
Oxalic Acid, a Versatile Coformer for Multicomponent Forms with 9-Ethyladenine



Abstract
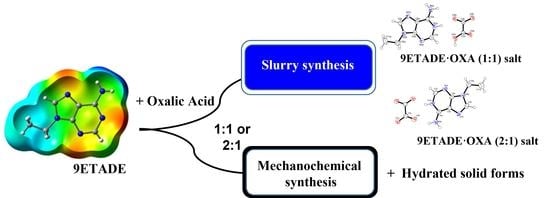
:
1. Introduction
2. Materials and Methods
2.1. Materials
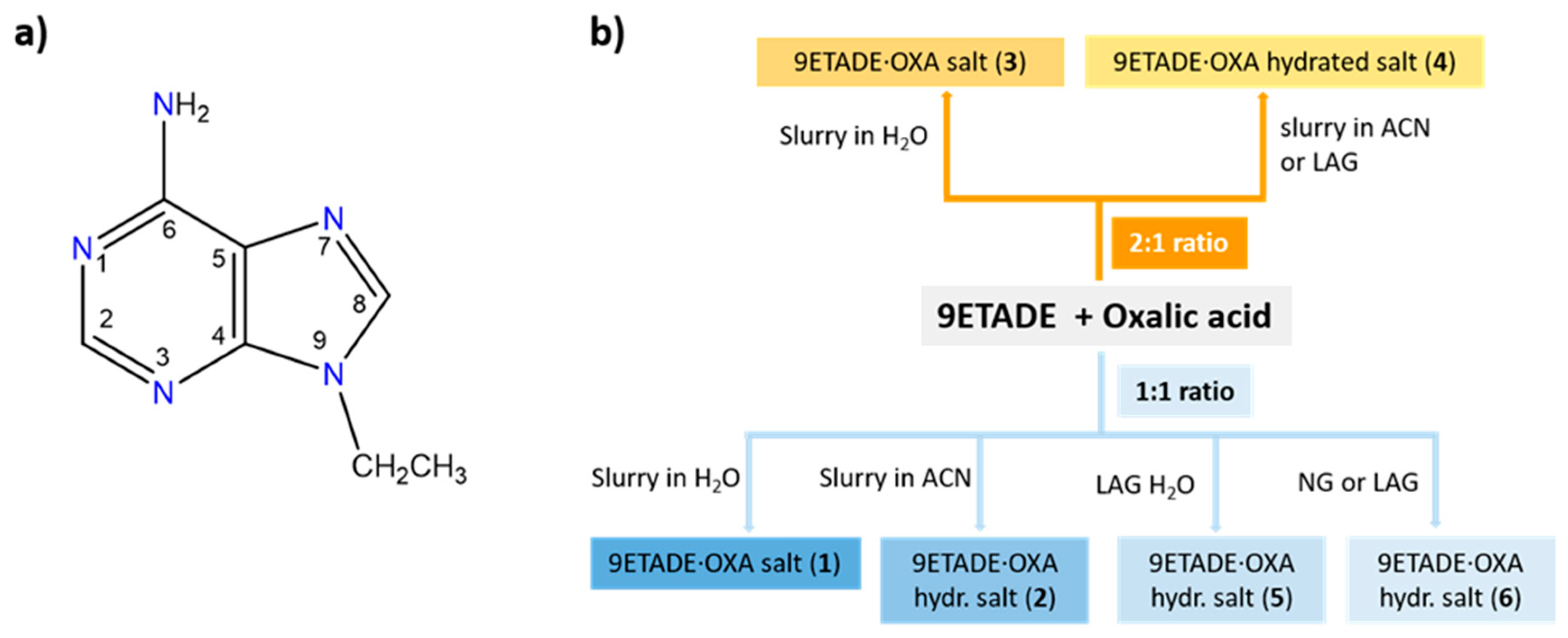
2.2. Syntheses of Multicomponent Solids
2.3. Characterization
2.4. Stability Studies
2.5. Theoretical Calculations
3. Results and Discussion
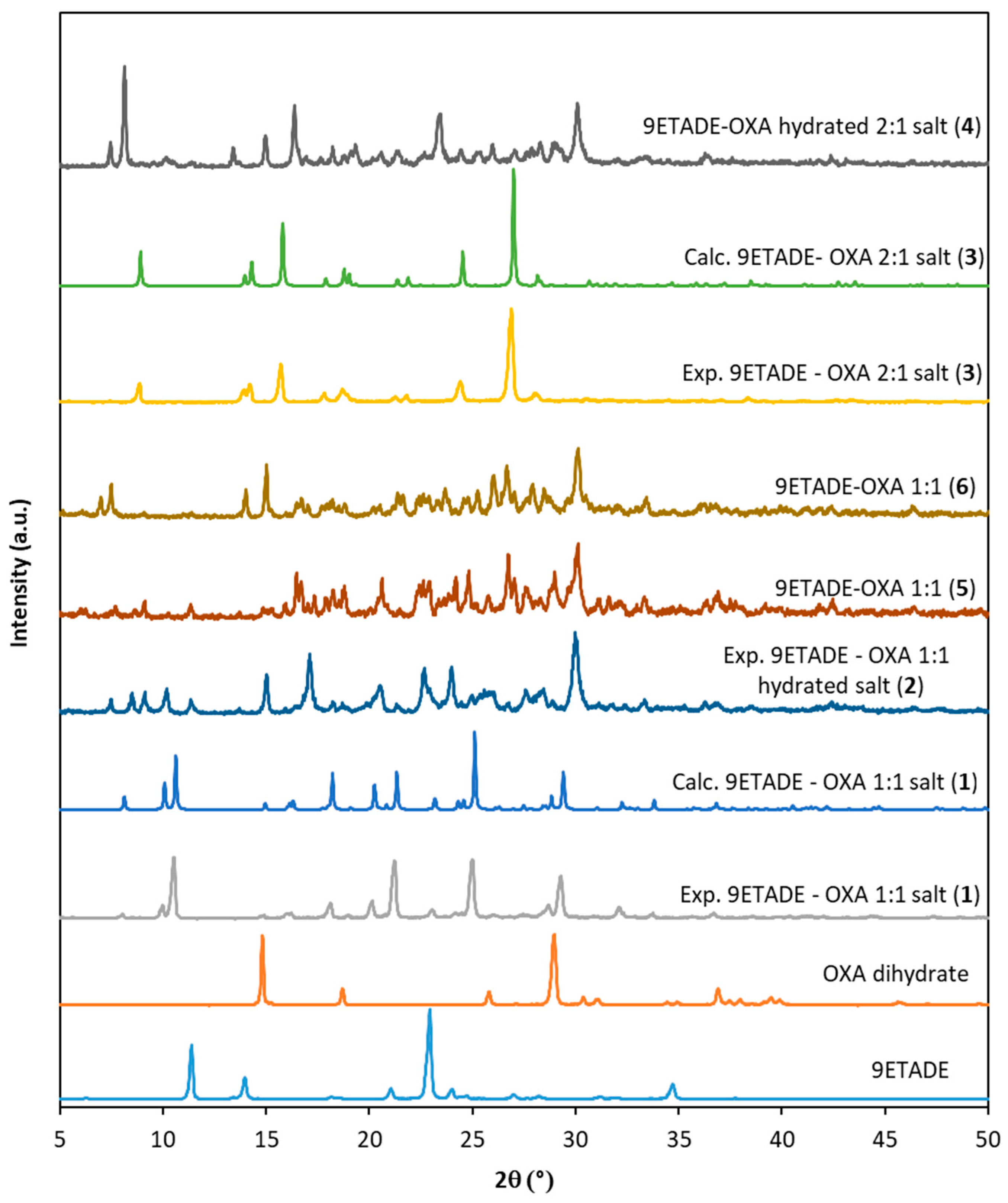
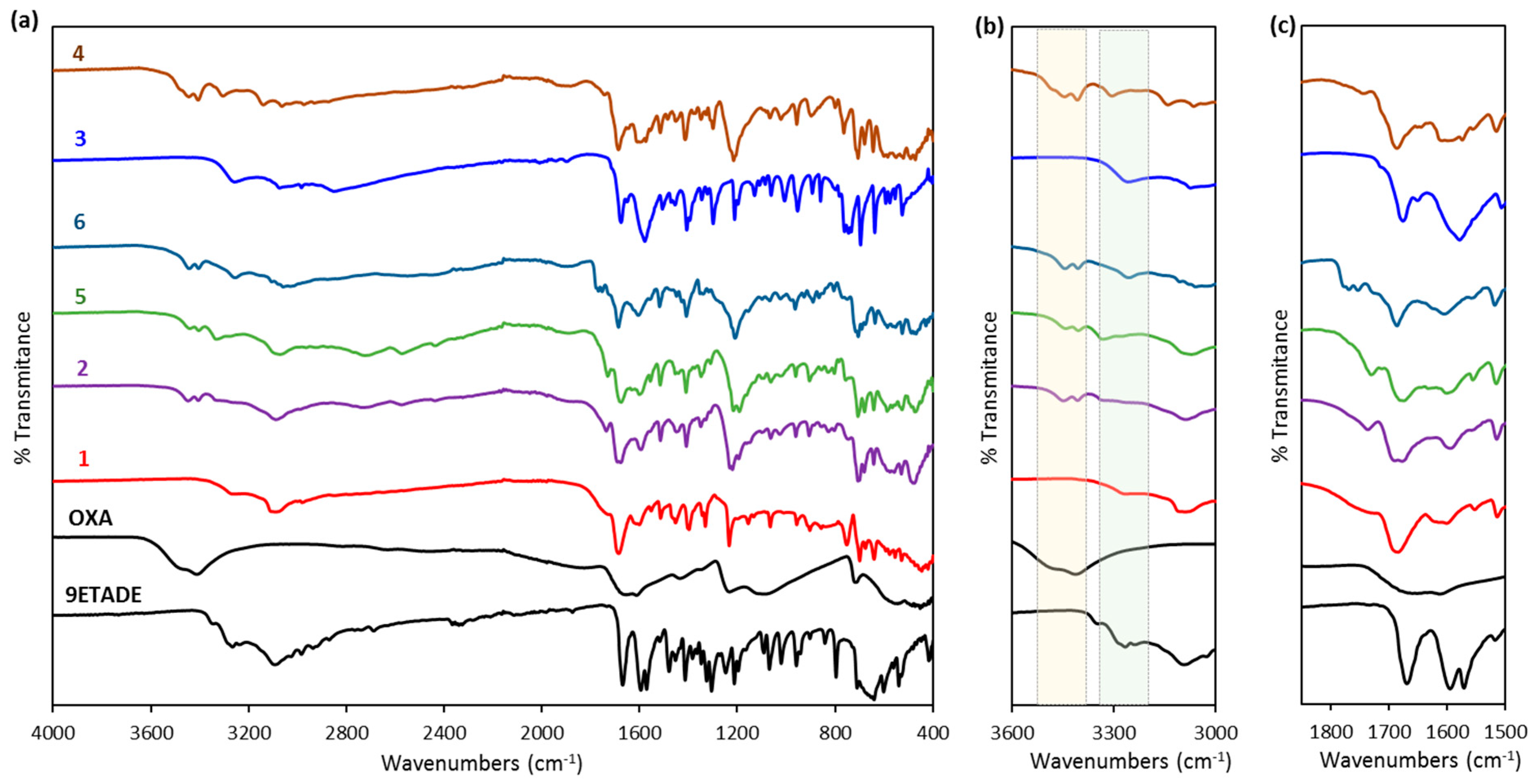
