
Au Capping Agent Removal Using Plasma at Mild Temperature

Abstract
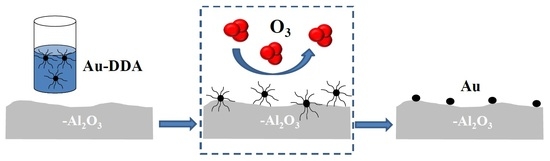
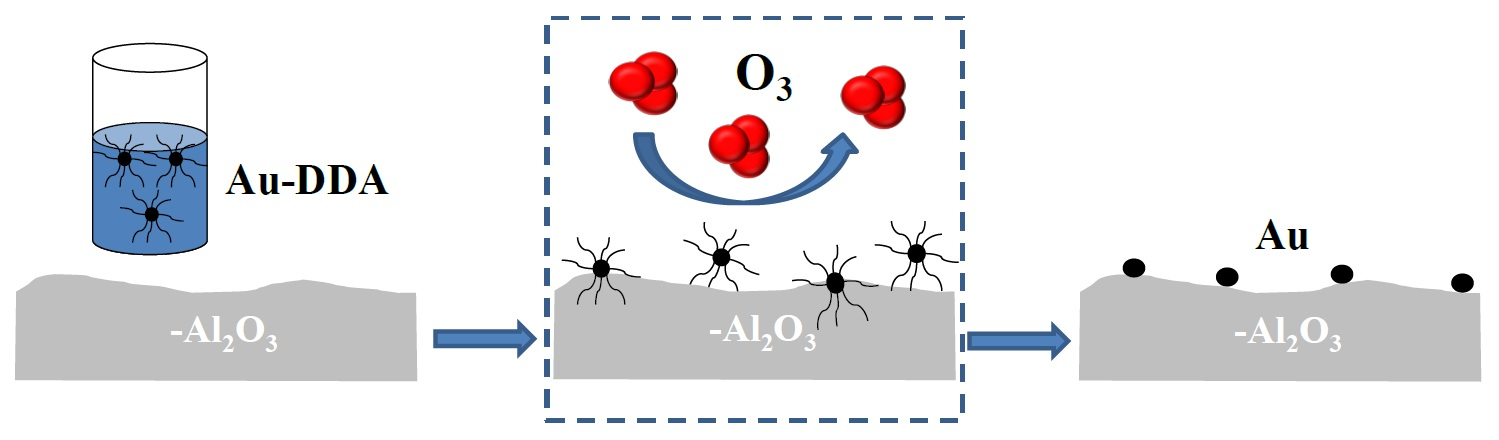
:
1. Introduction
2. Results and Discussion
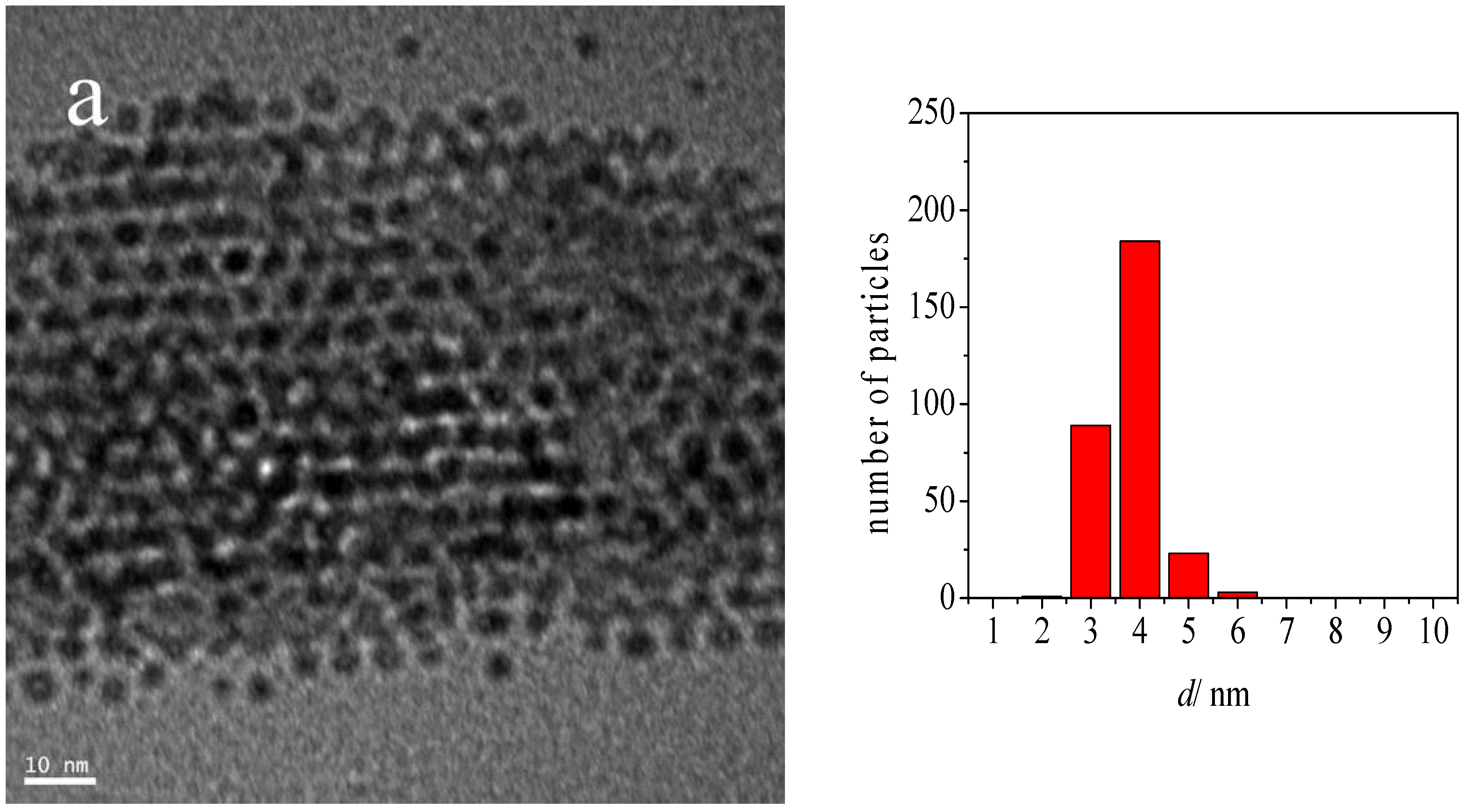
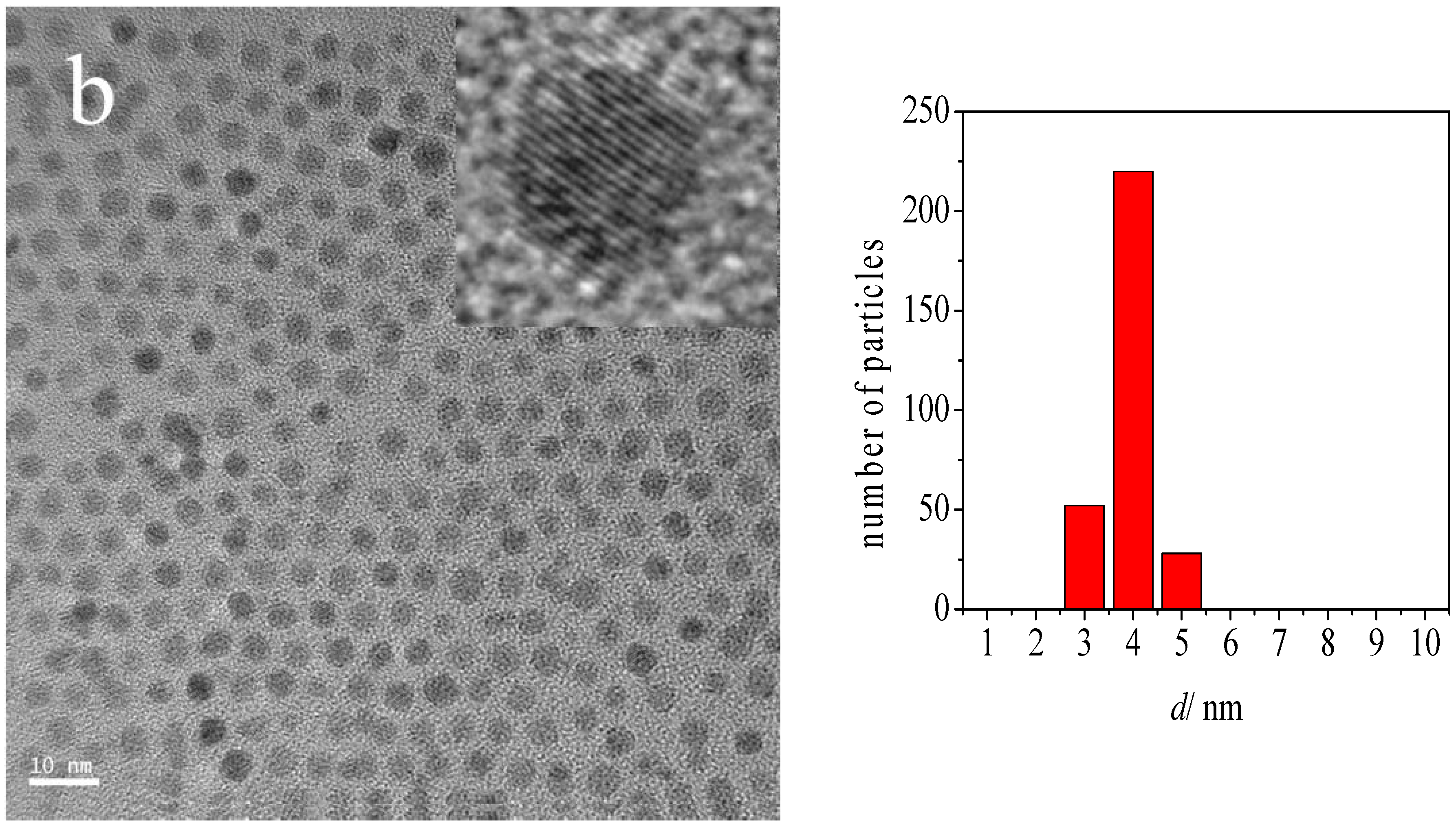
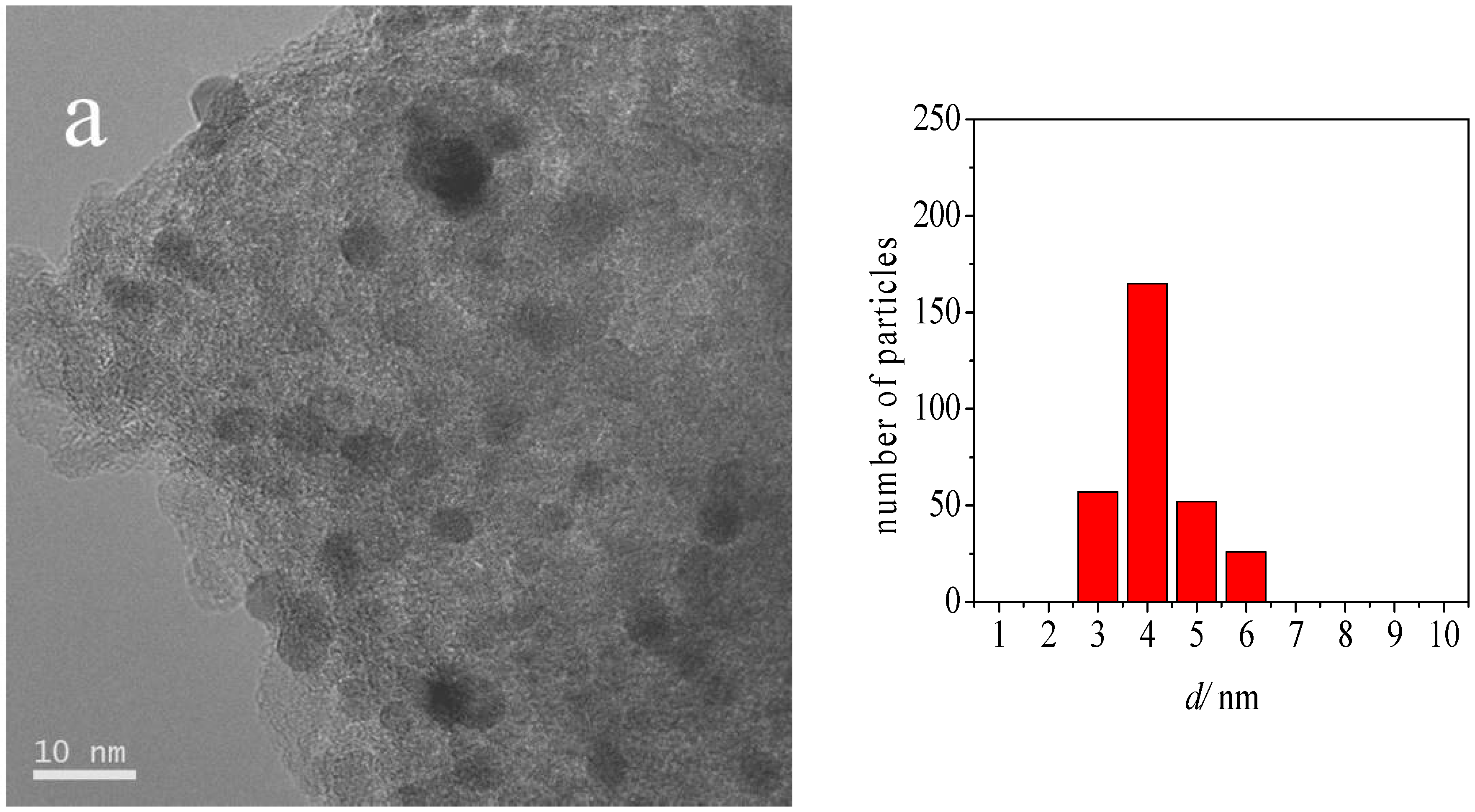
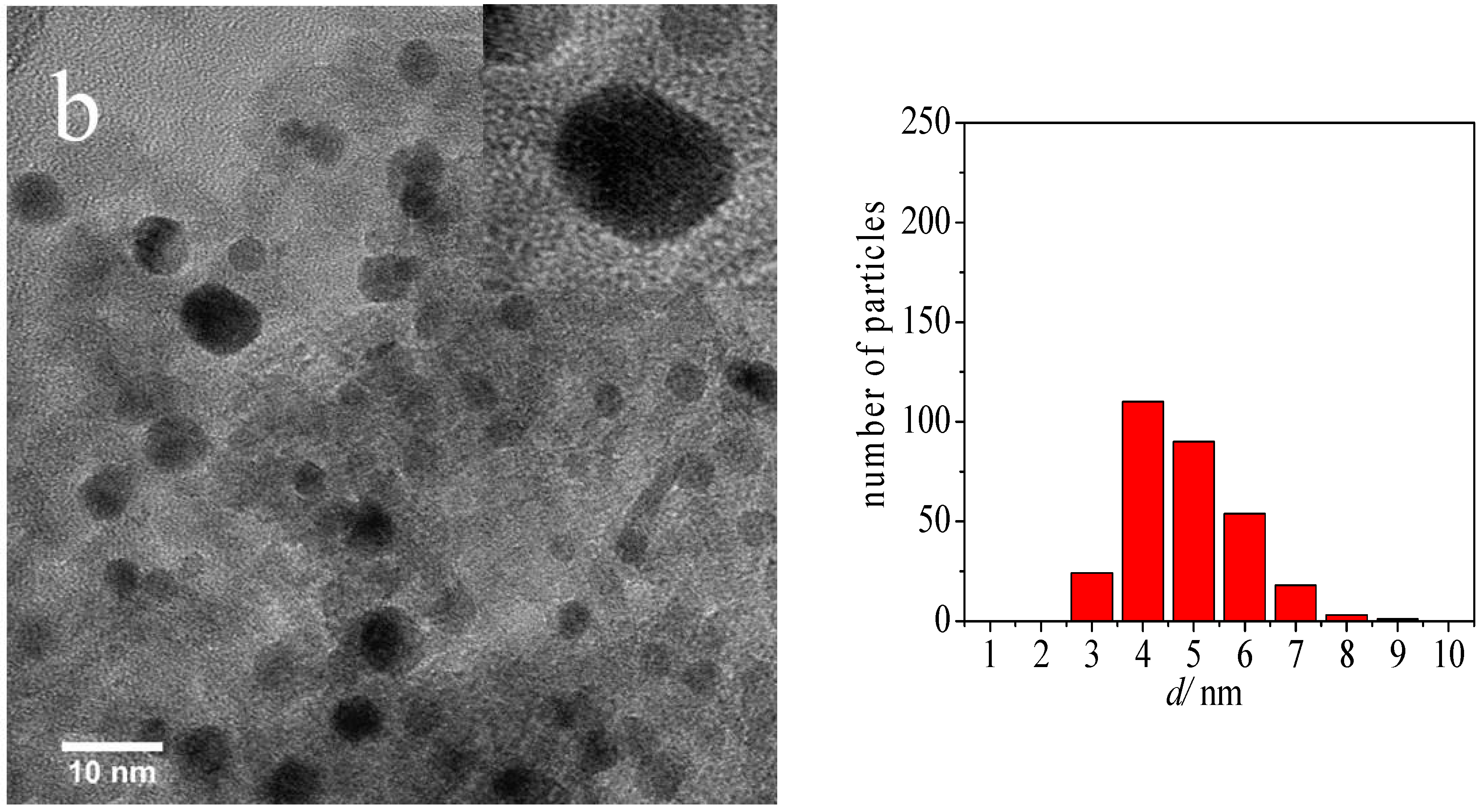
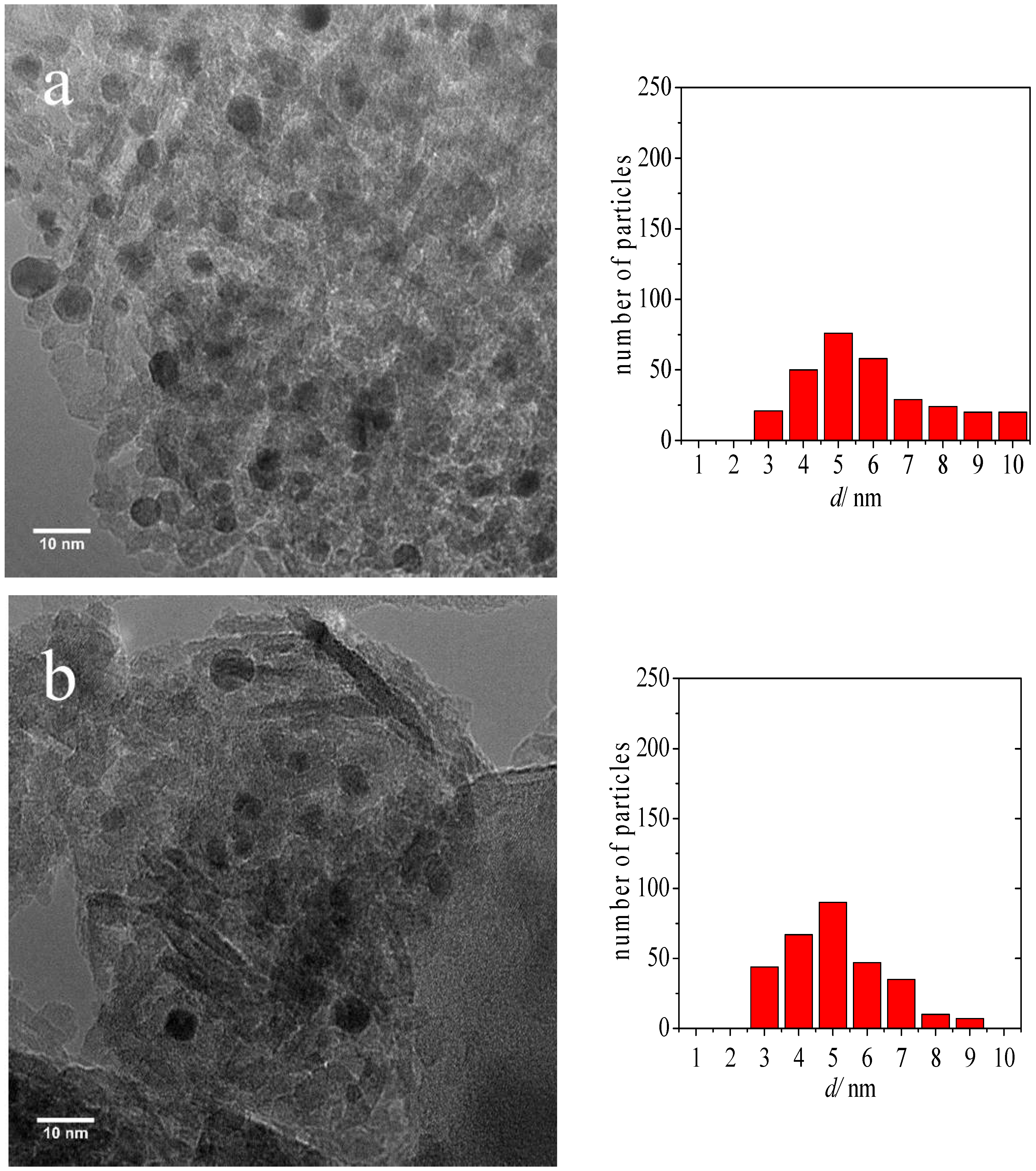
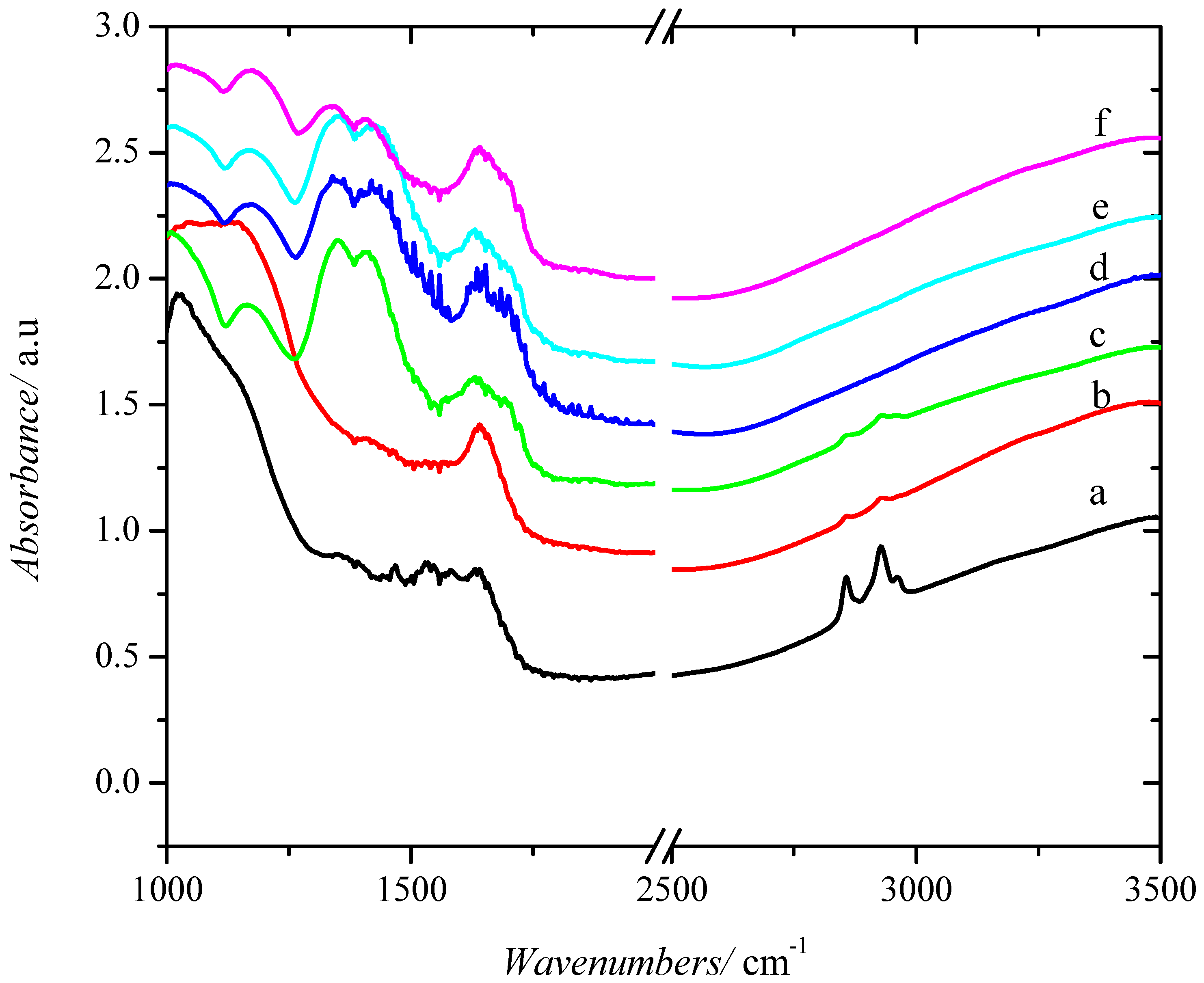
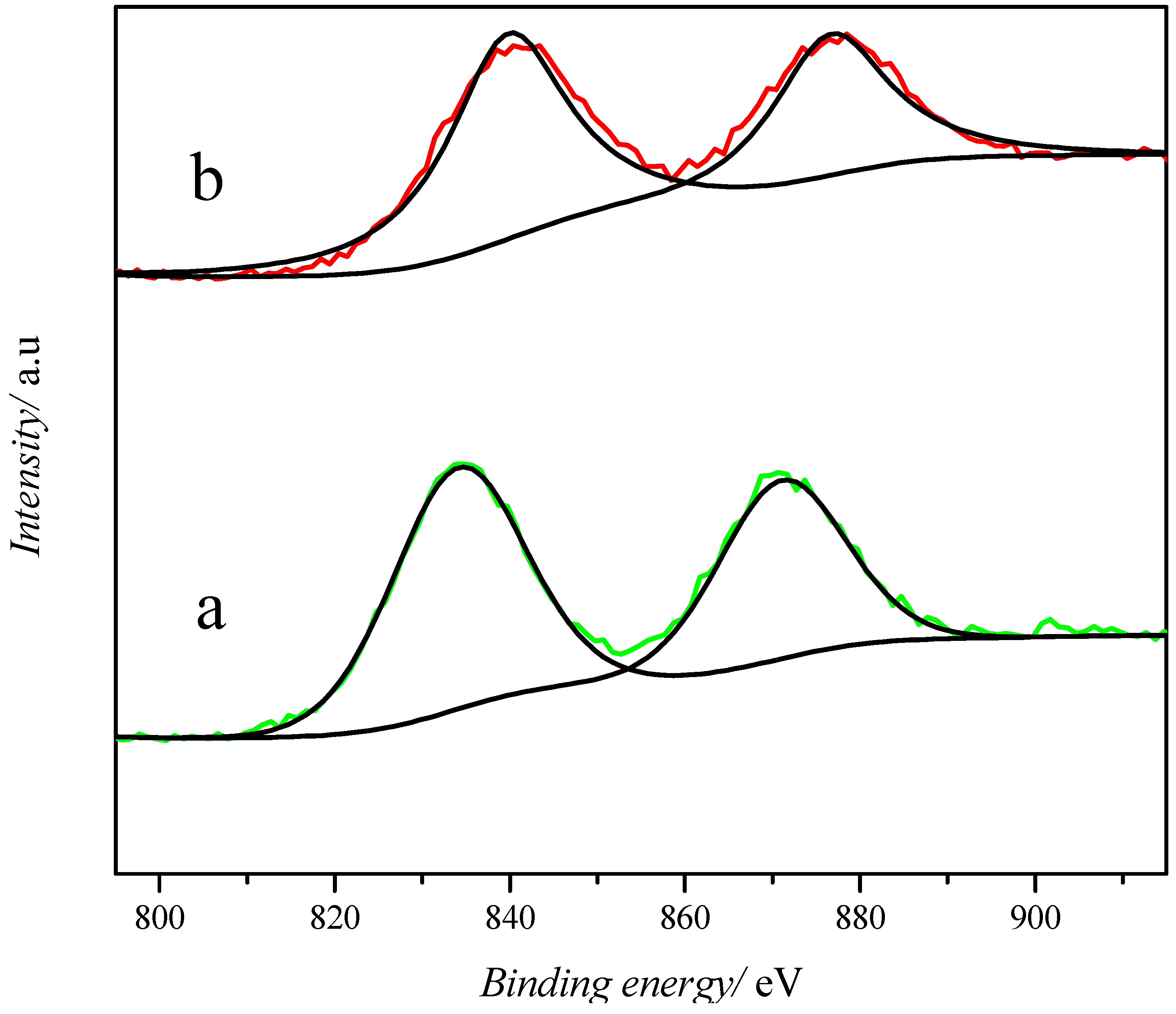
2.1. Catalyst Characterization
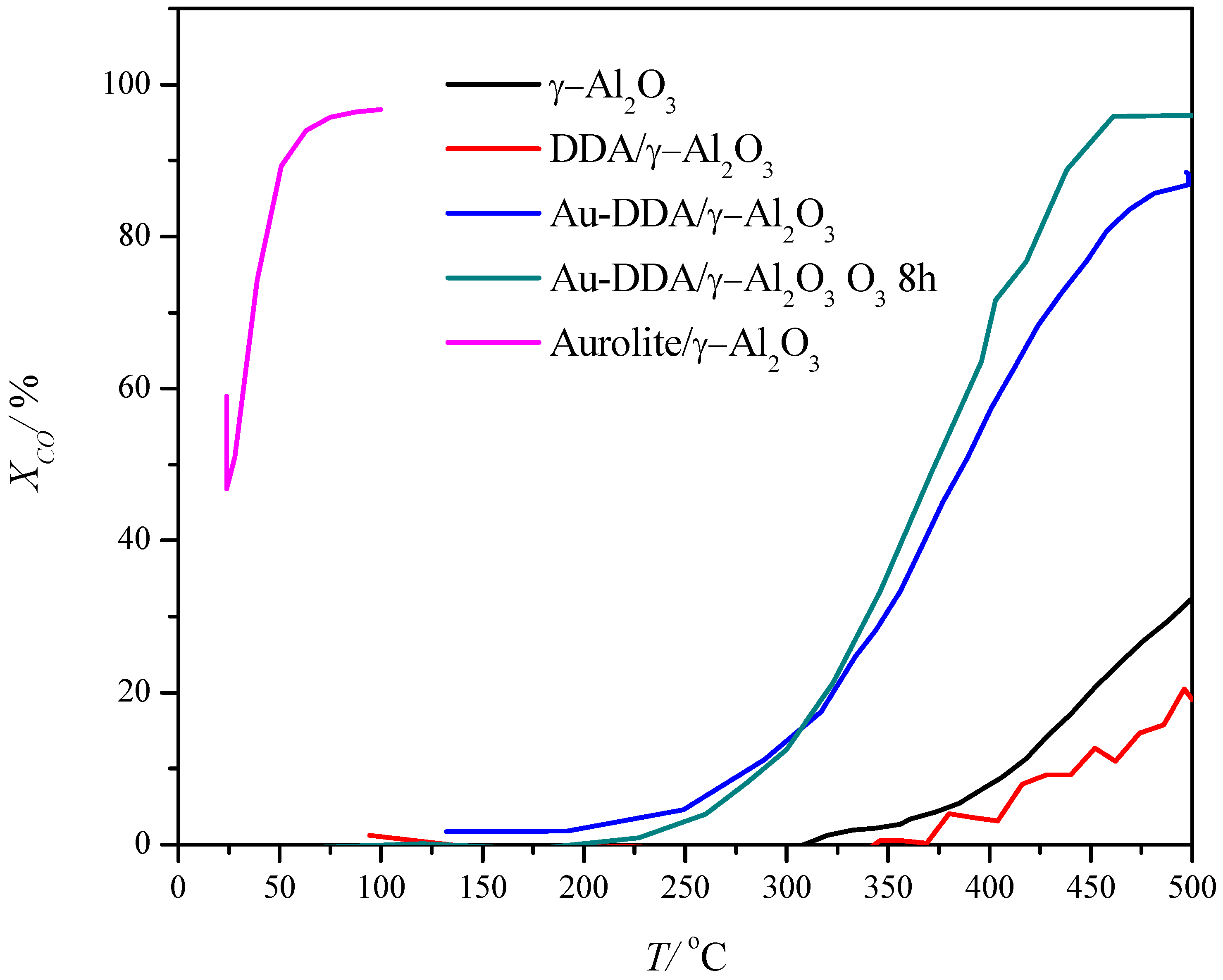
2.2. Catalytic Performance
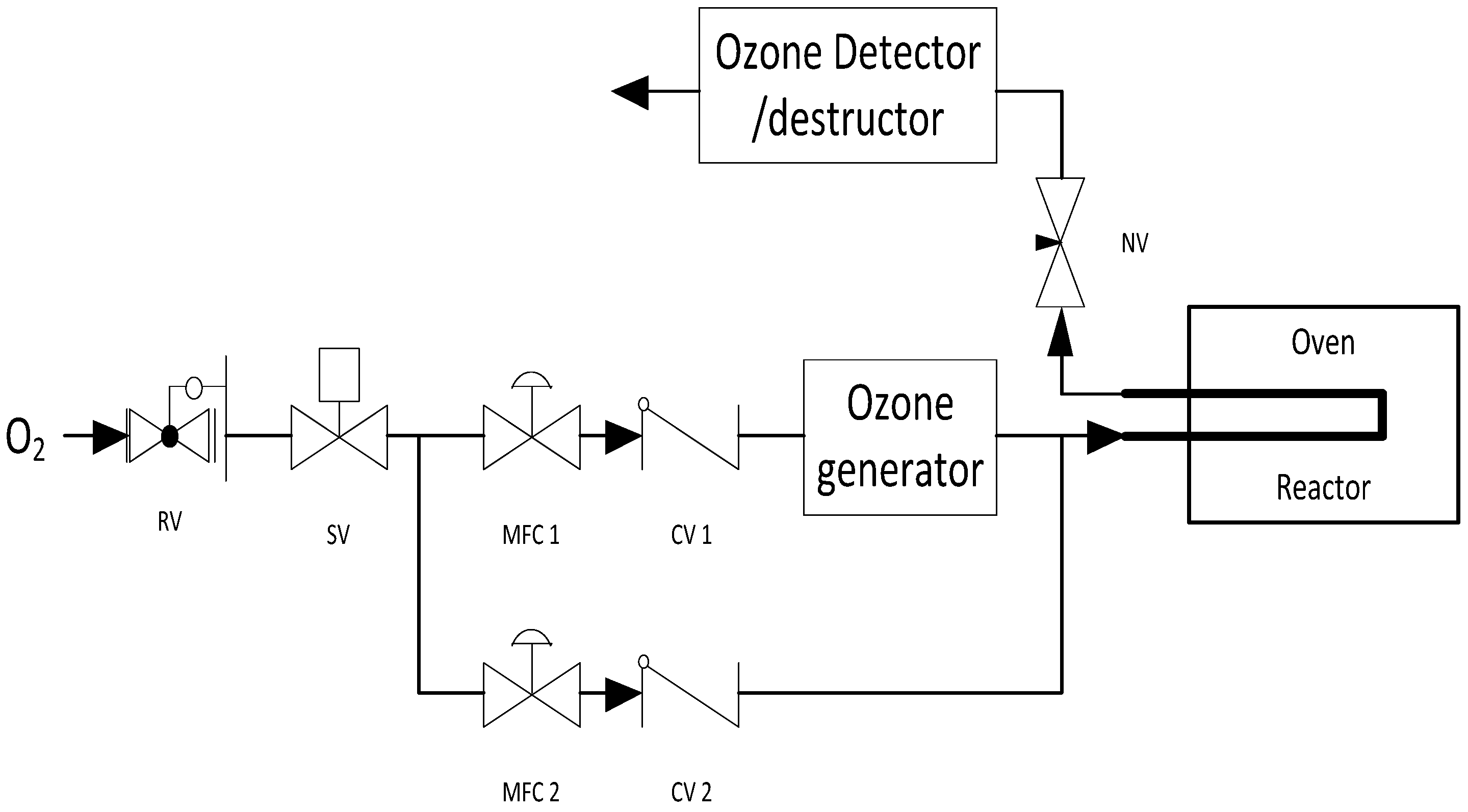
3. Materials and Methods
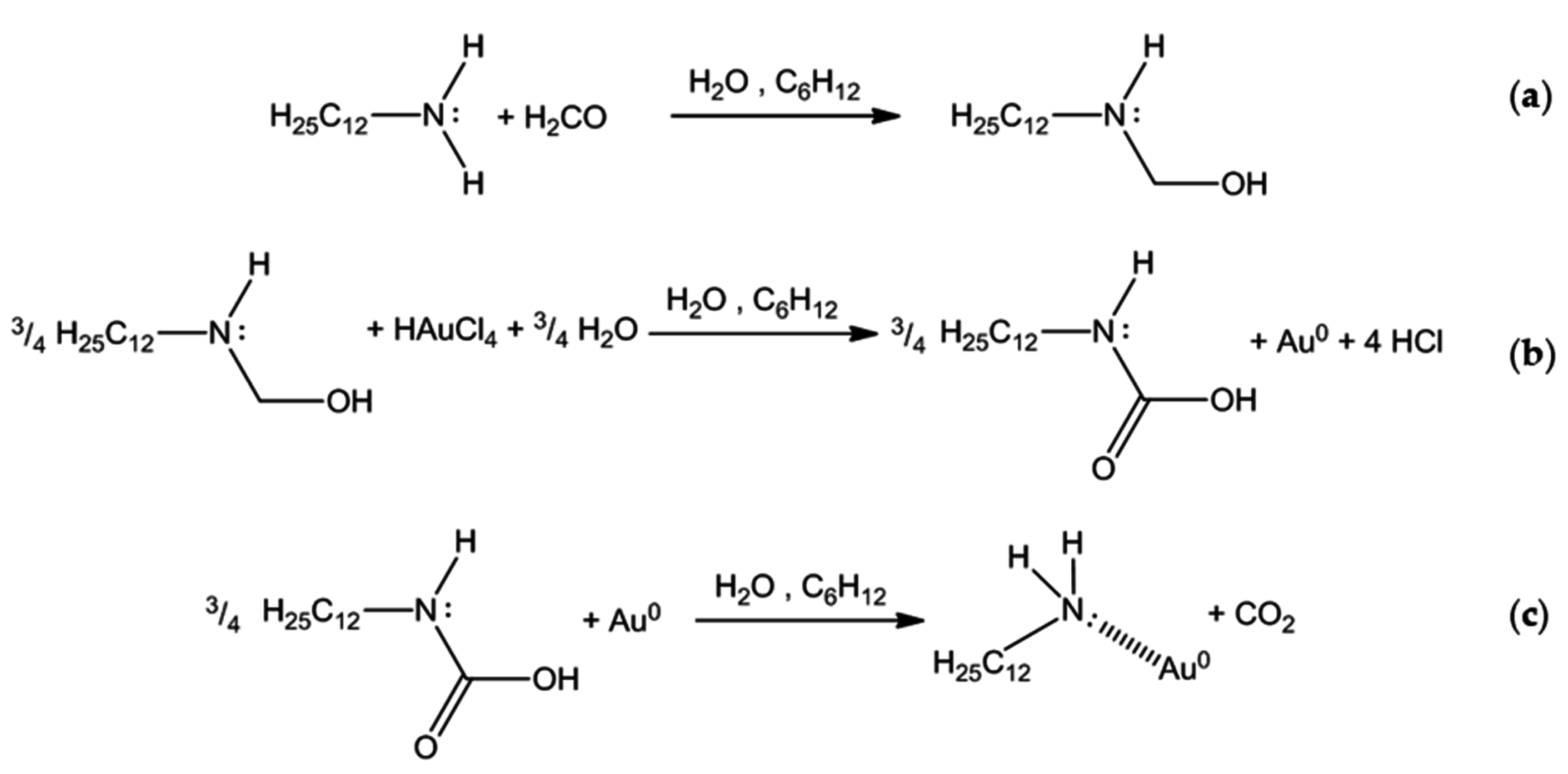
3.1. Catalyst Preparation
3.2. Capping Agent Removal
3.3. Catalyst Characterization
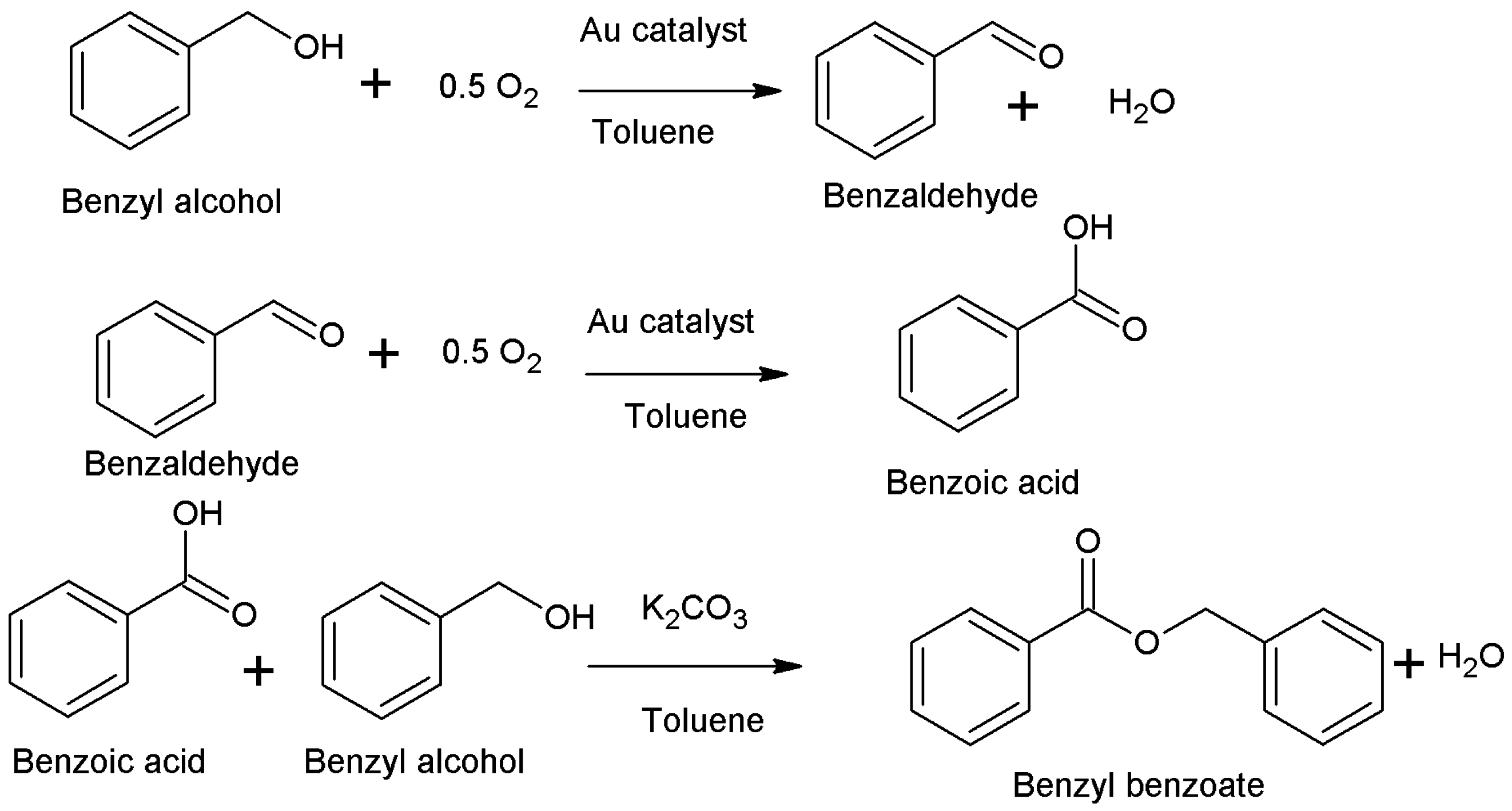
3.4. Catalytic Reactions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hutchings, G.J.; Haruta, M. A golden age of catalysis: A perspective. Appl. Catal. A 2005, 291, 2–5. [Google Scholar] [CrossRef]
- Geoffrey, C.B.; Catherine, L.; David, T.T. Catalysis by Gold; Imperial College Press: London, UK, 2006. [Google Scholar]
- Otieno, B.A.; Krause, C.E.; Latus, A.; Chikkaveeraiah, B.V.; Faria, R.C.; Rusling, J.F. On-line protein capture on magnetic beads for ultrasensitive microfluidic immunoassays of cancer biomarkers. Biosens. Bioelectron. 2014, 53, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Nagelli, E.; Naik, R.; Xue, Y.; Gao, Y.; Zhang, M.; Dai, L. Sensor arrays from multicomponent micropatterned nanoparticles and graphene. Nanotechnology 2013, 24, 444010. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Morales, R.; Albiter, M.A.; Zaera, F. Synthesis of heterogeneous catalysts with well shaped platinum particles to control reaction selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 15241–15246. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, L.; Xu, Q.H. A simple method for large scale synthesis of highly monodisperse gold nanoparticles at room temperature and their electron relaxation properties. Nanotechnology 2009, 20, 185606. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. Novel phase-transfer preparation of monodisperse silver and gold nanoparticles at room temperature. Mater. Lett. 2008, 62, 2215–2218. [Google Scholar] [CrossRef]
- Sun, Y.; Mayers, B.; Herricks, T.; Xia, Y. Polyol synthesis of uniform silver nanowires: A plausible growth mechanism and the supporting evidence. Nano Lett. 2003, 3, 955–960. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, Y. Shape-controlled synthesis of gold and silver nanoparticles. Science 2002, 298, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Bratlie, K.M.; Lee, H.; Komvopoulos, K.; Yang, P.; Somorjai, G.A. Platinum nanoparticle shape effects on benzene hydrogenation selectivity. Nano Lett. 2007, 7, 3097–3101. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.R.; Habas, S.; Yang, P. Shape control of colloidal metal nanocrystals. Small 2008, 4, 310–325. [Google Scholar] [CrossRef]
- Quintanilla, A.; Butselaar-Orthlieb, V.C.L.; Kwakernaak, C.; Sloof, W.G.; Kreutzer, M.T.; Kapteijn, F. Weakly bound capping agents on gold nanoparticles in catalysis: Surface poison? J. Catal. 2010, 271, 104–114. [Google Scholar] [CrossRef]
- Lange, C.; De Caro, D.; Gamez, A.; Storck, S.; Bradley, J.S.; Maier, W.F. Polymer-induced selectivity enhancement in the hydrogenation of 2-hexyne catalyzed by poly(vinylpyrrolidone)-stabilized platinum colloids in an amorphous mixed metal oxide support. Langmuir 1999, 15, 5333–5338. [Google Scholar] [CrossRef]
- Imura, Y.; Furukawa, S.; Ozawa, K.; Morita-Imura, C.; Kawaib, T.; Komatsu, T. Surface clean gold nanoflower obtained by complete removal of capping agents: An active catalyst for alcohol oxidation. RCS Adv. 2016, 6, 17222–17227. [Google Scholar] [CrossRef]
- Zhong, Z.; Teo, J.; Lin, M.; Ho, J. Synthesis of Porous α-Fe2O3 Nanorods as Catalyst Support and a Novel Method to Deposit Small Gold Colloids on them. Top. Catal. 2008, 49, 216–226. [Google Scholar] [CrossRef]
- Chen, Y.; Hassel, A.W.; Erbe, A. Enhancement of the Electrocatalytic Activity of Gold Nanoparticles towards Methanol Oxidation. Electrocatal 2011, 2, 106–113. [Google Scholar] [CrossRef]
- Naresh, N.; Wasim, F.G.S.; Ladewig, B.P.; Neergat, M. Removal of surfactant and capping agent from Pd nanocubes (Pd-NCs) using tert-butylamine: Its effect on electrochemical characteristics. J. Mater. Chem. A 2013, 1, 8553–8559. [Google Scholar] [CrossRef]
