
On the Importance of σ–Hole Interactions in Crystal Structures



Abstract
:1. Introduction
2. Results and Discussion
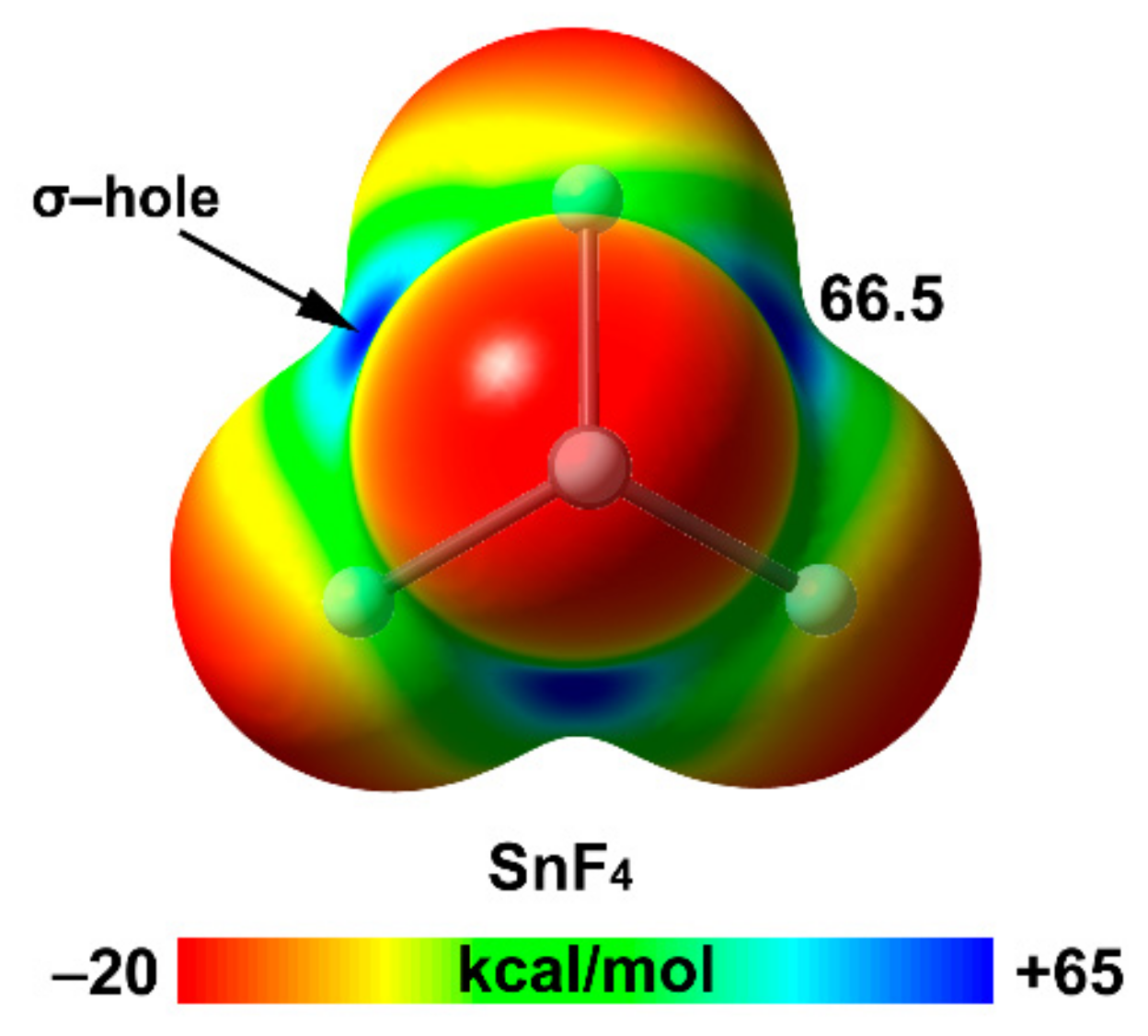
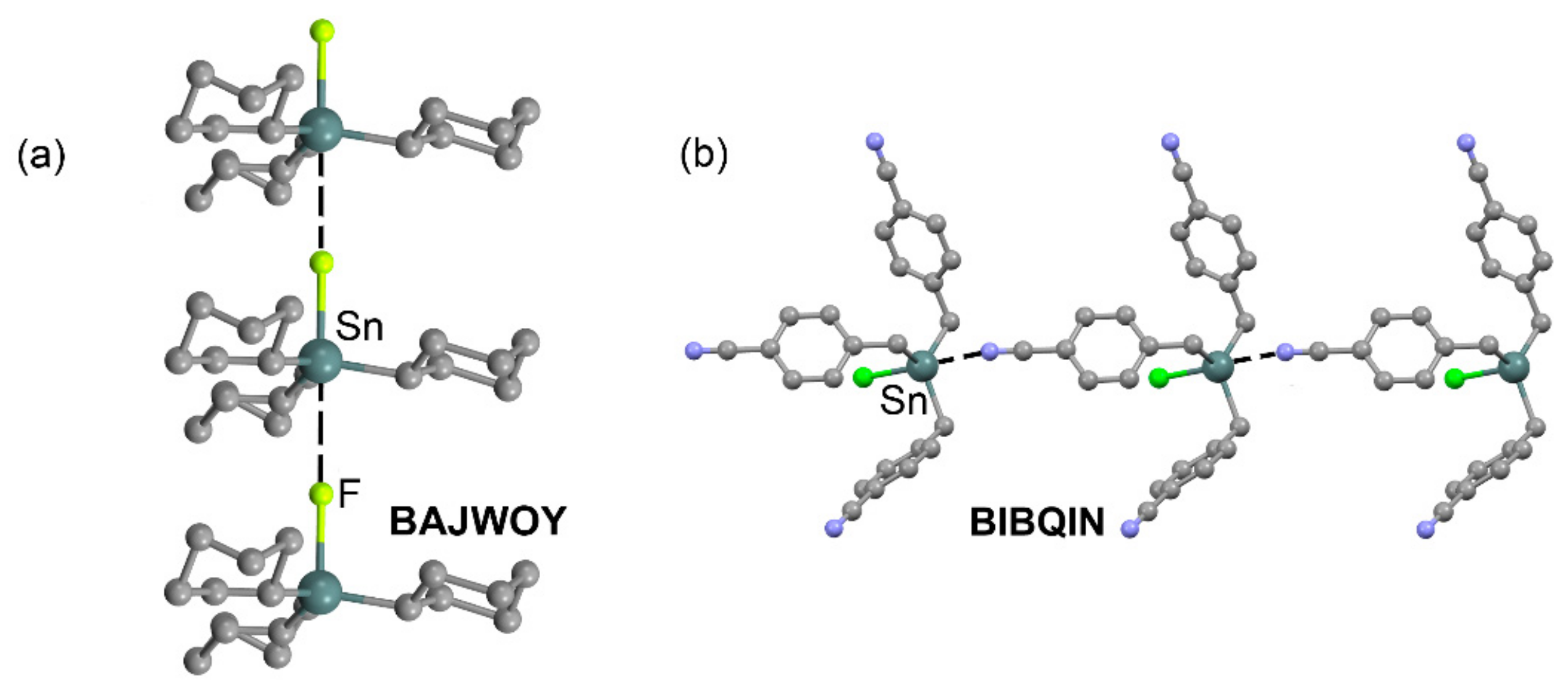
2.1. Tetrel Bonding
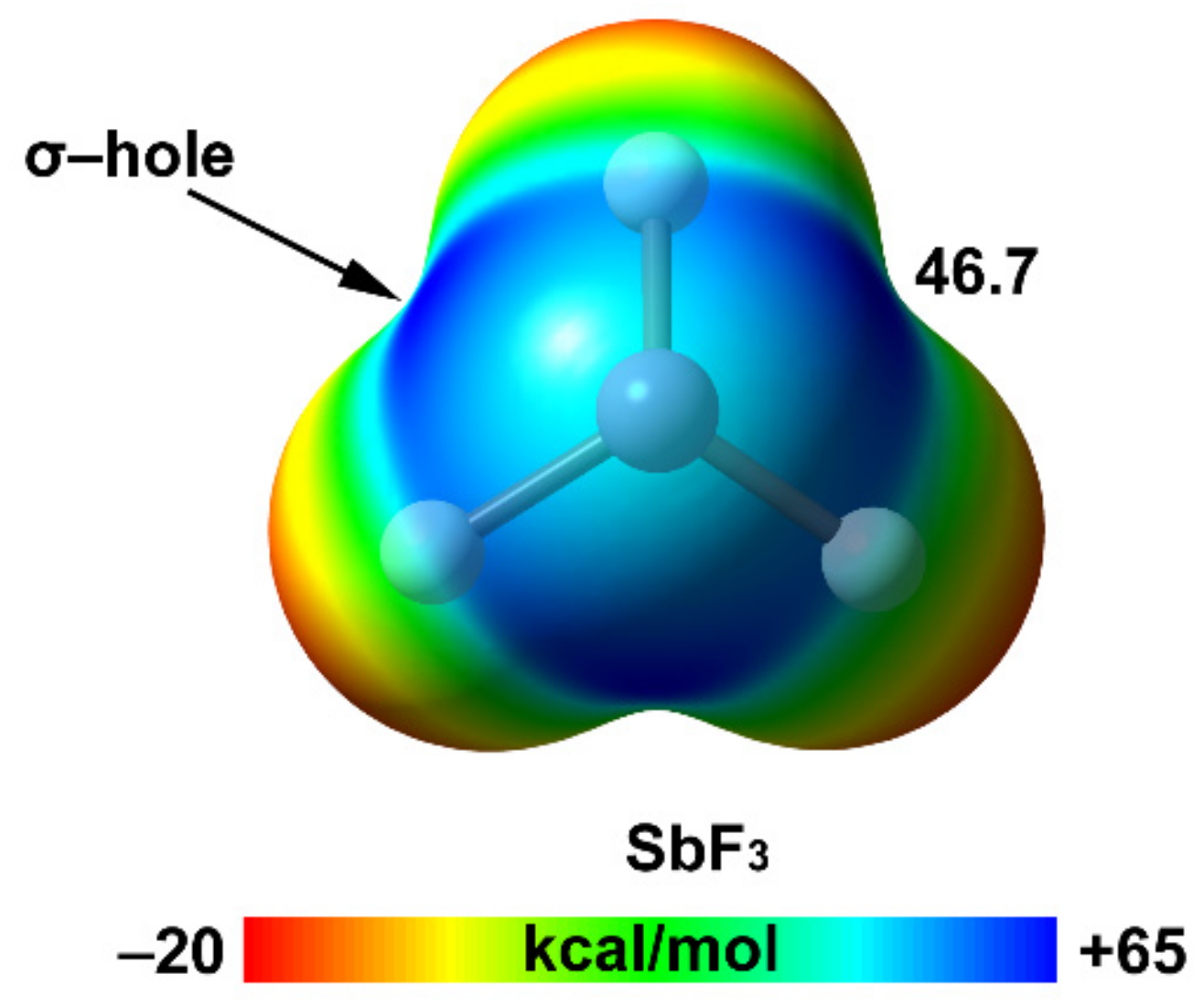
2.2. Pnictogen Bonding
2.3. Chalcogen Bonding
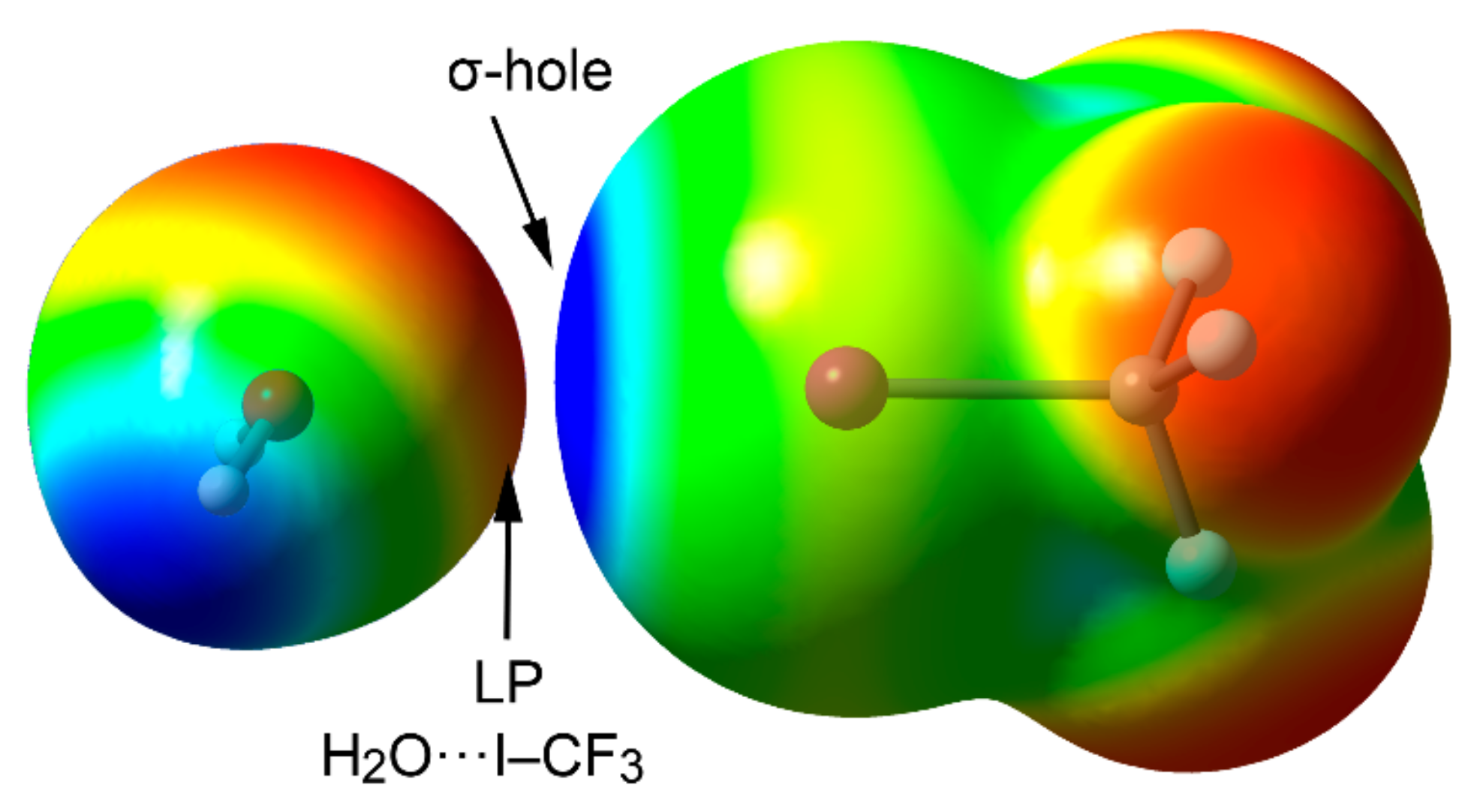
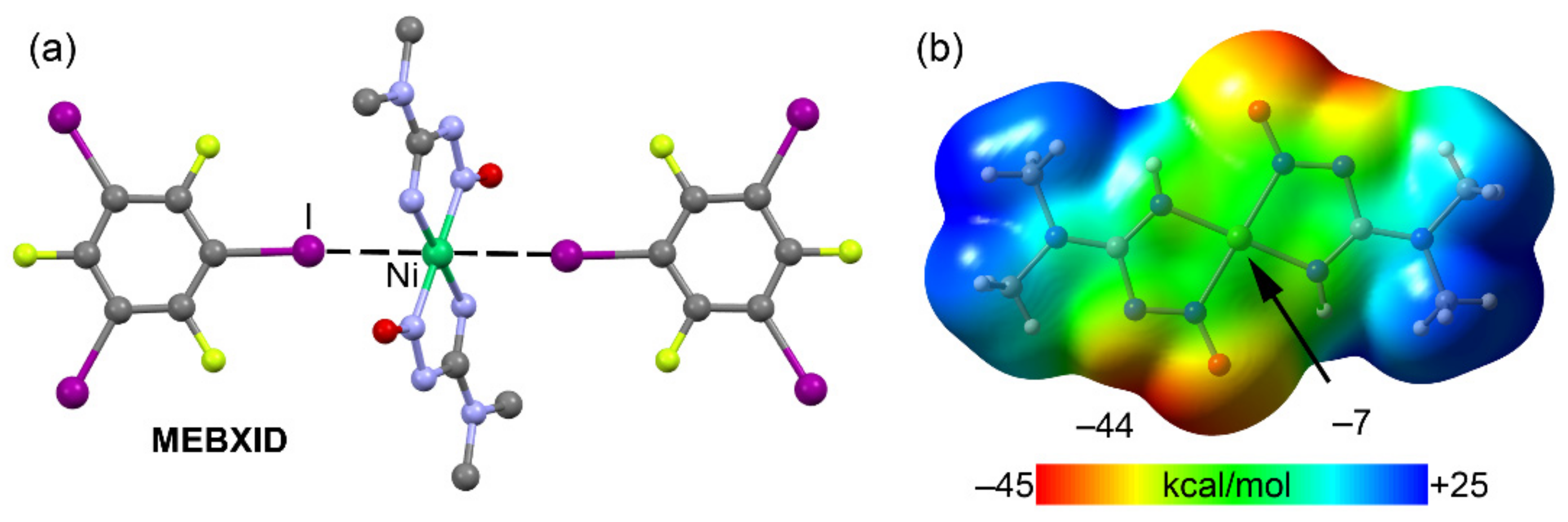
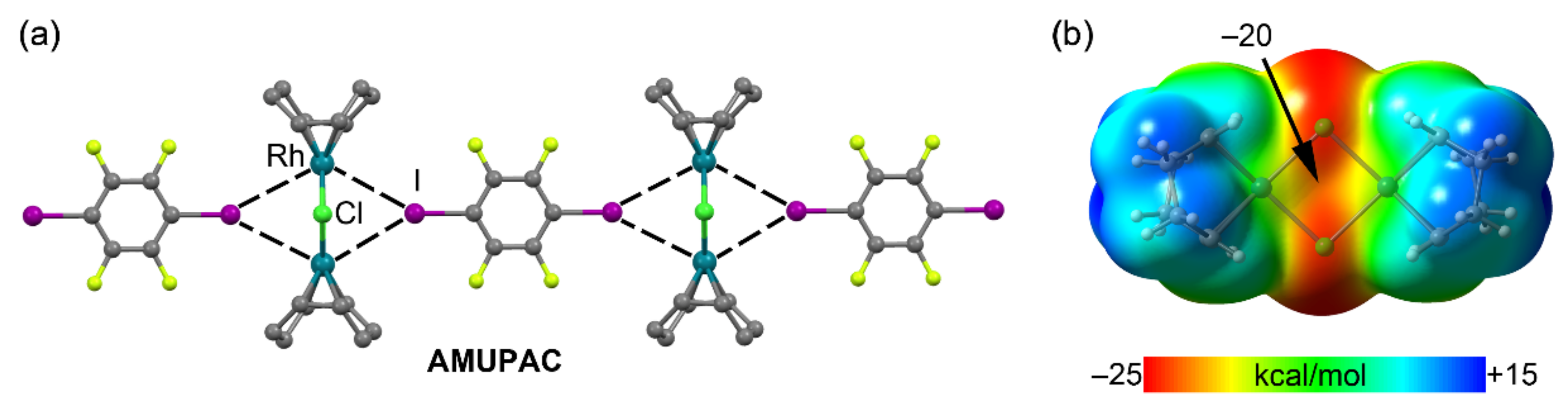
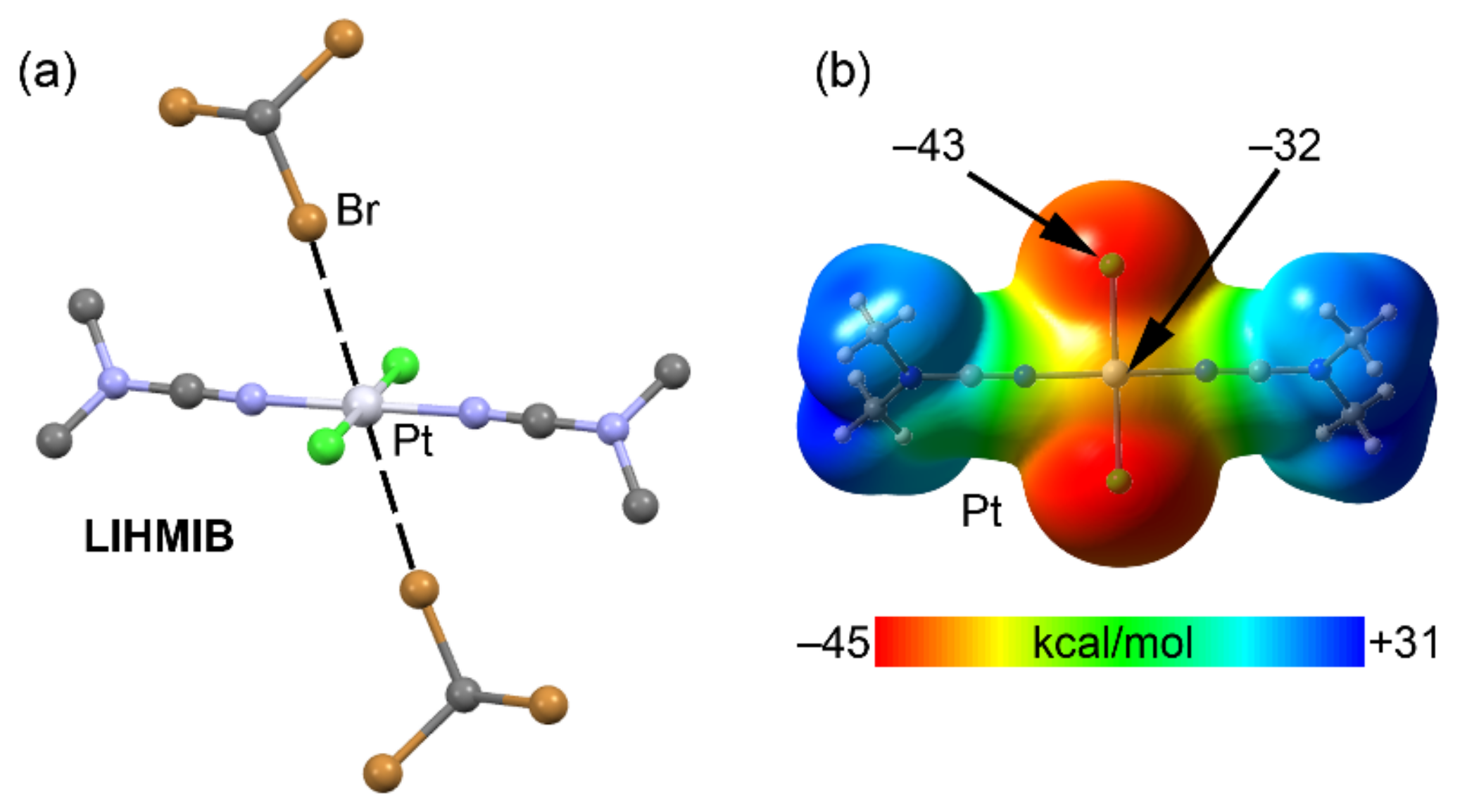
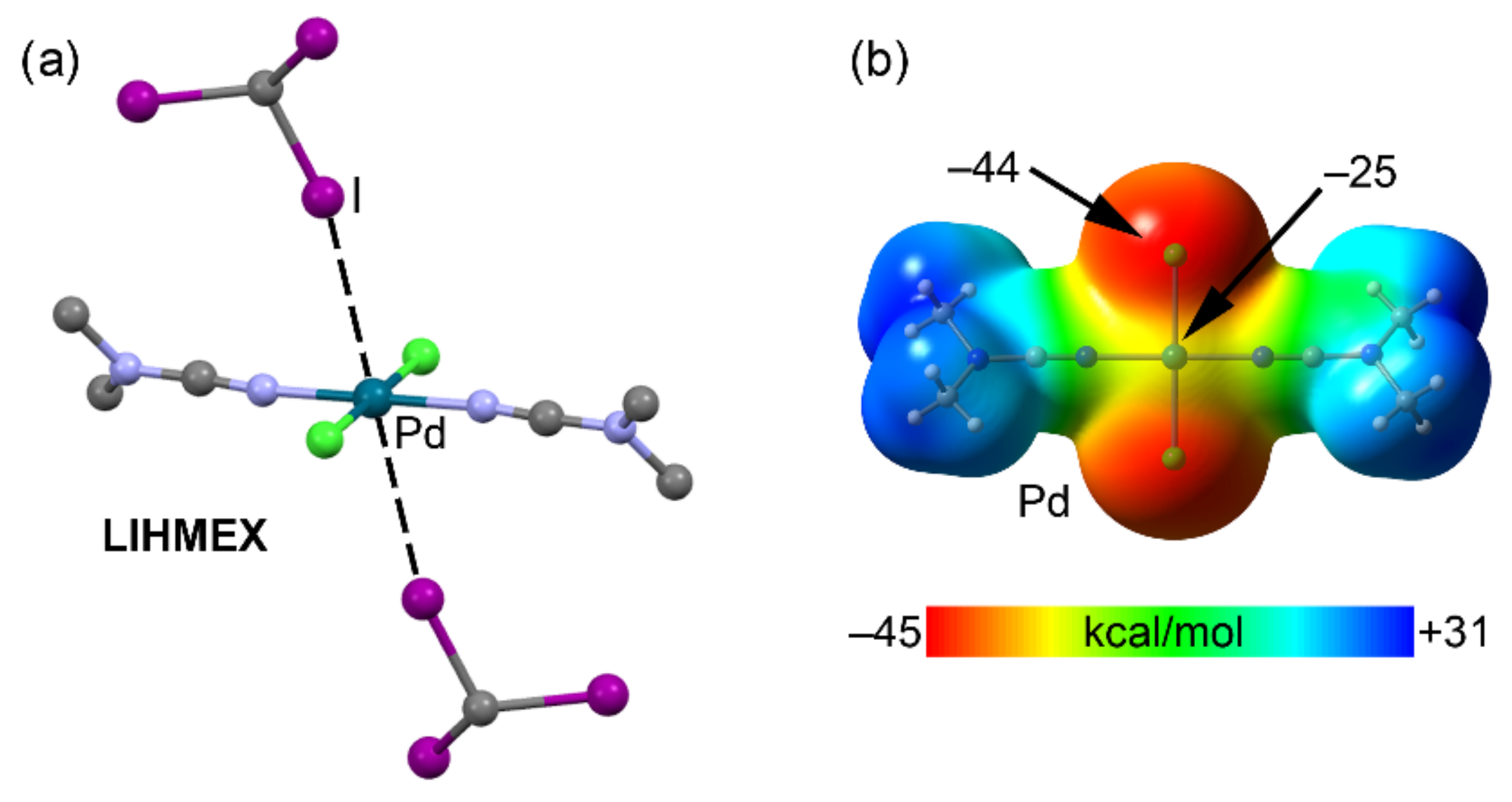
2.4. Halogen Bonding
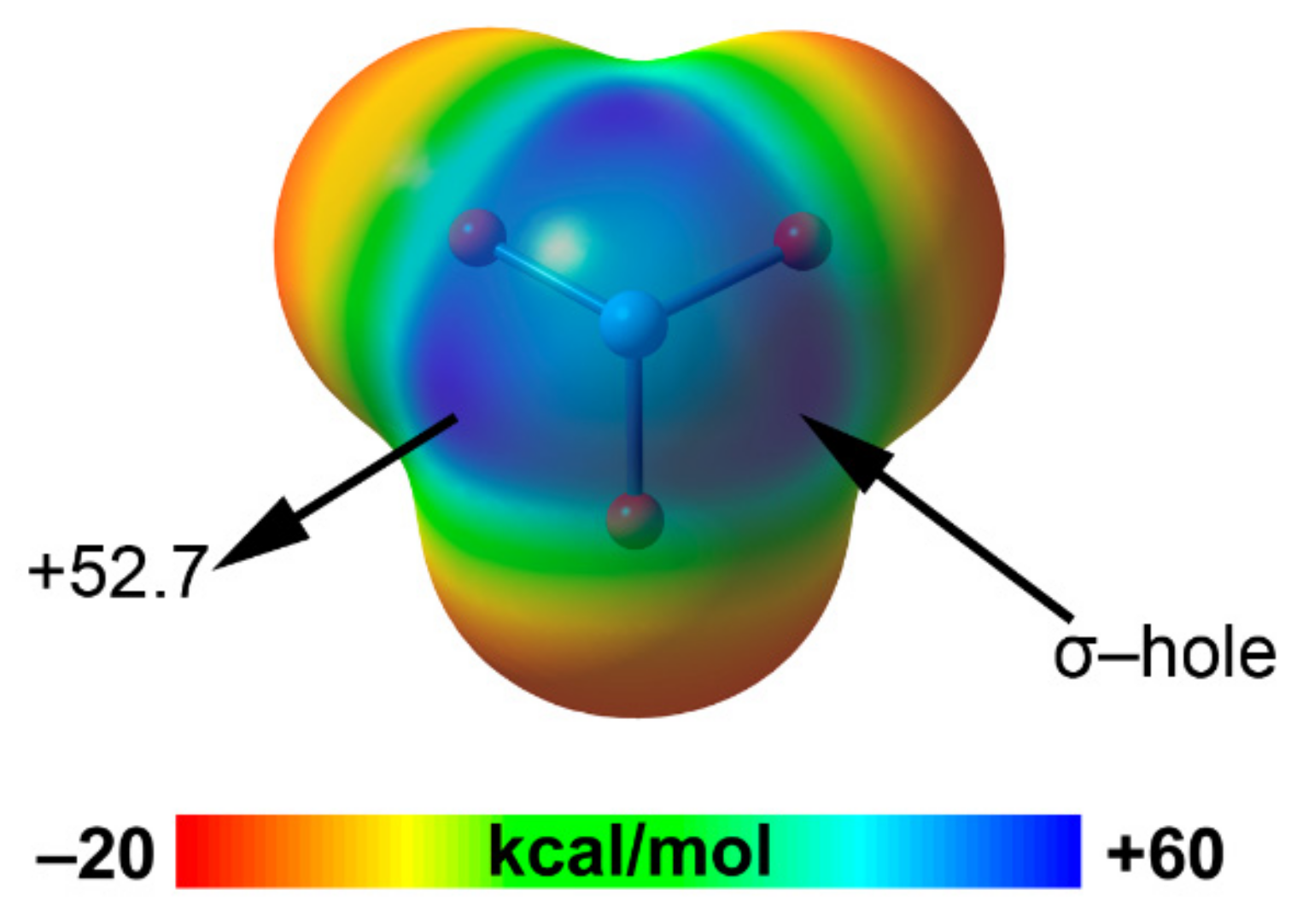
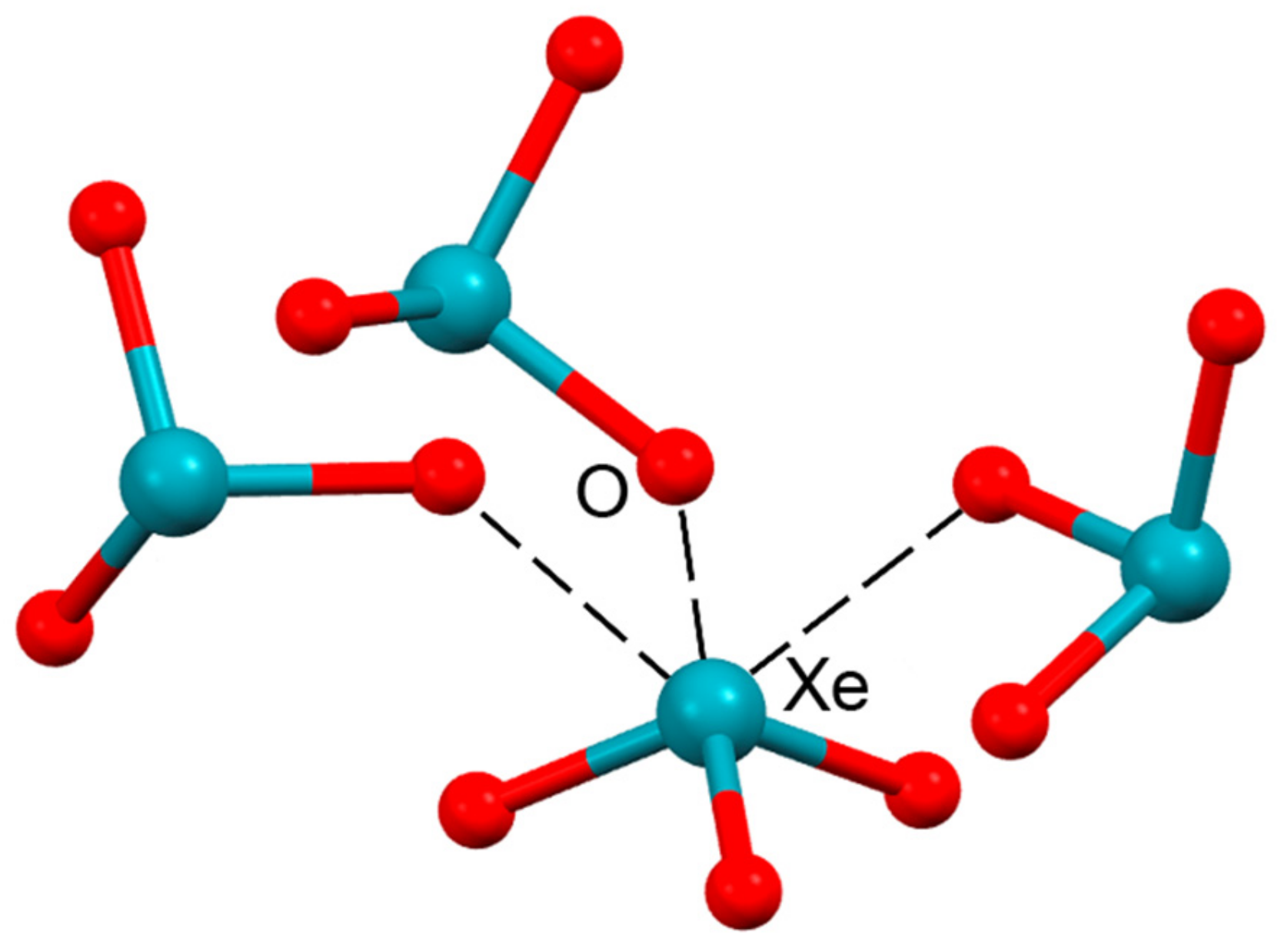
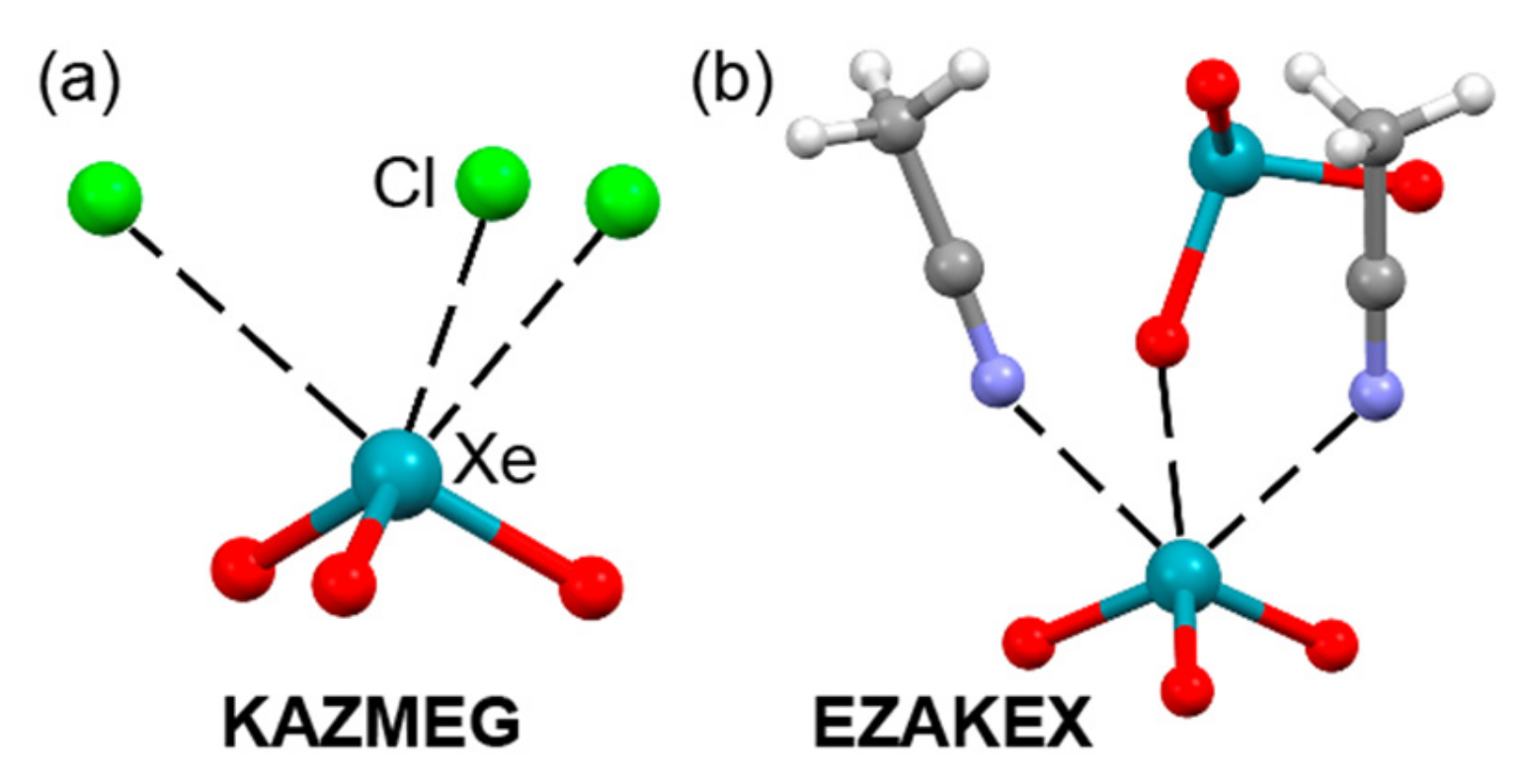
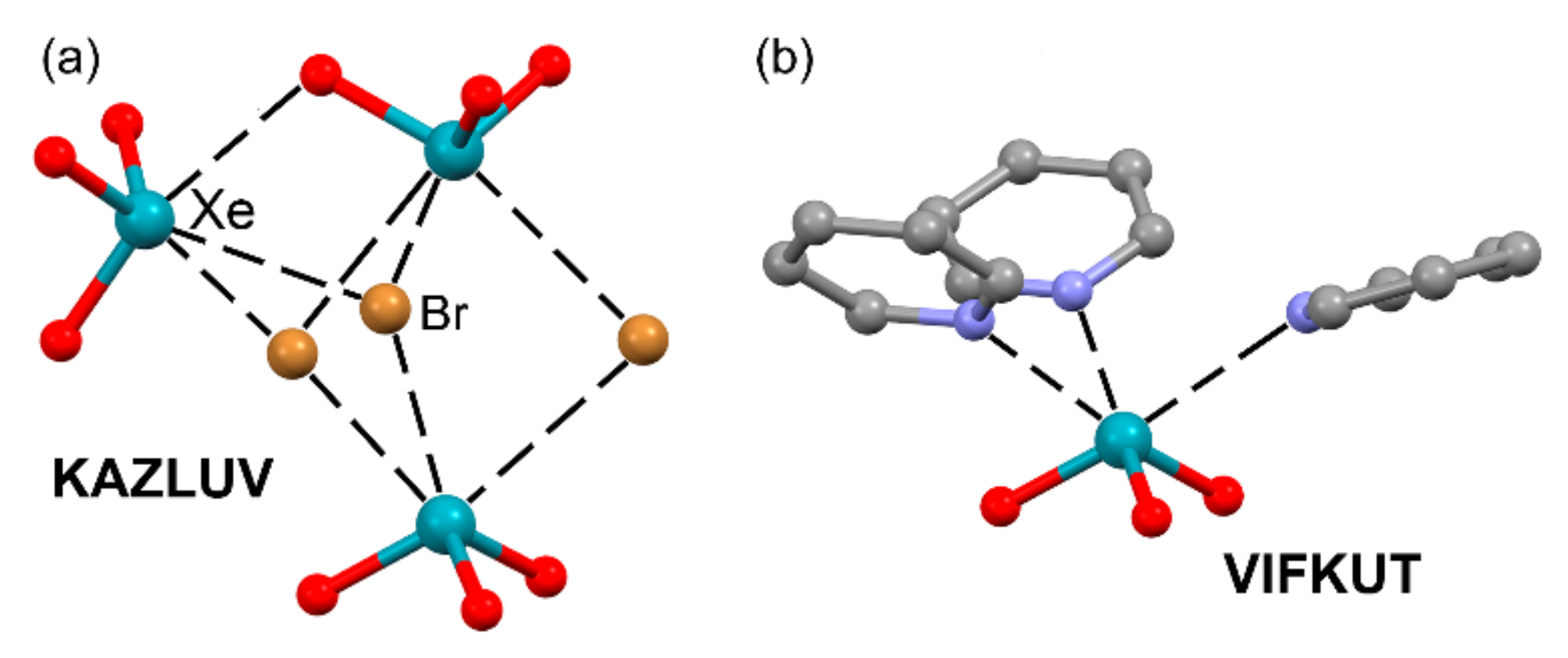
2.5. Noble Gas Bonding
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pizzi, A.; Pigliacelli, C.; Bergamaschi, G.; Gori, A.; Metrangolo, P. Biomimetic engineering of the molecular recognition and self-assembly of peptides and proteins via halogenation. Coord. Chem. Rev. 2020, 411, 213242. [Google Scholar] [CrossRef]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(sp3) atoms as tetrel bond donors: A crystallographic survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Von der Heiden, D.; Vanderkooy, A.; Erdélyi, M. Halogen bonding in solution: NMR spectroscopic approaches. Coord. Chem. Rev. 2020, 407, 213147. [Google Scholar] [CrossRef]
- Gill, H.; Gokel, M.R.; McKeever, M.; Negin, S.; Patel, M.B.; Yin, S.; Gokel, G.W. Supramolecular pore formation as an antimicrobial strategy. Coord. Chem. Rev. 2020, 412, 213264. [Google Scholar] [CrossRef]