3.1. Solid-State Characterization of the Solids Obtained from Slurry or Mechanochemical Methods
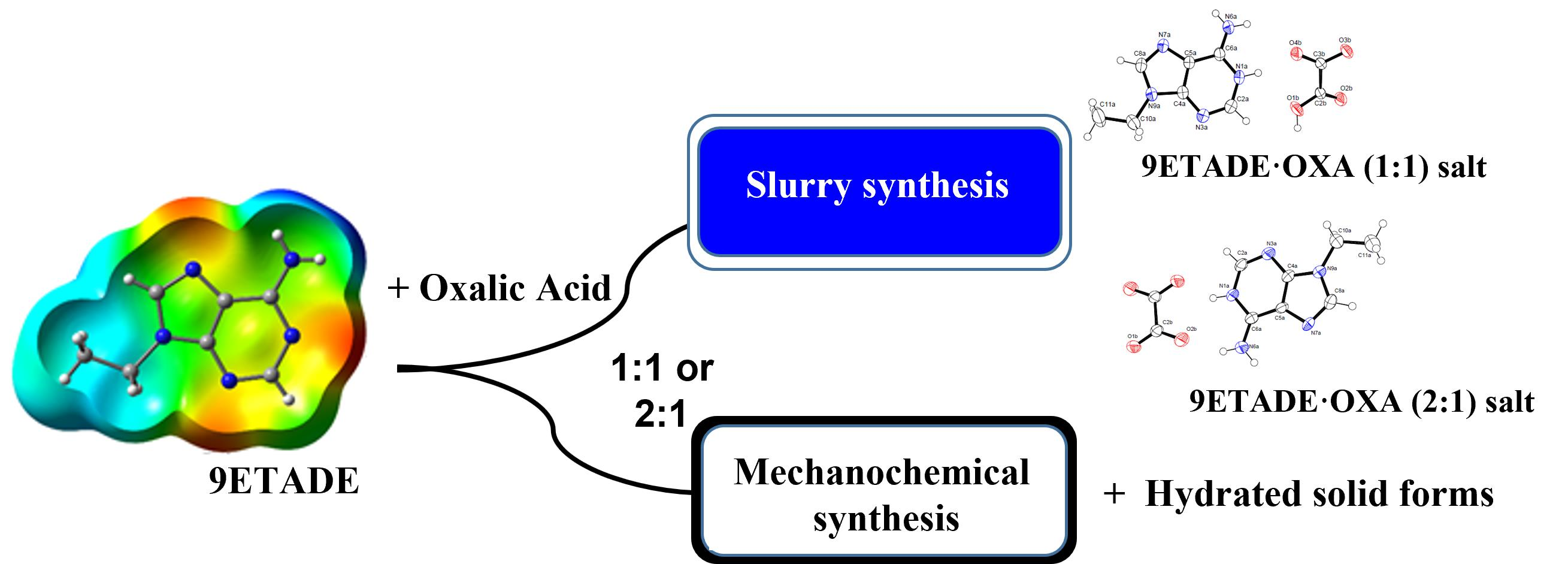
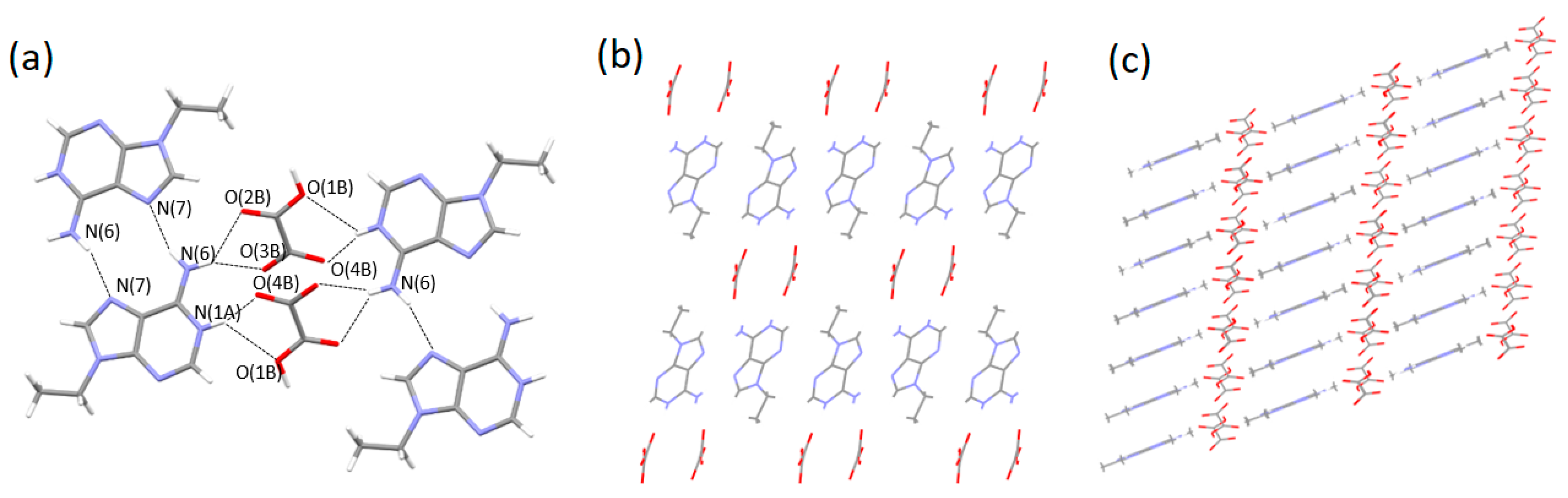
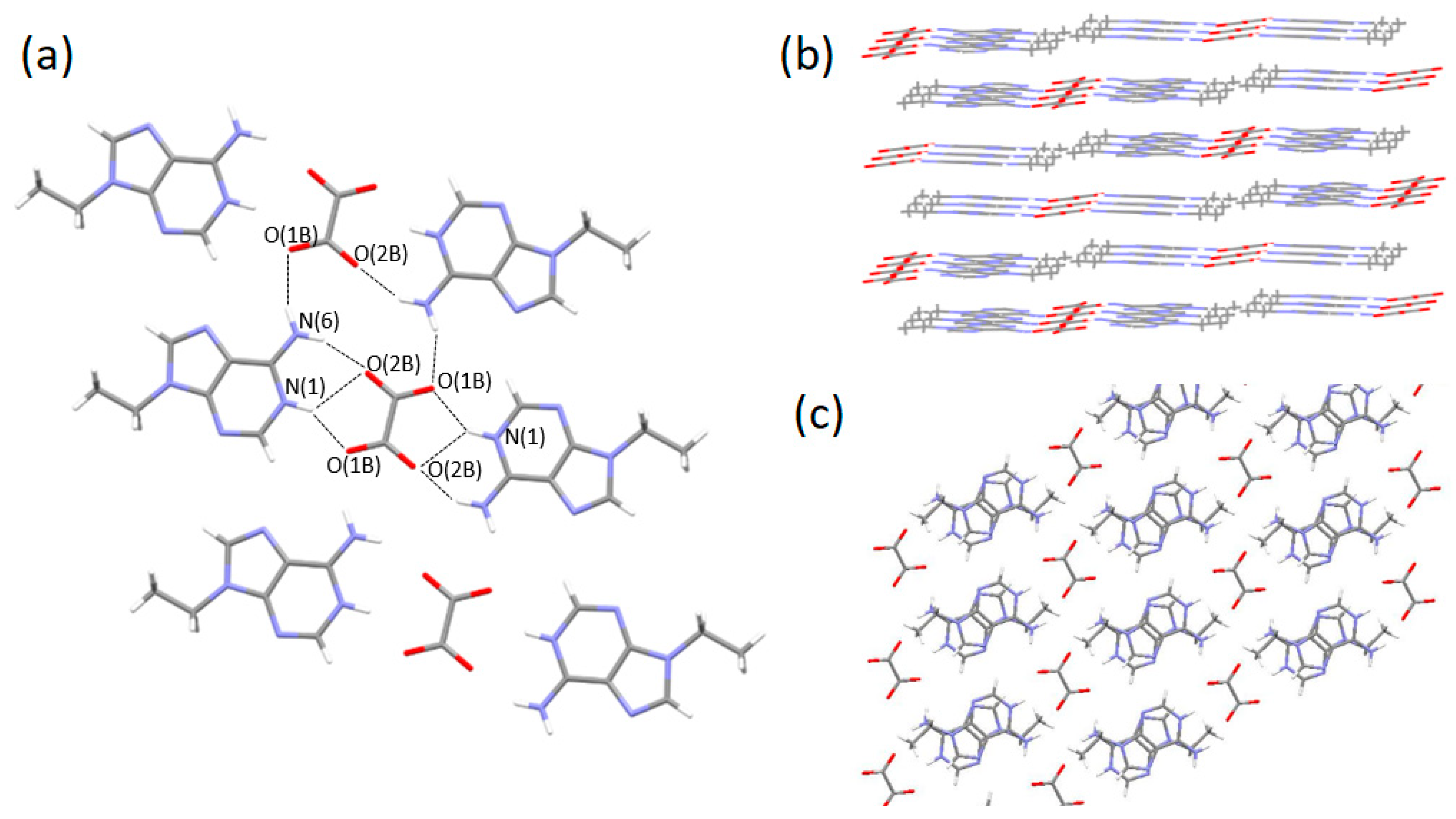
3.2. Single-Crystal Structures
3.3. Aqueous Solubilities
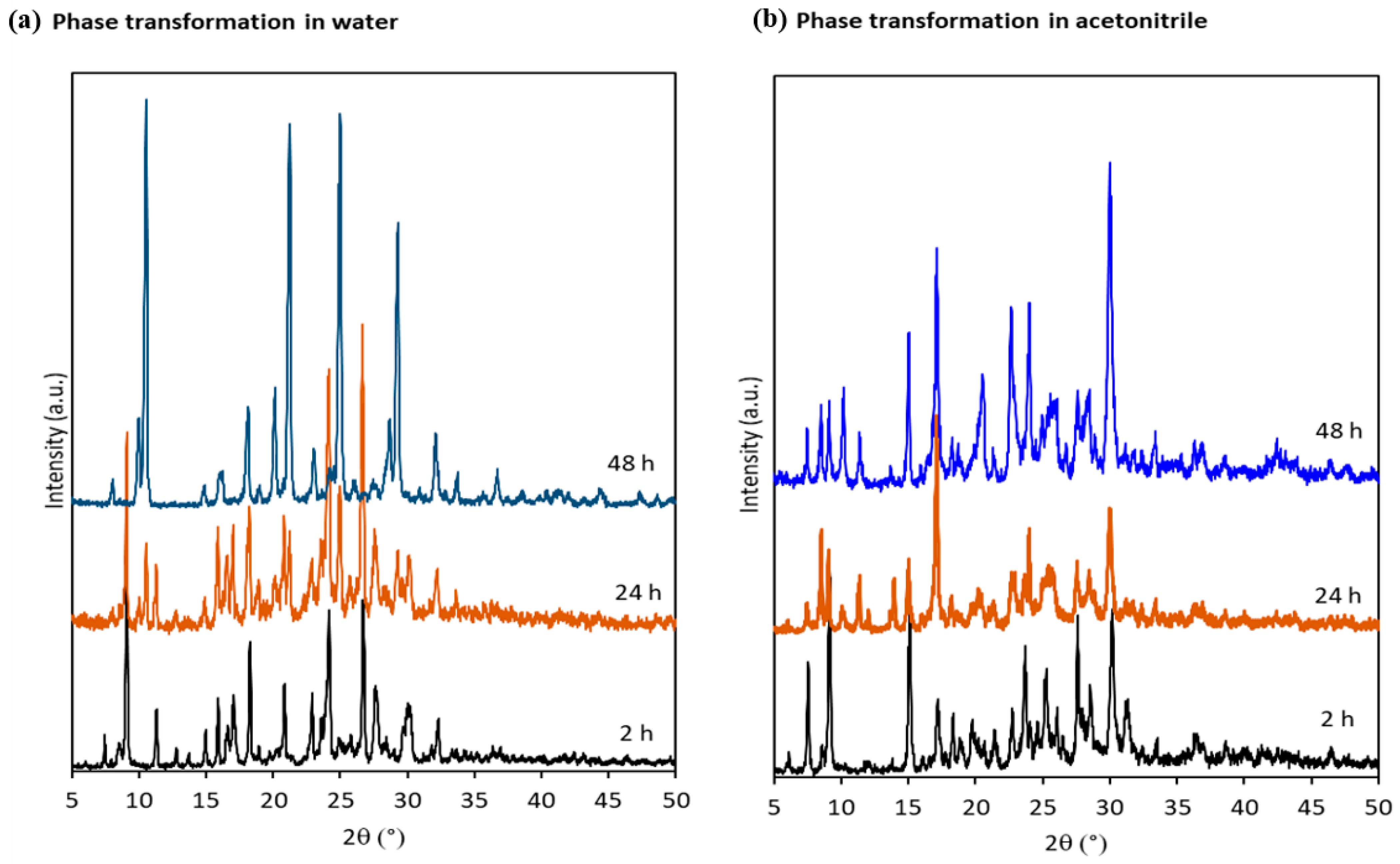
3.4. Stability Studies in Solution and Solid State
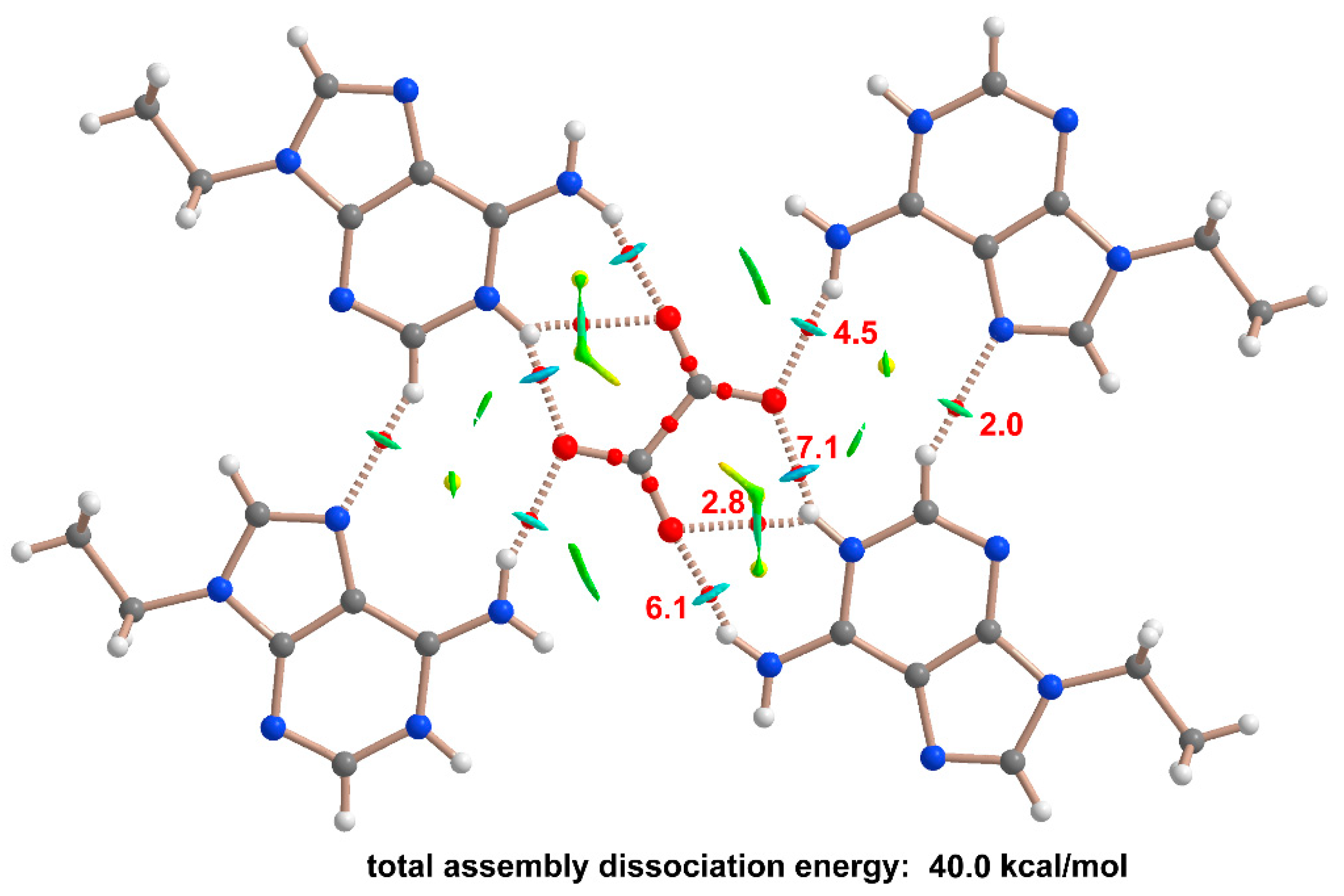
3.5. Computational Studies
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stahl, P.H.; Wermuth, C.G. (Eds.) Handbook of Pharmaceutical