Heterogeneous Catalysis on Metal Oxides


Laboratoire de Réactivité de Surface, Université P. & M. Curie, Sorbonne Université, UMR-CNRS 7197, 4 Place Jussieu, F-75252 Paris, France
Catalysts 2017, 7(11), 341; https://doi.org/10.3390/catal7110341
Submission received: 8 October 2017
/
Revised: 25 October 2017
/
Accepted: 27 October 2017
/
Published: 10 November 2017
(This article belongs to the Special Issue C1 Chemistry—C1-Platform Chemicals as Cornerstone for a Sustainable Energy)
Abstract
:This review article contains a reminder of the fundamentals of heterogeneous catalysis and a description of the main domains of heterogeneous catalysis and main families of metal oxide catalysts, which cover acid-base reactions, selective partial oxidation reactions, total oxidation reactions, depollution, biomass conversion, green chemistry and photocatalysis. Metal oxide catalysts are essential components in most refining and petrochemical processes. These catalysts are also critical to improving environmental quality. This paper attempts to review the major current industrial applications of supported and unsupported metal oxide catalysts. Viewpoints for understanding the catalysts’ action are given, while applications and several case studies from academia and industry are given. Emphases are on catalyst description from synthesis to reaction conditions, on main industrial applications in the different domains and on views for the future, mainly regulated by environmental issues. Following a review of the major types of metal oxide catalysts and the processes that use these catalysts, this paper considers current and prospective major applications, where recent advances in the science of metal oxide catalysts have major economic and environmental impacts.
1. Introduction to Catalysis
Catalysis is an important domain of chemistry [1,2,3]. Berzelius first used the word “catalysis” in 1836, taken from the Greek word “καταλεινν” (=loose down, dissolve) by analogy to the word “analysis” in order to rationalise well-known experimental observations such as wine and beer fermentation, soap and sulfuric acid (oil of vitriol) synthesis, starch transformation to sugar by acids, decomposition of H2O2 by metals, ethanol oxidation to acetic acid on Pt, etc. By definition, catalysis is a process by which a reaction rate is enhanced by a small amount of the so-called catalyst, which supposedly does not undergo any change during the reaction, at variance with surface or stoichiometric reactions. However, as any manager of industrial plants knows, this is a very optimistic definition as real catalysts change in structure, activity and selectivity with time on stream (activation step) and deactivate more or less rapidly. Some 60 years after Berzelius, Oswald established the kinetic nature of this phenomenon and gave in 1895 the definition: “a catalyst is a substance that changes the rate of a chemical reaction without itself appearing in the products”. According to IUPAC (1976), a catalyst is a substance that, being present in small proportions, increases the rate of attainment of chemical equilibrium without itself undergoing chemical change.
It has been recognised that the catalyst acts by reducing the energy necessary to proceed along the reaction pathway, i.e., the activation energy Ea that needs to be surmounted to yield products. This activation energy is the energy required to overcome the reaction barrier and determines how fast a reaction occurs. The lower the activation barrier, the faster the reaction will be. Note that the thermodynamics of the reaction remains unchanged under catalyst action and that the main effect is that the catalyst influences the reaction rate.
Heterogeneous catalysis (gas or liquid phase and solid catalyst) proceeds via adsorption of one or two reactant molecule(s) on the solid surface, enhancing the reactant(s) concentration on the surface and favouring its (their) activation. The first step of the reaction is thus the reactant(s) adsorption, whereas the reaction energy includes the activation barrier energies of adsorbed reactants (Aads), of adsorbed intermediates (Iads) and of desorption of products (Pads). In other words, the degree of catalytic efficiency gained in following a given path is governed by the energetics of the various intermediates, which encompass adsorbed reactant, the activation energy required to convert the bound reactant into a surface intermediate and finally to a product and its desorption.
Catalysis plays a prominent role in modern life. The majority of actual industrial chemical processes, involving the manufacturing of commodity-, petro-, pharmaceutical- and fine-chemicals, clean fuels, etc., as well as pollution abatement technologies, have a common catalytic origin. It is well recognised that 85–90% of industrial chemical processes involve at least one catalytic step. The main objectives of using a catalyst are to get high activity, i.e., high conversion of reactants and overall high selectivity to a desired product, the latter property avoiding or limiting separation/purification procedure, which involves important steps to take into account, particularly for economical and overall environmental issues.
Solid catalysts are classified as (i) conductors, (metals and alloys); (ii) semiconductors (oxides and sulphides); and (iii) insulators (metal oxides and solid acids or bases, including heteropolyacids, natural clays, silica–alumina and zeolites). Oxidation reactions are catalysed by oxides, while desulphurisation reactions occur on a sulphide catalyst. Insulator oxides catalyse dehydration, and acid/base solids act in processes with carbocationic/carbanionic intermediates. These features have led to the idea that there is a kind of compatibility between the catalyst and the reactant molecules. We will discuss this point when mentioning the structure sensitivity of catalytic reactions on metal oxides and describing some active sites (vide infra).
The major domains of heterogeneous catalysis, applied industrially, concern:
- Oil refining, energy and transport,
- Bulk chemicals,
- Polymers materials and detergents textiles,
- Fine chemicals, pharmaceutical medical chemicals and food feed,
- Plant design/engineering and realisation, catalyst design, subsequent development of catalysts and of catalytic processes,
- Commercial production of catalysts in sufficient quantities,
- Monitoring and control of chemical reactions and plant operations,
- Environmental issues.
2. Introduction to Metal Oxide Catalysts
Among the different fields of heterogeneous catalysis, catalysis by metal oxides is one of the most important, as it covers the majority of processes and of catalyst families used industrially, such as silica, alumina, clays, zeolites, TiO2, ZnO, ZrO2, porous and mesoporous metal oxides, polyoxometallates (POMs) of Keggin or Dawson type, the phosphates family (e.g., VPO, FePO4, silica phosphoric acid (SPA)), multicomponent mixed oxides (molybdates, antimonates, tungstates, MoVTe(Sb)Nb-O, etc.), perovskites, hexaaluminates, etc.
Metal oxides became prominent in the mid-1950s when they were found to effectively catalyse a wide variety of reactions, in particular oxidation and acid-base reactions. They are involved in many petrochemicals, intermediates, fine and pharmaceutical chemicals and biomass transformation reactions. They are also the basis for metallic (mono- or pluri-metallic) catalysts, for hydrodesulphurisation catalysts (CoMoO4-, NiMoO4-, NiWO4-based), for deNOx, deSOx, and bulk single or mixed metal oxide catalysts. The main catalytic domains cover oxidation (selective or total), acid and base catalyses, photocatalysis, depollution and biomass conversion, as described below.
This paper, being devoted to gas–solid heterogeneous catalysis, considers that the reaction occurs at the interface of two media, namely the solid catalyst and the medium of the reactants and products, and thus involves fluid-phase transport of reactants to and products away from the catalyst surface. In heterogeneous catalysis the catalysts are in the form of powders, pellets or extrudates, in order to permit the reactant flow to cross the full catalytic bed in the reactor. For model catalysts, films or single crystals may also be considered.
Metal oxides [4,5] constitute a class of inorganic materials that have peculiar and various properties and applications as sensors, catalysts, fuel cells, etc. Oxide surfaces terminate by oxide O2− anions, as their size is much larger than that of Mn+ cations. It follows that the symmetry and coordination of Mn+ cations are lost at the surface. Moreover, the surface of an oxide may contain different types of defects and environments (kinks, steps, terraces), which play a determining role in the catalytic phenomenon [6,7]. This surface unsaturation is usually compensated for by a reaction with water vapour, leading to the formation of surface hydroxyls according to: O2− + H2O → 2OH−. OH groups are conjugated acids of lattice oxygen ions O2−, which are strong bases and conjugated bases of water molecules.
Among the solid state of metal oxides [8], important parameters or features act on catalytic properties. One may distinguish point and extended defect structures, the atomic composition and structure of a crystalline phase, and electronic defects, which correspond to the probability that an electron occupies an energy state, given by the Fermi–Dirac function: F(E) = ne/N = 1/(exp[(E − EF)/RT], where ne is the number of electrons, N is the number of available energy states, and EF is the Fermi level energy. In an intrinsic semiconductor, the number of electrons in the conduction band equals the number of holes in the valence band and is given by: n = ne = nh = n0 exp (−EG/2kT), where n0 is constant and EG is the energy gap. Given that most oxides have a large band gap and often high contents of impurities, most electronic defects are extrinsic. Extrinsic defects can introduce carriers into localised energy levels within the band gap and, in such cases, are electrically active. An electronic defect in an energy level just below the conduction band edge is considered a donor as it may give electrons to the conduction band and increase the n-type conductivity. For instance, when a P atom of +5 oxidation state is added to silicon in silica where Si is at +4 oxidation state, the additional electron for charge compensation introduces a donor state (exciting the electron necessitates ED rather than EG), while introducing a Ga ion at +3 oxidation state creates a hole in the valence band and can accept an electron, increasing the p-type conductivity. For a donor-doped material, the total number of the charge carrier equals: ntotal = ne (dopant) + ne (intrinsic) + nh (intrinsic) = n0D exp(−ED/kT) + 2n0 exp(−EG/2kT). This aspect is illustrated in Figure 1.
As heterogeneous catalysis occurs at the interface of the two media, the fluid-phase transport of reactants to and of products away from the catalyst has to be considered. In the case of solid metal oxide catalysts, solid-phase transport of defects, oxide anions and electronic conductivity should also be taken into consideration. Important properties of a metal oxide, such as electrical conductivity, lattice oxygen anions’ mobility, atoms/ions’ diffusion acting on sintering or phase separation, catalytic activity, melting point and various optical properties, depend on the presence of defects vs. an ideal ionic crystal. There are different types of defects: electrons and positive holes, excitons, vacant lattice sites (designated V), interstitial atoms, impurity atoms at interstitial or substitution location, dislocations and stacking faults. The subscript denotes the site that the defect occupies (lattice atom site or interstitial atom), the dot • denotes a positive charge, and x represents neutrality. Point defects are missing, substituted, or interstitial ions. They are electronically charged and can be intrinsic (thermally generated in a crystal) or extrinsic (impurity or dopant). The most common point defects are Schottky-type, i.e., vacant cationic or anionic lattice site, or Frenkel-type, i.e., cations or anions displaced to interstitial sites. One also has electronic defects (electrons e− and electron holes h• influencing electric, ionic and protonic conductivities) and non-stoichiometry in oxides, such as perovskites with an unfilled 3d electron shell and cation and lattice oxygen anion deficiency or excess.
Single or complex metal oxides based on the first transition series present a wide variety of non-stoichiometric phenomena, which originate from the unfilled 3d electron shell. For instance, Fe sites can be vacant in FeO of rocksalt framework (Fe1−xO with 0.05 x 0.18). In ABO3 perovskite oxides, non-stoichiometry comes from a cation deficiency in A or B sites (AxWO3, A being an alkali ion) or oxygen anions excess. Cation A has a great influence on electrical properties. For instance, WO3 is an insulator, while AxWO3 is a semiconductor at low x and metallic at high x. Oxygen-deficient perovskites have attracted much attention because of their oxygen storage ability and their redox properties, quite useful for total oxidation reactions. In fully oxidised CaMnO3−δ and CaFeO3−δ perovskites, Mn and Fe are at +4 oxidation state and δ = 0, the material can accommodate up to 17% oxygen vacancies without losing its structure. The Sr1−xLaxCo1−yFeyO3−δ series, with brownmillerite-type oxygen defects, exhibits high electronic/oxygen ion mobility. In some perovskites a small oxygen excess can be accommodated by the formation of cation vacancies at A- or B-sites, leaving the oxygen sub-lattice intact. Electronic defects may be created upon reduction and oxidation of metal cations at different oxygen partial pressures (pO2). At low pO2, the material loses oxygen, which generates electrons, enhancing the n-type conductivity, according to: O0 ↔ (1/2) O2 + VO•• + 2e−. At high pO2, oxygen is incorporated into an oxygen vacancy and takes two electrons from the valence band, leading to holes contributing to the p-type conduction, according to (1/2) O2 + VO•• ↔ O0 + 2h•.
A consequence of the trapping of electronic defects is that the solid becomes insulating at low temperatures. Defect associations between oxygen vacancies and acceptor dopants are observed in Y-ZrO2, where VO•• are trapped by YZr• defects leading to a drastic decrease in oxide ion conductivity. Protonic defects, associated with acceptor dopants, limit protonic conduction in metal oxides. As all metal oxides contain impurities, the defect mechanism of “pure” oxides is similar to that of doped oxides. For instance, strontium titanate, SrTiO3, has a similar defect chemistry to acceptor-doped SrTiO3, because the impurities are ions at lower oxidation states such as Fe3+, Al3+, Mg2+, Na+, etc., and their concentrations, typically ca. 102–103 ppm, are sufficient to dominate the defect chemistry, especially at high pO2. All these aspects should be taken into account when considering the reaction mechanisms and kinetics on metal oxide catalysts, for instance in selective or total oxidation reactions on mixed metal oxides, where electrons and oxygen anions mobilities and lattice oxygen diffusion or storage capacity are important, for instance in a redox catalytic oxidation reaction. Lattice defects can lead to an electron transfer from the solid toward an adsorbed molecule (anionic chemisorption) or, by contrast, from the adsorbed species toward the solid (cationic chemisorption) in the case of non-stoichiometric oxides or of oxides doped with an ion of different valence. In the first case and for n-type semi-conduction, one observes a decrease in conductivity and the reverse for semi-conductors of p-type, while in the second case, conductivity increases for n-type semiconductors and decreases for p-type semiconductors.
The development of descriptors that may correlate the activity of catalysts and their physical properties is an important goal in catalysis. For instance, it has been shown [9] that the apparent activation energy for propene oxidation to acrolein over scheelite-structured, multicomponent, mixed metal oxides (Bi3FeMo2O12, Bi2Mo2.5W0.5O12, Bi1−x/3V1−xMoxO4, with 0 ≤ x ≤ 1) is related to the band gap of the catalyst measured at reaction temperature, as supported by theoretical calculations.
As we have seen above, catalysis starts with chemisorption, which should be not too strong to permit intermediate formation, and the intermediate desorption not too weak to permit adsorbed species to be activated, which is known as Sabatier’s principle. Later, Taylor suggested [10] that activation energy was a key factor during the adsorption stage. He has also suggested that preferential adsorption on a catalyst surface should take place at those atoms or ensemble of atoms, depending on the reaction and reactant size, situated at the peaks, fissures, kinks, edges, etc., of a crystallite rather than on flat surfaces. The notion of active sites or active centres was then suggested to be the locus of catalytic conversion.
However, depending on the reactant molecule size and reaction, more than one surface atom may be involved in the reaction. This is what was defined for metals by Boudart as a structure-sensitive reaction [11,12]. In the case of metal oxides, the active sites are often composed of surface ensembles of atoms to permit the reaction to occur. For instance, for oxidation reactions where both the reactant molecule and the gaseous oxygen have to be activated, the surface active site should be large enough to accommodate the reactant molecule and the oxygen from the lattice to diffuse through the solid catalyst to permit oxidation. This diffusion should be fast enough to renew lattice oxygen anions, leaving the surface with the product in a Mars and van Krevelen mechanism but not too high to avoid over-oxidation to CO2 [13]. This is illustrated in Figure 2 and Figure 3 for butane oxidation to maleic anhydride on a vanadyl pyrophosphate catalyst, designated as a VPO catalyst [14,15], and corresponds to the structure sensitivity of metal oxide for selective oxidation reactions, as demonstrated by Volta [16] for MoO3 and extended to other metal oxides [17,18].