- Mazumder, V.; Sun, S. Oleylamine-mediated synthesis of Pd nanoparticles for catalytic formic acid oxidation. JACS 2009, 131, 4588–4589. [Google Scholar] [CrossRef] [PubMed]
- Nalajala, N.; Gooty Saleha, W.F.; Ladewig, B.P.; Neergat, M. Sodium borohydride treatment: A simple and effective process for the removal of stabilizer and capping agents from shape-controlled palladium nanoparticles. Chem. Commun. 2014, 50, 9365–9368. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Li, Y.P. Removal and utilization of capping agents in nanocatalysis. Chem. Mater. 2014, 26, 72–83. [Google Scholar] [CrossRef]
- Kuhn, J.; Gascon, J.; Gross, J.; Kapteijn, F. Detemplation of DDR type zeolites by ozonication. Micropor. Mesopor Mat. 2009, 120, 12–18. [Google Scholar] [CrossRef]
- Heng, S.; Lau, P.P.S.; Yeung, K.L.; Djafer, M.; Schrotter, J.C. Low-temperature ozone treatment for organic template removal from zeolite membrane. J. Membr. Sci. 2004, 243, 69–78. [Google Scholar] [CrossRef]
- Benson, S.W.; Axworthy, A.E. Reconsideration of the rate constants from the thermal decomposition of ozone. J. Chem. Phys. 1965, 42, 2614–2615. [Google Scholar] [CrossRef]
- Benson, S.W.; Axworthy, A.E., Jr. Mechanism of the gas phase, thermal decomposition of ozone. J. Chem. Phys. 1957, 26, 1727–1733. [Google Scholar] [CrossRef]
- Menard, L.D.; Xu, F.; Nuzzo, R.G.; Yang, J.C. Preparation of TiO2-supported Au nanoparticle catalysts from a Au13 cluster precursor: Ligand removal using ozone exposure versus a rapid thermal treatment. J. Catal. 2006, 243, 64–73. [Google Scholar] [CrossRef]