Salts: Properties, Selection and Use; Wiley-VCH: Zurich, Switzerland, 2008. [Google Scholar]
- Harrison, W.T.A.; Yathirajan, H.S.; Bindya, S.; Anilkumar, H.G. Devaraju Escitalopram oxalate: Co-existence of oxalate dianions and oxalic acid molecules in the same crystal. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2007, 63, o129–o131. [Google Scholar] [CrossRef]
- Åaslund, B.L.; Aurell, C.J.; Bohlin, M.H.; Sebhatu, T.; Ymen, B.I.; Healy, E.T.; Jensen, D.R.; Jonaitis, D.T.; Parent, S. Crystalline Naloxol-Peg Conjugate. Patent WO2012044243A1, 5 April 2012. [Google Scholar]
- Gelbrich, T.; Langes, C.; Stefinovic, M.; Griesser, U.J. Naloxegol hydrogen oxalate displaying a hydrogen-bonded layer structure. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 474–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database. J. Med. Chem. 2007, 50, 6665–6672. [Google Scholar] [CrossRef] [PubMed]
- Owoyemi, B.C.D.; da Silva, C.C.P.; Diniz, L.F.; Souza, M.S.; Ellena, J.; Carneiro, R.L. Fluconazolium oxalate: Synthesis and structural characterization of a highly soluble crystalline form. CrystEngComm 2019, 21, 1114–1121. [Google Scholar] [CrossRef]
- Chen, Y.; Li, L.; Yao, J.; Ma, Y.-Y.; Chen, J.-M.; Lu, T.-B. Improving the Solubility and Bioavailability of Apixaban via Apixaban–Oxalic Acid Cocrystal. Cryst. Growth Des. 2016, 16, 2923–2930. [Google Scholar] [CrossRef]
- Dichiarante, E.; Curzi, M.; Giaffreda, S.L.; Grepioni, F.; Maini, L.; Braga, D. Crystal forms of the hydrogen oxalate salt of o-desmethylvenlafaxine. J. Pharm. Pharmacol. 2015, 67, 823–829. [Google Scholar] [CrossRef]
- Perumalla, S.R.; Sun, C.C. Design and Preparation of a 4:1 Lamivudine–Oxalic Acid CAB Cocrystal for Improving the Lamivudine Purification Process. Cryst. Growth Des. 2014, 14, 3990–3995. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/news-events/press-announcements/fda-hold-advisory-committee-meeting-discuss-merck-and-ridgebacks-eua-application-covid-19-oral (accessed on 1 December 2021).