Taylor [10] has suggested that chemisorption may be an activated process, and may occur slowly. The idea that adsorbed species and intermediates in catalysis may require ensembles composed of several adjacent free-surface atoms was first expressed by Balandin [17,19], who suggested that a reacting molecule may be simultaneously adsorbed on several atoms (the multiplets theory). The typical concentration of active sites has been evaluated to be in the domain of 1019 m−2 for metals and 1015 m−2 for metal oxides.
The above description considers active sites as rather static. However, a metal oxide catalyst surface has been observed to change under working conditions. Moreover, it is worth recalling that oscillations on the surface occur during a catalytic reaction, which is important for kinetics studies; a theory was proposed [20]. It was shown [21] that stationary heterogeneous catalytic reactions involve a gas/catalyst equilibrium, which acts on the rate-determining step (RDS) of the reaction. Moreover, during stationary catalytic processes, the surface is covered with many two-dimensional chemisorbed crystal-like islands of different sizes rather than with isolated chemisorbed molecules. Depending on the chemical nature of the catalyst and of the reacting mixture, and on the ambient conditions, these islands can be of the same or of two or more chemical compositions. Each ensemble of these islands of any one chemical composition is considered a two-dimensional chemically adsorbed phase (chemadphase, ChPh), which oscillates, i.e., moves relative to other islands the surface of a crystal in such a way that, under stationary catalysis, the total surface area of each ChPh remains unchanged. Most of the two-dimensional ChPh crystals consist of a great number of chemadmolecules and represent an element of surface metalloinorganic or metalloorganic entities that are connected with the solid. Following such an approach, a revisited view of the active sites in heterogeneous catalysis on metal oxides is given in [22] and an example of active sites composed of four dimers for VPO catalyst is given above in Figure 4.
Another important aspect of the surface of metal oxides is the appearance of a segregated phase on the surface as a consequence of the inability of the surface to homogeneously accommodate all cations in its lattice and to be altered when exposed to an external environment. This particularly holds true for complex mixed oxides (e.g., multicomponent catalysts in selective oxidation reactions) and depends on the preparation method and activation medium and procedure, and often occurs during a catalytic reaction, as illustrated in Figure 4 for MoVTeNb-O (so-called M1 phase) catalyst. In the case of supported oxides, this is even more drastic as mass transport on the surface or overall, within the pores, limits regular deposition far from equilibrium, leading to a concentration gradient of the deposited oxide within the pores.
In a study using microcalorimetry, Schlögl et al. have shown [23] that, on an activated mixed oxide MoVTeNb-O catalyst, active for propene and propane selective oxidation and ammoxidation to acrylic acid and acrylonitrile, respectively, the adsorption energy of propene and propane are about constant vs. coverage (57 kJ·mol−1). However, after reaction the energy decreases and its distribution in strength becomes heterogeneous, as illustrated in Figure 4.
The first part of this curve is thus characteristic of a “single-site heterogeneous catalyst” (SSHC site, as defined by Thomas [24]), i.e., with an active site of similar activity. However, under reaction conditions the surface is modified and the sites are heterogeneous in strength (no longer satisfying the SSHC principle). Moreover, in situ X-ray photoelectron spectroscopy (XPS) analysis has shown that depletion of Mo and V enrichment did occur during the reaction. This observation characterises element segregation or the formation of a sublayer of different composition than the bulk, as shown by Millet et al. [25]. It was observed [26] that the surface composition of the M1 phase differs significantly from the bulk, implying that the catalytically active sites are not part of the M1 crystal structure and rather part of all terminating planes. It then appeared that the active sites are formed under propane oxidation conditions and are embedded in a thin layer enriched in V, Te, and Nb on the surface of the stable self-supporting M1 phase [23]. This example shows how complex a catalytic surface can be and how the surface structure and chemical composition may change under reaction (working) conditions.
This aspect of changes and reconstructions in the surface composition of metal oxide catalysts under reaction conditions is well known. For instance, for propane oxidative dehydrogenation on VMgO catalyst, the active surface was shown to be amorphous and composed of VO43− units scattered over magnesia and polymeric VOx species. During the reaction a completely reversible phenomenon of order/disorder of the V overlayer was observed in connection with the redox state of the surface [27]. This aspect is therefore recognised as occurring very often and shows that characterising a catalyst surface under working conditions with analysis of reactants and products on line (so-called real in situ or operando) is compulsory for a reliable description of active sites in metal oxide catalysts.
3. Theoretical Approach and Calculations on Metal Oxide-Based Heterogeneous Catalysts
The description of chemical catalytic reactions occurring at the surface of a solid catalyst has led to many studies by chemists and physicists all over the world since the 1960s, favoured by the huge increase of computational power [28,29,30]. A wide set of theoretical approaches has been developed from configuration interactions [31] to coupled cluster methods [32] and quantum chemistry Monte Carlo [33]. At present, the method of choice is the density functional theory (DFT) [34] to describe adsorption, surface properties and reactions at surfaces of metals, semi-conductors and insulator surfaces. The theory permits us to compute and predict reaction enthalpies and entropies, transition state structures and identify reaction mechanisms and surface properties [35]. For oxide catalysts, one needs an acceptable description of the electronic structure of the oxide, the energy of its defects and localised states, and the position of the valence and conduction bands to obtain a reliable prediction of the thermodynamic and kinetic aspects of the reaction. It is important to determine theoretically the oxide band gap and alignment of occupied and unoccupied levels vs. vacuum level for redox properties and calculation of the reaction energies at the oxide surface. Standard implementations of DFT are based on the Kohn–Sham (KS) equation [36] and on the use of local density or a generalised gradient approximation for the exchange correlation functional. In a study of a catalytic system, a crucial problem is to determine the band gap of an oxide as the positions of the top of the valence band (VB) and bottom of the conduction band (CB) determine the redox properties of the catalyst, as they are electronic levels involved in charge transfer from or to the catalyst [9]. The KS band gap is the difference between eigenvalues of CB minimum and VB maximum. Also, the role of oxygen vacancies is important, as discussed above, and can be clarified with theoretical calculations [37].
We have seen above (paragraph 2) that one can modify an oxide surface by substituting some metal atoms with another metal, M. The presence of the other metal atom changes the bonding in the oxide, and then either the oxygen atoms near M become chemically active or M becomes reactive and able to activate oxygen. It may also happen that both M and O are activated and participate in reactions in which an incoming molecule is dissociatively adsorbed.
However, it has been shown that DFT may have serious troubles when it is used to study catalysis by some oxides [34]. It was shown that the energies of the Kohn–Sham orbitals are not correct, as there is a large difference between the molecular orbitals and the excitation energy. One has to consider the shortcomings of gradient approximation and density function theory (GGA-DFT) when one extends it to doped oxides [38]. For instance, for Li-doped MgO, some of the Mg atoms in the surface layer are replaced with Li, which creates an electron deficit in the oxide, which allows it to be a catalyst for methane, ethane, or propane activation. GGA-DFT calculations show that the electron deficit is distributed evenly on the surrounding oxygen atoms, and B3LYP and higher-quality methods find that it is localised on one oxygen atom.
4. Metal Oxide Catalyst Preparation
The objective of this section is to address the broad topic of metal oxide syntheses for heterogeneous catalysis by considering the main preparation procedures for bulk and supported metal oxide catalysts [39,40,41,42]. It deals with three main areas: (1) synthesis of bulk simple metal oxides; (2) synthesis of bulk mixed oxides; and (3) elaboration of supported metal oxides.
For simple oxides, it is important to control the following key parameters: the nature of the polymorph, the morphology, the textural properties (surface area, porosity), as well as the thermal, chemical and mechanical stability. Besides purely thermal methods, which do not give access to fine control of these parameters, three main families of usual preparation routes based on the reactions between water or an organic solvent and inorganic precursors are usually considered, namely gas-phase polymerisation, aqueous-phase precipitation, and sol-gel (hydrolytic and non-hydrolytic) chemistry. Solvothermal syntheses in the presence of organic molecules and templating approaches yield materials with controlled morphology and textural properties, in particular organised microporous, mesoporous, macroporous and hierarchically organised oxides. Some novel preparation routes, such as complexation and solvent-free activated reactive synthesis, allow one to overcome the limited surface area reached after crystallisation at high temperature. Organisation of the porosity by hard or colloidal templating is a recent approach to maximise the specific surface area, leading to mesoporous and hierarchical porosity materials.
For mixed oxides, the most common technique is co-precipitation by mixing two metal salts in a solution and forcing them to precipitate using a suitable base at a given pH. The obtained precipitate is then calcined under specific atmosphere depending on the catalyst used.
Bulk catalysts mainly comprise active substances, but some inert binder is often added to facilitate the catalyst particles’ formation and limit attrition, particularly in moving bed reactors. However, in some cases, bulk catalysts are used as prepared, without binder, when prepared by high-temperature fusion, as for the iron-based catalysts used in ammonia synthesis.
The preparation of mixed-oxide catalysts involves co-precipitation of both components at a given pH, followed by the classical treatments schematised above. They can also be prepared by solid–solid interaction between both salts at high temperature.
The preparation of supported metal oxide catalysts [43] is based on ambient, aqueous-phase methods (selective adsorption, impregnation and deposition–precipitation), which predominate at the industrial scale for environmental and economic reasons. The impregnation and drying steps are important, as are the physical–chemical parameters that control the surface chemistry at the solid/liquid interface during active phase deposition, which control the speciation and distribution of the active phase in a porous support. Surface speciation of the active phase depends on the interfacial pH, which is mainly controlled by the buffering effect of the support (nature and density of surface ionisable groups) but weakly dependent on the initial impregnation pH. Distribution of the active phase in a support body can be adjusted both at the impregnation step (by using complexing or non-complexing additives) and at the drying step by limiting the liquid convection toward the external surface. The support can also be chemically activated by attaching chemical groups, such as acid or amino groups, able to bind easily with the salt to be attached.
The synthesis strategies for mesoporous silica-based materials [44] with improved physicochemical and textural properties and optimised catalytic features are based on the adjustment of the surface functionality by the incorporation of active sites in the silica walls or by the deposition of active species on the inner surface of the material. The incorporation of active sites is possible directly (from mixtures containing both silicon and a hetero-element) or indirectly via post-synthesis procedures using a multitude of pathways. The results of these two different methods are not identical. While the direct method typically results in a relatively homogeneous incorporation of the hetero-element, post-synthesis treatment modifies the wall surface and leads to increased concentrations of the hetero-element on the surface.
5. Acid-Base Catalysis by Metal Oxides
Solid acid and base metal oxide catalysts are mainly used in refining and petrochemical processes [45,46,47]. Many of them are replacing homogeneous catalysts to decrease the E-factor, defined by Sheldon [48] as “weight of waste/weight of product”. This was the origin, in the early 1990s, of green chemistry, by a simple metric for judging the environmental impact of chemical processes. The E-factors for refining and bulk chemicals are ca. 0.1 and 1–5, respectively, while those for fine chemicals and pharmaceuticals are 5–50 and 25–100, respectively. Although the annual productions of fine chemicals and pharmaceuticals are not as large (102–104 tonnes and 10–103 tonnes, respectively) as those of refining (106–108) and bulk chemicals (104–106), the amounts of waste produced are comparable to those produced in refining and bulk chemicals. The number of processes that use heterogeneous acid and base catalysts for fine chemical synthesis is unfortunately limited at present, but increasing with time.
Acid-base properties of metal oxides are characterised by many physical techniques [49] such as IR of hydroxyl groups and of CO adsorption (the shift in O-H stretching frequency caused by CO adsorption reflects the acid strength of the OH groups) or of adsorbed acid (CO2, SO2, phenol, etc.) or basic (NH3, pyridine, amines, water, etc.) probe molecules, microcalorimetry for the determination of such probe molecules adsorption energy, their desorption by temperature programmed desorption (TPD) [50,51], 1H proton [52] and 31P [53] nuclear magnetic resonance spectroscopy (NMR), etc.
Catalytic reactions catalysed by metal oxides are hydrocarbon transformations where carbenium ions participate in hydrocarbon conversions according to the following principles: (i) β-scission; C-C bond cleavage occurs at β position to cationic C; (ii) stability order is tertiary (−130) secondary (−75) protonated cyclopropane (−62) primary (0), where the numbers in parentheses are the approximate energy differences in kJ·mol−1, referring to primary carbocation. The reactions involving the formation of primary carbocations are slow.
For acid-type reactions, one may cite cracking, synthesis of ethylbenzene and cumene and isomerisation, synthesis of organic chemicals, such as methanol to hydrocarbons, alkylation of aromatics with alcohols (e.g., toluene + methanol gives xylenes), acylation, esterification, biodiesel synthesis, Meerwein–Ponndorf–Verley reduction and Oppenauer oxidation, dehydration of alcohols, hydration of alkenes and alkynes, and Beckmann rearrangement. For solid base catalysts one may cite isomerisation of alkenes, aldol and aldol-type reactions, Knoevenagel condensation, Michael addition, Tishchenko reaction, side chain aromatic alkylation (e.g., toluene + methanol gives ethyl benzene), hydrogenation, etc.
The main solid acid catalysts are protonic zeolites, heteropolyacids (Keggin and Dawson types), silica-alumina, sulphated zirconia and tungsta, WO3-ZrO2 catalysts, ion exchange resins, and solid phosphoric acid. The main solid base catalysts are basic zeolites, alkaline earth oxides, zirconia, alumina, hydrotalcite and mixed oxides derived from hydrotalcite, basic clays, layered oxides, and alkaline salts supported on oxides.
For many reactions in aqueous solutions or involving water as a reactant or product, water- and temperature-tolerant solid acid or base catalysts such as ion-exchanged resins are used at low reaction temperatures. Water-tolerant solid acid and base catalysts, which can be used and regenerated at a high temperature, are also now replaced by ion-exchange resins.
Multifunctional catalysts with acidic and basic properties behave as either bifunctional or dual functional catalysts. A classic example is alkane isomerisation, in which hydrogenation/dehydrogenation function and acidic function operate successively. Another example is aldol condensation, which results from successive reactions of aldol addition by base sites and dehydration by acid sites. Another example is glucose conversion to fructose by Lewis acid, followed by fructose transformation to 2-hydroxymethlfurfural by Brønsted acid sites. There should be many cases in which two or more functions are required such as acidic, basic, redox, and metallic functions. For taking advantage of heterogeneous catalysts, where the active sites may have different functions co-existing on the surface, design and application of such multifunctional catalysts are promising.
