

Non-Noble Metal Aromatic Oxidation Catalysis: From Metalloenzymes to Synthetic Complexes

Abstract
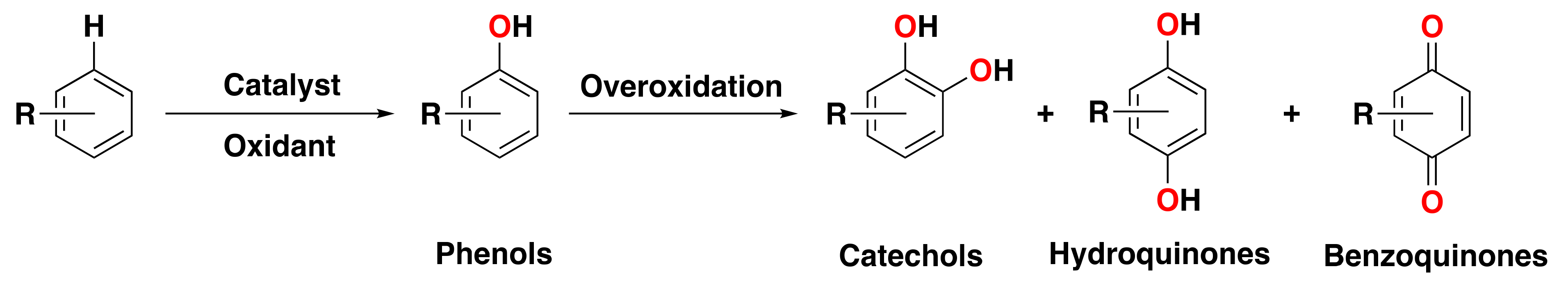
:1. Overview of Arene Oxidations
1.1. Relevance and Challenges
