

Comparative Study of the Compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) Orthovanadates

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
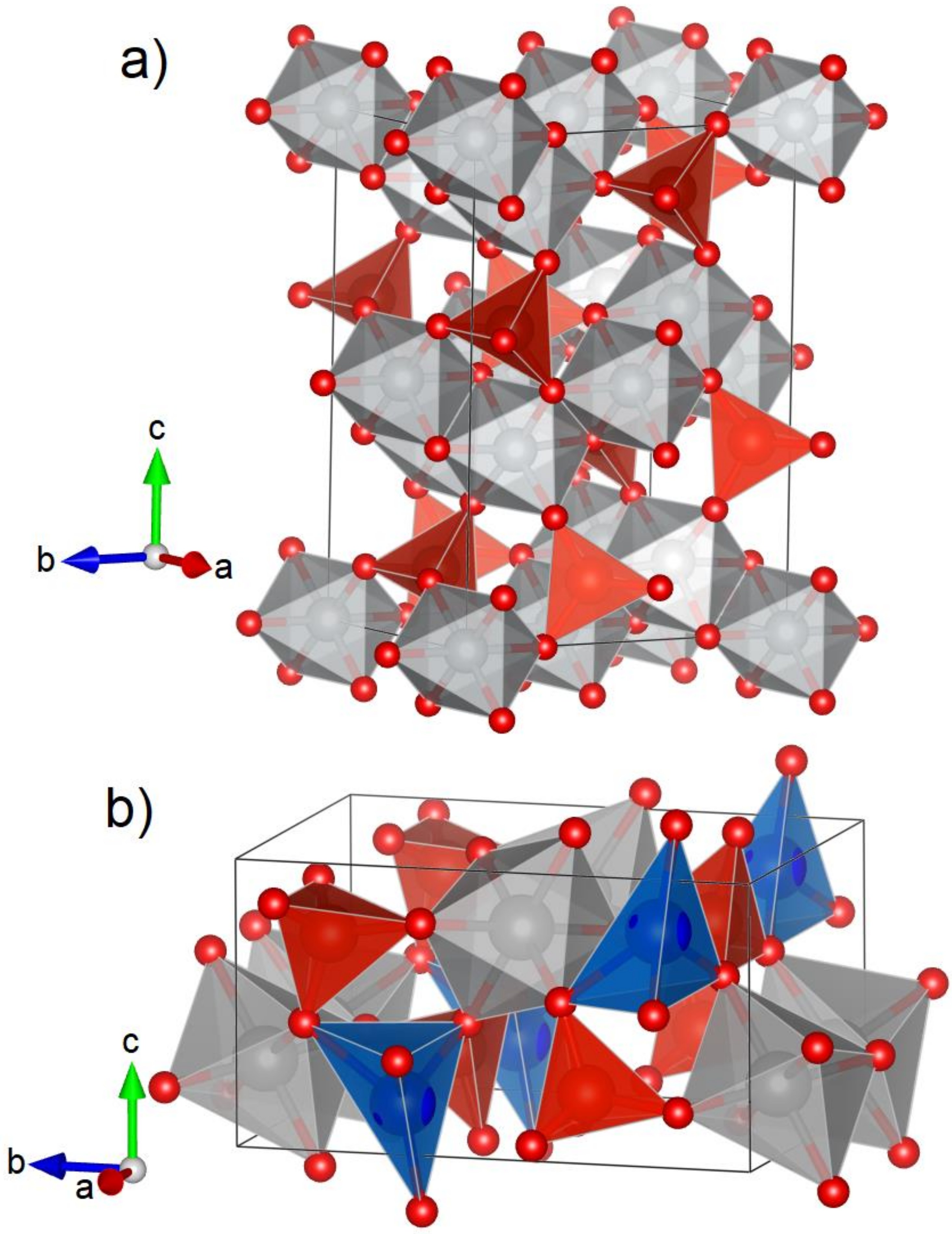
3.1. Crystal Structure
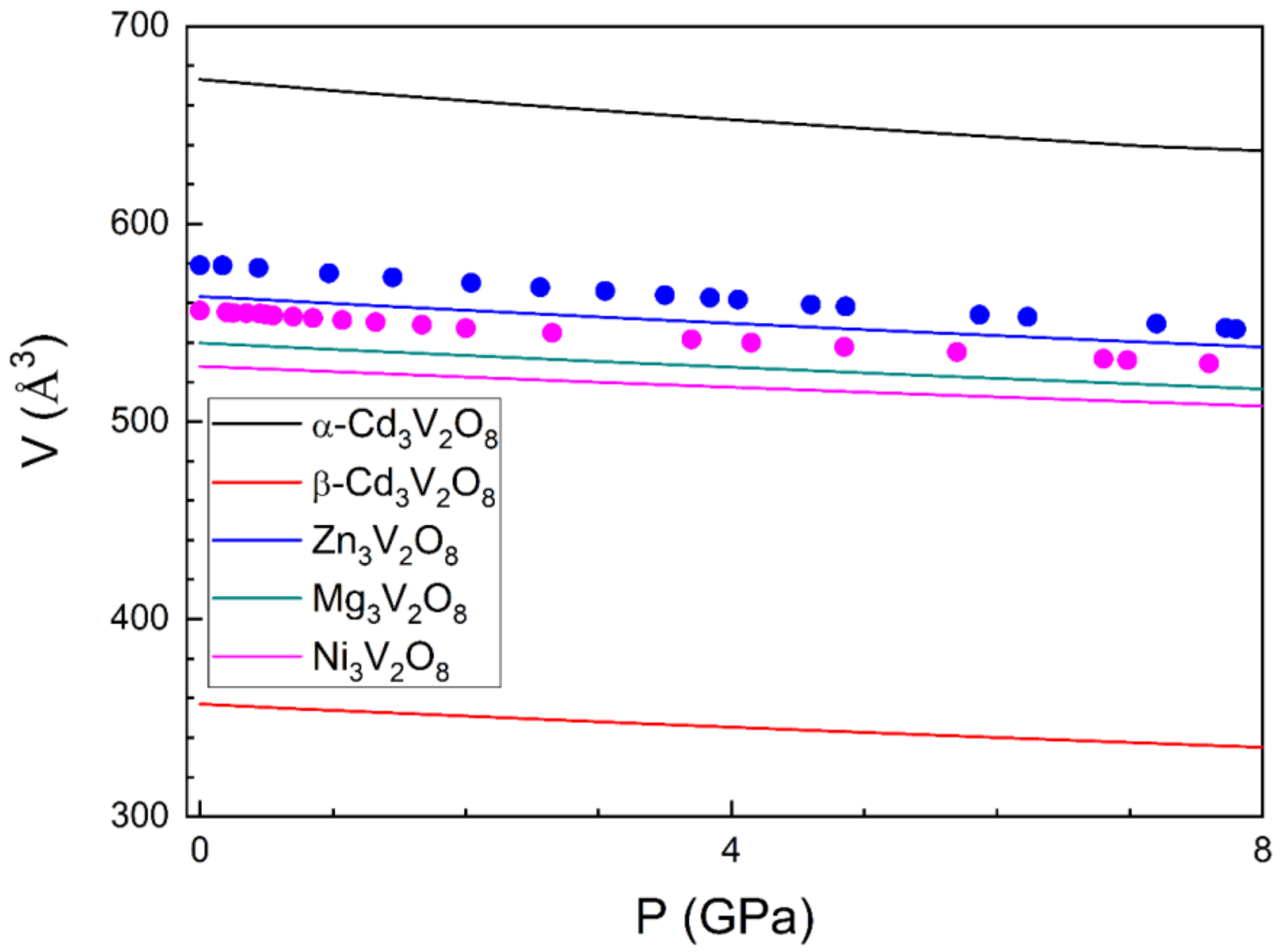
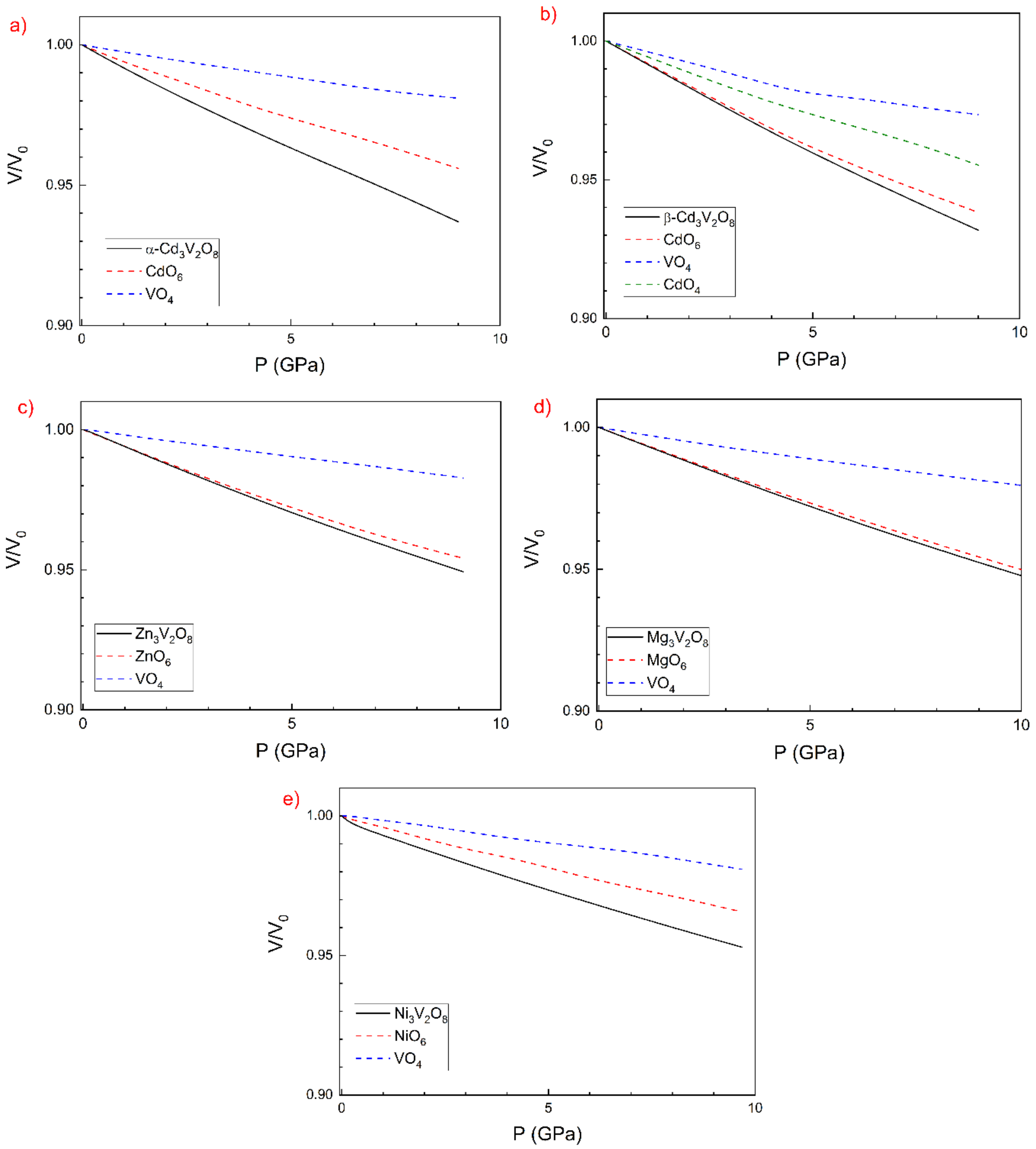
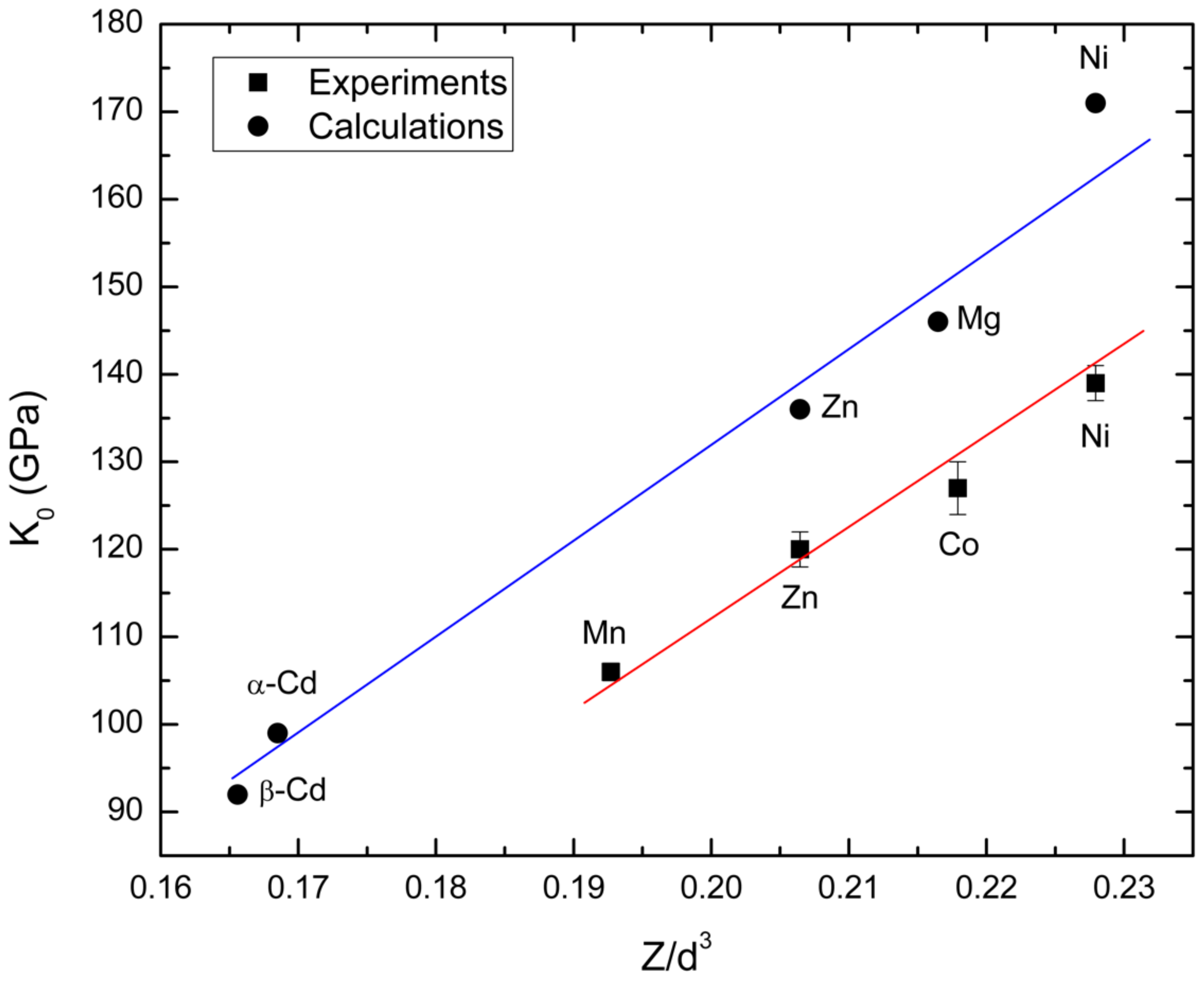
3.2. Pressure–Volume Equation of State and Compressibility
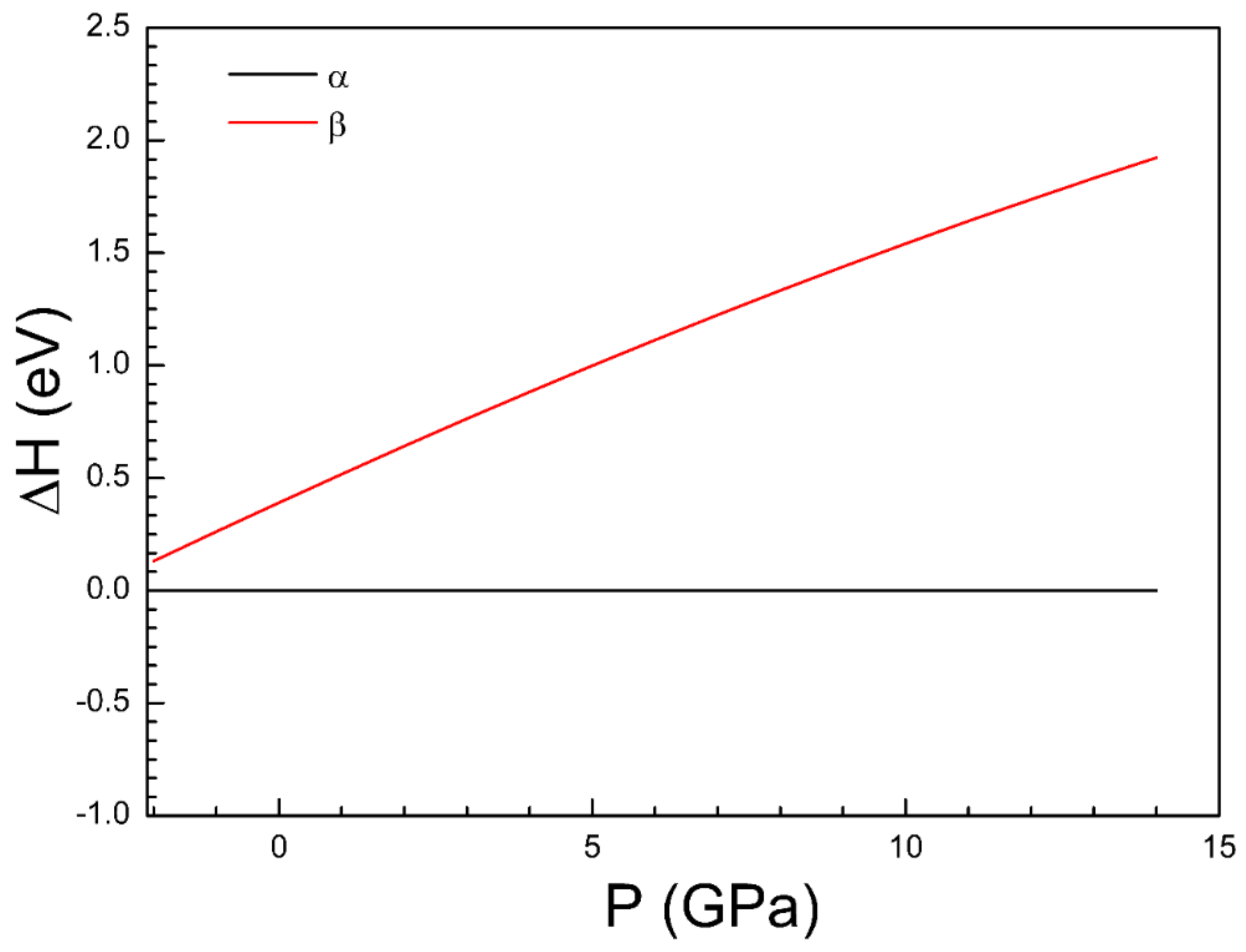
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Cui, Y. Microwave-assisted hydrothermal synthesis of hollow flower-like Zn2V2O7 with enhanced cycling stability as electrode for lithium ion batteries. Mater. Lett. 2018, 228, 369–371. [Google Scholar] [CrossRef]
- Sameie, H.; Sabbagh Alvani, A.A.; Naseri, N.; Du, S.; Rosei, F. First-principles study on ZnV2O6 and Zn2V2O7: Two new photoanode candidates for photoelectrochemical water oxidation. Ceram. Int. 2018, 44, 6607–6613. [Google Scholar] [CrossRef]