Further application of solid acid and base catalysts covers fine chemical synthesis. Solid acid catalysts have mainly been used in the field of refinery and petrochemical production. Application of solid acids and bases to the synthesis of fine chemicals is a promising area for the discovery of new synthetic strategy for the synthesis of fine chemicals and biomass-derived chemicals. There are many benefits to utilising solid acids and bases: (i) the reduction of waste, i.e., salt formation, deposition or purification of used solvents; (ii) free choice of solvent, if any; (iii) one-pot reactions. Tandem reactions, in which more than one step is involved, can be carried out in one pot over solid acids/bases. The reactions can be catalysed by solid acids, solid bases and solids having both acid and base sites. One-pot reactions reduce the number of procedures to result in eco-friendly reaction systems.
6. Selective Oxidation Reactions
Catalytic selective oxidation of hydrocarbons [54,55,56] is an attractive reaction since the reaction produces valuable chemicals such as alcohols, aldehydes, acids, amino-acids, amino-aldehydes, oxynitriles, etc., and accordingly ca. 25% of the chemicals and intermediates are industrially produced by the catalytic oxidation processes [57,58]. Recently, the selective catalytic oxidation of light alkanes has attracted much attention due to the shale gas revolution and concerns about petroleum. Success in the reactions, however, has been limited so far despite tremendous efforts. This is due to the difficulty of the light alkane catalytic selective oxidation, which is derived from the chemical properties of light alkanes. Generally, the C-H bonding of hydrocarbons is the most effective parameter for its reactivity in catalytic oxidation and, in the case of light alkanes, severe reaction conditions like high reaction temperature are usually required because of the low reactivity due to the strong C-H bond. The severe reaction conditions, however, often cause undesirable side reactions and consecutive reactions of the desired products, which makes it difficult to achieve high selectivity toward the desired products at a high conversion rate. One usually proceeds either by oxidative dehydrogenation (ODH)—such as, for example, the dehydrogenation of allyl alcohol to acrolein over bulk or supported silver-based catalysts [59] or of lower alkanes to the corresponding olefins [54]—or by oxidation/ammoxidation with gaseous oxygen in controlled conditions (lower conversion leading to higher selectivity but necessitating the recycling of the reactant) [60].
Mixed-metal oxides have been used industrially for a long time (since the 1960s) as catalysts in the gas phase partial oxidation of hydrocarbons to yield oxygenates (acids, alcohols, aminoacids, oxynitriles, aldehydes, etc.) (see e.g., [61]). In the majority of cases, the reaction mechanism is of redox-type so-called Mars and van Krevelen [62]. Representative reactions are: (i) oxidative dehydrogenation (ODH) of short chain alkanes (from C1 to C6) [63,64]; (ii) propene partial oxidation/ammoxidation on bismuth molybdate/antimonate based catalysts; (iii) n-butane direct oxidation to maleic anhydride on VPO type catalysts (vanadyl pyrophosphate (VO)2P2O7-based); (iv) propane direct oxidation/ammoxidation on mixed MoVTe(Sb)Nb-O catalysts [65,66]. The actual trends in partial oxidation processes are especially focusing on the use of biomass as a raw material.
As a general rule of selective partial oxidation reactions involving a redox mechanism, the concept of “seven pillars”, as proposed by Grasselli [13], has to be satisfied: (i) nature of lattice oxygen anions, namely: nucleophilic (selective) rather than electrophilic (total oxidation); (ii) redox properties of the metal oxide (removal of lattice oxygen anions and its rapid reinsertion); (iii) host structure (permits redox mechanism to take place without structure collapse), see the examples given above in Figure 2 and Figure 3 and in Figure 5; (iv) phase cooperation in multicomponent catalyst or supported catalyst (epitaxial growth and synergetic effects); (v) multi-functionality (e.g., α-H abstraction and O-/NH-insertion); (vi) active site isolation (to avoid a too high lattice O surface mobility and thus over-oxidation); (vii) M-O bond strength (not too weak (total oxidation) nor too strong (inactivity) (known as Sabatier’s principle).
Catalytic partial oxidation reactions are favoured on mixed metal oxides based mainly on V and Mo cations attached to other redox cations such as Sb, Nb, Te, and Ta. V2O5 and MoO6 octahedra are assembled as shown in Figure 6 and are often composed of several phases acting synergetically between them and giving complex chemical compositions, as in the case of the mixture of M1 and M2 phases in the MoVTeNb-O catalyst used for propane amm/oxidation to acrylonitrile/acrylic acid. The selectivity depends on the nature of surface oxygen species (O2−) associated to cation(s) Men+, on Me-O bond energy and their distribution on the surface. This may come from the fact that oxidation reactions are demanding, i.e., are structure-sensitive when the number of electrons involved in the reaction is high. For instance, many catalysts are able to transform propane to propylene (reaction with 2e−), whereas only three (Cu-O, Bi-Mo-O, Sn-Sb-O) are able to transform propylene to acrolein (4e− reaction) and only one ((VO)2P2O7) to oxidise butane to maleic anhydride (a 14e− reaction) or (VTi-O) for o-xylene oxidation to phthalic anhydride (a 12e−) reaction.
It has been proposed that the selectivity in olefins or oxygenated product depends on the conversion extent (Figure 7) and atomic arrangements around the active transition metal (Mo or V), as shown in Figure 8.
In summary, heterogeneous oxidation reactions are favoured on metal oxides exhibiting high surface flexibility under catalytic conditions without structure collapse, redox properties, i.e., cations with variable oxidation state as V, Mo, Te, Nb, Sb, Fe, etc., high electrons and oxygen anions diffusivity.
7. Total Oxidation Reactions
Transition metal oxides (M = Mn, Co, Fe, Ni) are potential catalysts for application in combustion and depollution processes [68]. Owing to huge improvements in their preparation, perovskites (LnBO3, in which Ln is a lanthanide (usually La) and the B cation possesses two valence states like Mn, Co, or Fe, e.g., La1−xMxMnO3+δ with M = Ce, Sr), spinels, hexaaluminates and some other oxide structures can replace noble metals in a number of processes. Hexaaluminates (ABxAl12−xO19−δ, where Ax+ and By+ are a lanthanide and a transition/noble metal cation, respectively, are formed of alternate layers of alumina spinel blocks and mirror planes in which a large A cation is located [69]).
The use of single oxides for total oxidation (CO, methane, COVs, wet air oxidation), as exemplified in Table 1, Table 2 and Table 3, and for treatment of nitrogen, chlorinated and sulphided compounds (NOx, NH3, urea), is an important topic. In every case, the most probable mechanism (Langmuir–Hinshelwood, Eley–Rideal or Mars–van Krevelen) and the nature of the active sites (Mn+/Mn+1 ion pairs, acid-base sites), as well as the role of reactive oxygen species, are the main parameters. The activity of single oxides for total oxidation is generally linked to the presence of Mn+/M(n+1)+ ion pairs associated with oxygen vacancies, essential for the adsorption or heterolytic splitting of the molecule (CO, hydrocarbons) and for O2 activation. This is why oxides such as Co3O4 or Fe3O4, Mn- and Cu-based oxides, which intrinsically possess two valences of the metal in their structure, are good candidates for oxidation reactions, even in the absence of a noble metal. The density of active sites depends on the crystal face; the oxide shape (nanorods, nanosheets, nanocubes) plays a major role in total oxidation reactions.
Mixed oxides M1M2Ox (perovskites, spinels, hexaaluminates) generally exhibit superior performance owing to the presence of very active M1n+/M2(n+1)+ ion pairs and thanks to the high surface area and improved stability. Redox supports, especially those based on ceria, reinforce the performance of oxide catalysts in oxidation reactions by stabilising the proper Mn+/M(n+1)+ balance required for good activity. Copper- or iron-exchanged zeolites are also used in DeNOx processes with zeolites stable above 900 °C. Hopcalites (CuMnO2 or CoFeOx) are good examples of such catalytic materials. In these materials, M1 and M2 may initially have the same charge but they adapt the valence to the reaction conditions: the reductant-to-oxygen ratio is then a critical parameter for good activity. In certain mixed oxides, the metal M1, not reducible and inactive per se, is able to induce a partial reduction of M2: the active site is a M2n+/M2(n+1)+ ion pair stabilised by the presence of M1. LaMnO3 and LaCoO3 perovskites are remarkable examples of this category of materials. Mn4+ and Mn3+ ions coexist in LaMnO3, while Co3+ and Co2+ ions are present together in LaCoO3. This is why the conventional nomenclature of these perovskites is LaMnO3+δ and LaCoO3−δ respectively, with δ = 0.15 for the manganite and δ = 0.1–0.3 for the cobaltite. Substitution of one or two cations of the binary oxides by other elements is a classic way to improve the activity of the oxide catalysts widely used in oxidation reactions (perovskites, hexaaluminates, spinels). Many studies deal with ternary and quaternary oxides, particularly for perovskite-based materials. In parallel with oxides and mixed oxides prepared by sophisticated methods, waste-derived materials were developed as active oxidation catalysts. A number of industrial waste products (for instance, red mud from the aluminium industry) contain several oxides (iron, manganese, etc.) that are currently used in oxidation.
Shape-structured oxides (nanocubes, nanowires, nanobelts) were widely investigated in recent years. The nature of the exposed faces and crystal defects (vacancies, interstitial atoms, shear planes, electronic defects) can have an effect on the catalyst performance. In most cases, oxygen mobility is a key factor in oxidation processes. 18O/16O exchange between the gas phase and surface or bulk O species has been observed to give useful information about the mechanism of reaction. In the future, more sophisticated oxides (mesoporous and nanocasting, core-shell, shaped-structured), with perfect control of the metal–metal and metal–oxygen interaction, should be developed for new applications, provided that these new oxides have proven their stability at high temperatures. Another method of improvement is to fractionate very stable oxides into nanoparticles with a high surface area by reactive grinding. Finally, new activation processes (plasma, ultrasound) may also be excellent ways of researching these materials.
8. Biomass Transformation Reactions
From the point of view of chemicals and energy resources, utilisation of resources other than fossil resources is important for overcoming environmental issues and for the construction of a sustainable society. One candidate for chemical resources is biomass [73,74], in particular cellulosic biomass. Solid acids and bases should play important roles in some steps of biomass conversions. Actually, the conversion of biomass and biomass-derived products has been studied extensively in recent years. Biodiesel synthesis and utilisation of glycerol are also important topics [75,76], though only one process has been industrialised for biodiesel production on ZnO-Al2O3 catalysts. Note that the solid acids and bases applicable to biomass conversions should be water-tolerant catalysts [77,78].
Currently, acrolein is produced primarily by the gas-phase oxidation of propylene. However, as the use of propylene derivatives, particularly polymers, has recently experienced rapid growth, and since this trend is likely to continue in the foreseeable future, the availability and cost of propylene is going to increase. One approach to substitute propylene relies on the use of new raw materials. For instance, catalysts and processes have been developed for the direct conversion of glycerol to acrolein [79]. However, in such processes catalysts face two major difficulties: (i) there is a more or less rapid deactivation; (ii) their selectivity is capped at an upper limit of less than 80%. There is thus a need for the development of novel routes for the production of acrolein. One approach could involve the conversion of allyl alcohol, which could be produced in an initial step either by fermentation or through the use of processes such as the dehydration of 1,3-propanediol or the dehydration/oxidation of glycerol [80,81].
9. Photocatalysis
Photocatalysis is a relatively young science [82,83,84,85], but is already of great interest for green and environmental chemistries as it operates at room temperature and does not need thermal energy, the catalyst being activated by photons. The technique is able to excite electrons e− from the conduction band or to generate electron holes h+ in the valence band. The catalysts, simple oxides such as TiO2, doped oxides, sulphides, etc., are polyphasic: solid catalyst, liquid or gaseous reactants and electromagnetic (photons from light or UV rays). The first example came from a thermogravimetric study of oxygen chemisorption on evacuated TiO2 [82], which was observed to lead to different weight gain and colour under oxygen, depending on the solar or unsolar day in the laboratory. The same results were obtained by irradiating the sample with UV light. The pioneers took advantage of this labile oxygen O* species to study at room temperature selective oxidation of alkanes CnH2n+2 (with n ≥ 2) to the corresponding aldehydes and extended further to cycloalkanes, alkenes, substituted aromatics, amines, etc. Electrical photoconductivity experiments have shown that oxygen is photoadsorbed as ionic O2− and O− species [86], O− being the precursor of the activated O* species according to O−ads + h+ → O*ads. In the presence of water (gas or liquid), adsorbed H2O molecules dissociate to OH−ads and H+ads and OH−ads + h+ → OHads, radical, which are very nucleophilic and thus non-selective in oxidation reactions. This aspect has led to the use of photocatalysis for pollution abatement of organic compounds such as pesticides, in particular organophosphorous compounds as phenitrothion [(CH3)2-P(=S)-O-C6H3(NO2)-CH3] or dyes.
Among all the semiconductor materials under investigation, metal oxides represent the most important candidates due to their unique physiochemical properties. Producing hydrogen fuel via photocatalytic water splitting on semiconductor materials has attracted intense interest in the past decades due to its potential to address important energy and environmental problems. As the main sector for implementing photocatalytic reactions, semiconductor materials have been the focus of research efforts.
Since the report of using TiO2 for photoelectrochemical water splitting, metal oxide-based semiconductors have received great attention in the field of solar energy conversion. In particular, significant advances have been achieved in using metal oxide semiconductors for photocatalytic water-splitting reactions. The objective is the development of novel metal oxide-based photocatalysts with appreciable light absorption and electronic properties, the tuning of the electronic structures of existing photocatalysts to obtain enhanced light absorption and more efficient charge separation and transportation, and the surface functionalisation of a photocatalyst with co-catalysts to promote surface electrocatalysis. However, there is still a huge gap between the actual solar-to-hydrogen (STH) efficiency achieved and the theoretical maximal value. Currently, the highest STH efficiency achieved on metal oxide photocatalysts is only ca. 1%. This value is far from what is required for any practical application. Therefore, there is still a long way to go before the large-scale application of photocatalytic water splitting for solar energy conversion. In the future, efforts should be devoted to the following aspects. Firstly, the development of metal oxides with wide light absorption and appropriate band edge positions for water splitting, especially those that can absorb light with wavelengths of up to 600–700 nm, is highly desirable. This presents the most challenging issue regarding the use of a metal oxide semiconductor for photocatalytic water splitting. Secondly, the development of new concepts and strategies that can promote charge separation, transportation and surface catalysis is critical for the more efficient utilisation of photogenerated charges. Thirdly, the fundamental issues involved in photocatalytic water splitting reactions need to be clarified with the assistance of new, advanced characterisation techniques such as ultrafast spectroscopy, in situ characterisation and theoretical prediction. These represent significant research opportunities in this highly challenging and exciting field.
The main constraint of the usual photocatalysts, in particular TiO2, is that they mainly absorb UV photons (5% of the solar spectrum). Metal oxides with suitable band gaps for efficient absorption of solar light, such as Fe2O3, have short carrier diffusion lengths and are ineffective at driving photocatalytic reactions. Recently, MIL-101 (Fe) particles have been coated with an amorphous shell of titania and were shown [87] to exhibit hydrogen production from water using visible light, while neither component alone was able to do it.