- Xu, Y.; Szell, P.M.J.; Kumar, V.; Bryce, D.L. Solid-state NMR spectroscopy for the analysis of element-based non-covalent interactions. Coord. Chem. Rev. 2020, 411, 213237. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Jin, W.J. Halogen bonding in room-temperature phosphorescent materials. Coord. Chem. Rev. 2020, 404, 213107. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bond and coordination of triel centres—Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Fromm, K.M. Chemistry of alkaline earth metals: It is not all ionic and definitely not boring! Coord. Chem. Rev. 2020, 408, 213192. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Coordination of anions by noncovalently bonded σ-hole ligands. Coord. Chem. Rev. 2020, 405, 213136. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. σ/π–Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming interactions from the electrophilic site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Daolio, A.; Pizzi, A.; Terraneo, G.; Frontera, A.; Resnati, G. σ-Holes Allow for Attractive Anion·anion Interactions Involving Perrhenate, Permanganate, and Pertechnetate Anions. ChemPhysChem 2021, 22. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Molecular Electrostatic Potential and Noncovalent Interactions in Derivatives of Group 8 Elements. Angew. Chem. Int. Ed. 2021, 60, 20723–20727. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Daolio, A.; Pizzi, A.; Terraneo, G.; Ursini, M.; Frontera, A.; Resnati, G. Anion⋅Anion Coinage Bonds: The Case of Tetrachloridoaurate. Angew. Chem. Int. Ed. 2021, 60, 14385–14389. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. Regium–π bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Terrón, A.; Buils, A.; Mooibroek, T.J.; Barceló, M.; García-Raso, A.; Fiol, J.J.; Frontera, A. Synthesis, X-ray characterization and regium bonding interactions of a trichlorido(1-hexylcytosine)gold(III) complex. Chem. Commun. 