- Elliott, E.W., III; Glover, R.D.; Hutchison, J.E. Removal of Thiol Ligands from surface-confined nanoparticles without particle growth or desorption. ACS Nano 2015, 9, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, T.A.; Sacaliuc, E.; Beale, A.M.; van der Eerden, A.M.J.; Schouten, J.C.; Weckhuysen, B.M. Spectroscopic evidence for the adsorption of propene on gold nanoparticles during the hydro-epoxidation of propene. J. Catal. 2008, 258, 256–264. [Google Scholar] [CrossRef]
- Delannoy, L.; Fajerwerg, K.; Lakshmanan, P.; Potvin, C.; Méthivier, C.; Louis, C. Supported gold catalysts for the decomposition of VOC: Total oxidation of propene in low concentration as model reaction. Appl. Catal. B 2010, 94, 117–124. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Go, M.J.; Lee, B.K.; Kumar, P.A.; Lee, W.K.; Joo, O.S.; Ha, H.P.; Lim, H.B.; Hur, N.H. Immobilization of nanocatalysts on cordierite honeycomb monoliths for low temperature NOx reduction. Appl. Catal. A 2009, 370, 102–107. [Google Scholar] [CrossRef]
- Miquel, P.; Granger, P.; Jagtap, N.; Umbarkar, S.; Dongare, M.; Dujardin, C. NO reduction under diesel exhaust conditions over Au/Al2O3 prepared by deposition-precipitation method. J. Mol. Catal. A Chem. 2010, 322, 90–97. [Google Scholar] [CrossRef]
- Sárkány, A. Acetylene hydrogenation on SiO2 supported gold nanoparticles. React. Kinet. Catal. Lett. 2009, 96, 43–54. [Google Scholar] [CrossRef]