Practical reduction of carbon dioxide (CO2) requires mediating multielectron and multiproton processes while achieving desired product selectivity in a cost-effective manner. Nanoporous materials, such as KIT-6, MCM-41, US-FAU, MFI, and mesoporous silica, can be used as a support for nanoparticles of metals (Ni, Ru, Rh, Co, Fe) for the catalytic and photocatalytic reduction of CO2 to methane [88]. The role of the support dominates the design in terms of developing an efficient methanation catalyst, specifically with respect to ensuring enhanced metal dispersion and a long catalyst lifetime, because such support can prevent sintering and deactivation through coking, which otherwise blocks the metal surface as carbon accumulates.
Heterogenisation of molecular rhodium catalysts is accomplished via the synthesis, post-synthetic linker exchange, and characterisation of a new metal–organic framework (MOF) Cp*Rh/UiO-67. While the catalytic activities of the homogeneous and heterogeneous systems are found to be comparable, the MOF-based system is more stable and selective. Furthermore, it can be recycled without loss of activity. For formate production, an optimal catalyst loading of ~10% molar Rh incorporation is determined. Increased incorporation of rhodium catalyst favours thermal decomposition of formate into H2. Formate has wide industrial applications and is seen as valuable within fuel cell technologies as well as being an interesting H2-storage compound.
Incorporating photo-sensibilisors as linkers constituting a MOF structure is a particular topic of study at present. Some porphyrine- or anthracene-based MOFs are able to reduce CO2 under irradiation. Other strategies aim at improving their photostability by incorporating an active ligand, known to favour the durability and recyclability of solids, in interpenetrated frameworks. In addition to new MOFs with intrinsic photocatalytic properties, MOFs with multiple ligands bearing different functionalities and spatially controlled are conceived.
10. Industrial Applications of Metal Oxides
Industrial applications of metal oxide catalysts [89] mainly cover the synthesis of petrochemicals (mainly, chemical intermediates such as acrylonitrile, maleic anhydride, acrolein, acrylic acid and substituted aromatic compounds) [90], in which V oxides appear as key elements and for the manufacture of fuels. Many processes have been developed, as summarised in Table 4.
Currently, most of the acrylic acid produced world-wide is made using the same process, namely the two-stage oxidation of propylene. In this process, propylene is oxidised to acrolein in a first reactor on scheelite Bi molybdate-based catalyst (defect structure of scheelite (Bi2/3□1/3Mo3O12 where □ represents a cation vacancy in the ideal ABO4 structure), containing Co and Fe molybdates and acrolein is further oxidised to acrylic acid in a second reactor without intermediate separation. The second stage catalysts are MoVWCu-O mixed oxides as key formulation. These catalysts are mostly X-ray amorphous and contain V in a reduced state.
It was observed that an oxide catalyst consisting of molybdenum, vanadium and niobium was uniquely capable of high conversion of ethane and high selectivity to both ethylene and acetic acid products. Furthermore, the work identified two defect oxide structures of molybdenum and vanadium as the likely catalysts for the selective oxidation of ethane. These two phases are uniquely described by the stoichiometry Mo4V6O25 and Mo6V9O40 with the inclusion of niobium to stabilise the structure and promote microcrystallinity.
Propane direct oxidation to acrylic acid has been studied [91] on mixed oxides such as MoVTe(Sb)Nb-O, as well as the dehydration of lactic acid and 3-hydroxypropionic, but has not been commercialised yet. Recently, a key invention [92] was the calcination of the mixed oxide, which was previously done in air at around 350 °C, in a non-oxygen containing (i.e., inert) atmosphere (nitrogen gas) at around 620 °C. This resulted in acrylonitrile yields of up to 59% (92% propane conversion; 64% acrylonitrile selectivity), which made the system competitive with the previous processes starting from propene. The active phase was identified as the so-called M1 phase [93]. Acrylonitrile hydrolysis, as well as the Reppe chemistry to get acrylic acid, have been used in the past, but are no longer economically attractive. However, direct ammoxidation of propane to acrylonitrile on the same MoVTe(Sb)Nb-O catalysts has been successively industrialised. Among the disclosed compositions is Mo1V0.4Te0.2Nb0.1Ox with a reported per pass yield of acrylonitrile of only around 14% (23% propane conversion; 61.5% acrylonitrile selectivity). Yields were improved up to 25% (58% propane conversion; 44% acrylonitrile selectivity) with the addition of various elements as promoters. Recently, glycerol dehydration to acrolein has received much attention as a potential route to renewable acrylic acid. A shift to new reactor technologies, such as a microstructured reactor or thermoplate reactors, better able to handle the reaction heat, could trigger the need for more active catalysts. However, currently incremental improvements of the catalyst and process are to be expected, since all the installed capacities are using the same type of technology: multitubular fixed bed reactors operating slightly above the upper flammability limit, over MoVWCu-mixed oxide catalysts.
As the nitrile functionality serves as a useful reactive centre for transformation into a wide spectrum of commercially valuable chemical products via polymerisation, hydrogenation, hydration and condensation, the production of aromatic nitriles using mixed metal oxide catalysts as chemical intermediates is widespread in commercial practice. Commercial application of alkyl aromatic ammoxidation is based on three categories of feedstocks—toluene, xylenes, and picolines (alkyl pyridines). Catalysts for alkyl aromatic ammoxidation are based on vanadium oxide. The ammoxidation reaction produces the corresponding aromatic nitrile. The nitrile products are intermediates for industrially important chemicals including amines, isocyanates for polyurethanes, phthalocyanine dyes and vitamin B compounds (niacin).
An interesting case is the upgrading of methane by oxidative coupling (OCM), which has led to many studies since the early 1970s by Lunsford on Li/MgO-based catalysts. Recently, Siluria [94] has developed a process to convert methane to ethylene by OCM. The catalyst is prepared with rare earth oxides, either unsupported or supported on MgO, CaO or AlPO4 and containing at least two dopants (a metal element, a semi-metal element and a non-metal element). A demonstration plant has been set up in La Porte, Texas and a commercial plant is scheduled for 2019.
11. Concluding Remarks
Advances in catalysis with metal oxides have allowed for the development of catalysts with high selectivity. To obtain high selectivity, new preparation approaches have been developed. Catalysts have been seen to greatly ameliorate catalytic performance over the last 40 years and should continue to be used, as 1% improvement in selectivity is known to lead to important savings and further improvements are becoming more and more challenging.
It has been considered above that the performance of a catalyst relies on its grain size, shape, chemical composition, preparation and activation. New strategies for the preparation of metal oxide catalysts have resulted in the development of catalysts with single sites that give 100% selectivity, such single sites being atoms or ensembles of atoms. Nanocrystals with sizes of only a few nm present the best catalytic efficiencies, yet their supports are very important for the synergistic activation of substrates, electron conductivity enhancement in redox reactions and thermal conductivity enhancement for exothermal reactions. The reduction of size to the nanoscale has prompted many studies over the past 30 years. The activity, selectivity, resistance to deactivation and possibility of regeneration are the main properties that characterise the usefulness of catalysts, which have been improved by advances in catalysis with metal oxides.
Advances in the development and application of metal oxide catalysts are driven by several factors:
- A need to focus on economics. Considerable RD should be focused on technology that uses ethane and natural gas as feedstock, as well as methanol and derivatives. In this regard, the search for energy improvement will continue, as it has in the past, even with low energy and oil prices.
- A need for greenhouse gas (GHG) reduction. Greenhouse gases can be reduced by several methods. Enhanced oil recovery (EOR) using CO2 is the least expensive way, but with the drop in the price of crude oil we are seeing less of it. Of course, this could change if the price of crude oil goes back up. Carbon capture and sequestration (CCS) is the next alternative and considerable work is being done in this area.
- The conversion of CO2 to chemicals. Catalytically transforming carbon dioxide to chemicals is challenging because of thermodynamic limitations—more energy may be required to produce these chemicals than from other feedstock, and if this causes more CO2 to be released into the atmosphere, it does not serve the purpose. However, there is an elegance to pursuing this route and we expect there to be considerable research in this area. Research projects are underway at several universities and industry-sponsored institutions. Among the most interesting are those that are developing catalyst systems and use sunlight as a source for at least part of the energy requirements.
- The use of biomass as feedstock. The catalytic conversion of biomass has gained tremendous attention in recent years as this utilises a renewable material and biomass as it grows, absorbing CO2 from the atmosphere. There is a tremendous amount of RD in this area and we would expect that many chemicals will ultimately be produced from bio-based technology. In this context, it will be important to focus on utilising non-edible biomass. Furthermore, research is likely to shift towards using catalytic processes rather than enzymatic processes, since reaction rates with enzymes are typically quite slow.
- With the success achieved by Siluria with their oxidative coupling technology, and the ability to produce ethylene from methane at a very low cost (as long as natural gas remains cheap), there will continue to be a strong emphasis on conducting increased RD in this area.
- Meanwhile, since so many chemicals are currently produced from syngas, particularly in China, there will also be continued interest and increasing RD in this area.
- Low alkane production from shale has modified oil needs and prices in the past 10 years, but may not last very long.
Let us take a few examples. For instance, one-step conversion of syngas to aromatics [95] with striking selectivity was achieved on Zn/ZrO2 nanoparticles and a H-MFI catalyst showed 80% aromatic selectivity at CO conversion of 20% at 673 K, with a selectivity in undesirable methane less than 3% in one step and 1000 h stability. Zn/Zr ratio, strong acidity and the proximity of catalyst components have been shown to be key factors. The fraction of benzene–toluene–xylenes (BTX) can be tuned to 60% by selective passivation of external Brønsted acidity of H-MFI. Methanol and dimethyl ether were formed as reaction intermediates, which were subsequently transformed into aromatics on H-MFI via olefins. This is a highly selective and stable non-petroleum route for the synthesis of aromatics as schematised in Figure 9.
Another example is found for liquid-phase selective oxidation catalysis with metal–organic frameworks, as shown in Figure 10.
As a general conclusion about the choice of a catalyst, one may say that a “good” catalyst must possess both high activity and long-term stability, but its single most important attribute is selectivity, which is more important than activity, as the unconverted reactants can be re-circulated while the selective transformation of reactants facilitates the final separation process. Many new processes and new catalysts have been developed in the second part of the 20th century. Since then, few new processes and catalysts have been discovered. On the other hand, during this period, huge improvements have been made with known reactions and known catalysts by studying and improving catalyst preparation, activation and characterisation, and catalytic reactor design. For the future, with the growing importance of biomass, durable raw materials and raw chemicals, new processes have already started to appear. Yet there is still plenty of room for improvement and discovering of new catalytic reactions, as in photocatalysis for water splitting as hydrogen source, membranes reactors, chemical engineering, etc. A recent example is given in Figure 9 to illustrate how new catalysts may lead to interesting catalytic reactions, while Figure 11 summarises the actual research trends in in the catalysis field.
When looking at the historic aspect of catalysis, it has appeared that catalysts have been used in the 20th century for bulk basic, industrial and inorganic chemicals and in the mid-20th century for petroleum industry and transportation fuels. In the 1990s, we saw the decline of long-term industrial research in many companies, except in some major petroleum ones, which then relied on large research centres, catalyst producers, institutes or academic groups to discover new catalysts and technologies. Moreover, the locus of research activity on catalysis has moved from Europe in the 19th–20th centuries to the USA in the 20th century and now in the 21st century to Asia (Japan, Korea and China). Today, we see a new evolution of alternative fuel and new raw materials based on biomass [97,98], water splitting, etc. It appears that although great improvements and new developments were observed in the second part of the 20th century, there remains plenty of room for young researchers from academia and industry to discover and develop new metal oxide catalysts, new preparation procedures, and new or improved processes. It can be expected that, in the first part of the 21st century, we will see the development of new catalysts and commercial processes related to environmental issues under legislation constraints. Major possibilities reside in: (i) the dehydrogenation of light alkanes (C1–C5); (ii) methanol synthesis; (iii) formaldehyde synthesis (mainly from methane); and (iv) the oxidative dehydrogenation of light alkanes (C1–C5).
Conflicts of Interest
The author declares no conflict of interest.