2020, 56, 3524–3527. [Google Scholar] [CrossRef]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. σ–Holes and σ–lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Kampes, R.; Zechel, S.; Hager, M.D.; Schubert, U.S. Halogen bonding in polymer science: Towards new smart materials. Chem. Sci. 2021, 12, 9275–9286. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. (Eds.) Halogen Bonding, Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. (Eds.) Halogen Bonding II Impact on Materials Chemistry and Life Sciences; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [Green Version]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From noncovalent chalcogen–chalcogen interactions to supramolecular aggregates: Experiments and calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin–spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Heidelberg/Berlin, Germany, 2015; pp. 191–264. [Google Scholar]
- Brammer, L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: Origins, current status and discussion. Faraday Discuss. 2017, 203, 485–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politzer, P.; Murray, J.S.; Lane, P. σ-Hole bonding and hydrogen bonding: Competitive interactions. Int. J. Quantum. Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Zhu, S.; Xing, C.; Xu, W.; Jin, G.; Li, Z. Halogen bonding and hydrogen bonding coexist in driving self-assembly process. Cryst. Growth Des. 2004, 4, 53–56. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Forni, A.; Gorynsztejn-Leben, M.; Kaivola, M.; Metrangolo, P.; Milani, R.; Shishido, A.; Pilati, T.; Resnati, G.; et al. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly and Performance of Light-Responsive Supramolecular Polymers. Adv. Funct. Mater. 2012, 22, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Scheiner, S. Forty years of progress in the study of the hydrogen bond. Struct. Chem. 2019, 30, 1119–1128. [Google Scholar] [CrossRef]
