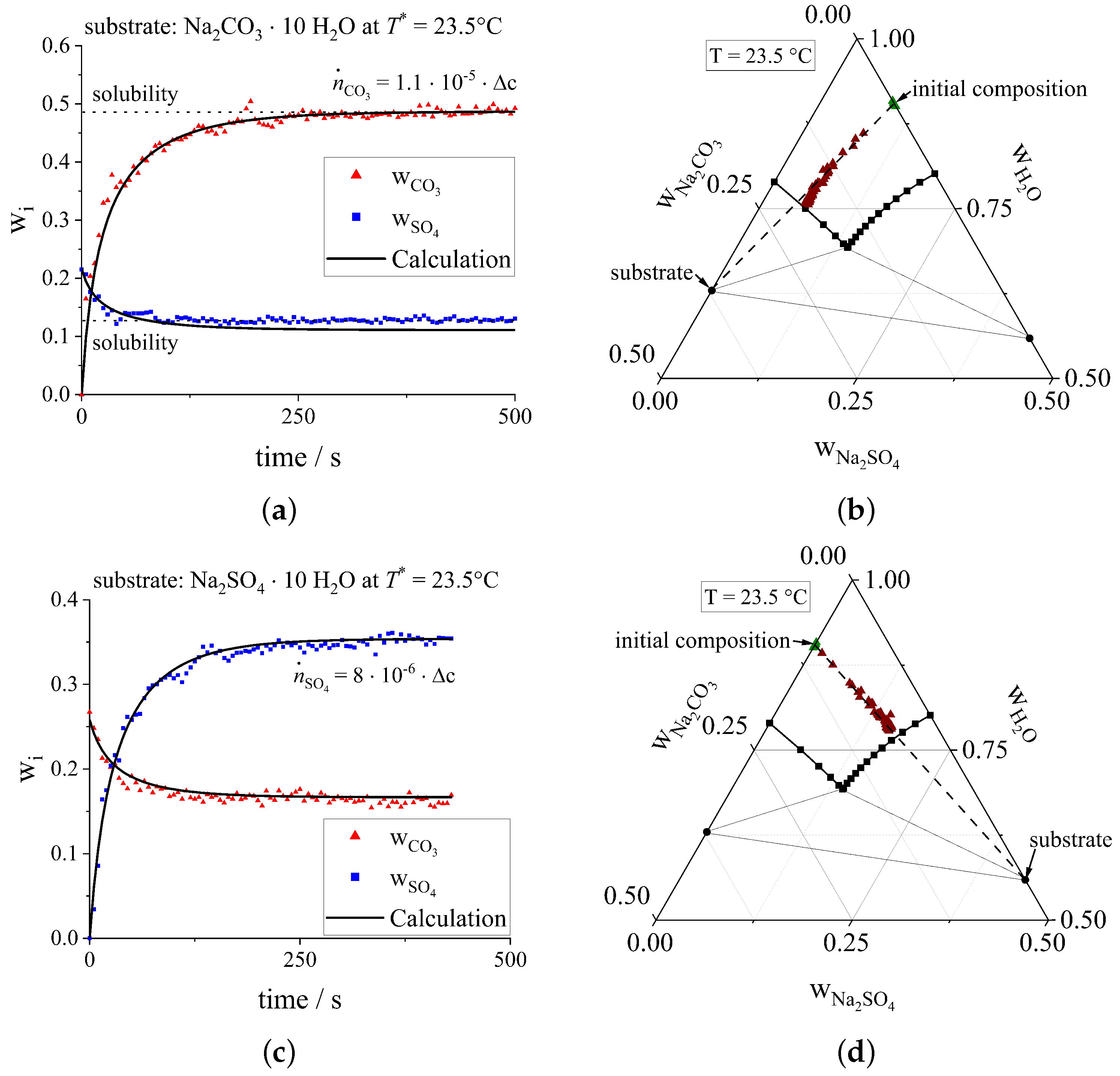
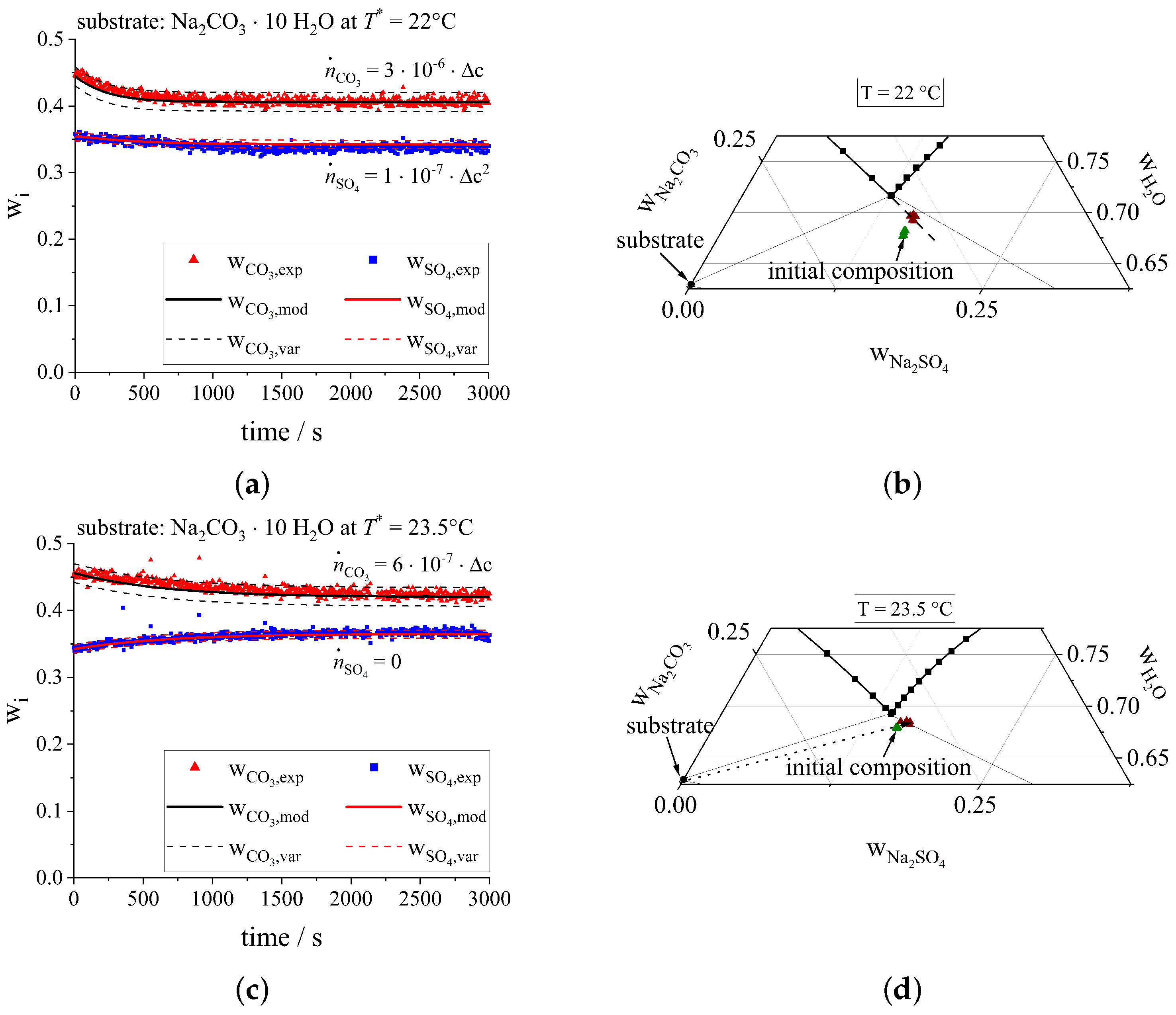
3.3. Ternary Crystallization
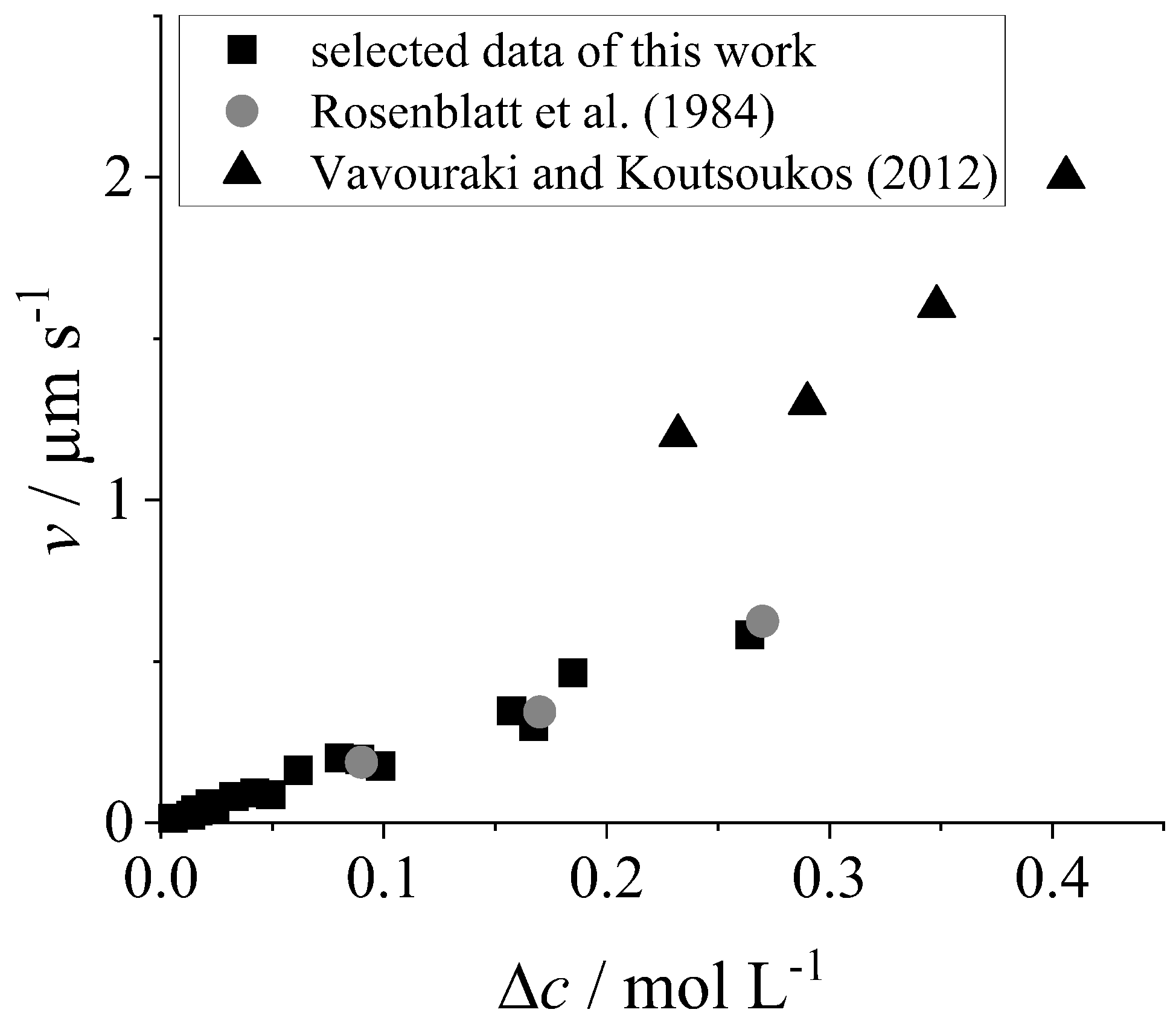
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guignon, B.; Duquenoy, A.; Dumoulin, E.D. Fluid bed encapsulation of particles: Principles and practice. Dry. Technol. 2002, 20, 419–447. [Google Scholar] [CrossRef]
- Degrève, J.; Baeyens, J.; van de Velden, M.; de Laet, S. Spray-agglomeration of NPK-fertilizer in a rotating drum granulator. Powder Technol. 2006, 163, 188–195. [Google Scholar] [CrossRef]
- Suresh, P.; Sreedhar, I.; Vaidhiswaran, R.; Venugopal, A. A comprehensive review on process and engineering aspects of pharmaceutical wet granulation. Chem. Eng. J. 2017, 328, 785–815. [Google Scholar] [CrossRef]
- Römbach, E.; Ulrich, J. Self-controlled coating process for drugs. Cryst. Growth Des. 2007, 7, 1618–1622. [Google Scholar] [CrossRef]
- Shin, G.S.; Choi, W.G.; Na, S.; Ryu, S.O.; Moon, T. Rapid crystallization in ambient air for planar heterojunction perovskite solar cells. Electron. Mater. Lett. 2017, 13, 72–76. [Google Scholar] [CrossRef]
- Yu, W.Z.; Zhang, Y.; Liu, X.; Xiang, Y.; Li, Z.; Wu, S. Synergistic antibacterial activity of multi components in lysozyme/chitosan/ silver/hydroxyapatite hybrid coating. Mater. Des. 2018, 139, 351–362. [Google Scholar] [CrossRef]
- Schönhoff, M. Self-assembled polyelectrolyte multilayers. Curr. Opin. Colloid Interface Sci. 2003, 8, 86–95. [Google Scholar] [CrossRef]
- Nijdam, J.; Trouillet, V.; Kachel, S.; Scharfer, P.; Schabel, W.; Kind, M. Coat formation of surface-active proteins on aqueous surfaces during drying. Colloids Surfaces B Biointerfaces 2014, 123, 53–60. [Google Scholar] [CrossRef]
- Seo, K.S.; Han, H.K. Multilayer-coated tablet of clopidogrel and rosuvastatin: Preparation and in vitro/in vivo characterization. Pharmaceutics 2019, 11, 313. [Google Scholar] [CrossRef] [Green Version]