References
- Bond, G.C. Heterogeneous Catalysis: Principles and Applications; Oxford University Press: Oxford, UK, 1974. [Google Scholar]
- Thomas, J.M.; Thomas, W.J. (Eds.) Principles and Practice of Heterogeneous Catalysis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Ertl, G.; Knözinger, H.; Schüth, F.; Weitkamp, J. (Eds.) Handbook of Heterogeneous Catalysis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Pawelec, B. Surface processes and composition of metal oxide surfaces. In Metal Oxides, Chemistry and Applications; Fierro, J.L.G., Ed.; CRC Taylor Francis: Boca Raton, FL, USA, 2006; pp. 111–193. [Google Scholar]
- Védrine, J.C. (Ed.) Heterogeneous Catalysis by Metal Oxides; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Collucia, S.; Tench, A.J. Spectroscopic studies of hydrogen adsorption on highly dispersed MgO. Stud. Surf. Sci. Catal. 1981, 7, 1154–1169. [Google Scholar]
- Chizallet, C.; Costentin, G.; Che, M.; Delbecq, F.; Sautet, P. Revisiting Acido-basicity of the MgO surface by periodic density functional theory calculations: Role of surface topology and ion coordination on water dissociation. J. Phys. Chem. B 2006, 110, 15878–15886. [Google Scholar] [CrossRef] [PubMed]
- Thursfield, A.; Melcalfe, I.S.; Kruth, A.; Irvine, J.T.S. Defect chemistry and transport in metal oxides. In Metal Oxides Chemistry and Applications; Fierro, J.L.G., Ed.; CRC Taylor Francis: Boca Raton, FL, USA, 2006; pp. 55–85. [Google Scholar]
- Getsoian, A.B.; Zhai, Z.; Bell, A. Band-gap energy as a descriptor of catalytic activity for propene oxidation over mixed metal oxide catalysts. J. Am. Chem. Soc. 2014, 136, 13684–13697. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S. A theory of the catalytic surface. Proc. R. Soc. Lond. 1925, A108, 105–111. [Google Scholar] [CrossRef]
- Boudart, M. Catalysis by supported metals. Adv. Catal. 1969, 20, 153–167. [Google Scholar]
- Boudart, M. Catalysis by supported metals. In Proceedings of the 6th International Congress on Catalysis, London, UK, 12–16 July 1976; Bond, G.C., Wells, P.B., Tompkins, F.C., Eds.; The Chemical Society: London, UK, 1977; pp. 1–9. [Google Scholar]
- Grasselli, R.K. Site isolation and phase cooperation: Two important concepts in selective oxidation catalysis: A retrospective. Catal. Today 2014, 238, 10–27. [Google Scholar] [CrossRef]
- Bordes, E.; Courtine, P. Influence of structural properties of catalysts at various stages of selective oxidation: From catalyst preparation to catalytic reactors. Top. Catal. 2000, 11, 61–65. [Google Scholar] [CrossRef]
- Ziółkowski, J.; Bordes, E.; Courtine, P. Dynamic description of the oxidation of n-butane on various faces of (VO)2P2O7 in terms of the crystallochemical model of active sites. J. Mol. Catal. 1993, 84, 307–326. [Google Scholar] [CrossRef]
- Volta, J.C.; Desquesnes, W.; Moraweck, B.; Coudurier, G. A new method to obtain supported oriented oxides: MoO3 graphite catalyst in propylene oxidation to acrolein. React. Kinet. Catal. Lett. 1979, 12, 241–245. [Google Scholar] [CrossRef]
- Volta, J.C.; Portefaix, J.L. Structure sensitivity of mild oxidation reactions on oxide catalysts: A review. Appl. Catal. 1985, 18, 1–32. [Google Scholar] [CrossRef]
- Haber, J. The concept of structure sensitivity in catalysis by oxides. Stud. Surf. Sci. Catal. 1989, 48, 447–467. [Google Scholar]
- Balandin, A.A. The nature of active centers and the kinetics of catalytic dehydrogenation. Adv. Catal. 1958, 10, 96–121. [Google Scholar]
- Ostrovski, V.E. Oscillation theory of heterogeneous catalysis and its use for identification of the reaction scheme and kinetics: Catalytic liquid-phase benzene-ring hydrogenation as an example. J. Sci. Isr. Technol. Adv. 2012, 14, 57–79. [Google Scholar]
- Hinshelwood, C.N. The Kinetics of Chemical Change; Clarendon: Oxford, UK, 1940. [Google Scholar]
- Védrine, J.C. Revisiting active sites in heterogeneous catalysis: Their structure and dynamical behaviour. Appl. Catal. A 2014, 474, 45–50. [Google Scholar] [CrossRef]
- Hävecker, M.; Wrabetz, S.; Kröhnert, J.; Csepei, L.I.; d’Alnoncourt, R.N.; Kolen’ko, Y.V.; Girgsdies, F.; Schlögl, R.; Trunschke, A. Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid. J. Catal. 2012, 285, 48–60. [Google Scholar] [CrossRef]
- Thomas, J.M. The societal significance of catalysis and the growing practical importance of single-site heterogeneous catalysts. Proc. R. Soc. A 2012, 468, 1884–1903. [Google Scholar] [CrossRef]
- Mehlomakulu, B.; Nguyen, T.T.N.; Delichère, P.; van Teen, E.; Millet, J.M.M. Identification of the active species in oxidation reactions on mixed oxide catalysts: Supra-surface or bulk-surface species. J. Catal. 2012, 289, 1–10. [Google Scholar] [CrossRef]
- Schlögl, R. Concepts in selective oxidation of small alkane molecules. In Modern Heterogeneous Oxidation Catalysis; Mizuno, N., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–42. [Google Scholar]
- Pantazidis, A.; Burrows, A.; Kiely, C.J.; Mirodatos, C. Direct evidence of active surface reconstruction during oxidative dehydrogenation of propane over VMgO catalyst. J. Catal. 1998, 177, 325–334. [Google Scholar] [CrossRef]
- Campbell, C.T.; Sauer, J. Surface chemistry of oxides. Chem. Rev. 2013, 113, 3859–4565. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Tosoni, S.; Illas, F. Theoretical approaches to excited related states phenomena in oxide surfaces. Chem. Rev. 2013, 113, 4456–4495. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Concepción, P. Combined theoretical and spectroscopic mechanistic studies for improving activity and selectivity in heterogeneous catalysis. Catal. Today 2017, 285, 166–178. [Google Scholar] [CrossRef]
- Akimov, A.V.; Neukirch, A.J.; Prezhdo, O.V. Theoretical insights into photoinduced charge transfer and catalysis at oxide interfaces. Chem. Rev. 2013, 113, 4496–4565. [Google Scholar] [CrossRef] [PubMed]
- Szalay, P.G.; Müller, T.; Gidofalvi, G.; Lischka, H.; Shepard, R. Multiconfiguration self-consistent field and multireference configuration interaction methods and applications. Chem. Rev. 2012, 112, 108–181. [Google Scholar] [CrossRef] [PubMed]
- Austin, B.M.; Zubarevand, D.Y.; Lester, W.A., Jr. Quantum Monte Carlo and related approaches. Chem. Rev. 2012, 112, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Pacchioni, G. Modelling doped and defective oxides in catalysis by DFT methods: Room for improvements. J. Chem. Phys. 2008, 128, 182505. [Google Scholar] [CrossRef] [PubMed]
- Mota, C.J.A.; Bhering, D.J.; Rosenbach, N. A DFT study of the acidity of ultrastable Y zeolite: Where is the Brønsted/Lewis acid synergism. Angew. Chem. Int. Ed. 2004, 43, 3050–3053. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-Consistent equations including exchange and correlation effects. Phys. Rev. 1965, A140, 1133–1138. [Google Scholar] [CrossRef]
- Gerosa, M.; Bottani, C.E.; Caramella, L.; Onida, G.; di Valentin, C.; Pacchioni, G. Defect calculations in semiconductors through a dielectric-dependent functional: The case of oxygen vacancies in metal oxides hybrid DFT. J. Chem. Phys. 2015, 143, 134702. [Google Scholar] [CrossRef] [PubMed]
- Metiu, H. Preface to special topic: A survey of some new developments in heterogeneous catalysis. J. Chem. Phys. 2008, 128, 182501. [Google Scholar] [CrossRef] [PubMed]
- Le Page, J.-F. Applied Heterogeneous Catalysis: Design-Manufacture-Use of Solid Catalysts; Technip: Paris, France, 1987. [Google Scholar]
- Perego, C.; Villa, P. Catalyst preparation methods. Catal. Today 1997, 34, 281–305. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Védrine, J.C. Catalyst preparation. In Basic Principles in Applied Catalysis; Baerns, M., Ed.; Springer Series in Chemical Physics #75; Springer: Berlin, Germany, 2004; pp. 215–258. [Google Scholar]
- Regalbuto, J.R. Catalyst Preparation Science and Engineering; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Lambert, J.F.; Che, M. The molecular approach to supported catalysts synthesis: State of the art and future challenges. J. Mol. Catal. A 2000, 162, 5–18. [Google Scholar] [CrossRef]
- Fechete, I.; Védrine, J.C. Nano-oxide mesoporous catalysts in heterogeneous catalysis. In Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection; Sels, B., Van de Voorde, M., Eds.; Wiley-VCH: Weinheim, Germany, 2017; pp. 50–89. [Google Scholar]
- Hattori, H.; Ono, Y. Solid Acid Catalysis; Pan Stanford Publishing: Singapore, 2015. [Google Scholar]
- Ono, Y.; Hattori, H. Solid Base Catalysis; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Hattori, H.; Ono, Y. Catalysts and catalysis for acid-base reactions. In Heterogeneous Catalysis by Metal Oxides; Chapter 4; Védrine, J.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Sheldon, R.A. The E factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Védrine, J.C. Acid base characterisation of heterogeneous catalysts: An up to date overview. Res. Chem. Intermed. 2015, 41, 9387–9423. [Google Scholar] [CrossRef]
- Niwa, M.; Katada, N.; Okumura, K. Characterization and Design of Zeolite Catalysts; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Cvetanovic, R.J.; Amenomiya, Y. Application of a temperature-programmed desorption technique to catalyst studies. Adv. Catal. 1967, 17, 103–149. [Google Scholar]
- Hunger, M. Brønsted acid sites in zeolites characterized by multi nuclear solid state NMR spectroscopy. Catal. Rev. 1997, 39, 345–393. [Google Scholar] [CrossRef]
- Rakiewic, E.F.; Peters, A.W.; Wormsbecher, R.F.; Sutovich, K.J.; Mueller, K.T. Characterization of acid sites in zeolitic and other inorganic systems using solid-state 31P NMR of the probe molecule trimethylphosphine oxide. J. Phys. Chem. B 1998, 102, 2890–2896. [Google Scholar] [CrossRef]
- Centi, G.; Cavani, F.; Trifiró, F. Selective Oxidation by Heterogeneous Catalysis; Kluwer Academic: New York, NY, USA, 2001. [Google Scholar]
- Ivars, F.; Nieto, J.M.L. Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry; Cavani, F., Duprez, D., Eds.; Imperial College Press: London, UK, 2014; pp. 767–833. [Google Scholar]
- Blasco, T.; Lopéz Nieto, J.M. Oxidative dehydrogenation of short chain alkanes on supported vanadium oxides catalysts. Appl. Catal. A: Gen. 1997, 157, 117–142. [Google Scholar]
- Chieregato, A.; Nieto, J.M.L.; Cavani, F. Mixed-oxide catalysts with vanadium as the key element for gas-phase reactions. Coord. Chem. Rev. 2015, 301–302, 3–23. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Burrington, J.D.; Buttrey, D.J.; DeSanto, P.; Lugmair, C.G.; Volpe, A.F.; Weingand, T. Multifunctionality of active centers in (amm)oxidation catalysts: From Bi-Mo-Ox to Mo-V-Nb–(Te, Sb)-Ox. Top. Catal. 2003, 23, 5–22. [Google Scholar] [CrossRef]
- Cant, N.W.; Hall, W.K. Catalytic oxidation: VI. Oxidation of labeled olefins over silver. J. Catal. 1978, 52, 81–94. [Google Scholar]
- Védrine, J.C. Heterogeneous partial (amm)oxidation and oxidative dehydrogenation catalysis on mixed metal oxides. Catalysts 2016, 6, 1–26. [Google Scholar] [CrossRef]
- Callahan, J.L.; Grasselli, R.K. A selectivity factor in vapor-phase hydrocarbon oxidation catalysis. AIChE J. 1963, 9, 755–760. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–57. [Google Scholar] [CrossRef]
- Védrine, J.C.; Fechete, I. Heterogeneous partial oxidation catalysis on metal oxides. CR Chim. 2016, 19, 1203–1225. [Google Scholar] [CrossRef]
- Védrine, J.C. Heterogeneous catalytic partial oxidation of lower alkanes (C1–C6) on mixed metal oxides. J. Energy Chem. 2016, 25, 936–946. [Google Scholar] [CrossRef]
- Ushikubo, T.; Oshima, K.; Kayou, A.; Hatano, M. Ammoxidation of propane over Mo-V-Nb-Te mixed oxide catalysts. Stud. Surf. Sci. Catal. 1997, 112, 473–480. [Google Scholar]
- Ushikubo, T. Activation of propane and butanes over niobium- and tantalum-based oxide catalysts. Catal. Today 2003, 78, 79–84. [Google Scholar] [CrossRef]
- Shiju, N.R.; Guliants, V. Recent developments in catalysis using nanostructured materials. Appl. Catal. A 2009, 356, 1–17. [Google Scholar] [CrossRef]
- Bion, N.; Can, F.; Courtois, X.; Duprez, D. Transition metal oxides for combustion and depollution processes. Chapter 6. In Heterogeneous Catalysis by Metal Oxides; Védrine, J.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Iyi, N.; Takekawa, S.; Kimura, S. Crystal chemistry of hexaaluminates: β-alumina and magnetoplumbite structures. J. Solid State Chem. 1989, 83, 8–19. [Google Scholar] [CrossRef]
- García, T.; Solsona, B.; Taylor, S.H. Naphthalene total oxidation over metal oxide catalysts. Appl. Catal. B 2006, 66, 92–99. [Google Scholar] [CrossRef]
- Assebban, M.; El Kasmi, A.; Harti, S.; Chafik, T. Intrinsic catalytic properties of extruded clay honeycomb monolith toward complete oxidation of air pollutants. J. Hazard. Mater. 2015, 300, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Neeft, J.P.A.; Makkee, M.; Moulijn, J.A. Catalytic oxidation of carbon black—1. Activity of catalysts and classification of oxidation profiles. Fuel 1998, 77, 111–119. [Google Scholar] [CrossRef]
- Corma, A.; Renz, M.; Susante, H. Transformation of biomass products into fine chemicals catalyzed by solid Lewis and Brønsted acid. Top. Catal. 2009, 52, 1182–1189. [Google Scholar] [CrossRef]
- Kubička, D.; Kubičova, I.; Čejka, J. Application of molecular sieves of biomass and biomass-derived feedstocks. Catal. Rev. Sci. Eng. 2013, 55, 1–78. [Google Scholar] [CrossRef]
- Chouhan, A.P.S.; Sarma, A.K. Modern heterogeneous catalysts for biodiesel production: A comprehensive review. Renew. Sustain. Energy Rev. 2011, 15, 4378–4399. [Google Scholar] [CrossRef]
- Sharma, Y.C.; Singh, B.; Korstad, J. Latest developments on application of heterogeneous basic catalysts for an efficient and eco-friendly synthesis of biodiesel: A review. Fuel 2011, 90, 1309–1324. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant acid catalysts. Chem. Rev. 2002, 46, 3641–3666. [Google Scholar] [CrossRef]
- Noma, R.; Nakajima, K.; Kamata, K.; Kitano, M.; Hayashi, S.; Hara, M. Formation of 5-(hydroxymethyl)furfural by stepwise dehydration over TiO2 with water-tolerant Lewis acid sites. J. Mol. Catal. A 2014, 387–389, 100–105. [Google Scholar] [CrossRef]
- Lauriol-Garbey, P.; Millet, J.M.M.; Loridant, S.; Bellière-Baca, V.; Rey, P. New efficient and long-life catalyst for gas-phase glycerol dehydration to acrolein. J. Catal. 2011, 280, 68–76. [Google Scholar] [CrossRef]
- Sato, S.; Takahashi, R.; Sodesawa, T.; Honda, N.; Shimizu, H. Selective dehydration of diols to allylic alcohols catalyzed by ceria. Catal. Commun. 2003, 4, 77–81. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y. Highly efficient process for the conversion of glycerol to acrylic acid via gas phase catalytic oxidation of an allyl alcohol intermediate. ACS Catal. 2016, 6, 143–150. [Google Scholar] [CrossRef]
- Formenti, M.; Juillet, F.; Teichner, S.J. Photo-oxydation ménagée de paraffines et oléfines sur anatase à température ambiante. CR Acad. Sci. 1970, 270, 138–141. [Google Scholar]
- Herrmann, J.M. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129. [Google Scholar] [CrossRef]
- Linsebigler, A.L.; Lu, G.Q.; Yates, J.T. Photocatalysis on TiO2 surfaces: Principles, mechanims, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Serpone, N.; Sauve, G.; Koch, R.; Tahiri, H.; Pichat, P.; Piccinini, P.; Pelizzetti, E.; Hidaka, H. Standardization protocol of process efficiencies and activation parameters in heterogeneous photocatalysis: Relative photonic efficiencies ζr. J. Photochem. Photobiol. A 1996, 94, 191–203. [Google Scholar] [CrossRef]
- Herrmann, J.M.; Disdier, J.; Pichat, P. Oxygen species ionosorbed on powder photocatalyst oxides from room temperature photoconductivity as a function of oxygen pressure. J. Chem. Soc. Faraday Trans. I 1981, 77, 2815–2826. [Google Scholar] [CrossRef]
- De Krafft, K.E.; Wang, C.; Lin, W. Metal organic framework templated synthesis of Fe2O3/TiO2 nanocomposite for hydrogen production. Adv. Mater. 2102, 24, 2014–2118. [Google Scholar] [CrossRef] [PubMed]
- Fechete, I.; Védrine, J.C. Nanoporous materials as new engineered catalysts for the Synthesis of green fuels. Molecules 2015, 20, 5638–5666. [Google Scholar] [CrossRef] [PubMed]
- Degnan, T.F. Industrial applications of metal oxide catalysts an impressive past and a promising future Chapter 8–3. In Heterogenous Catalysis by Metal Oxides; Védrine, J.V., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Brazdil, J.F. Selective oxidation in industry: Applications of metal oxides in the petrochemical industry, Chapter 8–2. In Heterogenous Catalysis by Metal Oxides; Védrine, J.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Dubois, J.L.; Patience, G.S.; Millet, J.M.M. Propane selective oxidation to acrylic acid. In Nanotechnology in Catalysis, Applications in the Chemical Industry, Energy Development and Environmental Protection; Sels, B., van de Voorde, M., Eds.; Wiley-VCH: Weinheim, Germany, 2017; pp. 503–536. [Google Scholar]
- Ushikubo, T.; Oshima, K.; Umezawa, T.; Kiyono, K.I. Process for Producing Nitriles. US Patent 5231214A, 27 July 1993. [Google Scholar]
- Aouine, M.; Dubois, J.L.; Millet, J.M.M. Crystal chemistry and phase composition of the MoVTeNbO catalysts for the ammoxidation of propane. Chem. Commun. 2001, 13, 1180–1181. [Google Scholar] [CrossRef]
- Siluria Technologies. Siluria Company Overview and Linde. Available online: http://siluria.com/about/company_overview (accessed on 1 November 2017).