- Corradi, E.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly Processes. Angew. Chem. Int. Ed. 2000, 39, 1782–1786. [Google Scholar] [CrossRef]
- Zingaro, R.A.; Hedges, M. Phosphine Oxide-Halogen Complexes: Effect on P–O and P–S Stretching Frequencies. J. Phys. Chem. 1961, 65, 1132–1138. [Google Scholar] [CrossRef]
- Martire, D.E.; Sheridan, J.P.; King, J.W.; O’Donnell, S.E. Thermodynamics of Molecular Association. 9. An NMR Study of Hydrogen Bonding of CHCl3 and CHBr3 to Di-N-Octyl Ether, Di-NOctyl Thioether and Di-N-Octylmethylamine. J. Am. Chem. Soc. 1976, 98, 3101–3106. [Google Scholar] [CrossRef]
- Dumas, J.-M.; Gomel, M.; Guerin, M. Molecular Interactions Involving Organic Halides. In Halides, Pseudo-Halides and Azides; John Wiley & Sons, Ltd.: Chichester, UK, 1983; Volume 2, pp. 985–1020. [Google Scholar]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coord. Chem. Rev. 2020, 416, 213281. [Google Scholar] [CrossRef]
- Costa, P.J. The Halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2, 81–106. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Zablotny, S.; Bryce, D.L. Halogen bonding as a supramolecular dynamics catalyst. Nat. Commun. 2019, 10, 916. [Google Scholar] [CrossRef] [Green Version]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. WIREs Comput. Molt. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Project No. 2016-001-2-300. Available online: https://iupac.org/project/2016-001-2-300 (accessed on 5 September 2021).