- Song, K.S.; Lim, J.; Yun, S.; Kim, D.; Kim, Y. Composite fouling characteristics of CaCO3 and CaSO4 in plate heat exchangers at various operating and geometric conditions. Int. J. Heat Mass Transf. 2019, 136, 555–562. [Google Scholar] [CrossRef]
- Lv, Y.; Lu, K.; Ren, Y. Composite crystallization fouling characteristics of normal solubility salt in double-pipe heat exchanger. Int. J. Heat Mass Transf. 2020, 156, 119883. [Google Scholar] [CrossRef]
- Nestler, B.; Garcke, H.; Stinner, B. Multicomponent alloy solidification: Phase-field modeling and simulations. Phys. Rev. E 2005, 71, 041609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.J.; Stepanski, M.; Ulrich, J. Fremdstoffeinflüsse auf Kristallwachstumsgeschwindigkeiten bei der Kristallisation aus Lösungen. Chem. Ing. Tech. 1990, 62, 645–646. [Google Scholar] [CrossRef]
- Rauls, M.; Bartosch, K.; Kind, M.; Lacmann, R.; Mersmann, A. The influence of impurities on crystallization kinetics—A case study on ammonium sulfate. J. Cryst. Growth 2000, 213, 116–128. [Google Scholar] [CrossRef]
- Vergara, A.; Paduano, L.; Vitagliano, V.; Sartorio, R. Multicomponent diffusion in solutions where crystals grow. Mater. Chem. Phys. 2000, 66, 126–131. [Google Scholar] [CrossRef]
- Kubota, N. Effect of impurities on the growth kinetics of crystals. Cryst. Res. Technol. J. Exp. Ind. Crystallogr. 2001, 36, 749–769. [Google Scholar] [CrossRef]
- Louhi-Kultanen, M.; Kallas, J.; Partanen, J.; Sha, Z.; Oinas, P.; Palosaari, S. The influence of multicomponent diffusion on crystal growth in electrolyte solutions. Chem. Eng. Sci. 2001, 56, 3505–3515. [Google Scholar]
- Astilleros, J.M.; Pina, C.M.; Fernández-Díaz, L.; Putnis, A. Nanoscale growth of solids crystallising from multicomponent aqueous solutions. Surf. Sci. 2003, 545, L767–L773. [Google Scholar]