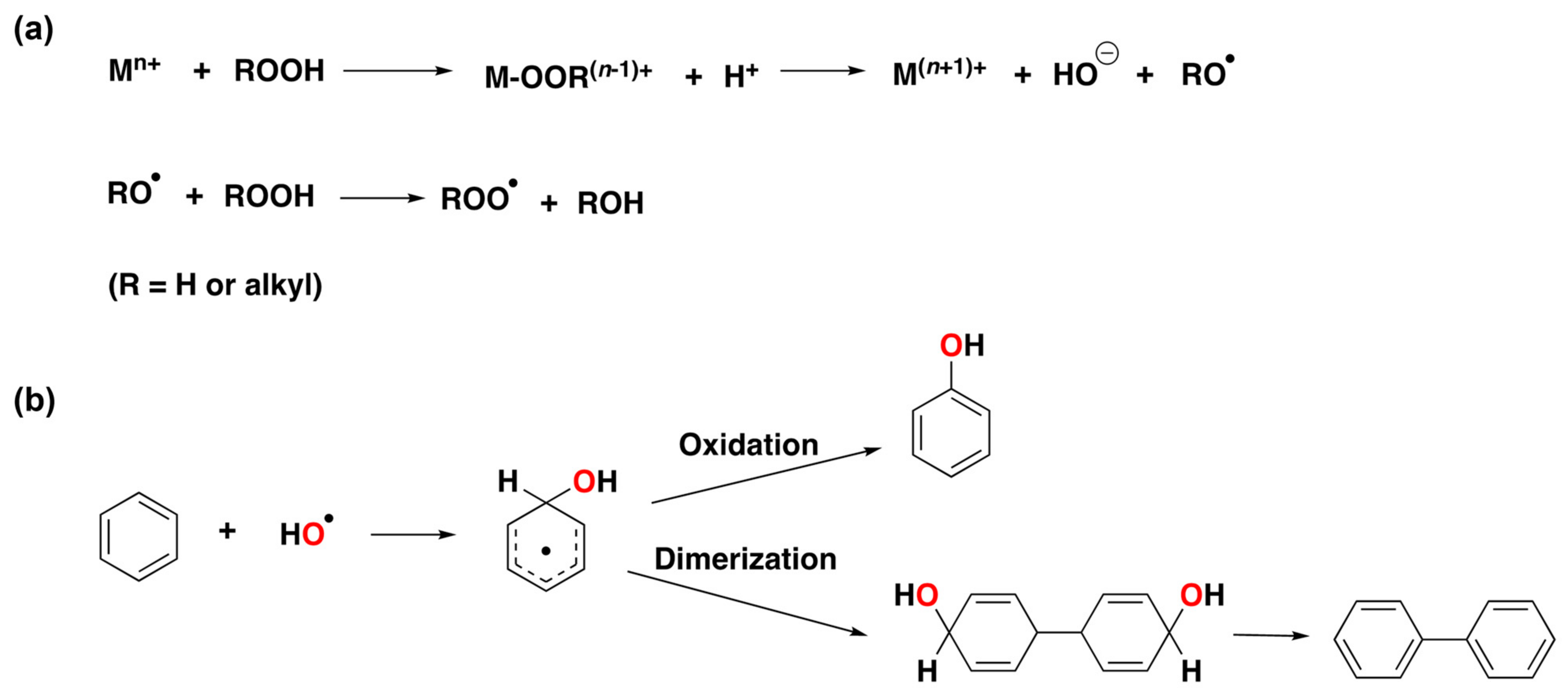
1.2. Hydroxyl Radicals vs. Metal-Based Oxidants
2. Iron in Biological and Synthetic Systems
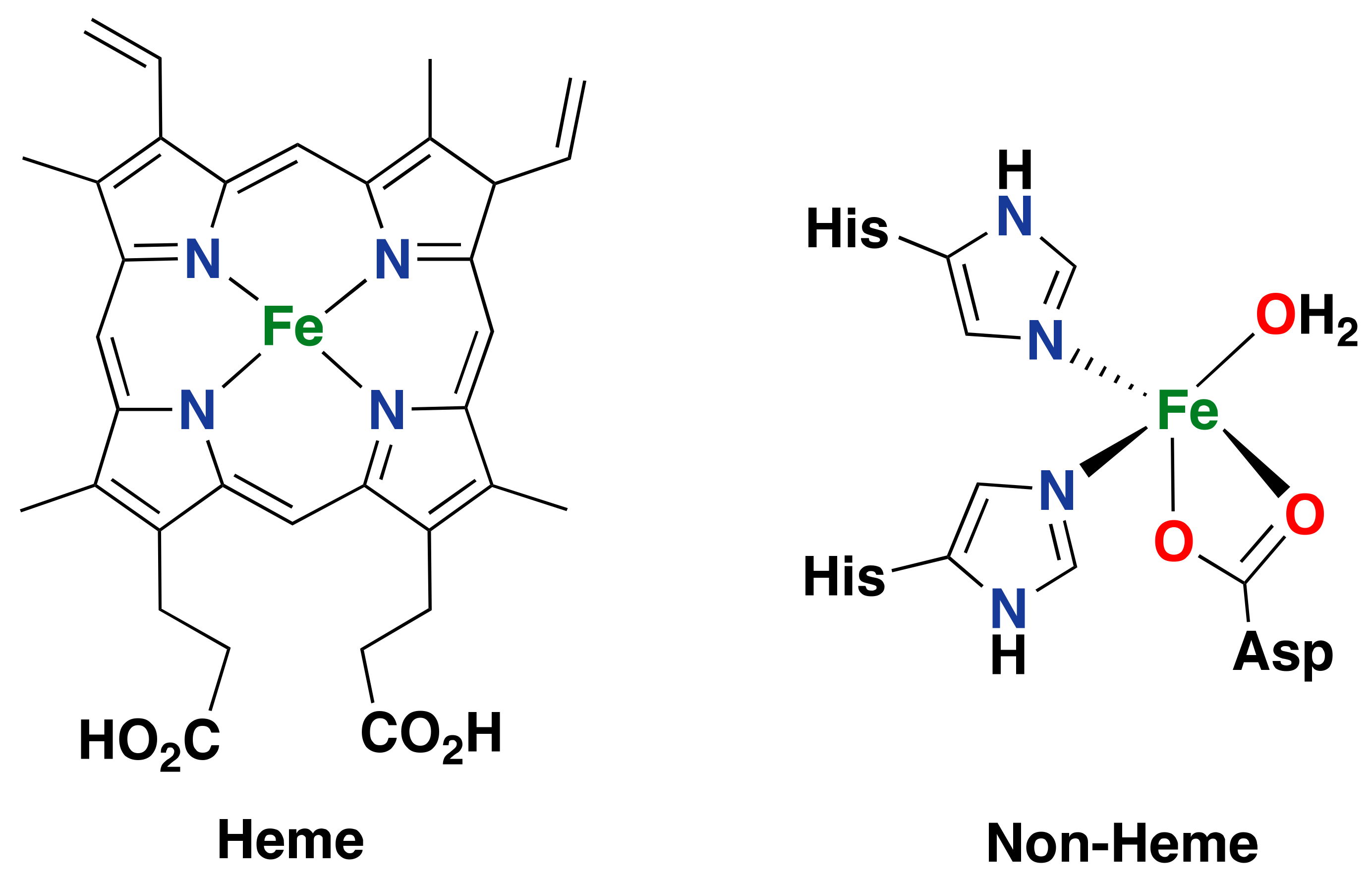
2.1. Iron-Containing Metalloenzymes