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical Investigations on Heteronuclear Chalcogen−Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical investigations on chalcogen−Chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An overview of strengths and directionalities of noncovalent interactions: σ-holes and π-holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Gomila, R.M.; Mooibroek, T.J.; Frontera, A. A Combined Theoretical and CSD Perspective on σ-Hole Interactions with Tetrels, Pnictogens, Chalcogens, Halogens, and Noble Gases. In Hot Topics in Crystal Engineering; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Scheiner, S. Competition between a Tetrel and Halogen Bond to a Common Lewis Acid. J. Phys. Chem. 2021, 125, 308–316. [Google Scholar] [CrossRef]
- Na, L.; Liu, J.; Li, Q.; Scheiner, S. Noncovalent bond between tetrel π–hole and hydride. Phys. Chem. Chem. Phys. 2021, 23, 10536–10544. [Google Scholar]
- Chen, Y.; Wang, F. Intermolecular Interactions Involving Heavy Alkenes H2Si=TH2 (T = C, Si, Ge, Sn, Pb) with H2O and HCl: Tetrel Bond and Hydrogen Bond. ACS Omega 2020, 5, 30210–30225. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Fruchier, A.; Macquarrie, D.J.; Virgili, A. Aminopropylsilanes versus silatranes: An experimental and theoretical study. J. Organomet. Chem. 2001, 625, 148–153. [Google Scholar] [CrossRef]
- Grabowski, S.J. Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ–Hole Concept with the QTAIM Approach. Crystals 2017, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, P.; Kumar, V.; Ursini, M.; Resnati, G. Close contacts involving germanium and tin in crystal structures: Experimental evidence of tetrel bonds. J. Mol. Model. 2018, 24, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Lukevics, E.; Arsenyan, P.; Belyakov, S.; Popelis, J.; Pudova, O. Cycloaddition reactions of nitrile oxides to silyl-and germyl-substituted thiophene-1,1-dioxides. Organometallics 1999, 18, 3187–3193. [Google Scholar] [CrossRef]
- Villaescusa, L.A.; Lightfoot, P.; Morris, R.E. Synthesis and structure of fluoride-containing GeO2 analogues of zeolite double four-ring building units. Chem. Commun. 2002, 1, 2220–2221. [Google Scholar] [CrossRef]
- Calogero, S.; Ganis, P.; Peruzzo, V.; Tagliavini, G.; Valle, G. X-ray and Mössbauer studies of tricyclohexyltin (IV) halides. The crystal structures of (cyclo-C6H11)3SnX (X = F, Br and I). J. Organomet. Chem. 1981, 220, 11–20. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Kuang, D.-Z.; Zhang, F.-X.; Feng, Y.-L.; Xu, Z.-F. CCDC 211279: Experimental Crystal Structure Determination. 2004. [Google Scholar] [CrossRef]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Halogen, chalcogen and pnictogen interactions in (XNO2)2 homodimers (X = F, Cl, Br, I). New J. Chem. 2015, 39, 6791–6802. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Grabowski, S.J. Pnicogen and hydrogen bonds: Complexes between PH3X+ and PH2X systems. Phys. Chem. Chem. Phys. 2015, 17, 3261–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esrafili, M.D.; Mohammadian-Sabet, F.; Baneshi, M.M. The dual role of halogen, chalcogen, and pnictogen atoms as Lewis acid and base: Triangular XBr:SHX:PH2X complexes (X = F, Cl, Br, CN, NC, OH, NH2, and OCH3). Int. J. Quantum Chem. 2015, 115, 1580–1586. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Bauzá, A.; Quinonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. Cryst. Eng. Comm. 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. Pnicogen Bonds between X–PH3 (X = O, S, NH, CH2) and Phosphorus and Nitrogen Bases. J. Phys. Chem. A 2014, 118, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.; Chopra, D. Characterization of N O non-covalent interactions involving σ–holes: “electrostatics” or “dispersion”. Phys. Chem. Chem. Phys. 2016, 18, 29946–29954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokrai, R.; Jaie Barrett, J.; Apperley, D.C.; Benkő, Z.; Heift, D. Tweaking the Charge Transfer: Bonding Analysis of Bismuth(III) Complexes with a Flexidentate Phosphane Ligand. Inorg. Chem. 2020, 59, 8916–8924. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Pavan, M.S.; Guru Row, T.N. Experimental validation of ‘pnicogen bonding’ in nitrogen by charge density analysis. Phys. Chem. Chem. Phys. 2015, 17, 2330–2334. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Ramanathan, N.; Sundararajan, K.; Sankaran, K. Evidence for phosphorus bonding in phosphorus trichloride–methanol adduct: A matrix isolation infrared and ab initio computational study. J. Phys. Chem. A 2015, 119, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Experimental charge density evidence for pnicogen bonding in a crystal of ammonium chloride. ChemPhysChem 2015, 16, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Andrea Gini, A.; Paraja, M.; Galmés, B.; Besnard, C.; Poblador-Bahamonde, A.I.; Sakai, N.; Frontera, A.; Matile, S. Pnictogen-bonding catalysis: Brevetoxin-type polyether cyclizations. Chem. Sci. 2020, 11, 7086–7091. [Google Scholar] [CrossRef] [PubMed]
- Scilabra, P.; Terraneo, G.; Resnati, G. Fluorinated elements of Group 15 as pnictogen bond donor sites. J. Fluor. Chem. 2017, 203, 62–67. [Google Scholar] [CrossRef]
- Prokudina, Y.V.; Davydova, E.I.; Virovets, A.; Stöger, B.; Peresypkina, E.; Pomogaeva, A.V.; Timoshkin, A.Y. Structures and Chemical Bonding in Antimony(III) Bromide Complexes with Pyridine. Chem. Eur. J. 2020, 26, 16338–16348. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Moaven, S.; Andrews, M.C.; Polaske, T.J.; Karl, B.M.; Unruh, D.K.; Bosch, E.; Bowling, N.P.; Cozzolino, A.F. Triple-Pnictogen Bonding as a Tool for Supramolecular Assembly. Inorg. Chem. 2019, 58, 16227–16235. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Beer, P.D. Sigma-hole interactions in anion recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Hirai, M.; Cho, J.; Gabbaï, F.P. Promoting the Hydrosilylation of Benzaldehyde by Using a Dicationic Antimony-Based Lewis Acid: Evidence for the Double Electrophilic Activation of the Carbonyl Substrate. Chem. Eur. J. 2016, 22, 6537–6541. [Google Scholar] [CrossRef]
- Qiu, J.; Unruh, D.K.; Cozzolino, A.F. Design, Synthesis, and Structural Characterization of a Bisantimony(III) Compound for Anion Binding and the Density Functional Theory Evaluation of Halide Binding through Antimony Secondary Bonding Interactions. J. Phys. Chem. A 2016, 120, 9257–9269. [Google Scholar] [CrossRef]
- Haiges, R.; Vij, A.; Boatz, J.A.; Schneider, S.; Schroer, T.; Gerken, M.; Christe, K.O. First Structural Characterization of Binary AsIII and SbIII Azides. Chem. Eur. J. 2004, 10, 508–517. [Google Scholar] [CrossRef]
- Deokar, P.; Leitz, D.; Stein, T.H.; Vasiliu, M.; Dixon, D.A.; Christe, K.O.; Haiges, R. Preparation and Characterization of Antimony and Arsenic Tricyanide and Their 2,2′-Bipyridine Adducts. Chem. Eur. J. 2016, 22, 13251–13257. [Google Scholar] [CrossRef]
- Levason, W.; Light, M.E.; Maheshwari, S.; Reid, G.; Zhang, W. Unusual neutral ligand coordination to arsenic and antimony trifluoride. Dalton Trans. 2011, 40, 5291–5297. [Google Scholar] [CrossRef]
- Burk, R.F. (Ed.) Selenium in Biology and Human Health; Springer: New York, NY, USA, 1994. [Google Scholar]
- Saha, U.K. Selenium: A vital element in soil-pant-animal/human continuum. J. Agric. Sci. Bot. 2017, 1, 1–3. [Google Scholar]
- Thomas, S.P.; Thomas, R.; Grønbech, T.B.E.; Bondesgaard, M.; Mamkhel, A.H.; Birkedal, V.; Iversen, B.B. Bandgap Tuning in Molecular Alloy Crystals Formed by Weak Chalcogen Interactions. J. Phys. Chem. Lett. 2021, 12, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Gritzenco, F.; Kazmierczak, J.C.; Anjos, T.; Sperança, A.; Peixoto, M.L.B.; Godoi, M.; Ledebuhr, K.N.B.; Brüning, C.A.; Zamin, L.L.; Godoi, B. Base-Free Synthesis and Synthetic Applications of Novel 3-(Organochalcogenyl) prop-2-yn-1-yl Esters: Promising Anticancer Agents. Synthesis 2021, 53, 2676–2688. [Google Scholar]
- Vasiliu, M.; Peterson, K.A.; Dixon, D.A. Bond Dissociation Energies in Heavy Element Chalcogen and Halogen Small Molecules. J. Phys. Chem. A. 2021, 125, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Popa, R.A.; Licarete, E.; Banciu, M.; Silvestru, A. Organoselenium compounds containing pyrazole or phenylthiazole groups: Synthesis, structure, tin (IV) complexes and antiproliferative activity. Appl. Organomet. Chem. 2018, 32, e4252. [Google Scholar] [CrossRef]