- Zago, G.P.; Penha, F.M.; Seckler, M.M. Product characteristics in simultaneous crystallization of NaCl and CaSO4 from aqueous solution with seeding. Desalination 2020, 474, 114180. [Google Scholar] [CrossRef]
- Penha, F.M.; Andrade, F.R.D.; Lanzotti, A.S.; Moreira Junior, P.F.; Zago, G.P.; Seckler, M.M. In Situ Observation of Epitaxial Growth during Evaporative Simultaneous Crystallization from Aqueous Electrolytes in Droplets. Crystals 2021, 11, 1122. [Google Scholar]
- Garside, J.; Mersmann, A.; Nývlt, J. Measurement of Crystal Growth and Nucleation Rates; IChemE: Rugby, UK, 2002. [Google Scholar]
- Cornel, J.; Mazzotti, M. Estimating crystal growth rates using in situ ATR-FTIR and Raman spectroscopy in a calibration-free manner. Ind. Eng. Chem. Res. 2009, 48, 10740–10745. [Google Scholar] [CrossRef]
- Qu, H.; Alatalo, H.; Hatakka, H.; Kohonen, J.; Louhi-Kultanen, M.; Reinikainen, S.P.; Kallas, J. Raman and ATR FTIR spectroscopy in reactive crystallization: Simultaneous monitoring of solute concentration and polymorphic state of the crystals. J. Cryst. Growth 2009, 311, 3466–3475. [Google Scholar] [CrossRef]
- Nanev, C.N.; Tonchev, V.D.; Hodzhaoglu, F.V. Protocol for growing insulin crystals of uniform size. J. Cryst. Growth 2013, 375, 10–15. [Google Scholar] [CrossRef]
- Eder, C.; Choscz, C.; Müller, V.; Briesen, H. Jamin-interferometer-setup for the determination of concentration and temperature dependent face-specific crystal growth rates from a single experiment. J. Cryst. Growth 2015, 426, 255–264. [Google Scholar] [CrossRef]
- Glade, H.; Ilyaskarov, A.M.; Ulrich, J. Determination of Crystal Growth Kinetics Using Ultrasonic Technique. Chem. Eng. Technol. 2004, 27, 736–740. [Google Scholar] [CrossRef]