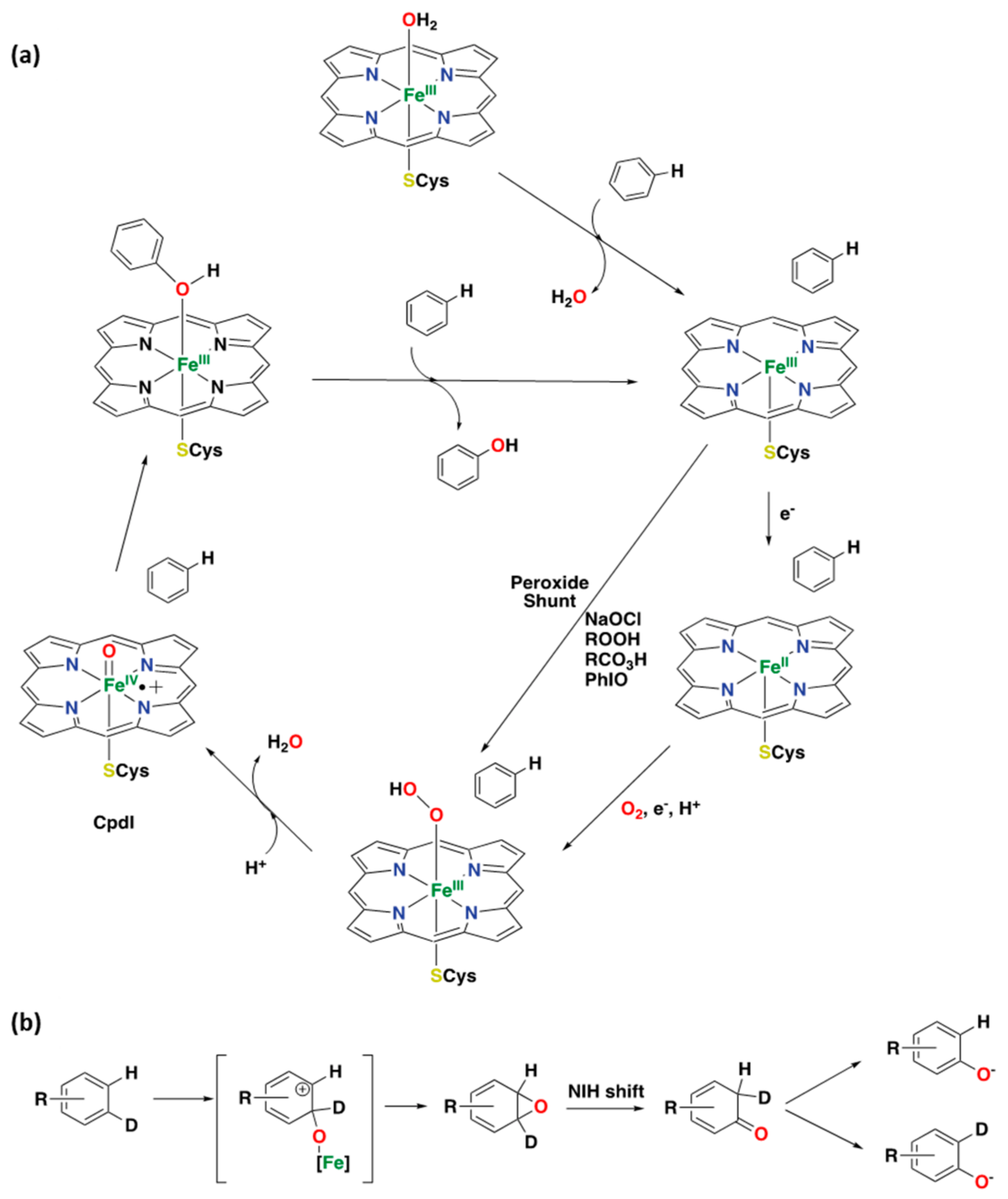
2.1.1. Cytochrome P450
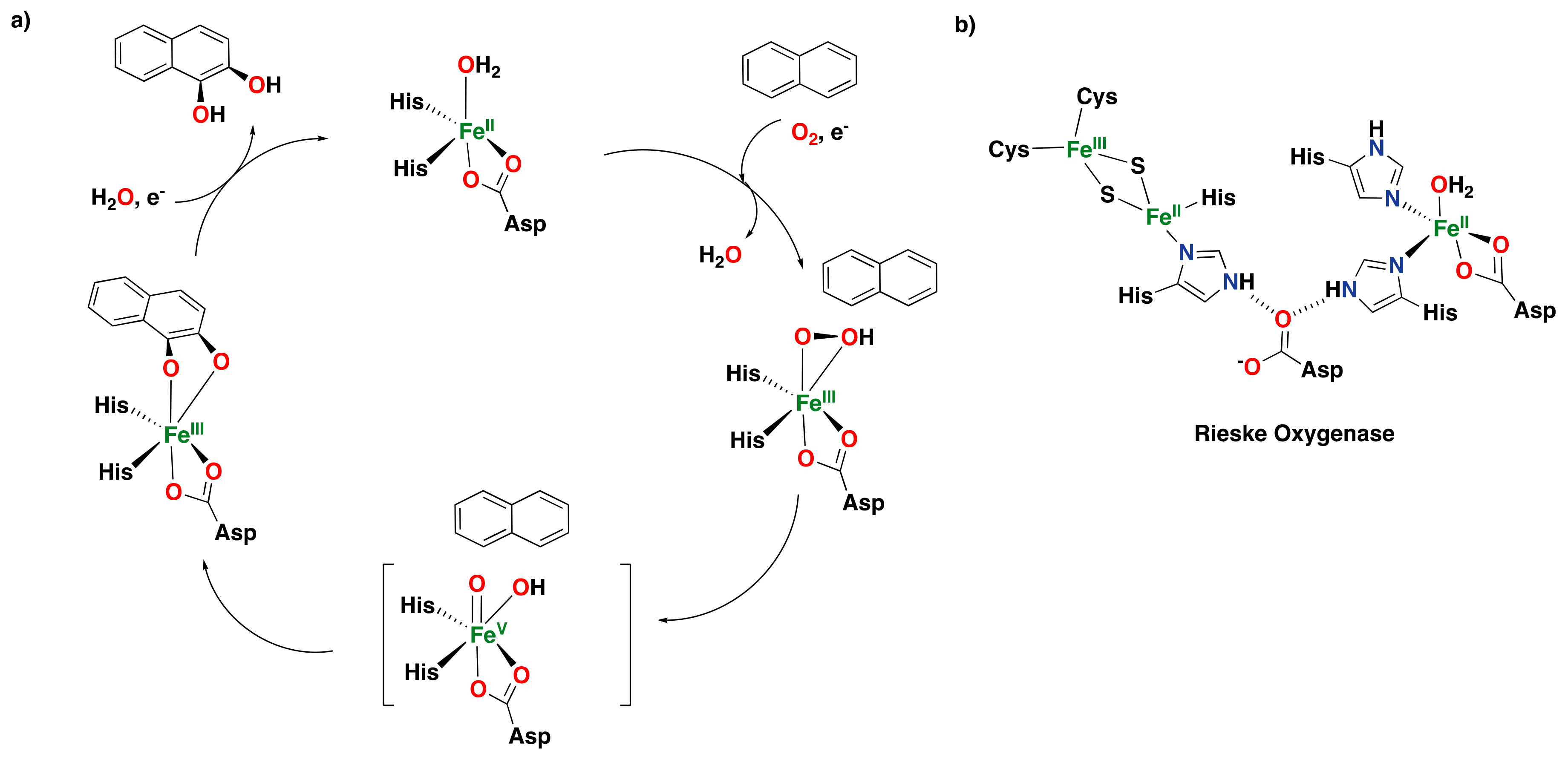
2.1.2. Rieske Oxygenases
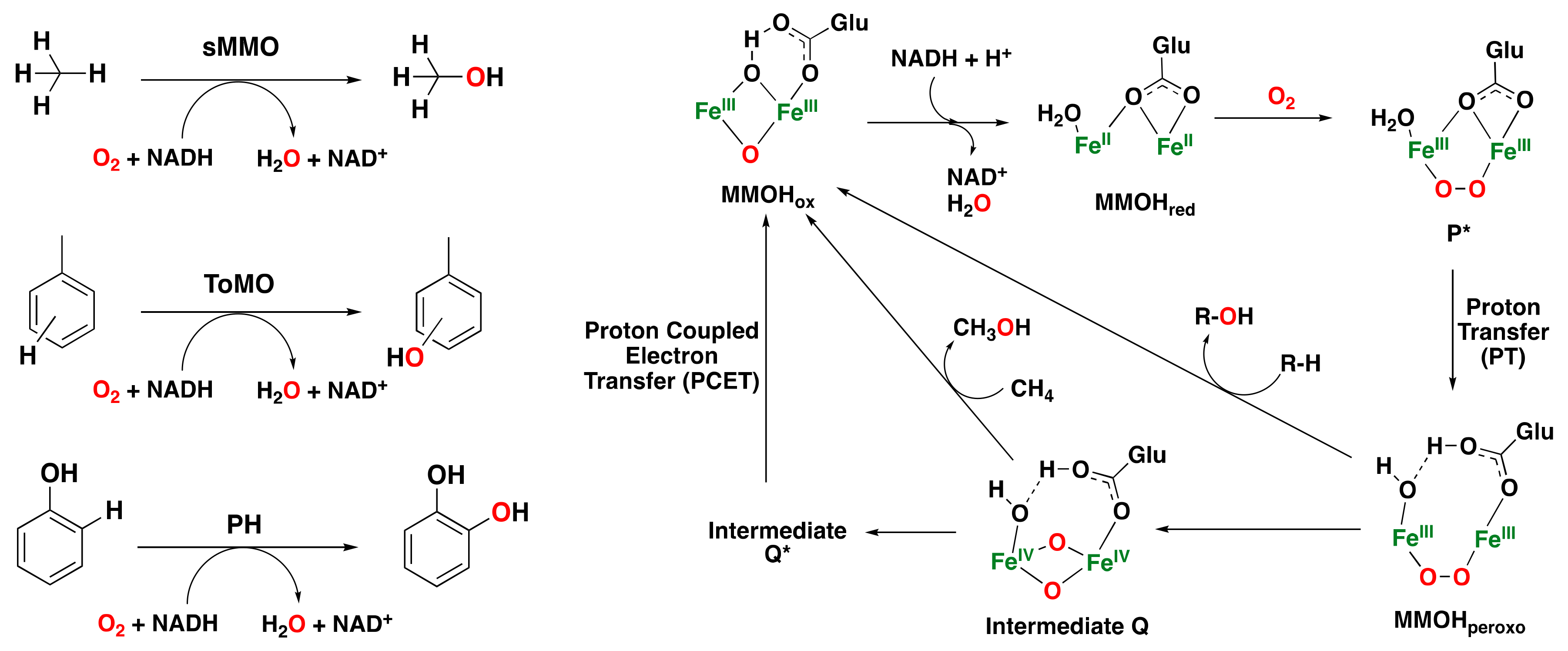
2.1.3. Bacterial Multicomponent Monooxygenases