- Kimura, T.; Nakahodo, T.; Fujihara, H.; Suzuki, E. 4,5-Dicyano-3,6-diethylbenzo-1,2-diselenete, a Highly Stable 1,2-Diselenete: Its Preparation, Structural Characterization, Calculated Molecular Orbitals, and Complexation with Tetrakis (triphenylphosphine) palladium. Inorg. Chem. 2014, 53, 4411–4417. [Google Scholar] [CrossRef]
- Xu, Y.; Kumar, V.; Bradshaw, M.J.Z.; Bryce, D.L. Chalcogen-Bonded Cocrystals of Substituted Pyridine N-Oxides and Chalcogenodiazoles: An X-ray Diffraction and Solid-State NMR Investigation. Cryst. Growth Des. 2020, 20, 7910–7920. [Google Scholar] [CrossRef]
- Suzuki, T.; Fujii, H.; Yamashita, Y.; Kabuto, C.; Tanaka, S.; Harasawa, M.; Mukai, T.; Miyashi, T. Clathrate formation and molecular recognition by novel chalcogen-cyano interactions in tetracyanoquinodimethanes fused with thiadiazole and selenadiazole rings. J. Am. Chem. Soc. 1992, 114, 3034–3043. [Google Scholar] [CrossRef]
- Fritz, S.; Ehm, C.; Lentz, D. Structure and Chemistry of SeFx(CN)4−x Compounds. Inorg. Chem. 2015, 54, 5220–5231. [Google Scholar] [CrossRef] [PubMed]
- Garrett, G.E.; Gibson, G.L.; Straus, R.N.; Seferos, D.S.; Taylor, M.S. Chalcogen bonding in solution: Interactions of benzotelluradiazoles with anionic and uncharged Lewis bases. J. Am. Chem. Soc. 2015, 137, 4126–4133. [Google Scholar] [CrossRef] [PubMed]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Frontera, A.; Caballero, A. Anion Recognition by Neutral Chalcogen Bonding Receptors: Experimental and Theoretical Investigations. Chem. Eur. J. 2020, 26, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, A.F.; Elder, P.J.W.; Vargas-Vaca, I. A survey of tellurium-centered secondary-bonding supramolecular synthons. Coord. Chem. Rev. 2011, 255, 1426–1438. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnictogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Nziko, V.P.N.; Scheiner, S. Chalcogen Bonding between Tetravalent SF4 and Amines. J. Phys. Chem. A 2014, 118, 10849–10856. [Google Scholar] [CrossRef]
- Subrahmanyan, I.; Aravamudan, G.; Rout, G.C.; Seshasayee, M. Synthesis, properties, and molecular structure of bis(thiobenzoato-S)tellurium(II), C14H10O2S2Te(II). J. Crystal. Spect. Res. 1984, 14, 239–248. [Google Scholar] [CrossRef]
- Neidlein, R.; Knecht, D.; Gieren, A.; Ruiz-Perez, C. Synthese und Röntgenstrukturanalyse des Phenanthro[9,10-c]-l,2,5-telluradiazols. Naturforschung, Chem. Sci. 1987, 42, 84–90. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl) dz2 [PtII] Halogen Bond. Chem. Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef] [PubMed]
- Bikbaeva, Z.M.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Electrophilic–Nucleophilic Dualism of Nickel(II) toward Ni·I Noncovalent Interactions: Semicoordination of Iodine Centers via Electron Belt and Halogen Bonding via σ-Hole. Inorg. Chem. 2017, 56, 13562–13578. [Google Scholar] [CrossRef] [PubMed]
- Rozhkov, A.V.; Eliseeva, A.A.; Baykov, S.V.; Galmés, B.; Frontera, A.; Kukushkin, V.Y. One-Pot Route to X-perfluoroarenes (X = Br, I) Based on FeIII-Assisted C–F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding. Cryst. Growth Des. 2020, 20, 5908–5921. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium (I) Centers as an Integrated σ–Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef]
- Gossage, R.A.; Ryabov, A.D.; Spek, A.L.; Stufkens, D.J.; van Beek, J.A.M.; van Eldik, R.; van Koten, G. Models for the Initial Stages of Oxidative Addition. Synthesis, Characterization, and Mechanistic Investigation of η1-I2 Organometallic “Pincer” Complexes of Platinum. X-ray Crystal Structures of [PtI(C6H3{CH2NMe2}2-2,6)(η1-I2)] and exo-meso-[Pt(η1-I3)(η1-I2)(C6H3{CH2N(t-Bu)Me}2-2,6)]. J. Am. Chem. Soc. 1999, 121, 2488–2497. [Google Scholar]
- Ivanov, D.M.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π–stacking with benzene provides 2D assembly of trans-[PtCl2 (p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Baykov, S.V.; Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Boyarskiy, V.P. Pt/Pd and I/Br Isostructural Exchange Provides Formation of C–I···Pd, C–Br···Pt, and C–Br···Pd Metal-Involving Halogen Bonding. Cryst. Growth Des. 2018, 18, 5973–5980. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- O’Toole, M. Miller-Keane Encyclopedia and Dictionary of Medicine, Nursing, and Allied Health, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π–hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Miao, J.; Xiong, Z.; Gao, Y.J. The effects of aerogen-bonding on the geometries and spectral properties of several small molecular clusters containing XeO3. Condens. Matter Phys. 2018, 30, 444001. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Sadr-Mousavi, A. A computational study on the strength and nature of bifurcated aerogen bonds. Chem. Phys. Lett. 2018, 698, 1–6. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Kiani, H. Cooperativity between the hydrogen bonding and σ-hole interaction in linear NCX···(NCH)n=2–5 and O3Z···(NCH)n = 2–5 complexes (X = Cl, Br; Z = Ar, Kr): A comparative study. Can. J. Chem. 2017, 95, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Goettel, J.T.; Haensch, V.G.; Schrobilgen, G.J. Stable chloro-and bromoxenate cage anions; [X3(XeO3)3]3− and [X4(XeO3)4]4− (X = Cl or Br). J. Am. Chem. Soc. 2017, 139, 8725–8733. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.T.; Matsumoto, K.; Mercier, H.P.A.; Schrobilgen, G.J. Syntheses and structures of xenon trioxide alkylnitrile adducts. Angew. Chem. Int. Ed. 2016, 55, 13780–13783. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.T.; Mercier, H.P.A.; Schrobilgen, G.J. XeO3 adducts of pyridine, 4-dimethylaminopyridine, and their pyridinium salts. J. Fluorine Chem. 2018, 121, 60–69. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tt | α | RvdW | MEP (TtF4) |
|---|---|---|---|
| C | 9.0 | 1.70 | 18.6 |
| Si | 26.1 | 2.10 | 39.0 |
| Ge | 28.4 | 2.11 | 50.2 |
| Sn | 37.3 | 2.17 | 66.5 |
| CSD Code | Tt | A | dTt···A | ∠ |
|---|---|---|---|---|
| QAHXIG | Ge | O | 3.520 | 179.8 |
| VUZVUH | Ge | F | 2.736–2.778 | 173.8–174.3 |
| BAJWOY | Sn | F | 3.321 | 178.9 |
| BIBQIN | Sn | N | 2.926 | 175.1 |
| Pn | α | RvdW | MEP (PnF3) |
|---|---|---|---|
| N | 5.3 | 1.55 | 15.9 |
| P | 16.9 | 1.80 | 27.4 |
| As | 21.6 | 1.85 | 38.5 |
| Sb | 30.8 | 2.06 | 46.7 |
| CSD Code | Pn | A | dPn···A | ∠ |
|---|---|---|---|---|
| ICSD413360 | As | N | 2.978–3.198 | 157.7–164.1 |
| ICSD413359 | Sb | N | 2.844 | 149.9 |
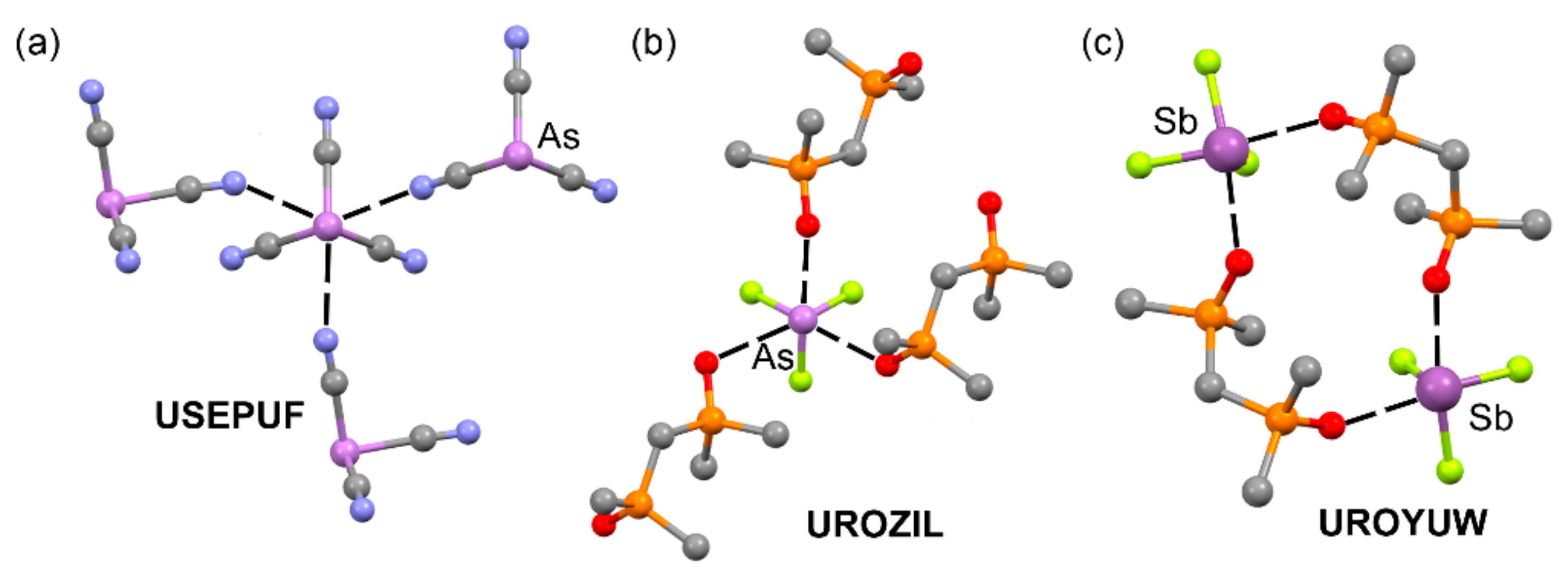
| USEPUF | As | N | 2.794–2.837 | 161.5–162.4 |
| UROZIL | As | O | 2.575–2.965 | 167.6–176.8 |
| UROYUW | Sb | O | 2.446–2.455 | 161.5–168.0 |
| USEQAM | Sb | N | 3.412 | 170.3 |
| Ch | α | RvdW | MEP (ChF2) |
|---|---|---|---|
| O | 3.0 | 1.52 | 16.8 |
| S | 11.8 | 1.80 | 35.6 |
| Se | 17.5 | 1.90 | 44.9 |
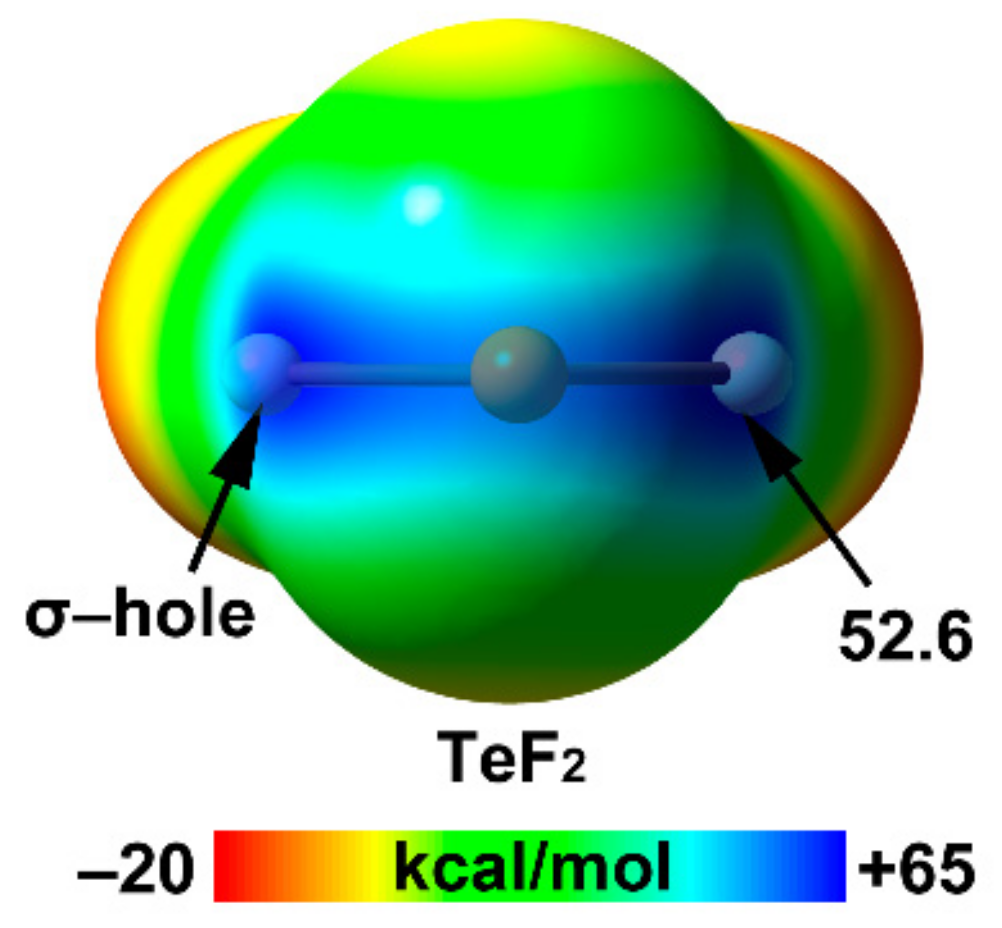
| Te | 25.9 | 2.06 | 52.6 |
| CSD Code | Ch | A | dCh···A | ∠ |
|---|---|---|---|---|
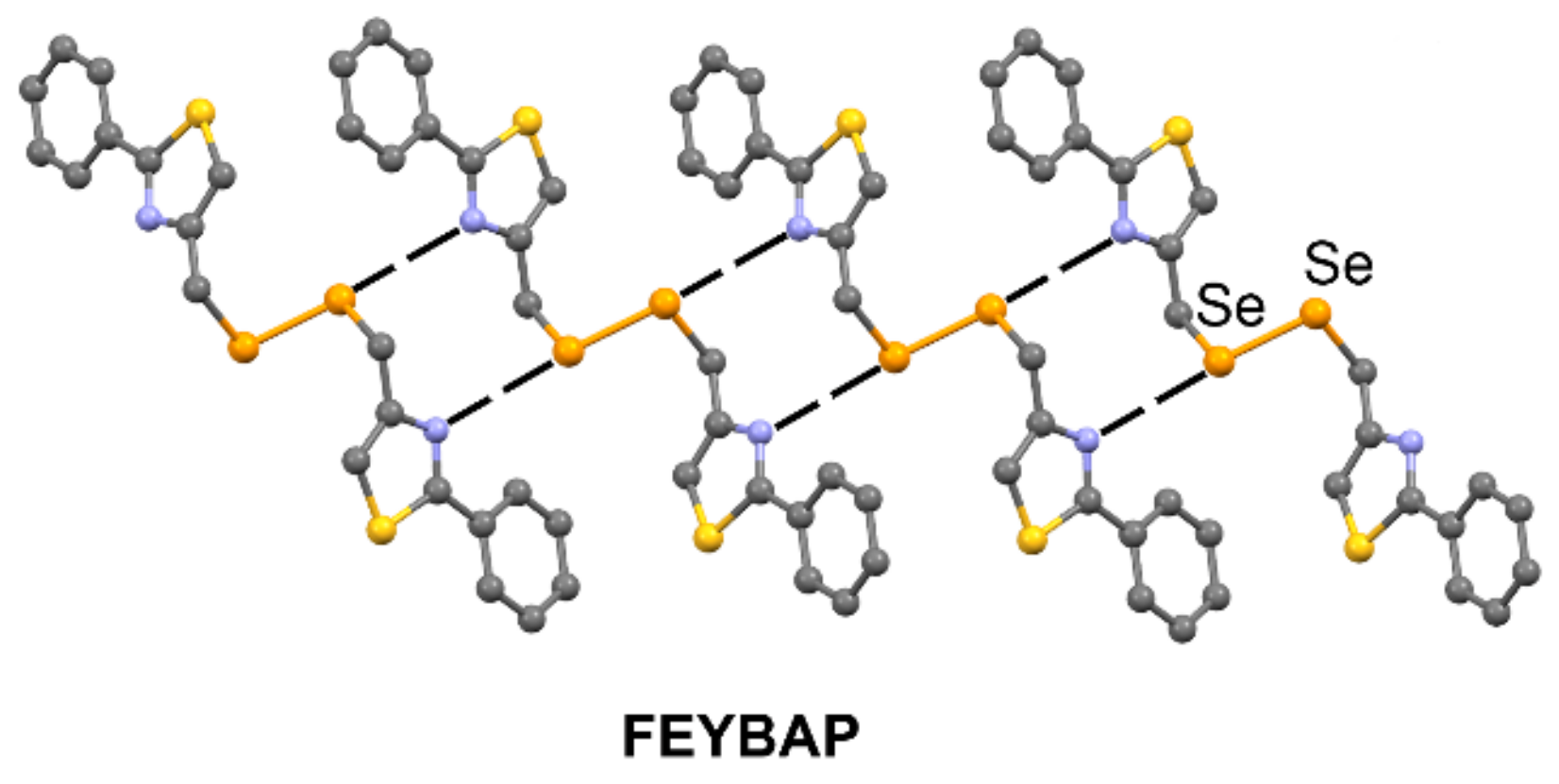
| FEYBAP | Se | N | 3.342 | 171.9 |
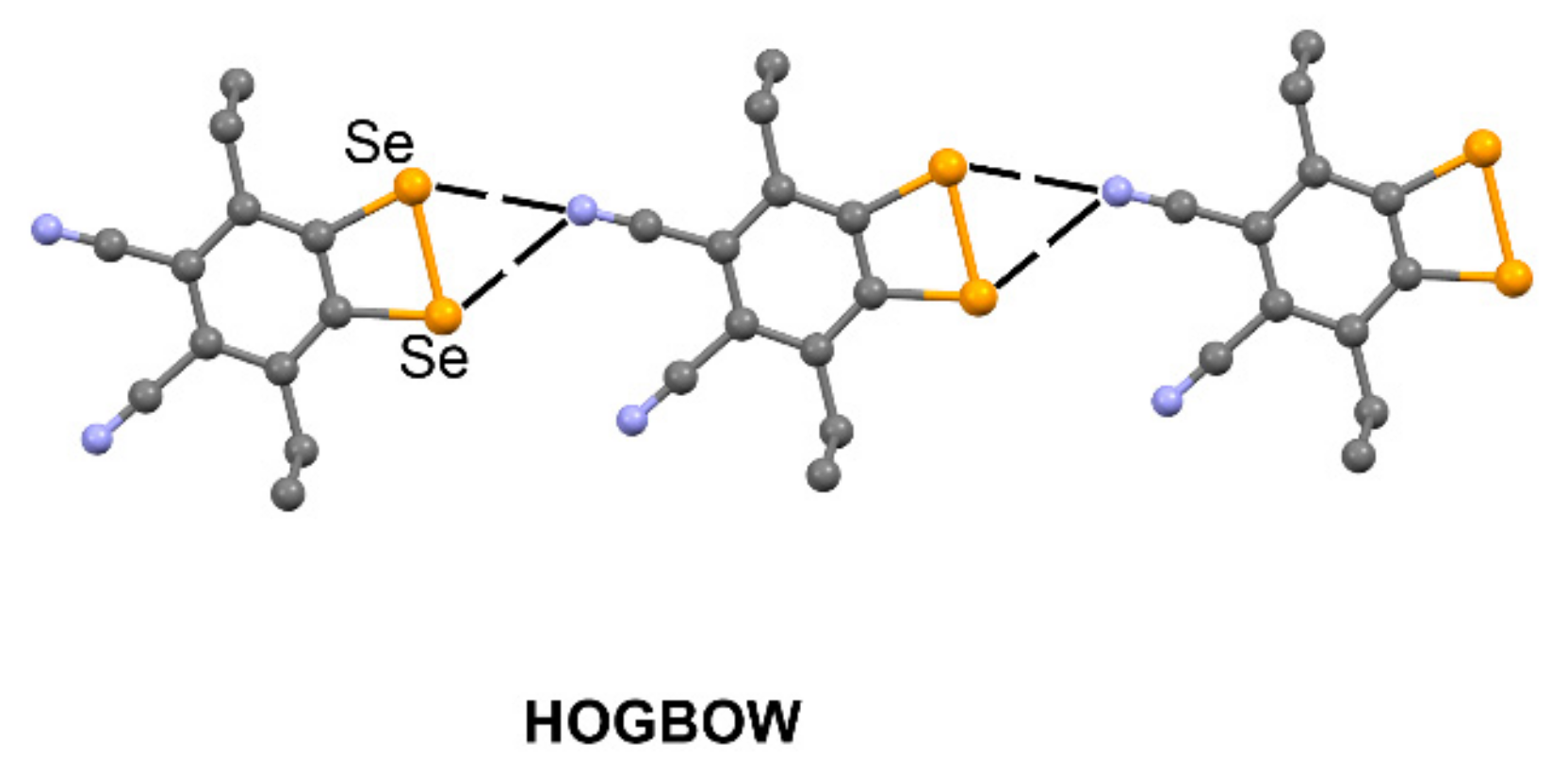
| HOGBOW | Se | N | 3.049–3.124 | 140.2–144.2 |
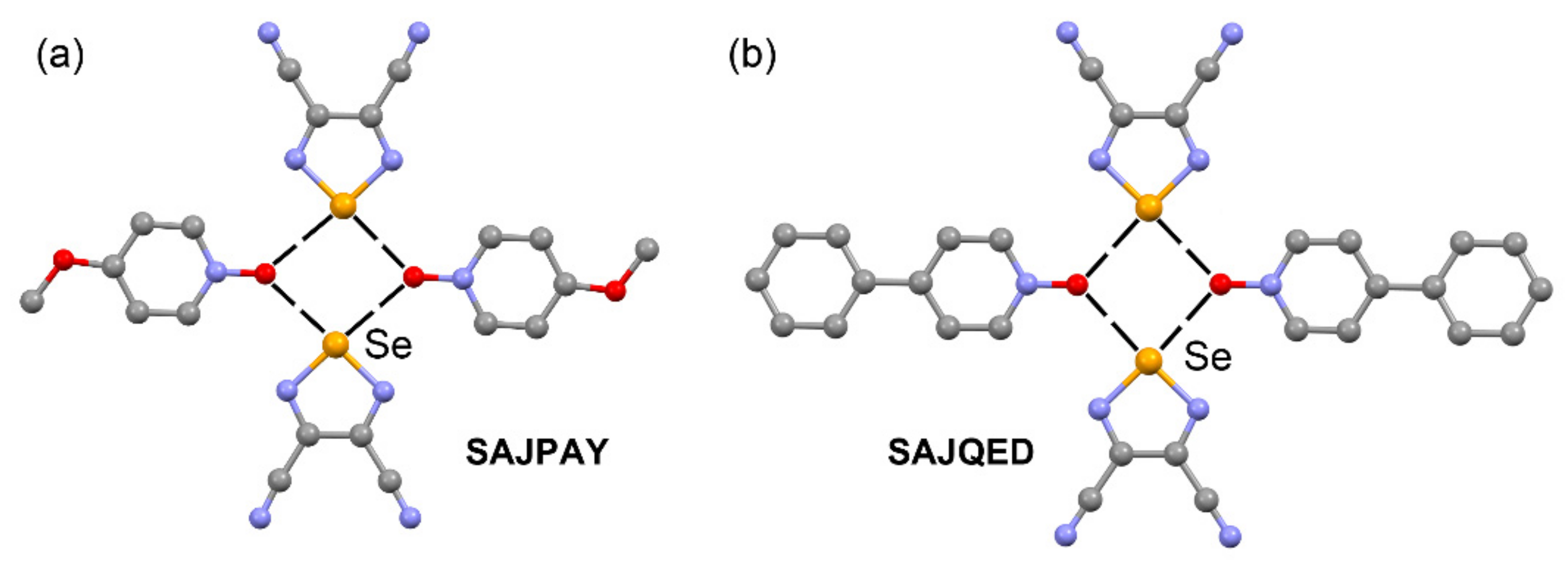
| SAJPAY | Te | O | 2.076–2.784 | 173.8–176.3 |
| SAJQED | Te | O | 2.756–2.758 | 172.6–174.7 |
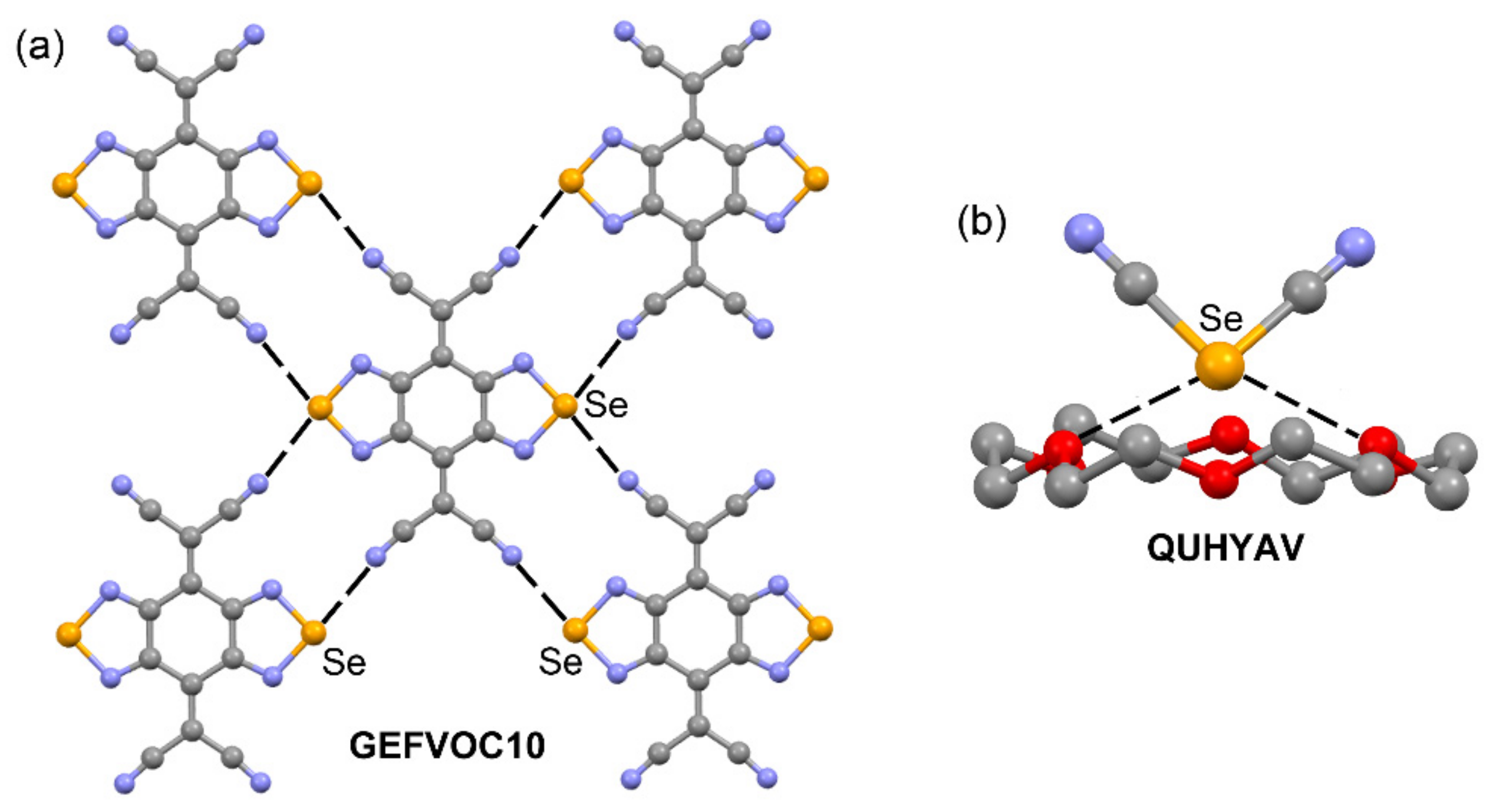
| GEFVOC10 | Se | N | 2.940 | 169.5 |
| QUHYAV | Se | O | 2.938 | 161.2–167.3 |
| CISPUP | Te | S | 3.409 | 161.5 |
| FELPUH | Te | N | 2.825 | 150.9 |
| Ha | α | RvdW | MEP (HaF) |
|---|---|---|---|
| F | 1.8 | 1.47 | 15.9 |
| Cl | 8.4 | 1.75 | 42.3 |
| Br | 14.0 | 1.89 | 49.7 |
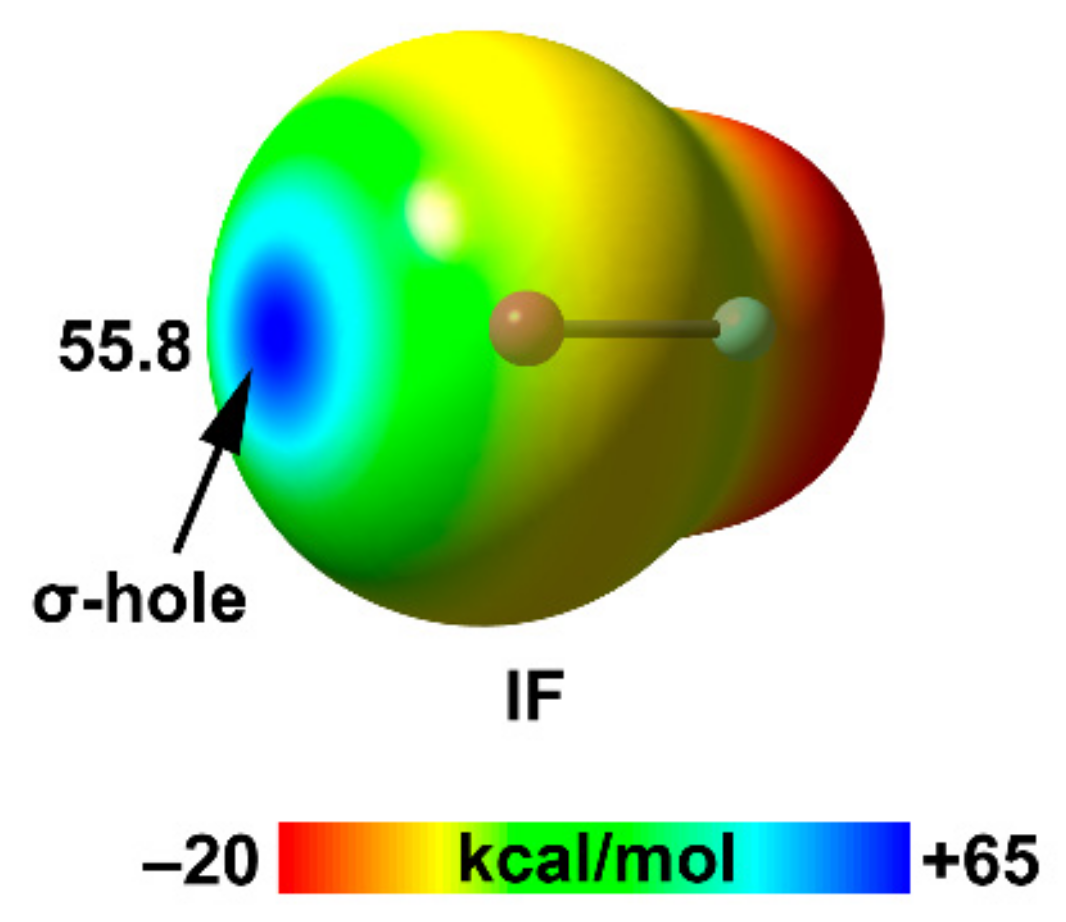
| I | 22.6 | 1.98 | 55.8 |
| CSD Code | Ha | A | dHa···A | ∠ |
|---|---|---|---|---|
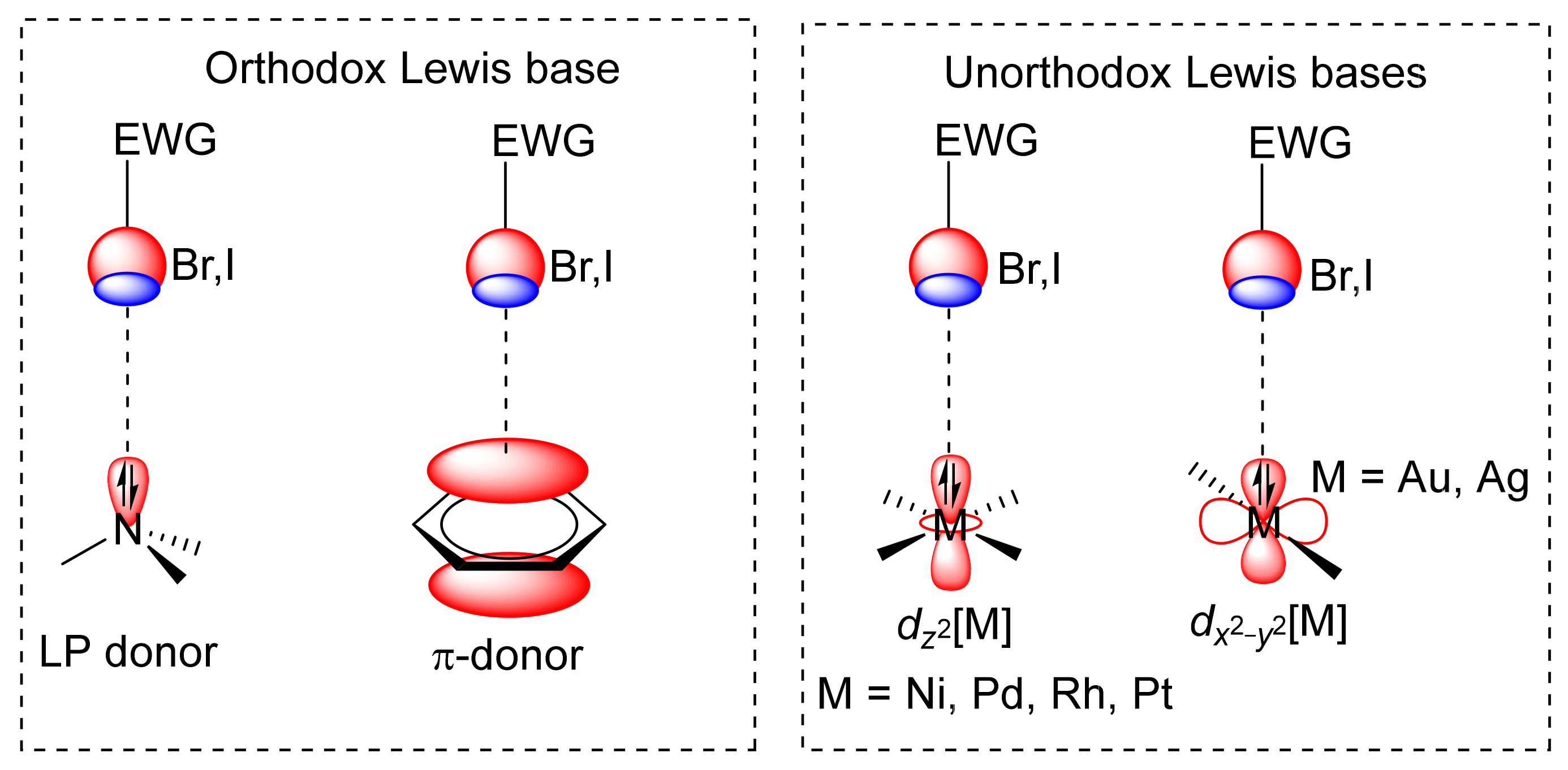
| MEBXID | I | Ni | 3.389 | 142.5 |
| AMUPAC | I | Rh | 3.693 | 147.2 |
| LIHMIB | Br | Pt | 3.365 | 165.9 |
| LIHMEX | I | Pd | 3.512 | 164.9 |
| Ng | A | RvdW | MEP (NgO3) |
|---|---|---|---|
| Ar | 6.3 | 1.88 | 37.7 |
| Kr | 11.5 | 2.02 | 43.9 |
| Xe | 19.9 | 2.16 | 52.7 |
| CSD/ICSD Code | Ng | A | dNg···A | ∠ |
|---|---|---|---|---|
| ICSD253590 | Xe | O | 2.80–2.90 | 159–172 |
| KAZMEG | Xe | Cl | 3.070–3.101 | 162–167.6 |
| EZAKEX | Xe | O | 2.758 | 164.4 |
| KAZLUV | Xe | Br | 3.104–3.274 | 158.5–165.8 |
| VIFKUT | Xe | N | 2.760–2.820 | 170.4–173.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. https://doi.org/10.3390/cryst11101205
Frontera A, Bauzá A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals. 2021; 11(10):1205. https://doi.org/10.3390/cryst11101205
Chicago/Turabian StyleFrontera, Antonio, and Antonio Bauzá. 2021. "On the Importance of σ–Hole Interactions in Crystal Structures" Crystals 11, no. 10: 1205. https://doi.org/10.3390/cryst11101205