- Bayat, A.; Mahjoub, A.R.; Amini, M.M. Optical properties of hydrophilic surfaced self-assembled Cd2V2O7 hollow sphere shape architecture. Mater. Lett. 2016, 186, 252–255. [Google Scholar] [CrossRef]
- Zhi, J. Study of MgV2O6 as Cathode Material for Secondary Magnesium Batteries. Asian J. Chem. 2011, 23, 1399–1400. [Google Scholar]
- Ni, S.; Liu, J.; Chao, D.; Mai, L. Vanadate-Based Materials for Li-Ion Batteries: The Search for Anodes for Practical Applications. Adv. Energy Mater. 2019, 9, 1803324. [Google Scholar] [CrossRef]
- Hassan, A.; Iqbal, T.; Tahir, M.B.; Afsheen, S. A review on copper vanadate-based nanostructures for photocatalysis energy production. Int. J. Energy Res. 2019, 43, 9–28. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, S.; Lee, S.-H.; Ryu, K.-S. Synthesis of Zn3V2O8 nanoplatelets for lithium-ion battery and supercapacitor applications. RSC Adv. 2015, 5, 91822–91828. [Google Scholar] [CrossRef]
- Cabrera, I.; Kenzelmann, M.; Lawes, G.; Chen, Y.; Chen, W.; Erwin, R.; Gentile, T.R.; Leao, J.B.; Lynn, J.W.; Rogado, N.; et al. Coupled Magnetic and Ferroelectric Domains in Multiferroic Ni3V2O8. Phys. Rev. Lett. 2009, 103, 087201. [Google Scholar] [CrossRef] [Green Version]
- Bîrdeanu, M.-I.; Vaida, M.; Ursu, D.; Fagadar-Cosma, E. Obtaining and characterization of Zn3V2O8 and Mg3V2O8 pseudo binary oxide nanomaterials by hydrothermal method. In High Energy Gamma-Ray Astronomy, Proceedings of the 6th International Meeting on High-Energy Gamma-Ray Astronomy, Heidelberg, Germany, 11–15 July 2016; AIP Publishing LLC: Melville, NY, USA, 2017; p. 030006. [Google Scholar]
- Mazloom, F.; Masjedi-Arani, M.; Salavati-Niasari, M. Novel size-controlled fabrication of pure Zn3V2O8 nanostructures via a simple precipitation approach. J. Mater. Sci. Mater. Electron. 2015, 27, 1974–1982. [Google Scholar] [CrossRef]