- Sridhar, B.; Ravikumar, K.; Varghese, B. Supramolecular hydrogen-bonded networks in adeninediium hemioxalate chloride and adeninium semioxalate hemi(oxalic acid) monohydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2009, 65, o202–o206. [Google Scholar] [CrossRef]
- McHugh, C.; Erxleben, A. Supramolecular Structures and Tautomerism of Carboxylate Salts of Adenine and Pharmaceutically Relevant N6-Substituted Adenine. Cryst. Growth Des. 2011, 11, 5096–5104. [Google Scholar] [CrossRef] [Green Version]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Pharmaceutical Cocrystallization: Engineering a Remedy for Caffeine Hydration. Cryst. Growth Des. 2005, 5, 1013–1021. [Google Scholar] [CrossRef]
- Otsuka, Y.; Ito, A.; Takeuchi, M.; Tanaka, H. Dry Mechanochemical Synthesis of Caffeine/Oxalic Acid Cocrystals and Their Evaluation by Powder X-ray Diffraction and Chemometrics. J. Pharm. Sci. 2017, 106, 3458–3464. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Scholz, G.; Batzdorf, L.; Wilke, M.; Emmerling, F. Synthesis, structure determination, and formation of a theobromine: Oxalic acid 2:1 cocrystal. CrystEngComm 2014, 17, 824–829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Rasmuson, C. The theophylline–oxalic acid co-crystal system: Solid phases, thermodynamics and crystallisation. CrystEngComm 2012, 14, 4644–4655. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Physical stability enhancement of theophylline via cocrystallization. Int. J. Pharm. 2006, 320, 114–123. [Google Scholar] [CrossRef]
- Roselló, Y.; Benito, M.; Bagués, N.; Martínez, N.; Moradell, A.; Mata, I.; Galcerà, J.; Barceló-Oliver, M.; Frontera, A.; Molins, E. 9-Ethyladenine: Mechanochemical Synthesis, Characterization, and DFT Calculations of Novel Cocrystals and Salts. Cryst. Growth Des. 2020, 20, 2985–2997. [Google Scholar] [CrossRef]
- García-Raso, A.; Fiol, J.J.; Bádenas, F.; Solans, X.; Font-Bardia, M. Reaction of trimethylene–bisadenine with d10 divalent cations. Polyhedron 1999, 18, 765–772. [Google Scholar] [CrossRef]
- SADABS, Bruker-AXS; Version 1; Bruker AXS Inc.: Madison, WI, USA, 2004.
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-2017/1, Program for the Solution of Crystal Structures; University of Göttingen: Göttingen, Germany, 2017. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Ahuja, D.; Svärd, M.; Rasmuson, C. Investigation of solid–liquid phase diagrams of the sulfamethazine–salicylic acid co-crystal. CrystEngComm 2019, 21, 2863–2874. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; Castiñeiras, A.; Frontera, A.; García-Santos, I.; González-Pérez, J.M.; Niclós-Gutiérrez, J.; Rodríguez-González, I.; Vílchez-Rodríguez, E.; Zaręba, J.K. Recurrent motifs in pharmaceutical cocrystals involving glycolic acid: X-ray characterization, Hirshfeld surface analysis and DFT calculations. CrystEngComm 2020, 22, 6674–6689. [Google Scholar] [CrossRef]
- Rockland, L.B. Saturated Salt Solutions for Static Control of Relative Humidity between 5° and 40° C. Anal. Chem. 1960, 32, 1375–1376. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian16; Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Gomila, R.M.; Frontera, A. Metalloid Chalcogen–pnictogen σ-hole bonding competition in stibanyl telluranes. J. Organomet. Chem. 2021, 954–955, 122092. [Google Scholar] [CrossRef]
- Mertsalov, D.F.; Gomila, R.M.; Zaytsev, V.P.; Grigoriev, M.S.; Nikitina, E.V.; Zubkov, F.I.; Frontera, A. On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms. Crystals 2021, 11, 1406. [Google Scholar] [CrossRef]
- García-Rubiño, M.; Matilla-Hernández, A.; Frontera, A.; Lezama, L.; Niclós-Gutiérrez, J.; Choquesillo-Lazarte, D. Dicopper(II)-EDTA Chelate as a Bicephalic Receptor Model for a Synthetic Adenine Nucleoside. Pharmaceuticals 2021, 14, 426. [Google Scholar] [CrossRef]