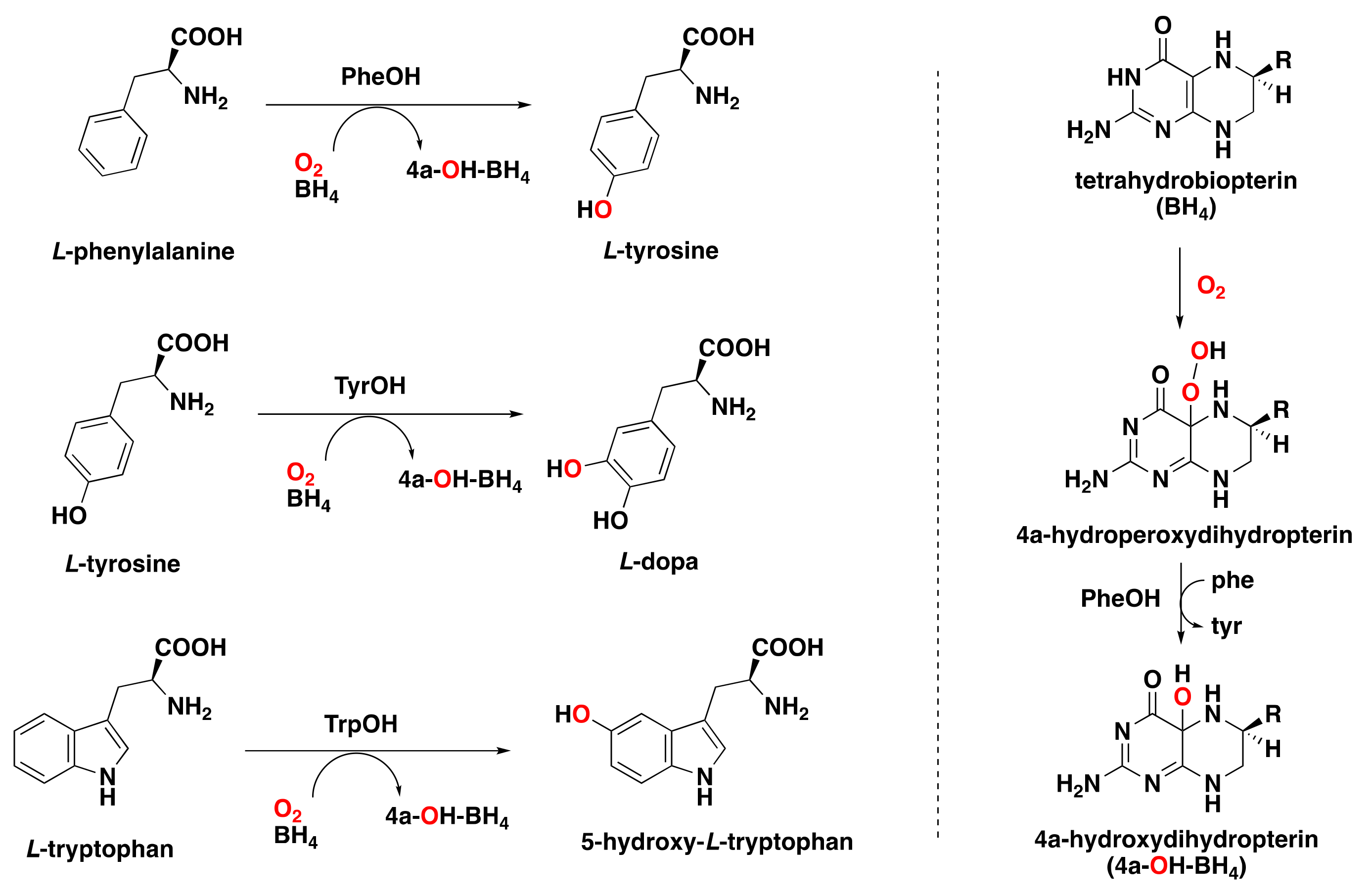
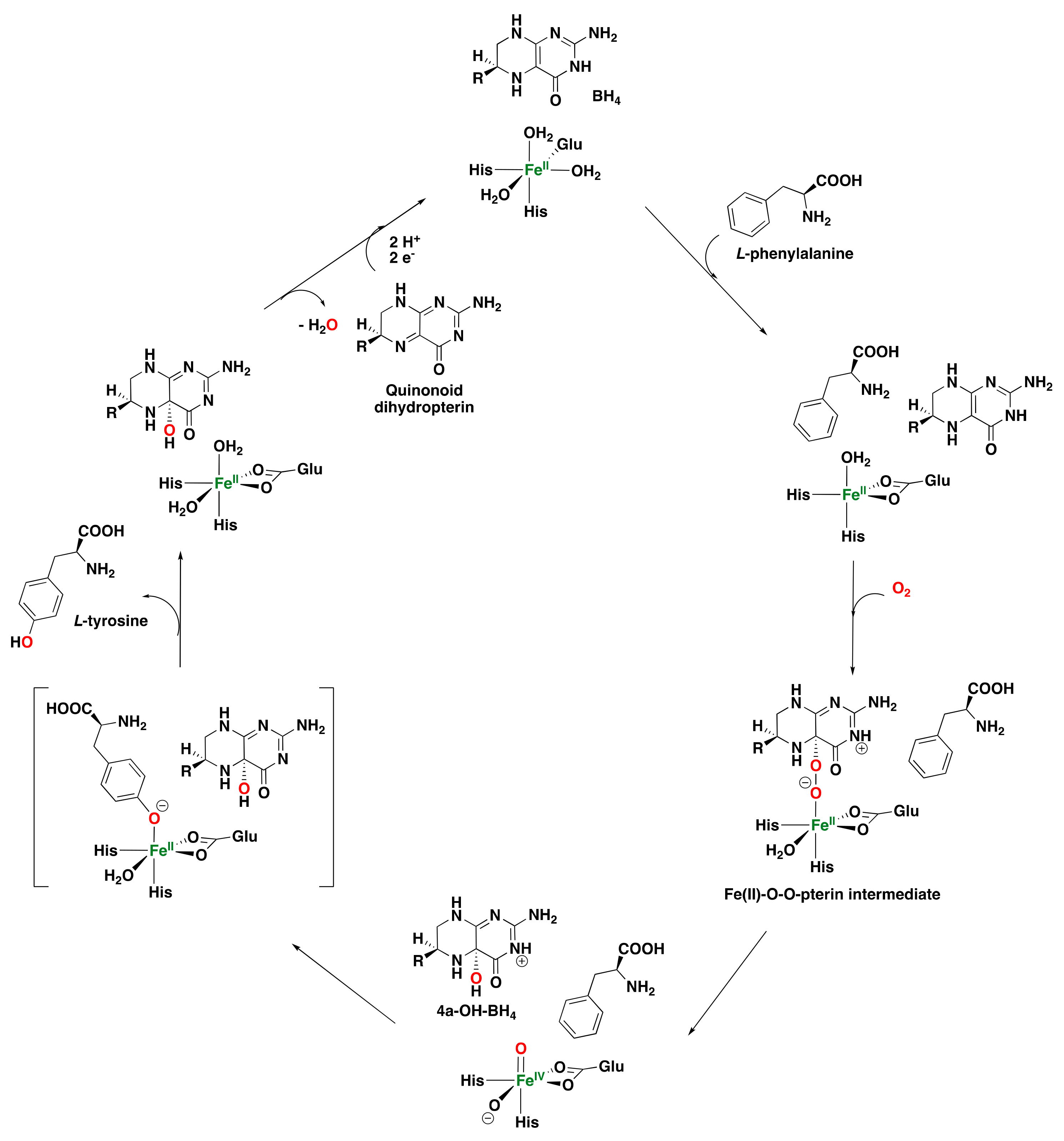
2.1.4. Pterin-Dependent Aromatic Amino Acid Hydroxylases
2.2. Synthetic Iron Systems
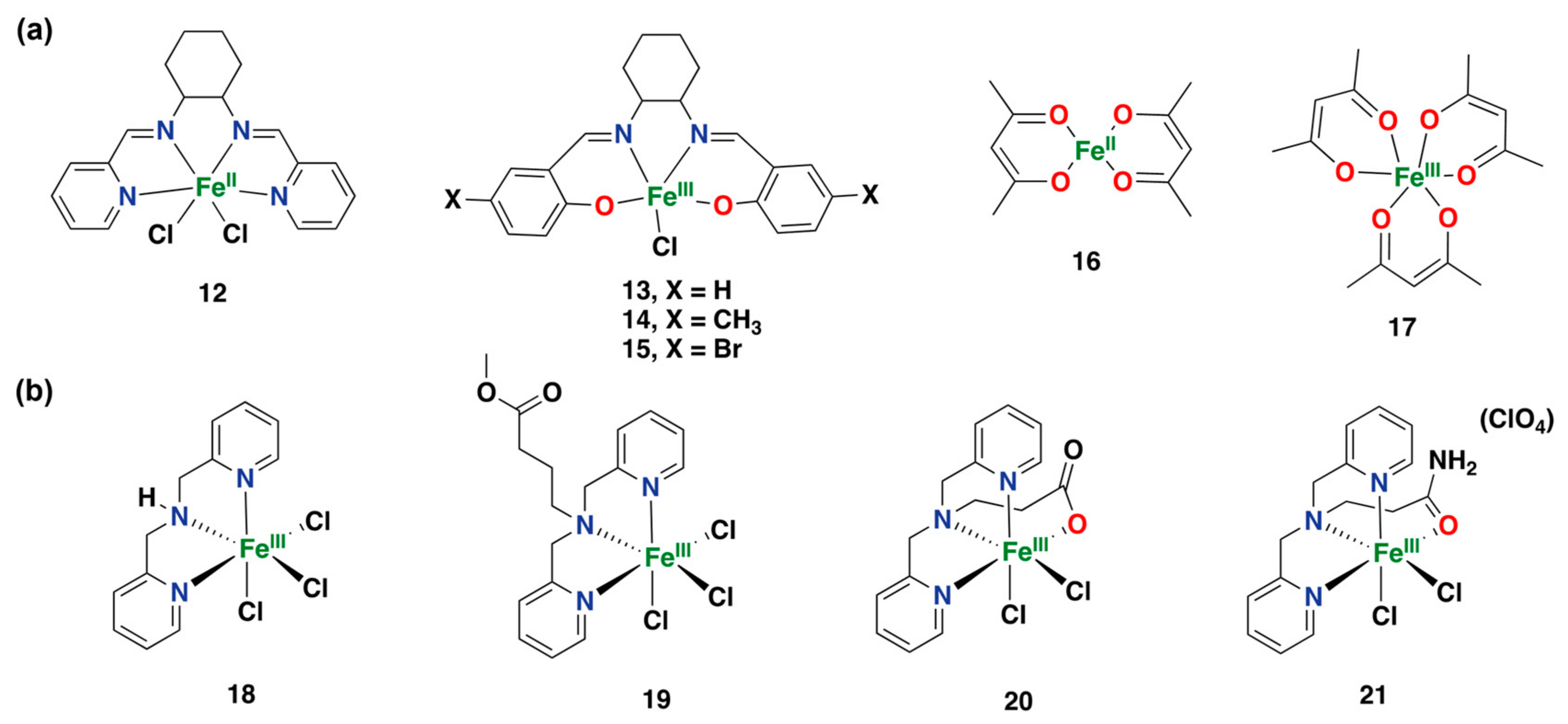