- Luo, J.; Chen, R.; Zhang, X. The Effect in the Production and Luminescence Property of Zn3V2O8 with Eu-doping and the First Principle Calculation of α-Zn3V2O8. Mater. Sci. Eng. 2017, 274, 012087. [Google Scholar] [CrossRef] [Green Version]
- Qian, T.; Fan, B.; Wang, H.; Zhu, S. Structure and luminescence properties of Zn3V2O8 yellow phosphor for white light emitting diodes. Chem. Phys. Lett. 2019, 715, 34–39. [Google Scholar] [CrossRef]
- Matsushima, Y.; Koide, T.; Hiro-Oka, M.; Shida, M.; Sato, A.; Sugiyama, S.; Ito, M. Self-activated vanadate compounds toward realization of rare-earth-free full-color phosphors. J. Am. Ceram. Soc. 2015, 98, 1236–1244. [Google Scholar] [CrossRef]
- Díaz-Anichtchenko, D.; Santamaria-Perez, D.; Marqueño, T.; Pellicer-Porres, J.; Ruiz-Fuertes, J.; Ribes, R.; Errandonea, D. Comparative study of the high-pressure behavior of ZnV2O6, Zn2V2O7, and Zn3V2O8. J. Alloys Compd. 2020, 837, 155505. [Google Scholar] [CrossRef]