- Egan, T.; Rodriguez-Pascual, M.; Lewis, A. In Situ Growth Measurements of Sodium Sulfate during Cooling Crystallization. Chem. Eng. Technol. 2014, 37, 1283–1290. [Google Scholar] [CrossRef]
- Garside, J.; Gibilaro, L.G.; Tavare, N.S. Evaluation of crystal growth kinetics from a desupersaturation curve using initial derivatives. Chem. Eng. Sci. 1982, 37, 1625–1628. [Google Scholar] [CrossRef]
- Montes-Hernandez, G.; Renard, F. Time-resolved in situ raman spectroscopy of the nucleation and growth of siderite, magnesite, and calcite and their precursors. Cryst. Growth Des. 2016, 16, 7218–7230. [Google Scholar] [CrossRef]
- Qu, H.; Kohonen, J.; Louhi-Kultanen, M.; Reinikainen, S.P.; Kallas, J. Spectroscopic Monitoring of Carbamazepine Crystallization and Phase Transformation in Ethanol–Water Solution. Ind. Eng. Chem. Res. 2008, 47, 6991–6998. [Google Scholar] [CrossRef]
- Offermann, H.; von Brachel, G.; Al-Sabbagh, A.; Farelo, F. Crystallization kinetics of NaCl in multicomponent solutions. Cryst. Res. Technol. 1995, 30, 651–658. [Google Scholar]
- Gurato, G.; Goatin, C.; Petrone, A.; Talamini, G.; Fenza, S.; Zannetti, R. Effect of impurities on the Na2SO4 crystallization. J. Cryst. Growth 1984, 66, 621–631. [Google Scholar] [CrossRef]
- Vavouraki, A.I.; Koutsoukos, P.G. The inhibition of crystal growth of mirabilite in aqueous solutions in the presence of phosphonates. J. Cryst. Growth 2016, 436, 92–98. [Google Scholar] [CrossRef]
- Su, N.; Wang, Y.; Xiao, Y.; Lu, H.; Lou, Y.; Huang, J.; He, M.; Li, Y.; Hao, H. Mechanism of influence of organic impurity on crystallization of sodium sulfate. Ind. Eng. Chem. Res. 2018, 57, 1705–1713. [Google Scholar] [CrossRef]
- Shi, B.; Rousseau, R.W. Crystal Properties and Nucleation Kinetics from Aqueous Solutions of Na2CO3 and Na2SO4. Ind. Eng. Chem. Res. 2001, 40, 1541–1547. [Google Scholar] [CrossRef]