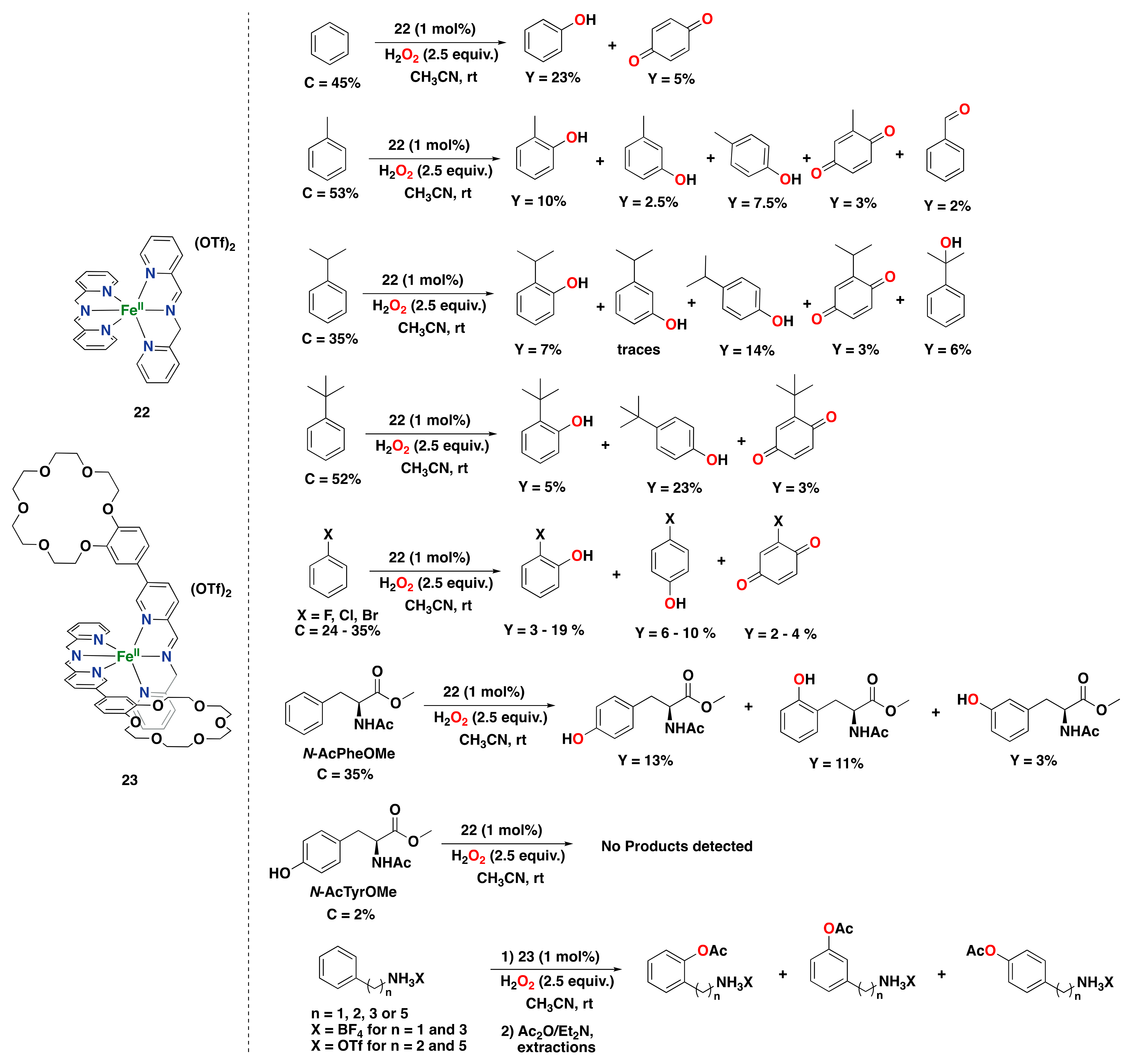
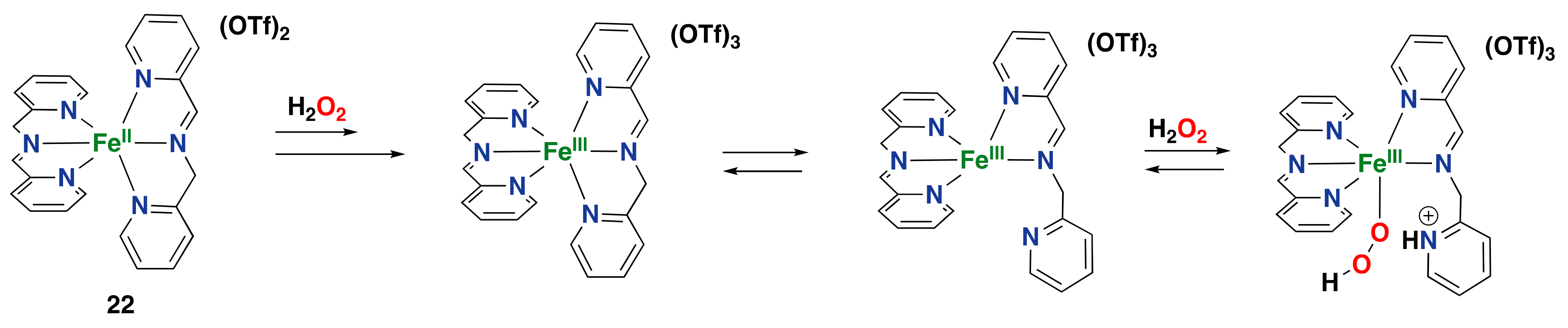
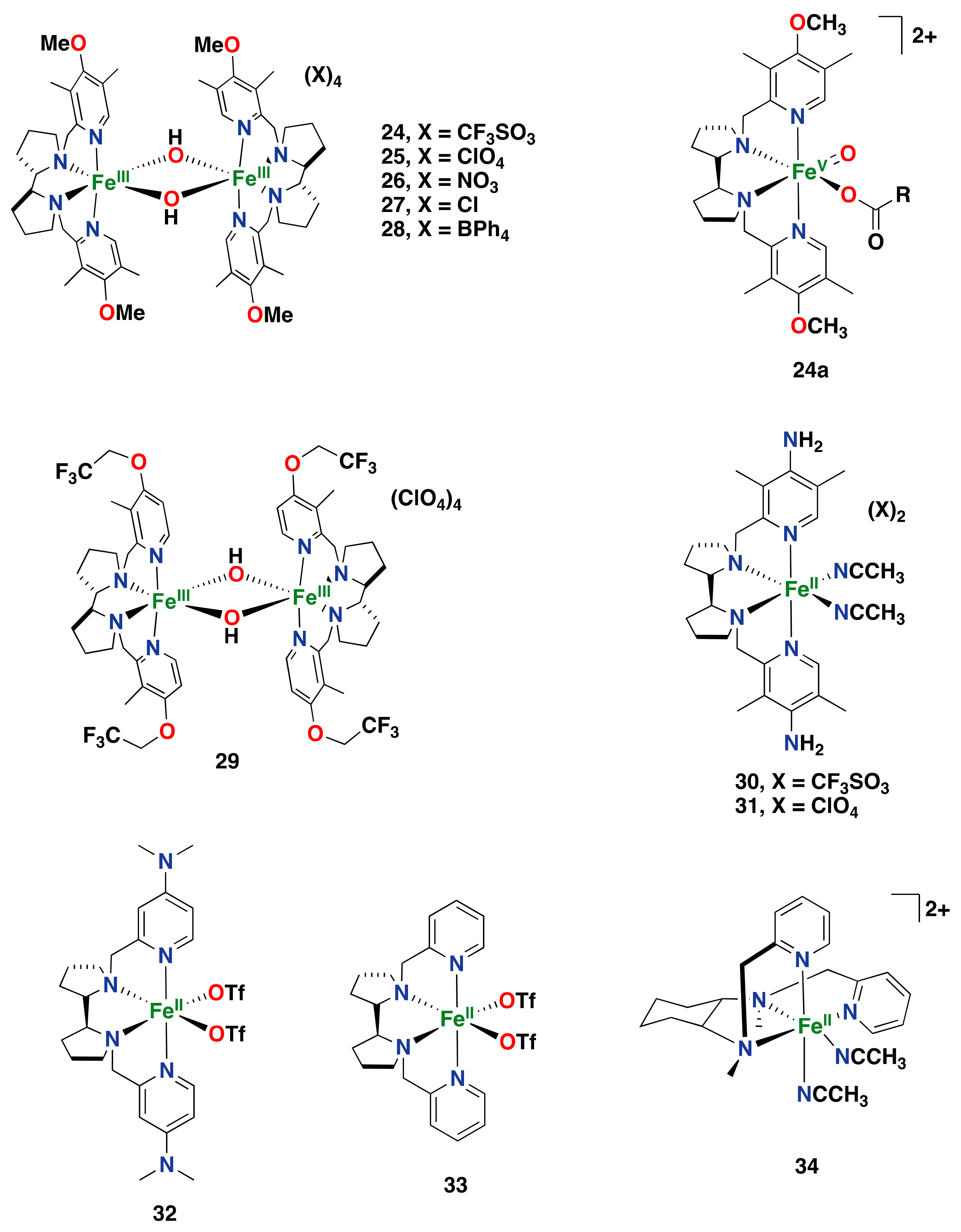
2.2.1. Iron-Based Systems and Oxidation Mechanism
2.2.2. Iron-Catalyzed Arene Oxidation