- Gluhoi, A.C.; Bakker, J.W.; Nieuwenhuys, B.E. Gold, still a surprising catalyst: Selective hydrogenation of acetylene to ethylene over Au nanoparticles. Catal. Today 2010, 154, 13–20. [Google Scholar] [CrossRef]
- Mohamed, M.; Khairou, K.S. Morphological characteristics of gold nanowires and nanoparticles: Structure elucidation and reactivity toward water-gas shift reaction. Energy Fuel 2009, 23, 4413–4419. [Google Scholar] [CrossRef]
- Rodriguez, J.A. Gold-based catalysts for the water-gas shift reaction: Active sites and reaction mechanism. Catal. Today 2011, 160, 3–10. [Google Scholar] [CrossRef]
- Lu, P.; Teranishi, T.; Asakura, K.; Miyake, M.; Toshima, N. Polymer-protected Ni/Pd bimetallic nano-clusters: Preparation, characterization and catalysis for hydrogenation of nitrobenzene. J. Phys. Chem. B 1999, 103, 9673–9682. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, Y. Large-scale synthesis of uniform silver nanowires through a soft, self-seeding, polyol process. Adv. Mater. 2002, 14, 833–837. [Google Scholar] [CrossRef]
- Comotti, M.; Li, W.C.; Spliethoff, B.; Schüth, F. Support effect in high activity gold catalysts for CO oxidation. JACS 2006, 128, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Liu, D.; Li, W.; Li, L.; Wang, L.; Zhou, X. Effects of dodecylamine and dodecanethiol on the conductive properties of nano-Ag films. Appl. Surf. Sci. 2011, 257, 5746–5753. [Google Scholar] [CrossRef]