- Diaz-Anichtchenko, D.; Turnbull, R.; Bandiello, E.; Anzellini, S.; Errandonea, D. High-Pressure Structural Behavior and Equation of State of Kagome Staircase Compound, Ni3V2O8. Crystals 2020, 10, 910. [Google Scholar] [CrossRef]
- Kesari, S.; Garg, A.B.; Clemens, O.; Joseph, B.; Rao, R. Pressure-Induced Structural Behavior of Orthorhombic Mn3(VO4)2: Raman Spectroscopic and X-ray Diffraction Investigations. ACS Omega 2022, 7, 3099–3108. [Google Scholar] [CrossRef]
- Beltrán, A.; Gracia, L.; Andrés, J. Polymorphs of ZnV2O6 under pressure: A first-principle investigation. J. Phys. Chem. C 2019, 123, 3239–3253. [Google Scholar] [CrossRef]
- Lopez-Moreno, S.; Errandonea, D.; Rodriguez-Hernandez, P.; Munoz, A. Polymorphs of CaSeO4 under pressure: A first-principles study of structural, electronic, and vibrational properties. Inorg. Chem. 2015, 54, 1765–1777. [Google Scholar] [CrossRef]
- Benmakhlouf, A.; Errandonea, D.; Bouchenafa, M.; Maabed, S.; Bouhemadou, A.; Bentabet, A. New pressure-induced polymorphic transitions of anhydrous magnesium sulfate. Dalton Trans. 2017, 46, 5058–5068. [Google Scholar] [CrossRef] [Green Version]
- Errandonea, D.; Gracia, L.; Lacomba-Perales, R.; Polian, A.; Chervin, J.C. Compression of scheelite-type SrMoO4 under quasi-hydrostatic conditions: Redefining the high-pressure structural sequence. J. Appl. Phys. 2013, 113, 123510. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL14 User’s Manual; University of Torino: Torino, Italy, 2014. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of theelectron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M.E. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analyticrepresentation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.crystal.unito.it/basis-sets.php/ (accessed on 14 October 2022).