3. Copper in Biological and Synthetic Systems
3.1. Copper-Containing Metalloenzymes
Tyrosinase and Catechol Oxidases
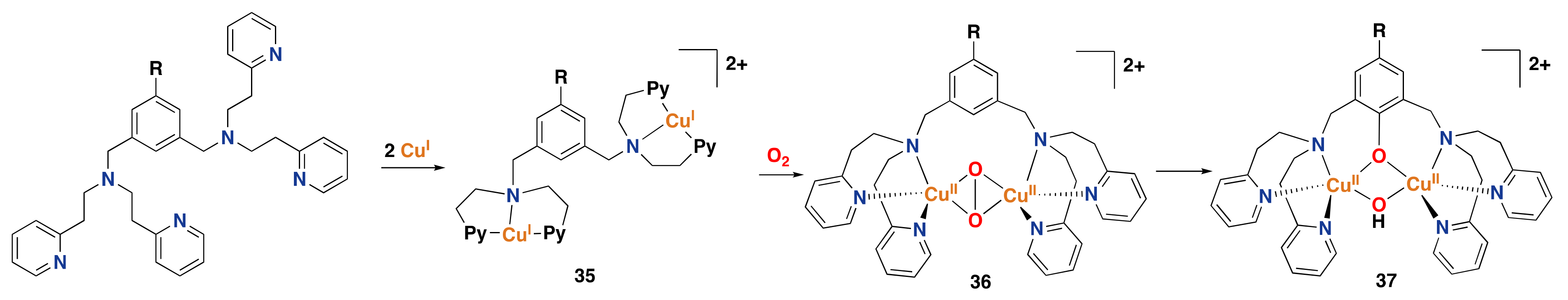
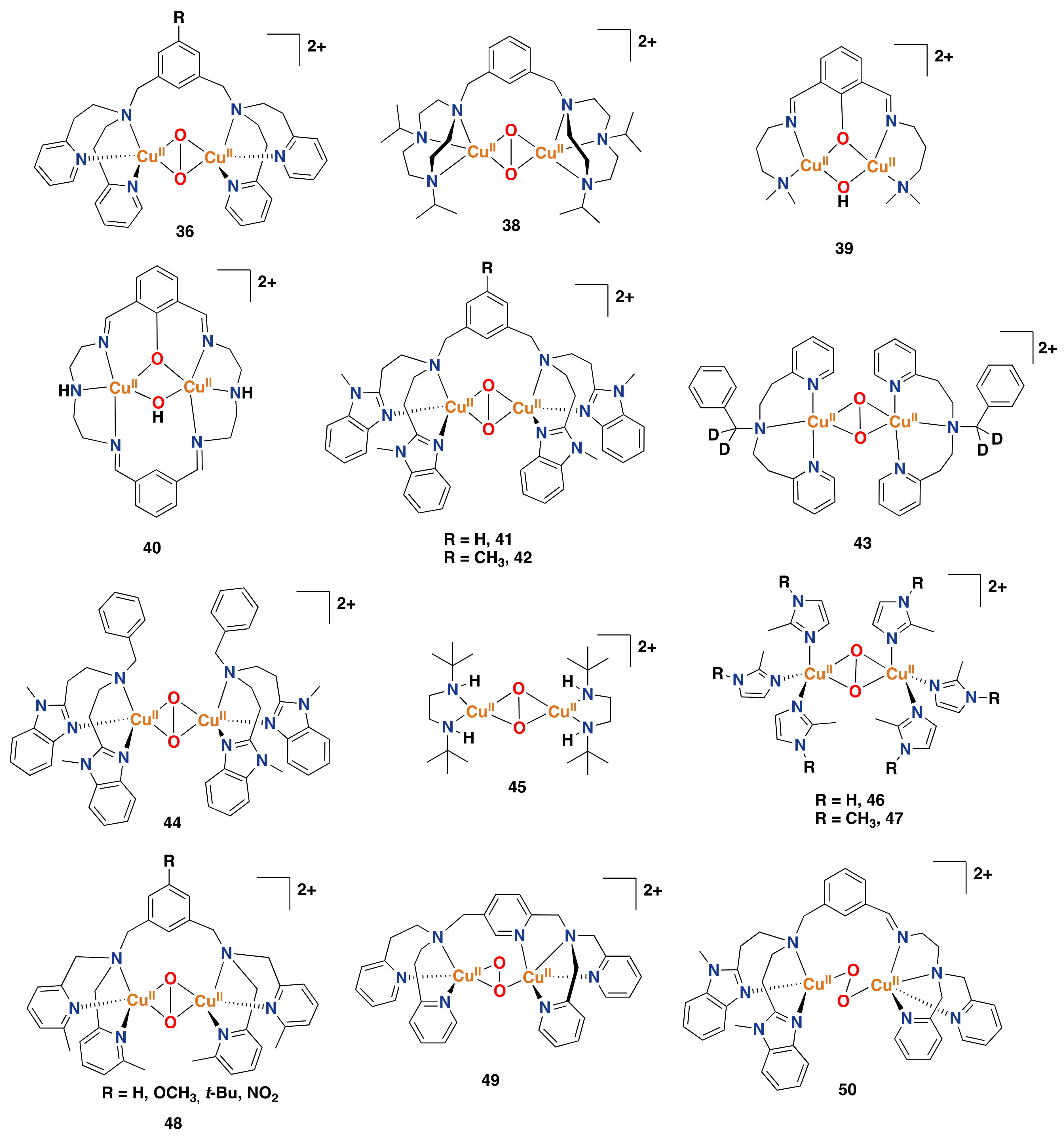
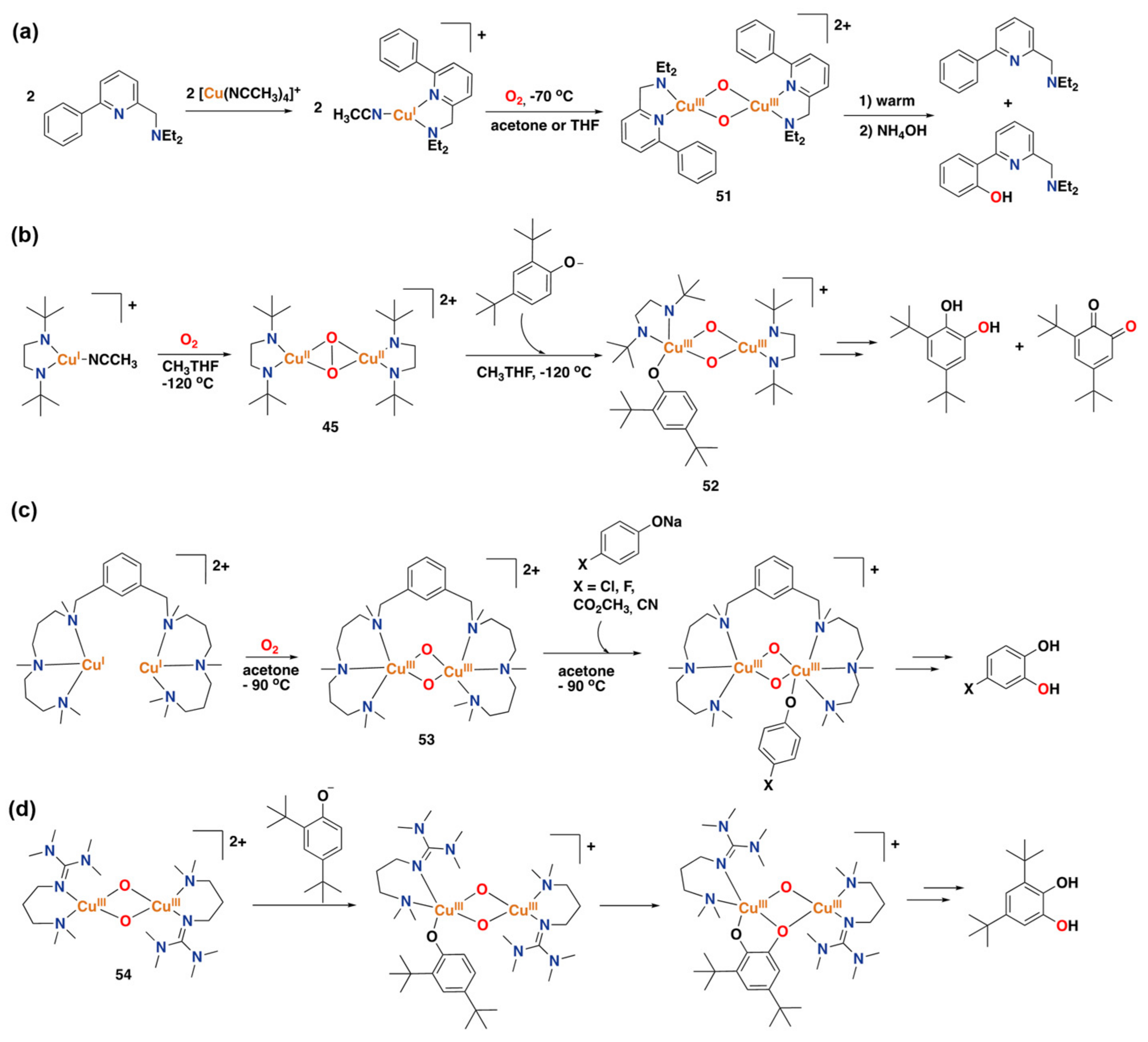