- Le, A.T.; Tran, V.T.T.; Le, D.T.; Gomila, R.M.; Frontera, A.; Zubkov, F.I. Synthesis, X-ray characterization and theoretical study of all-cis 1,4:2,3:5,8:6,7-tetraepoxynaphthalenes: On the importance of the through-space α-effect. CrystEngComm 2021, 23, 7462–7470. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMALL; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Carvalho, P.S.; Diniz, L.F.; Tenorio, J.C.; Souza, M.S.; Franco, C.H.J.; Rial, R.; De Oliveira, K.R.W.; Nazario, C.E.D.; Ellena, J. Pharmaceutical paroxetine-based organic salts of carboxylic acids with optimized properties: The identification and characterization of potential novel API solid forms. CrystEngComm 2019, 21, 3668–3678. [Google Scholar] [CrossRef]
- García-Raso, A.; Terrón, A.; López-Zafra, A.; García-Viada, A.; Barta, A.; Frontera, A.; Lorenzo, J.; Rodríguez-Calado, S.; Vázquez-López, E.M.; Fiol, J.J. Crystal structures of N6-modified-amino acid related nucleobase analogs (II): Hybrid adenine-β-alanine and adenine-GABA molecules. New J. Chem. 2019, 43, 9680–9688. [Google Scholar] [CrossRef]
- García-Raso, A.; Terrón, A.; Bauzá, A.; Frontera, A.; Molina, J.J.; Vázquez-López, E.M.; Fiol, J.J. Crystal structures of N6-modified-aminoacid/peptide nucleobase analogs: Hybrid adenine-glycine and adenine-glycylglycine molecules. New J. Chem. 2018, 42, 14742–14750. [Google Scholar] [CrossRef]
- García-Raso, A.; Terrón, A.; Balle, B.; López-Zafra, A.; Frontera, A.; Barceló-Oliver, M.; Fiol, J.J. Crystal structures of N6-modified-amino acid nucleobase analogs(iii): Adenine-valeric acid, adenine-hexanoic acid and adenine-gabapentine. New J. Chem. 2020, 44, 12236–12246. [Google Scholar] [CrossRef]
- Martínez, D.; Pérez, A.; Cañellas, S.; Silió, I.; Lancho, A.; García-Raso, A.; Fiol, J.J.; Terrón, A.; Barceló-Oliver, M.; Orte-ga-Castro, J.; et al. Synthesis, reactivity, X-ray characterization and docking studies of N7/N9-(2-pyrimidyl)-adenine derivatives. J. Inorg. Biochem. 2020, 203, 110879. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | 1 | 3 |
|---|---|---|
| Empirical Formula | C9H11N5O4 | C8H10N5O2 |
| Mr | 253.23 | 208.21 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P ī | P21/c |
| a/Å | 5.5478 (19) | 10.325 (4) |
| b/Å | 9.413 (3) | 7.0783 (3) |
| c/Å | 11.535 (4) | 13.207 (5) |
| α/° | 70.272 (5) | 90 |
| β/° | 87.851 (5) | 106.457 (5) |
| γ/° | 81.231 (6) | 90 |
| V/Å3 | 560.3 (3) | 925.6 (6) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| μ/mm−1 | 0.121 | 0.113 |
| Temperature/K | 300 (2) | 300 (2) |
| Crystal size/mm | 0.500 × 0.230 × 0.180 | 0.390 × 0.120 × 0.080 |
| Dcalc/g·cm−3 | 1.501 | 1.494 |
| Reflections collected | 6719 | 1676 |
| Independent Reflections | 2581 [R(int) = 0.0277] | 1676 [R(int) = 0.062] |
| Completeness to theta = 24.996° | 99.8% | 99.2% |
| F(000) | 264 | 436 |
| Data/restraints/parameters | 2581/0/170 | 1676/0/140 |
| Goodness-of-fit | 1.056 | 1.090 |
| Final R indices [I < 2d(I)] | R1 = 0.0420, wR2 = 0.1147 | R1 = 0.0610, wR2 = 0.1465 |
| R indices (all data) | R1 = 0.0478, wR2 = 0.1192 | R1 = 0.0863, wR2 = 0.1540 |
| Largest diff. peak and hole/e·Å−3 | 0.318 and −0.222 | 0.235 and −0.275 |
| CCDC nº | 2126070 | 2126069 |
| Compound | Solubility (mg/mL) | Coformer | Solubility * (mg/mL) |
|---|---|---|---|
| 9ETADE | 533 | ||
| 9ETADE-OXA (1:1) salt (1) | 25 | Oxalic acid dihydrate | 100 |
| 9ETADE-OXA (2:1) salt (3) | 10 | ||
| 9ETADE-SUC (1:1) salt | 7 | Succinic acid | 83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benito, M.; Barceló-Oliver, M.; Frontera, A.; Molins, E. Oxalic Acid, a Versatile Coformer for Multicomponent Forms with 9-Ethyladenine. Crystals 2022, 12, 89. https://doi.org/10.3390/cryst12010089
Benito M, Barceló-Oliver M, Frontera A, Molins E. Oxalic Acid, a Versatile Coformer for Multicomponent Forms with 9-Ethyladenine. Crystals. 2022; 12(1):89. https://doi.org/10.3390/cryst12010089
Chicago/Turabian StyleBenito, Mónica, Miquel Barceló-Oliver, Antonio Frontera, and Elies Molins. 2022. "Oxalic Acid, a Versatile Coformer for Multicomponent Forms with 9-Ethyladenine" Crystals 12, no. 1: 89. https://doi.org/10.3390/cryst12010089