- Cheng, K.; Zhou, W.; Kang, J.; He, S.; Shi, S.; Zhang, Q.; Pan, Y.; Wen, W.; Wang, Y. Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability. Chem 2017, 3, 334–347. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Liquid-phase selective oxidation catalysis with metal-organic frameworks. Catal. Today 2016, 278, 22–29. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Thomas, H.J.; Climent, M.J.; Romanelli, G.P.; Iborra, S. Heteropoly compounds as catalysts for biomass product transformations. Catal. Rev. Sci. Eng. 2016, 58, 497–586. [Google Scholar] [CrossRef]
- Ampelli, C.; Centi, G.; Genovese, C.; Papanikolaou, G.; Pizzi, R.; Perathoner, S.; van Putten, R.-J.; Schouten, K.J.P.; Gluhoi, A.C.; van der Waal, J.C. A comparative catalyst evaluation for the selective oxidative esterification of furfural. Top. Catal. 2016, 59, 1659–1667. [Google Scholar] [CrossRef]
Figure 1.
Creation of a donor band below the conduction band CB (left) (exciting the electron necessitates ED) and acceptor band (exciting the electron necessitates EA) above the valence band VB (right) upon doping. EG = energy gap.
Figure 1.
Creation of a donor band below the conduction band CB (left) (exciting the electron necessitates ED) and acceptor band (exciting the electron necessitates EA) above the valence band VB (right) upon doping. EG = energy gap.

Figure 2.
Site isolation by structure and composition: schematic of (VO)2P2O7 surface structure. Facile exchange of surface oxygens exists within domains (represented by arrows), but not between domains. Site isolation established between domains by picket fence of pyrophosphate groups posing oxygen diffusion barriers. Schematic representation of a surface structure of a type of polytype of (VO)2P2O7. The arrows represent the facile pathways for surface oxygen mobility. This illustrates the site isolation principle by surface P2O7 entities groups that constitute a barrier to oxygen diffusion. Reproduced with permission from [13]. Copyright Elsevier, 2014.
Figure 2.
Site isolation by structure and composition: schematic of (VO)2P2O7 surface structure. Facile exchange of surface oxygens exists within domains (represented by arrows), but not between domains. Site isolation established between domains by picket fence of pyrophosphate groups posing oxygen diffusion barriers. Schematic representation of a surface structure of a type of polytype of (VO)2P2O7. The arrows represent the facile pathways for surface oxygen mobility. This illustrates the site isolation principle by surface P2O7 entities groups that constitute a barrier to oxygen diffusion. Reproduced with permission from [13]. Copyright Elsevier, 2014.

Figure 3.
Adsorption of n-butane on (100) and (021) (VO)2P2O7. Anchoring of butane on (100) and one possible way of adsorption of butane on (021). Medium shaded and small black circles are C and H, respectively (carbon, oxygens and lattice at scale). Reproduced with permission from [14]. Copyright Springer, 2000.
Figure 3.
Adsorption of n-butane on (100) and (021) (VO)2P2O7. Anchoring of butane on (100) and one possible way of adsorption of butane on (021). Medium shaded and small black circles are C and H, respectively (carbon, oxygens and lattice at scale). Reproduced with permission from [14]. Copyright Springer, 2000.

Figure 4.
(A) Adsorption of propene on MoVTeNb oxide (so called M1 phase) before (filled circles) and after propane oxidation (open circles; reaction conditions of propane oxidation: 400 °C, C3/O2/H2O/N2 = 3/6/40/51); (B) adsorption of propane on activated (fresh) MoVTeNb oxide (M1 phase), (filled squares) and after adsorption and subsequent desorption of propene at 40 °C (open triangles). The differential heat of adsorption is plotted as a function of coverage normalised to the specific surface area. Reproduced with permission from [23]. Copyright Elsevier, 2012.
Figure 4.
(A) Adsorption of propene on MoVTeNb oxide (so called M1 phase) before (filled circles) and after propane oxidation (open circles; reaction conditions of propane oxidation: 400 °C, C3/O2/H2O/N2 = 3/6/40/51); (B) adsorption of propane on activated (fresh) MoVTeNb oxide (M1 phase), (filled squares) and after adsorption and subsequent desorption of propene at 40 °C (open triangles). The differential heat of adsorption is plotted as a function of coverage normalised to the specific surface area. Reproduced with permission from [23]. Copyright Elsevier, 2012.

Figure 5.
Bulk ab planes of M1 phase (right). Refined orthogonal structure had the space group Pba2, formula unit Mo7.8V1.2NbTe0.94O28.9 (M1), and lattice parameters, a = 2.1134 nm, b = 2.6658 nm, and c = 0.40146 nm with z = 4, from [67]. Active site: 2 × 2 unit cell structure model of M1 in [001] projection showing four isolated catalytically active centres (left). The active sites are isolated from each other by four Nb-bipyramids that are surrounded by five MO6 octahedra. Reproduced with permission from [58]. Copyright Springer, 2003.
Figure 5.
Bulk ab planes of M1 phase (right). Refined orthogonal structure had the space group Pba2, formula unit Mo7.8V1.2NbTe0.94O28.9 (M1), and lattice parameters, a = 2.1134 nm, b = 2.6658 nm, and c = 0.40146 nm with z = 4, from [67]. Active site: 2 × 2 unit cell structure model of M1 in [001] projection showing four isolated catalytically active centres (left). The active sites are isolated from each other by four Nb-bipyramids that are surrounded by five MO6 octahedra. Reproduced with permission from [58]. Copyright Springer, 2003.