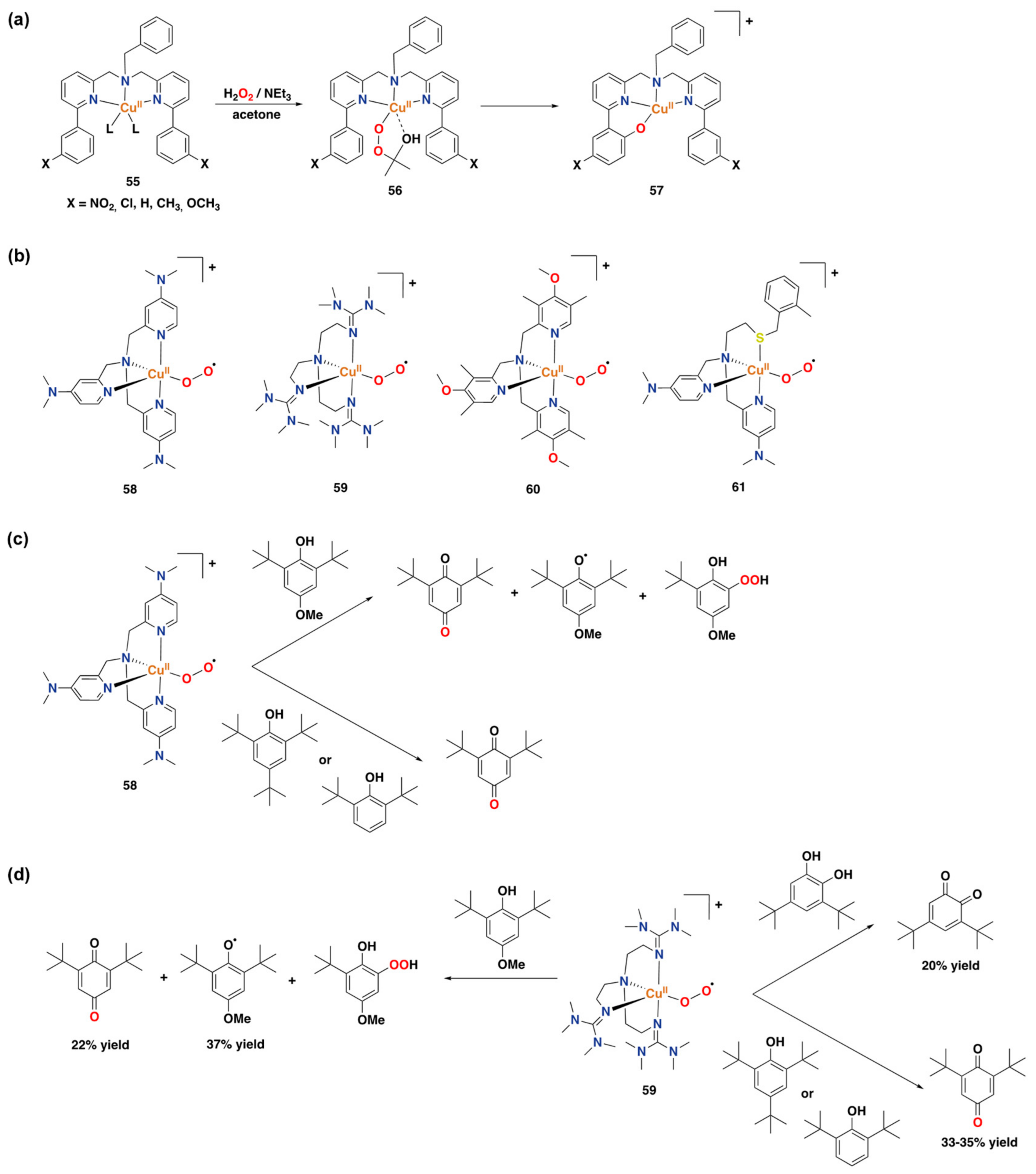
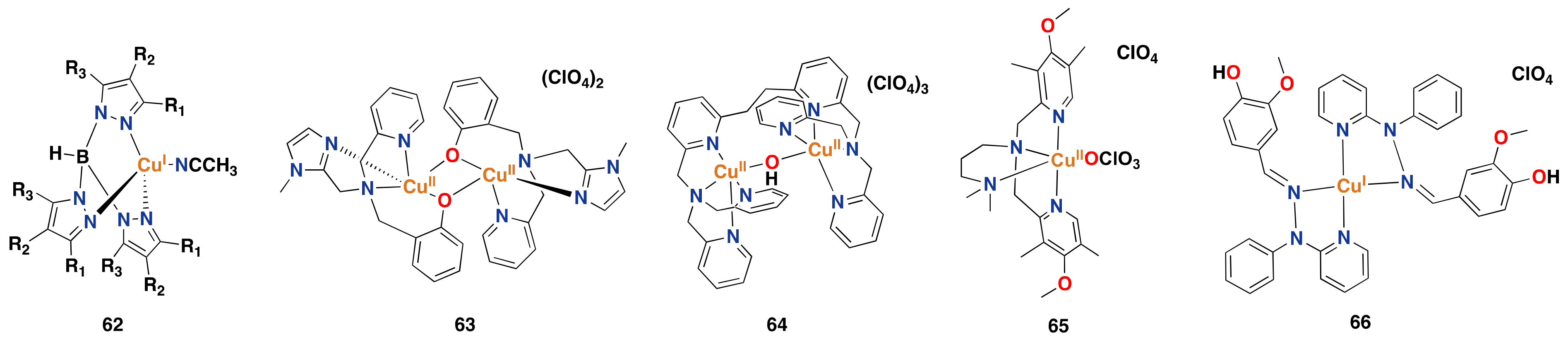
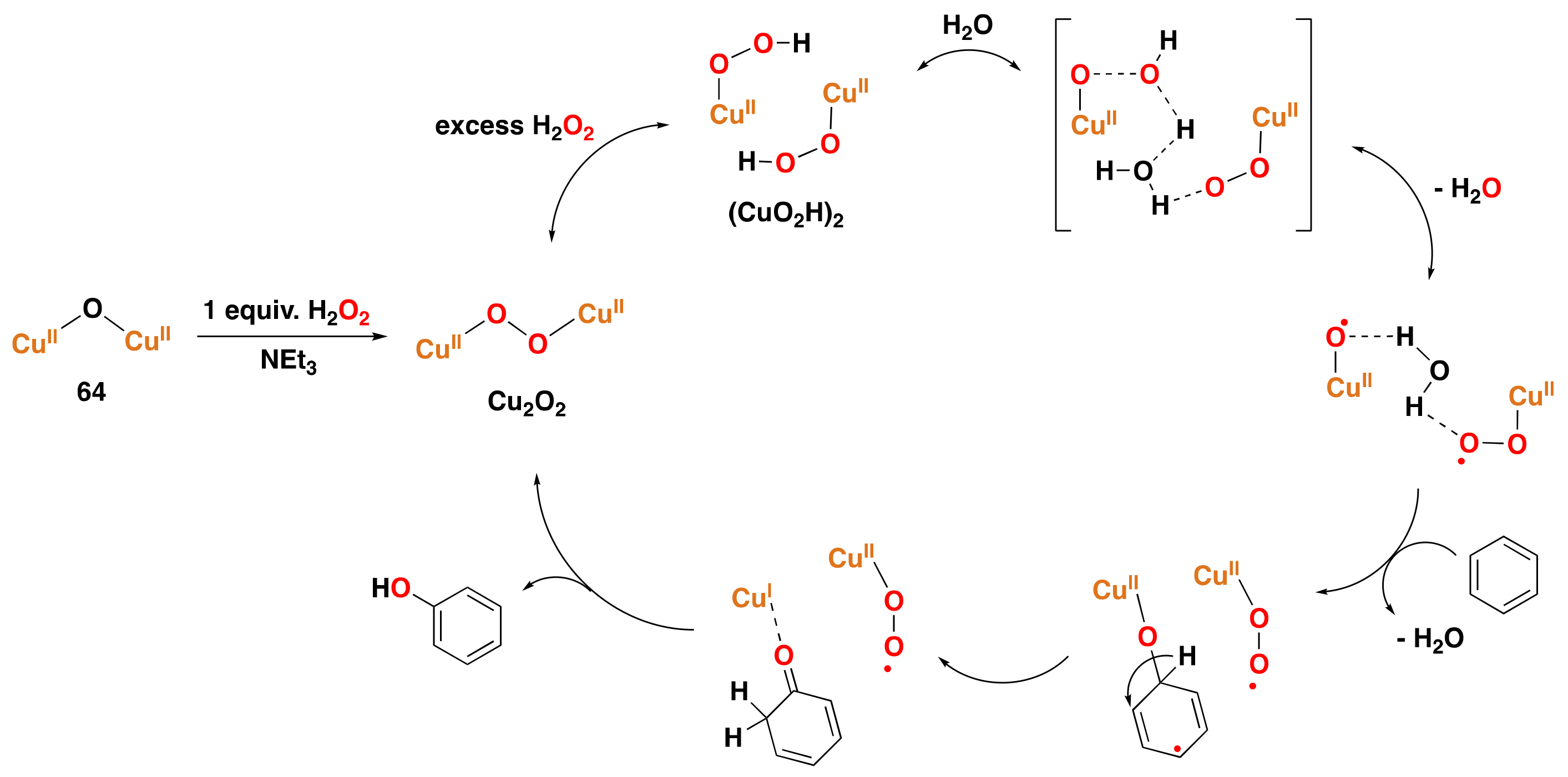
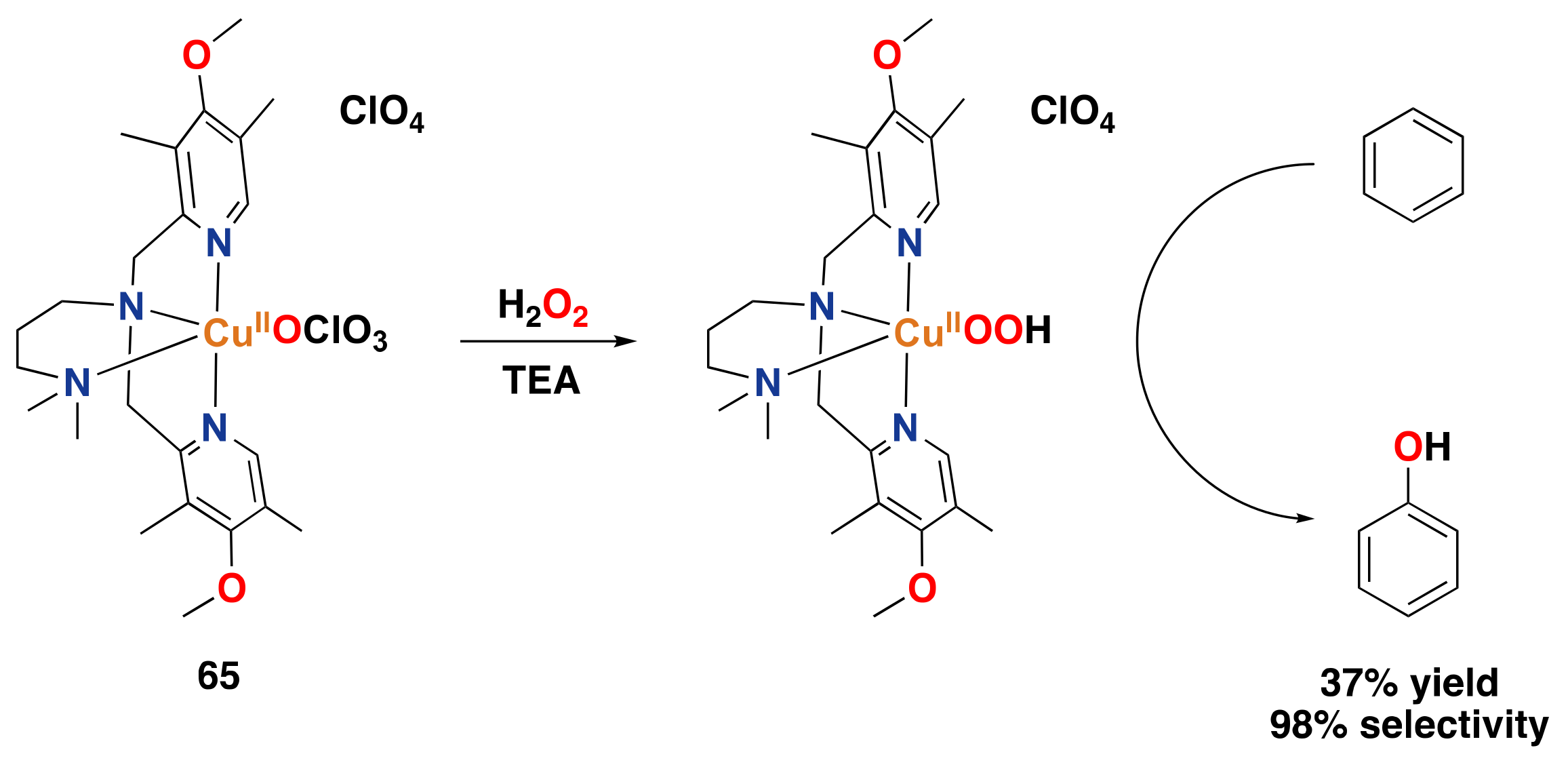
3.2. Synthetic Copper Systems