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Diaz-Anichtchenko, D.; Gracia, L.; Errandonea, D. Density-functional study of pressure-induced phase transitions and electronic properties of Zn2V2O7. RSC Adv. 2021, 11, 10401–10415. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Anichtchenko, D.; Errandonea, D. Pressure-induced phase transitions and electronic properties of Cd2V2O7. RSC Adv. 2022, 12, 14827–14837. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Gopal, R.; Calvo, C. Crystal structure of α-Zn3V2O8. Can. J. Chem. 1971, 49, 3056–3059. [Google Scholar] [CrossRef]
- Krishnamachari, N.; Calvo, C. Refinement of the Structure of Mg3V2O8. Can. J. Chem. 1971, 49, 1629–1637. [Google Scholar] [CrossRef]
- Saurbrei, E.E.; Faggiani, R.; Calvo, C. Refinement of the Crystal structures of Cd3V2O8 and Ni3V2O8. Acta Crist. 1973, B29, 2304–2306. [Google Scholar] [CrossRef]
- Yahia, H.B.; Gaudin, E.; Feral-Martin, C.; Darriet, J. Structural study of the NaCdVO4–Cd3V2O8 and CdO–V2O5 sections of the ternary system Na2O–CdO–V2O5. J. Solid State Chem. 2010, 183, 776–783. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Available online: https://materialsproject.org/ (accessed on 14 October 2022).
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Laverock, J.; Piper, L.F.J.; Preston, A.R.H.; Chen, B.; McNulty, J.; Smith, K.E.; Guo, J.H. Strain dependence of bonding and hybridization across the metal-insulator transition of VO2. Phys. Rev. B 2012, 85, 081104. [Google Scholar] [CrossRef] [Green Version]
- Errandonea, D. High pressure crystal structures of orthovanadates and their properties. J. Appl. Phys. 2020, 128, 040903. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R.J. EosFit7-GUI: A new GUI tool for equation of state calculations, analyses and teaching. J. Appl. Cryst. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Anzellini, S.; Burakovsky, L.; Turnbull, R.; Bandiello, E.; Errandonea, D. P–V–T Equation of State of Iridium Up to 80 GPa and 3100 K. Crystals 2021, 11, 452. [Google Scholar] [CrossRef]
- Angel, R.J. Equations of state. Rev. Mineral. Geochem. 2000, 41, 35–59. [Google Scholar] [CrossRef]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Errandonea, D.; Manjon, F.J. Pressure effects on the structural and electronic properties of ABX4 scintillating crystals. Prog. Mater. Sci. 1997, 53, 711–773. [Google Scholar] [CrossRef]
- Díaz-Anichtchenko, D.; Turnbull, R.; Bandiello, E.; Anzellini, S.; Achary, S.N.; Errandonea, D. Pressure-induced chemical decomposition of copper orthovanadate (α-Cu3V2O8). J. Mater. Chem. C 2021, 9, 13402–13409. [Google Scholar] [CrossRef]
- She, J.; Sawamura, S.; Wondraczek, L. Scratch hardness of rare-earth substituted calcium aluminosilicate glasses. J. Non-Cryst. Solids 2019, 1, 100010. [Google Scholar] [CrossRef]