- Shi, B.; Frederick, W.J.; Rousseau, R.W. Nucleation, growth, and composition of crystals obtained from solutions of Na2CO3 and Na2SO4. Ind. Eng. Chem. Res. 2003, 42, 6343–6347. [Google Scholar] [CrossRef]
- Helfenritter, C.; Kind, M. Multi-Component Diffusion in the Vicinity of a Growing Crystal. Crystals 2022, 12, 872. [Google Scholar] [CrossRef]
- Linnow, K.; Steiger, M.; Lemster, C.; de Clercq, H.; Jovanović, M. In situ Raman observation of the crystallization in NaNO3–Na2SO4–H2O solution droplets. Environ. Earth Sci. 2013, 69, 1609–1620. [Google Scholar] [CrossRef]
- Müller, M.; Kind, M.; Cairncross, R.; Schabel, W. Diffusion in multi-component polymeric systems: Diffusion of non-volatile species in thin films. Eur. Phys. J. Spec. Top. 2009, 166, 103–106. [Google Scholar] [CrossRef]
- Scharfer, P.; Schabel, W.; Kind, M. Mass transport measurements in membranes by means of in situ Raman spectroscopy—First results of methanol and water profiles in fuel cell membranes. J. Membr. Sci. 2007, 303, 37–42. [Google Scholar] [CrossRef]
- Caspari, W.A. CCCXXIV.—The system sodium carbonate–sodium sulphate–water. J. Chem. Soc. Trans. 1924, 125, 2381–2387. [Google Scholar] [CrossRef]
- Marion, G.M.; Mironenko, M.V.; Roberts, M.W. FREZCHEM: A geochemical model for cold aqueous solutions. Comput. Geosci. 2010, 36, 10–15. [Google Scholar] [CrossRef]
- Ludwig, I.; Schabel, W.; Kind, M.; Castaing, J.C.; Ferlin, P. Drying and film formation of industrial waterborne latices. AIChE J. 2007, 53, 549–560. [Google Scholar] [CrossRef]
- Nijdam, J.; Kachel, S.; Scharfer, P.; Schabel, W.; Kind, M. Effect of Diffusion on Component Segregation During Drying of Aqueous Solutions Containing Protein and Sugar. Dry. Technol. 2015, 33, 288–300. [Google Scholar] [CrossRef]
- Ben Mabrouk, K.; Kauffmann, T.H.; Aroui, H.; Fontana, M.D. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 2013, 44, 1603–1608. [Google Scholar] [CrossRef]