- Dablemont, C.; Lang, P.; Mangeney, C.; Piquemal, J.Y.; Petkov, V.; Herbst, F.; Viau, G. FTIR and XPS study of Pt nanoparticle functionalization and interaction with alumina. Langmuir 2008, 24, 5832–5841. [Google Scholar] [CrossRef] [PubMed]
- Leff, D.V.; Brandt, L.; Heath, J.R. Synthesis and characterization of hydrophobic, organically-soluble gold nanocrystals functionalized with primary amines. Langmuir 1996, 12, 4723–4730. [Google Scholar] [CrossRef]
- Borodko, Y.; Habas, S.E.; Koebel, M.; Yang, P.; Frei, H.; Somorjai, G.A. Probing the interaction of poly(vinylpyrrolidone) with platinum nanocrystals by UV-Raman and FTIR. J. Phys. Chem. B 2006, 110, 23052–23059. [Google Scholar] [CrossRef] [PubMed]
- Calla, J.T.; Davis, R.J. X-ray absorption spectroscopy and CO oxidation activity of Au/Al2O3 treated with NaCN. Catal. Lett. 2005, 99, 21–26. [Google Scholar] [CrossRef]
- Liu, X.; Liu, M.H.; Luo, Y.C.; Mou, C.Y.; Lin, S.D.; Cheng, H.; Chen, J.M.; Lee, J.F.; Lin, T.S. Strong metal-support interactions between gold nanoparticles and ZnO nanorods in CO oxidation. JACS 2012, 134, 10251–10258. [Google Scholar] [CrossRef] [PubMed]
- Oxford, S.M.; Henao, J.D.; Yang, J.H.; Kung, M.C.; Kung, H.H. Understanding the effect of halide poisoning in CO oxidation over Au/TiO2. Appl. Catal. A 2008, 339, 180–186. [Google Scholar] [CrossRef]