Figure 6.
Structures (composed of MO6 octahedra) of the most studied catalysts in alkane partial oxidation: (a) vanadyl pyrophosphate (VPO); (b) VSbO rutile phase; (c) M1 phase, MoVTe(Sb)NbO; and (d) Keggin molybdophophoric acid.
Figure 6.
Structures (composed of MO6 octahedra) of the most studied catalysts in alkane partial oxidation: (a) vanadyl pyrophosphate (VPO); (b) VSbO rutile phase; (c) M1 phase, MoVTe(Sb)NbO; and (d) Keggin molybdophophoric acid.

Figure 7.
Selectivity to the main partial oxidation product achieved at low conversion (XT = 2%) and high conversion (XT = 20%) during propane oxidation at 400 °C on VOx/Al2O3; Mo-V-Nb mixed oxides calcined in air at 450 °C (amorphous); Mo-V-Te-Nb mixed oxides heat-treated at 600 °C in N2 presenting M1 phase (MoVTeNb-O). The main adsorbed species observed during the adsorption of propylene on each catalyst is shown. Reproduced with permission from Figure 24.4 in [55]. Copyright Imperial College Press, 2014.
Figure 7.
Selectivity to the main partial oxidation product achieved at low conversion (XT = 2%) and high conversion (XT = 20%) during propane oxidation at 400 °C on VOx/Al2O3; Mo-V-Nb mixed oxides calcined in air at 450 °C (amorphous); Mo-V-Te-Nb mixed oxides heat-treated at 600 °C in N2 presenting M1 phase (MoVTeNb-O). The main adsorbed species observed during the adsorption of propylene on each catalyst is shown. Reproduced with permission from Figure 24.4 in [55]. Copyright Imperial College Press, 2014.

Figure 8.
Importance of the V-structure in V-catalysts for H-abstraction and O-insertion. Reproduced with permission from Figure 24.3 in [55] copyright Imperial College Press 2014 and adapted from table 5 in [56] Copyright Elsevier, 1997.

Figure 9.
Typical reaction processes related to syngas chemistry (Reproduced with permission from [95]. Copyright Elsevier, 2017). The red-line route demonstrates the SMA process for the direct synthesis of aromatics via reaction coupling. FT = Fischer Tropsch, MTO = methanol to olefins, MTA = methanol to aromatics, SMA = syngas-methanol-aromatics.
Figure 9.
Typical reaction processes related to syngas chemistry (Reproduced with permission from [95]. Copyright Elsevier, 2017). The red-line route demonstrates the SMA process for the direct synthesis of aromatics via reaction coupling. FT = Fischer Tropsch, MTO = methanol to olefins, MTA = methanol to aromatics, SMA = syngas-methanol-aromatics.

Figure 10.
Some possible oxidation reactions to be performed on Cr-MIL-101 MOF catalysts. Reproduced with permission from [96]. Copyright Elsevier, 2016.
Figure 10.
Some possible oxidation reactions to be performed on Cr-MIL-101 MOF catalysts. Reproduced with permission from [96]. Copyright Elsevier, 2016.

Figure 11.
Schemes of main procedures (left) to follow for future applications of heterogeneous catalysis on metal oxides and lower alkanes (C1–C3) upgrading (right).
Figure 11.
Schemes of main procedures (left) to follow for future applications of heterogeneous catalysis on metal oxides and lower alkanes (C1–C3) upgrading (right).


{kind=link}
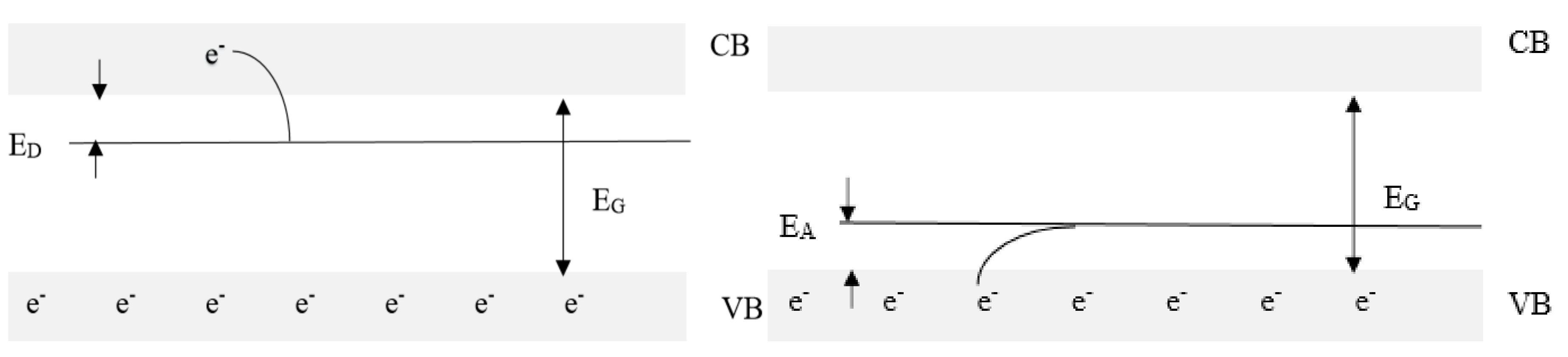{kind=link}
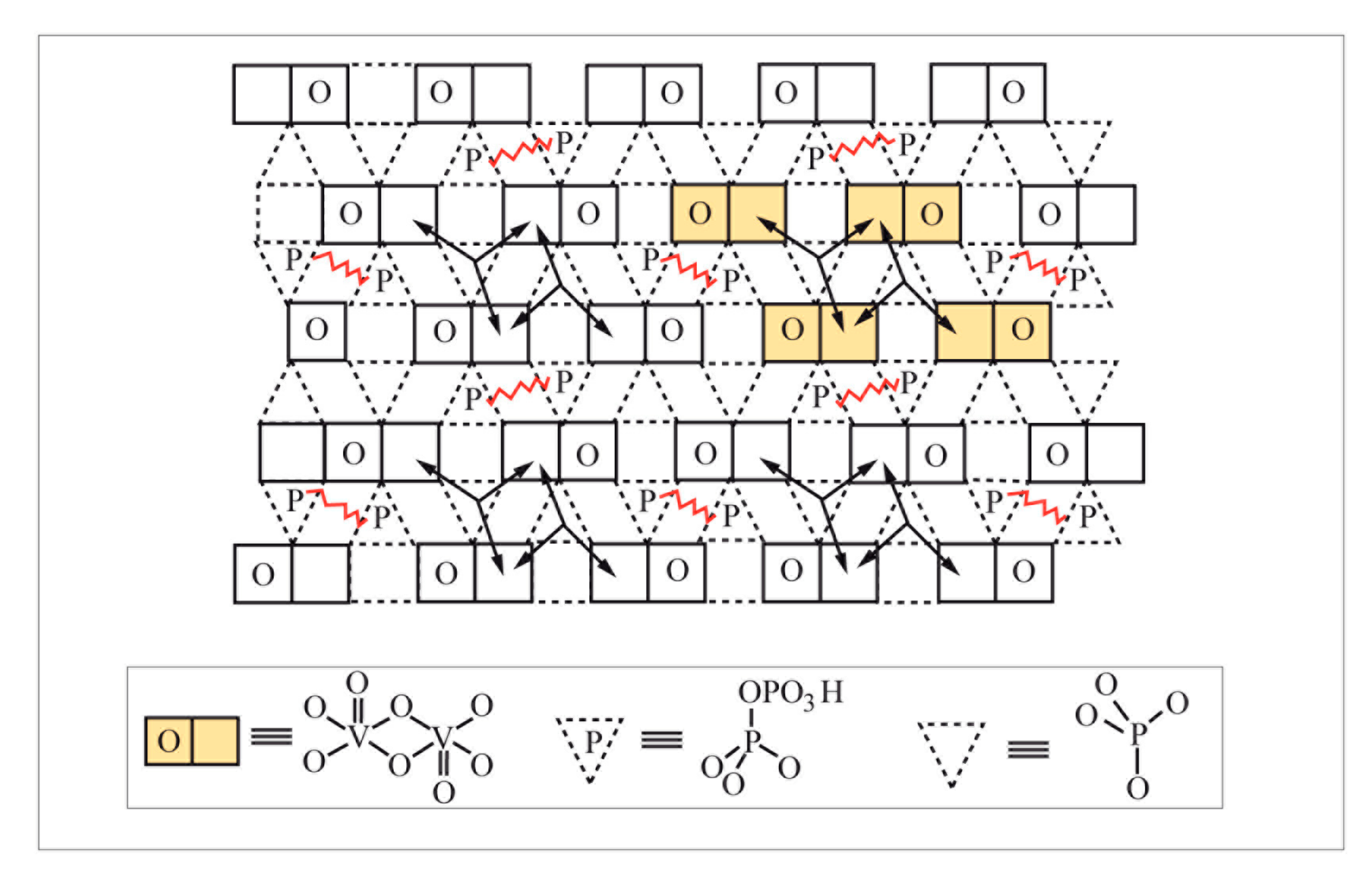{kind=link}
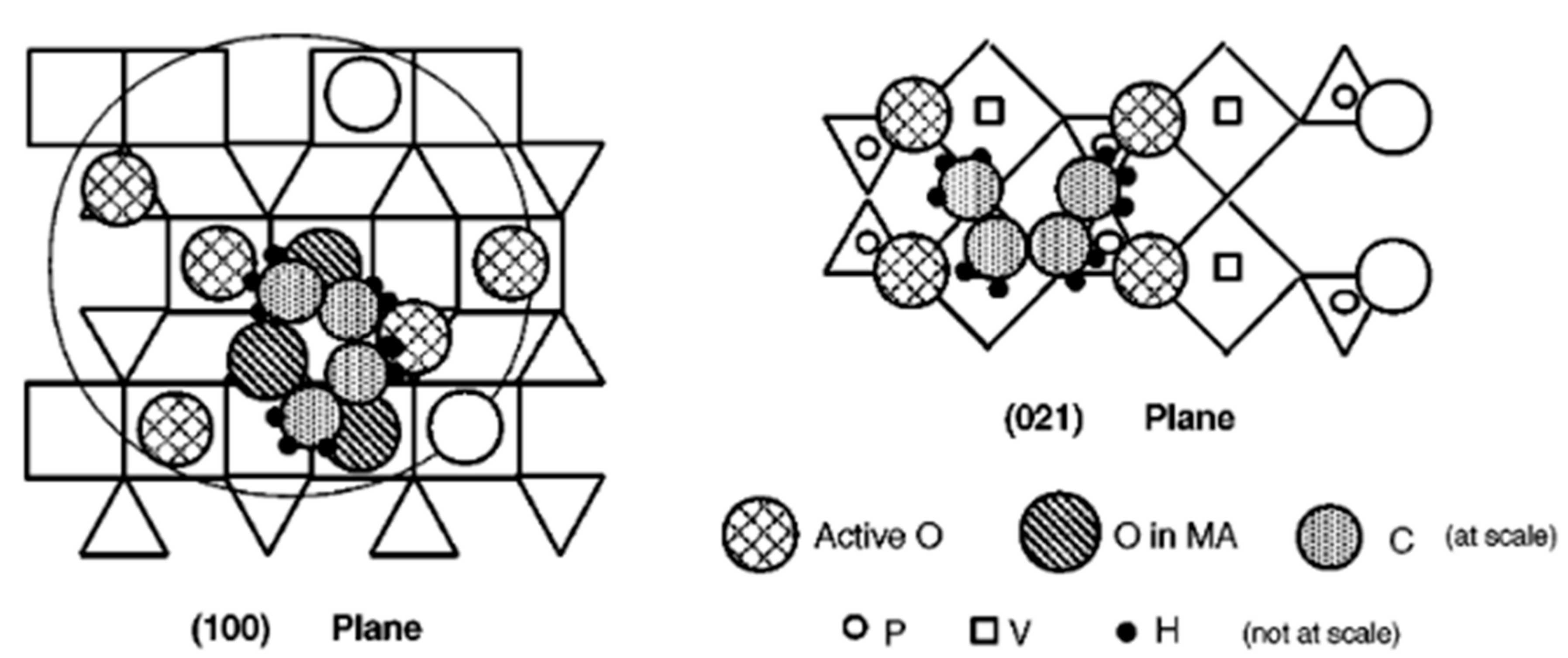{kind=link}
{kind=link}
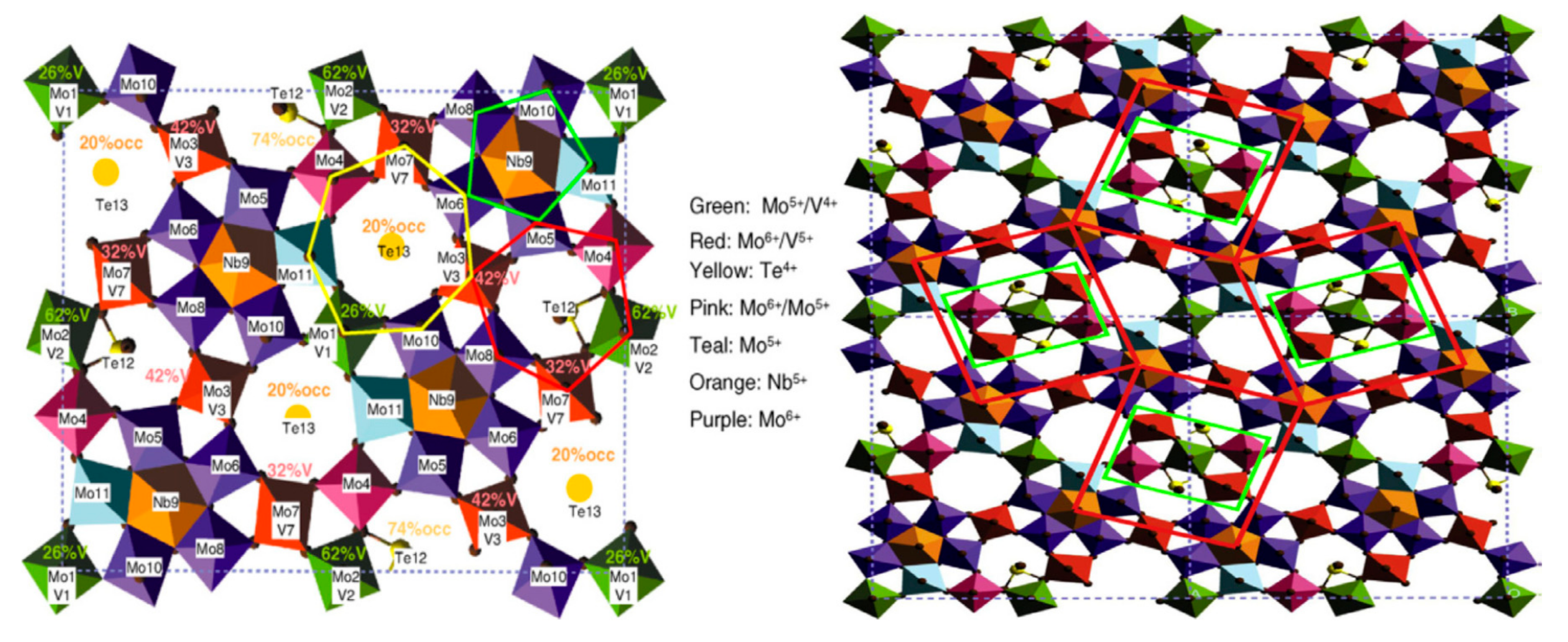{kind=link}
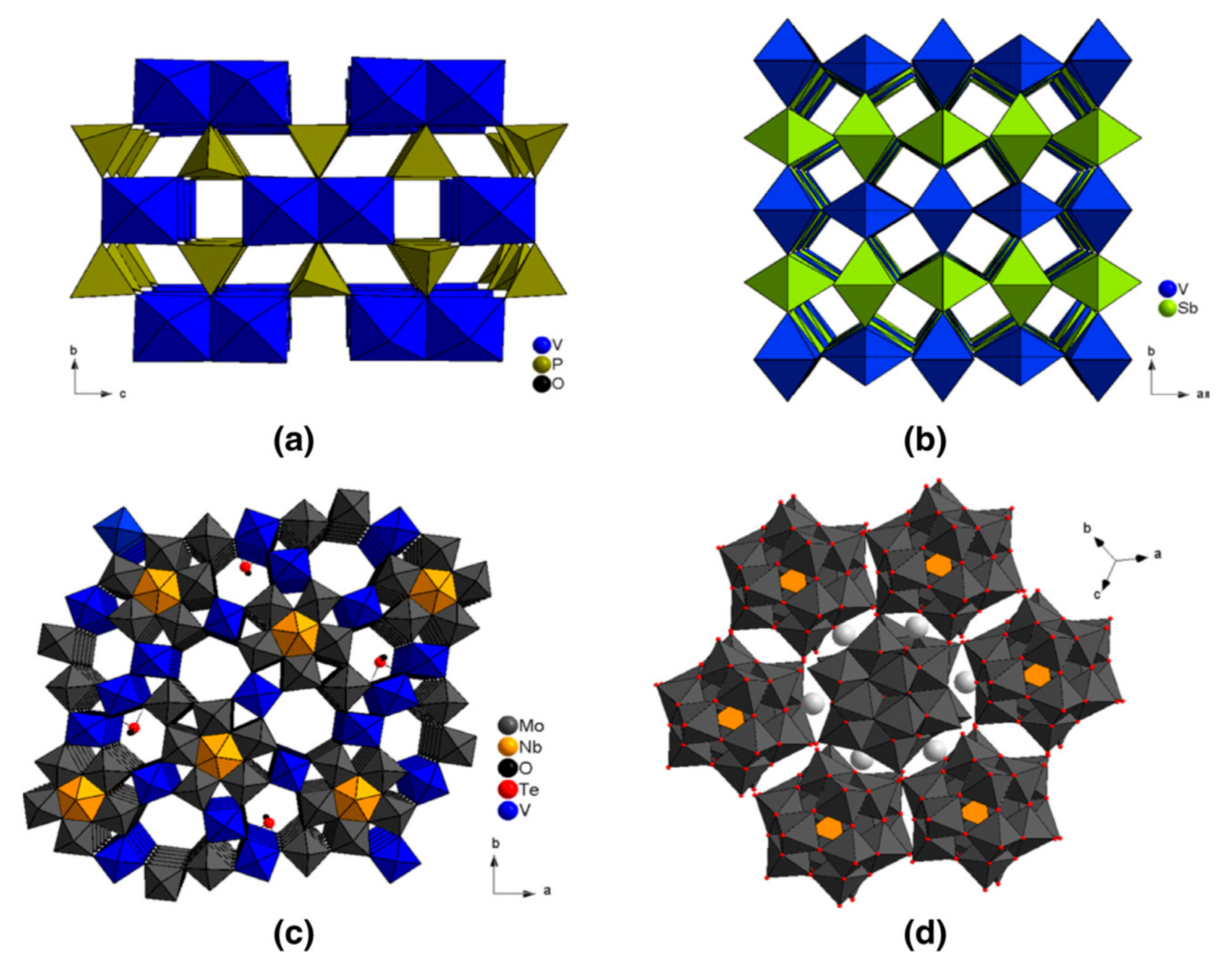{kind=link}
{kind=link}
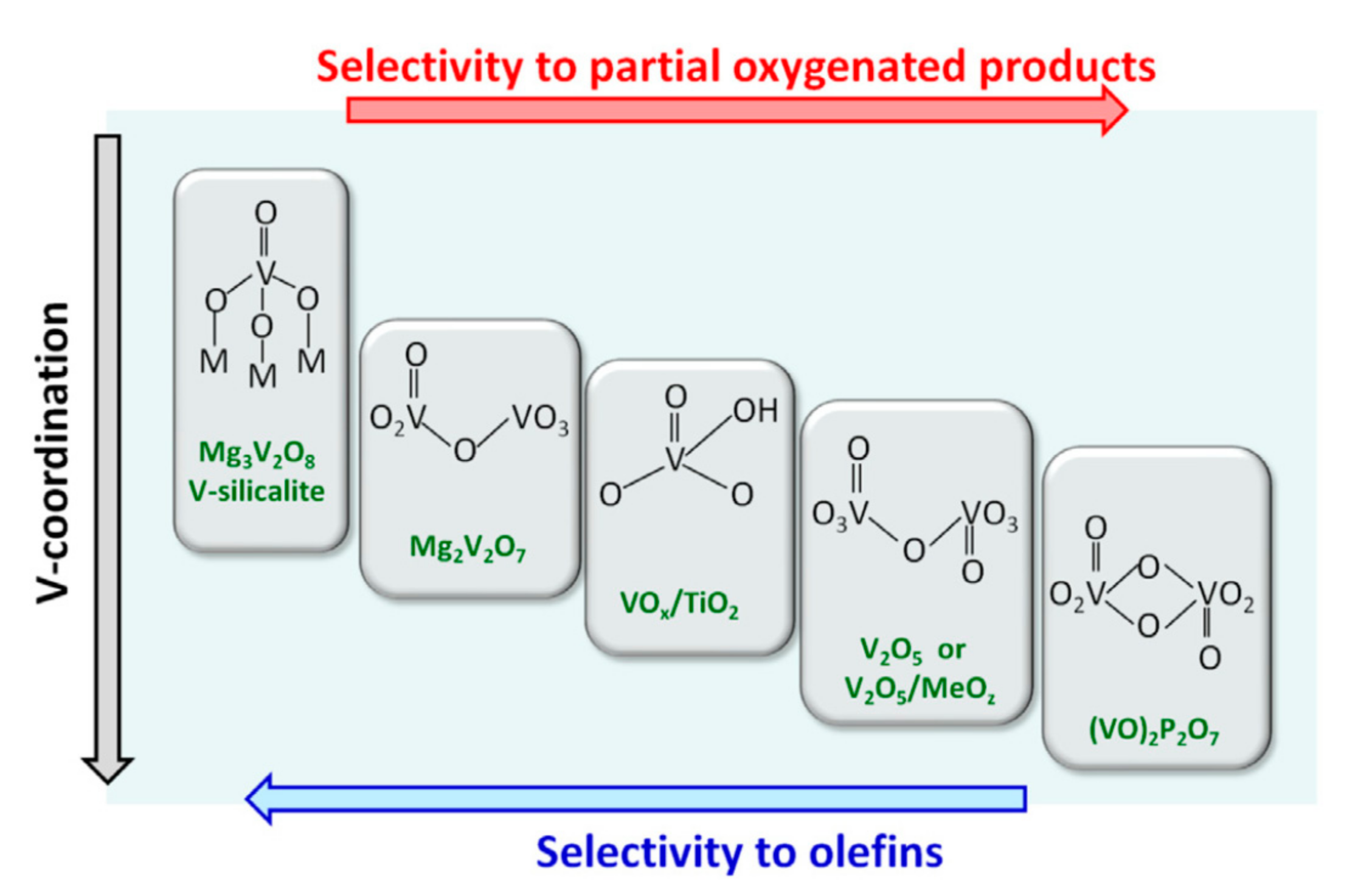{kind=link}
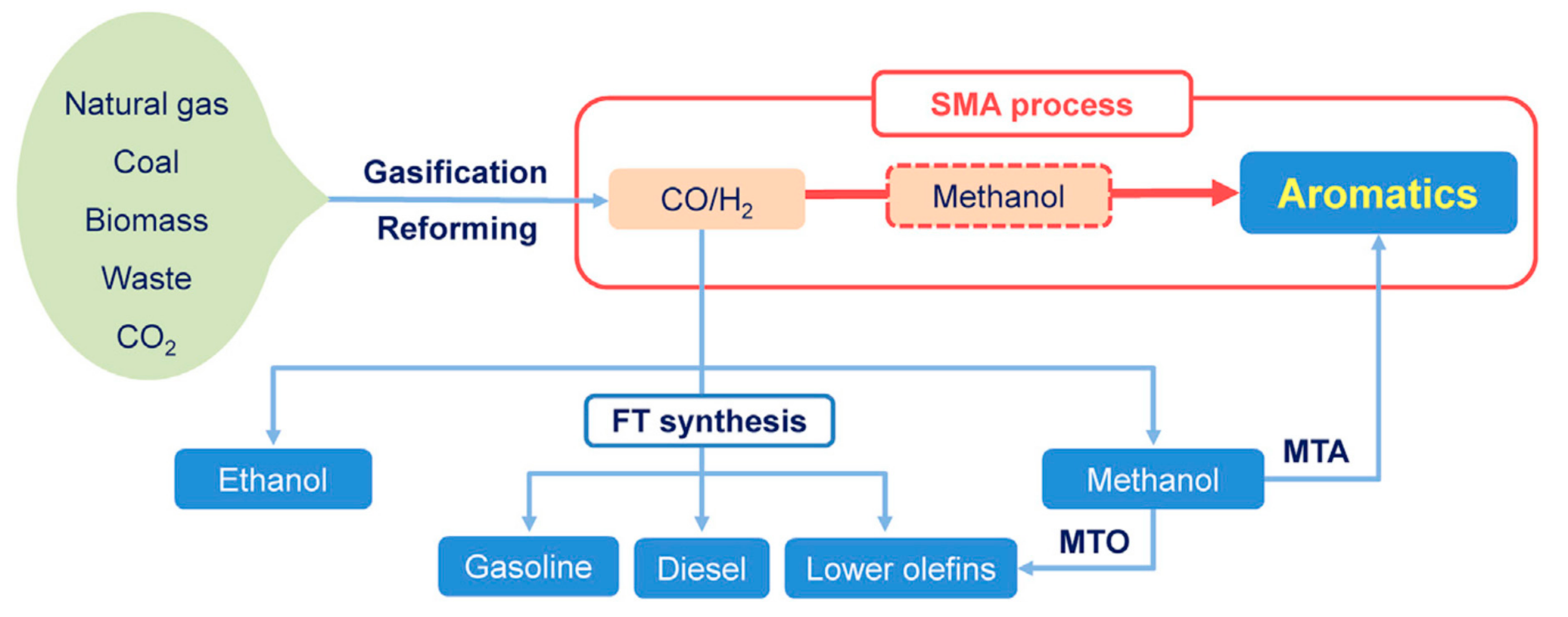{kind=link}
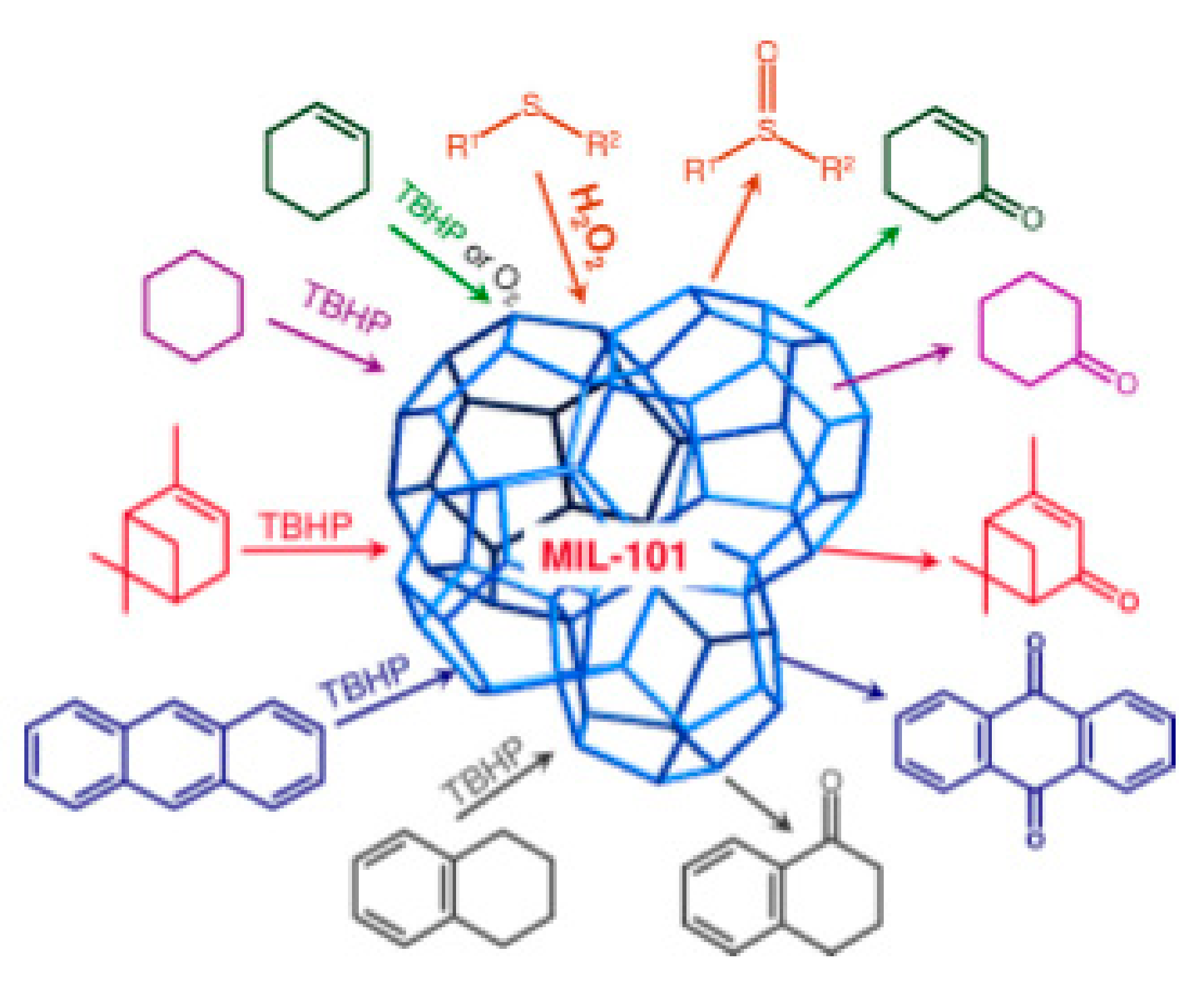{kind=link}
{kind=link}
Table 1.
Total oxidation of propane and naphthalene over various oxides. T90 is the temperature for a 90% conversion of hydrocarbon. All oxides are prepared by precipitation of nitrates by sodium carbonate, except CeO2-U, which is prepared by urea precipitation. Reaction conditions: 0.5% C3H8 in air (propane) at a gas hourly space velocity (GHSV) of 15,000 h−1 and 100 vpm C10H8 in air (naphthalene) at a GHSV of 60,000 h−1. Adapted with permission from [70]. Copyright Elsevier, 2006.
Table 1.
Total oxidation of propane and naphthalene over various oxides. T90 is the temperature for a 90% conversion of hydrocarbon. All oxides are prepared by precipitation of nitrates by sodium carbonate, except CeO2-U, which is prepared by urea precipitation. Reaction conditions: 0.5% C3H8 in air (propane) at a gas hourly space velocity (GHSV) of 15,000 h−1 and 100 vpm C10H8 in air (naphthalene) at a GHSV of 60,000 h−1. Adapted with permission from [70]. Copyright Elsevier, 2006.
| Oxide | BET Area (m2 g−1) | Main Phase | T90 Propane (°C) | T90 Naphthalene (°C) |
|---|---|---|---|---|
| Al2O3 | 180 | γ-Al2O3 | - | 38% conv. at 350 °C |
| CeO2 | 86 | CeO2 | - | 315 |
| CeO2-U | 171 | CeO2 | 360 | 210 |
| CoOx | 69 | Co3O4 | 180 | 270 |
| CuOx | 20 | CuO | 350 | 270 |
| CuOx–ZnOx | 29 | CuO, ZnO | 335 | 250 |
| Fe2O3 | 51 | Fe2O3 (hematite) | - | 295 |
| MnOx | 76 | Mn2O3 | 320 | 240 |
| TiO2 | 50 | Anatase + rutile | - | 66% conv. at 350 °C |
| ZnO | 19 | ZnO | - | 335 |
Table 2.