- Carey, D.M.; Korenowski, G.M. Measurement of the Raman spectrum of liquid water. J. Chem. Phys. 1998, 108, 2669–2675. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G.; Königsberger, E. Speciation studies in aqueous — solutions. A combined Raman spectroscopic and thermodynamic study. Dalton Trans. 2008, 900–908. [Google Scholar] [CrossRef]
- Rosenblatt, D.; Marks, S.B.; Pigford, R.L. Kinetics of phase transitions in the system sodium sulfate-water. Ind. Eng. Chem. Fundam. 1984, 23, 143–147. [Google Scholar] [CrossRef]
- Vavouraki, A.I.; Koutsoukos, P.G. Kinetics of crystal growth of mirabilite in aqueous supersaturated solutions. J. Cryst. Growth 2012, 338, 189–194. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| From Dissolution | From Desupersaturation | ||
|---|---|---|---|
| /°C | / | / | /- |
| 17 | |||
| 18.5 | |||
| 22 | |||
| 23.5 | |||
| From Dissolution | From Desupersaturation | ||
|---|---|---|---|
| /°C | / | / | /- |
| 17 | |||
| 18.5 | |||
| 22 | |||
| 23.5 | |||
| From Dissolution | From Desupersaturation | ||
|---|---|---|---|
| / | / | /- | |
| substrate component for | |||
| concurrent | 1 | ||
| separation | 1 | ||
| foreign component for | |||
| concurrent | 2 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helfenritter, C.; Kind, M. Determination of Crystal Growth Rates in Multi-Component Solutions. Crystals 2022, 12, 1568. https://doi.org/10.3390/cryst12111568
Helfenritter C, Kind M. Determination of Crystal Growth Rates in Multi-Component Solutions. Crystals. 2022; 12(11):1568. https://doi.org/10.3390/cryst12111568
Chicago/Turabian StyleHelfenritter, Christoph, and Matthias Kind. 2022. "Determination of Crystal Growth Rates in Multi-Component Solutions" Crystals 12, no. 11: 1568. https://doi.org/10.3390/cryst12111568