- Huang, Z.; Feng, J.; Pan, W. Theoretical investigations of the physical properties of zircon-type YVO4. J. Solid State Chem. 2012, 185, 42–48. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Site | x | y | z |
|---|---|---|---|---|
| Cd1 | 4a | 0 | 0 | 0 |
| Cd2 | 8e | 0.25 | 0.1554 | 0.25 |
| V1 | 8f | 0 | 0.3902 | 0.1247 |
| O1 | 8f | 0 | 0.2784 | 0.2491 |
| O2 | 8g | 0 | 0.0121 | 0.247 |
| O3 | 16g | 0.2134 | 0.3815 | 0.9847 |
| α-Cd3V2O8 | B3LYP | ||
| a (Å) | 6.5837 | ||
| b (Å) | 11.9604 | ||
| c (Å) | 8.5473 | ||
| V (Å3) | 673.046 | ||
| β-Cd3V2O8 | B3LYP | Exp. [34] | ε(%) |
| a (Å) | 6.8744 | 6.9882 | −1.6 |
| b (Å) | 5.3107 | 5.3251 | −0.3 |
| c (Å) | 9.7796 | 9.8133 | −0.3 |
| V (Å3) | 357.032 | 365.18 | −2.2 |
| Zn3V2O8 | B3LYP | Exp. [32] | ε(%) |
| a (Å) | 6.0332 | 6.088 | −0.9 |
| b (Å) | 11.4247 | 11.489 | −0.6 |
| c (Å) | 8.1709 | 8.280 | −1.3 |
| V (Å3) | 563.200 | 579.145 | −2.8 |
| Mg3V2O8 | B3LYP | Exp. [33] | ε(%) |
| a (Å) | 5.9061 | 6.053 | −2.4 |
| b (Å) | 11.2516 | 11.442 | −1.7 |
| c (Å) | 8.1216 | 8.33 | −2.5 |
| V (Å3) | 539.705 | 576.923 | −6.4 |
| Ni3V2O8 | B3LYP | Exp. [34] | ε(%) |
| a (Å) | 5.685 | 5.936 | −4.2 |
| b (Å) | 11.4508 | 11.42 | −0.3 |
| c (Å) | 8.1093 | 8.24 | −1.5 |
| V (Å3) | 527.903 | 558.582 | −5.5 |
| Compound | VHSE06 (Å3) | VPBE (Å3) | εHSE06(%) | εPBE(%) | VMP (Å3) | εMP(%) |
|---|---|---|---|---|---|---|
| α-Cd3V2O8 | 656.413 | 669.242 | ||||
| β-Cd3V2O8 | 348.866 | 355.061 | −4.5 | −2.8 | 384.85 | 5.4 |
| Zn3V2O8 | 549.367 | 559.630 | −5.1 | −3.4 | 609.12 | 5.2 |
| Mg3V2O8 | 530.461 | 541.487 | −8.1 | −6.1 | 599.51 | 3.9 |
| Ni3V2O8 | 503.762 | 504.320 | −9.8 | −8.7 | 579.38 | 3.7 |
| GPa−1 | ||
| α-Cd3V2O8 | GPa−1 | |
| GPa−1 | ||
| GPa−1 | ||
| β-Cd3V2O8 | GPa−1 | |
| GPa−1 | ||
| GPa−1 | GPa−1 [14] | |
| Zn3V2O8 | GPa−1 | GPa−1 |
| GPa−1 | GPa−1 | |
| GPa−1 | ||
| Mg3V2O8 | GPa−1 | |
| GPa−1 | ||
| GPa−1 | GPa−1 [15] | |
| Ni3V2O8 | GPa−1 | GPa−1 |
| GPa−1 | GPa−1 |
| Phase | |||
|---|---|---|---|
| α-Cd3V2O8 | 673.0 | 112 | 5.0 |
| β-Cd3V2O8 | 357.0 | 92 | 4.0 |
| Zn3V2O8 | 563.2 | 136 | 5.4 |
| Mg3V2O8 | 539.7 | 146 | 4.4 |
| Ni3V2O8 | 527.9 | 171 | 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Anichtchenko, D.; Errandonea, D. Comparative Study of the Compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) Orthovanadates. Crystals 2022, 12, 1544. https://doi.org/10.3390/cryst12111544
Díaz-Anichtchenko D, Errandonea D. Comparative Study of the Compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) Orthovanadates. Crystals. 2022; 12(11):1544. https://doi.org/10.3390/cryst12111544
Chicago/Turabian StyleDíaz-Anichtchenko, Daniel, and Daniel Errandonea. 2022. "Comparative Study of the Compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) Orthovanadates" Crystals 12, no. 11: 1544. https://doi.org/10.3390/cryst12111544