Reactivity of various VOC compounds on an extruded clay honeycomb monolith (11.1% Fe2O3). Reaction conditions: 1% VOC + 10% O2 at a GHSV (gas hourly space velocity) of 2300 h−1 with respect to the volume of the monolith. T50 is the temperature for a 50% conversion of hydrocarbon. Variable amounts of CO are observed in addition to CO2 for all VOCs. Aldehydes are the main reaction intermediate of alcohol oxidation (formaldehyde, acetaldehyde, propanal, butanal in oxidation of methanol, ethanol, propanol, butanol, respectively). Adapted with permission from [71]. Copyright Elsevier, 2015.
Table 2.
Reactivity of various VOC compounds on an extruded clay honeycomb monolith (11.1% Fe2O3). Reaction conditions: 1% VOC + 10% O2 at a GHSV (gas hourly space velocity) of 2300 h−1 with respect to the volume of the monolith. T50 is the temperature for a 50% conversion of hydrocarbon. Variable amounts of CO are observed in addition to CO2 for all VOCs. Aldehydes are the main reaction intermediate of alcohol oxidation (formaldehyde, acetaldehyde, propanal, butanal in oxidation of methanol, ethanol, propanol, butanol, respectively). Adapted with permission from [71]. Copyright Elsevier, 2015.
| VOC Compounds (CO HC) | T50 (°C) | VOC Compounds (Oxygenates Cl Compounds) | T50 (°C) |
|---|---|---|---|
| CO | 295 | Methanol | 261 |
| Methane | 512 | Ethanol | 272 |
| Propane | 420 | n-Propanol | 271 |
| Propene | 355 | n-Butanol | 278 |
| Acetylene | 299 | Acetone | 228 |
| Benzene | 345 | Dimethylether (DME) | 286 |
| Toluene | 329 | Chlorobenzene | 395 |
| o-Xylene | 321 | 1,2-Dichlorobenzene | 414 |
Table 3.
Combustion rates at 60% conversion of carbon and mean CO2/CO ratio in the 40–80% range of conversion. Close contact between carbon (U-printex Degussa) and catalyst (ratio 1:1). Measurements are made at 383 °C for oxides and 279 °C for carbonates. Adapted with permission from [72]. Copyright Elsevier, 1998.
Table 3.
Combustion rates at 60% conversion of carbon and mean CO2/CO ratio in the 40–80% range of conversion. Close contact between carbon (U-printex Degussa) and catalyst (ratio 1:1). Measurements are made at 383 °C for oxides and 279 °C for carbonates. Adapted with permission from [72]. Copyright Elsevier, 1998.
| Oxide | Reaction Rate at 383 °C (µg·gcarbon initial−1·s−1) | Mean CO2/CO Ratio | Carbonate | Reaction Rate at 279 °C (µg·gcarbon initial−1·s−1) | Mean CO2/CO Ratio |
|---|---|---|---|---|---|
| PbO | 240 | 20 | Cs2CO3 | 182 | 15 |
| Co3O4 | 233 | 500 | K2CO3 | 101 | 14 |
| V2O5 | 203 | 6 | Na2CO3 | 65 | 18 |
| Ag2O | 95 | 15 | Li2CO3 | 6 | 17 |
| MoO3 | 66 | 5 | none | 0.003 | nd |
| CuO | 27 | 6 | |||
| Bi2O3 | 22 | 8 | |||
| MnO2 | 21 | 150 | |||
| Sb2O3 | 20 | 8 | |||
| CaO | 16 | 7 | |||
| La2O3 | 13 | 8 | |||
| Cr2O3 | 12 | 3 | |||
| Fe2O3 | 7 | 6 | |||
| NiO | 1 | 5 | |||
| none | 0.9 | 2 |
Table 4.
Some industrial processes using metal oxide catalysts or supports.
| Reaction | Catalyst |
|---|---|
| Steam reforming of hydrocarbons to CO + H2 | Ni/Al2O3 |
| Water gas shift (CO + H2O → CO2 + H2) | Fe oxide or mixed oxides Zn, Cu, Cr |
| Methane dry reforming (CO2 + CH4 → 2CO + 2H2) | Ni/Al2O3 |
| Methanol synthesis from CO + CO2 + H2 | Cu-Zn-O/Al2O3 |
| Methanol steam reforming (CO2 + 3H2) | Cu-Zn-O/Al2O3 |
| Methanol to hydrocarbons (MTG process) | H-MFI zeolite |
| Methanol to light olefins (MTO process) | SAPO-34 or Chabazite zeolite |
| Methanol oxidation to formaldehyde | Fe2(MoO4)3 |
| SO2 → SO3 for H2SO4 | V2O5/diatomaceous earth |
| H2S oxidation to SO2 and H2SO4 | Fe2O3/SiO2 or α-Al2O3 |
| O-xylene + O2 → phthalic anhydride | V2O5/TiO2 (anatase) |
| Metathesis | Re-O, Ru-O W-O |
| C3–C4 alkane dehydrocyclisation to aromatics “Cyclar” | Ga-MFI zeolite |
| Selective catalytic reduction of NOx | V2O5 + (WO3 or MoO3)/TiO2 |
| SCR of NOx at high T | Cu/chabazite zeolite |
| Propene ammoxidation to acrylonitrile | Bismuth molybdate |
| Propane ammoxidation to acrylonitrile | MoVTeNb-O (M1 phase) |
| Propene oxidation to acrolein | Bismuth molybdate |
| Acrolein oxidation to acrylic acid | Mo-V-O |
| Aromatic carboxylic acids hydrogenation to aldehydes | Zirconia |
| Hydrodesulphurisation of oil distillates | CoMo-O, NiMo-O, Ni-W-O/γ-Al2O3 |
| Synthesis gas (CO + H2) to hydrocarbons (Fuel range) | Supported Co and Fe catalyts (FT) |
| Parafins (C5–C12) isomerisation | Supported W-O and sulfated ZrO2 |
| Olefin epoxidation | Titanosilicate (TS1) |
| Alkanes (C2–C5) dehydrogenation to olefins | Cr2O3/Al2O3 or Pt/Al2O3 |
| Alkanes oxidative dehydrogenation to olefins | V based catalysts |
| Butane to maleic anhydride | (VO)2P2O7 |
| Methane oxidative coupling to ethylene | Doped rare earth oxides |
| Ethylene + HCl + O2 to dichloroethane | ZnO, Cr2O3, CuO |
| Exhaust gas elimination | Pt-Rh-Pd alloys on oxides |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Védrine, J.C. Heterogeneous Catalysis on Metal Oxides. Catalysts 2017, 7, 341. https://doi.org/10.3390/catal7110341
AMA Style
Védrine JC. Heterogeneous Catalysis on Metal Oxides. Catalysts. 2017; 7(11):341. https://doi.org/10.3390/catal7110341
Chicago/Turabian StyleVédrine, Jacques C. 2017. "Heterogeneous Catalysis on Metal Oxides" Catalysts 7, no. 11: 341. https://doi.org/10.3390/catal7110341
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.