- Oh, H.S.; Yang, J.H.; Costello, C.K.; Wang, Y.M.; Bare, S.R.; Kung, H.H.; Kung, M.C. Selective catalytic oxidation of CO: Effect of chloride on supported Au catalysts. J. Catal. 2002, 210, 375–386. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, S.D.; Lee, J.F. Chlorine residue in the Au/γ-Al2O3 prepared by AuCl3 impregnation—An EXAFS analysis. Catal. Lett. 2003, 89, 235–242. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Macrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Skupien, E.; Berger, R.; Santos, V.; Gascon, J.; Makkee, M.; Kreutzer, M.T.; Kooyman, P.J.; Moulijn, J.A.; Kapteijn, F. Inhibition of a gold-based catalyst in benzyl alcohol oxidation: Understanding and remediation. Catalysts 2014, 4, 89–115. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Average Au Particle Size (nm) | Standard Deviation (nm) |
|---|---|---|
| Au-DDA colloid | 3.3 | 0.5 |
| Au-DDA colloid (w) | 3.8 | 0.5 |
| Au-DDA/γ-Al2O3 | 4.0 | 0.8 |
| Au-DDA/γ-Al2O3 (O3 8 h) | 4.3 | 1.0 |
| Post-CO ox Au-DDA/γ-Al2O3 | 5.5 | 1.9 |
| Post-CO ox Au-DDA/γ-Al2O3 (O3 8 h) | 4.7 | 1.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puspitasari, I.; Skupien, E.; Kapteijn, F.; Kooyman, P.J. Au Capping Agent Removal Using Plasma at Mild Temperature. Catalysts 2016, 6, 179. https://doi.org/10.3390/catal6110179
Puspitasari I, Skupien E, Kapteijn F, Kooyman PJ. Au Capping Agent Removal Using Plasma at Mild Temperature. Catalysts. 2016; 6(11):179. https://doi.org/10.3390/catal6110179
Chicago/Turabian StylePuspitasari, Indra, Emmanuel Skupien, Freek Kapteijn, and Patricia J. Kooyman. 2016. "Au Capping Agent Removal Using Plasma at Mild Temperature" Catalysts 6, no. 11: 179. https://doi.org/10.3390/catal6110179