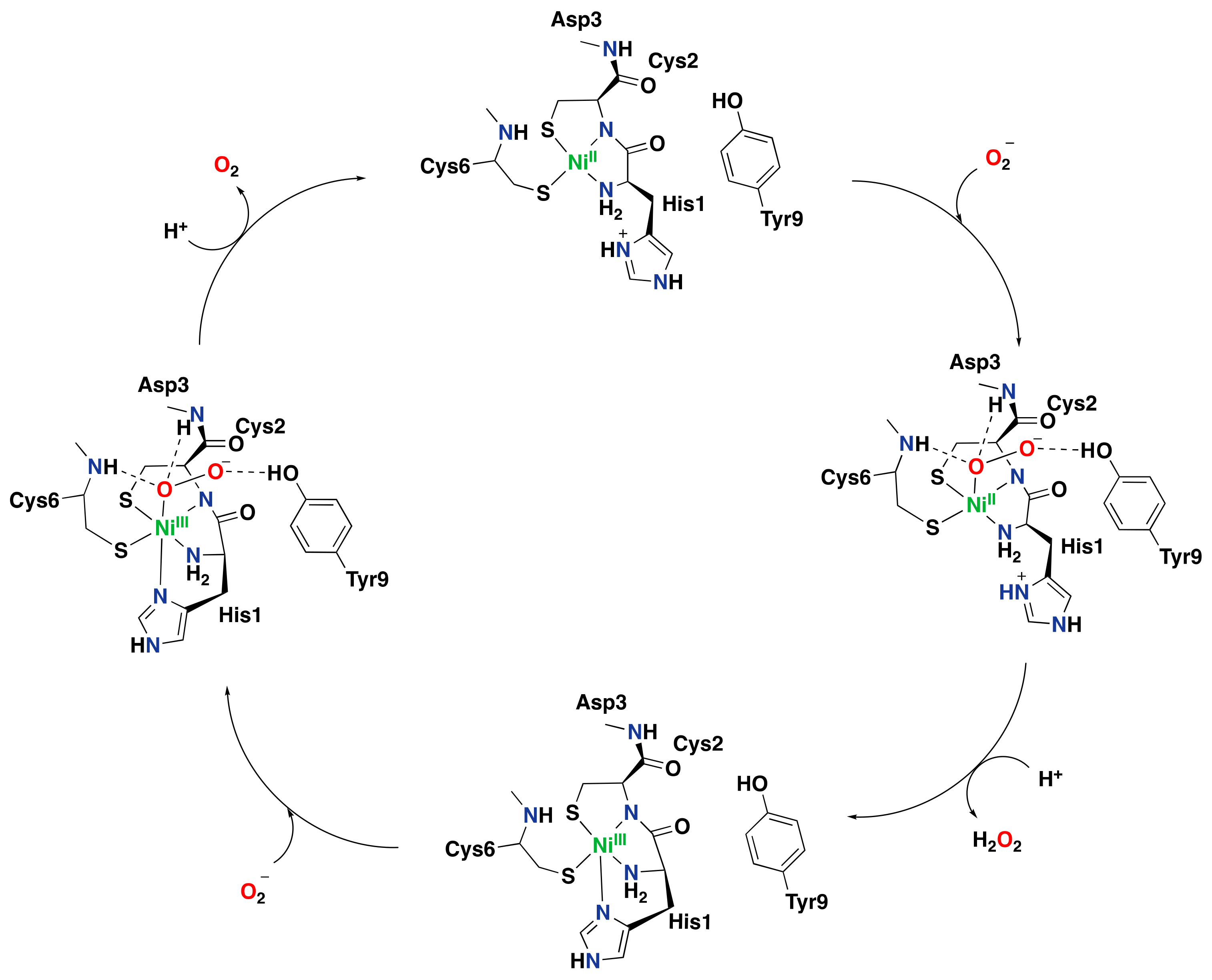
4. Nickel in Biological and Synthetic Systems
4.1. Nickel-Containing Metalloenzymes
Nickel Superoxide Dismutase
4.2. Synthetic Aromatic Oxidation Systems Based on Nickel
5. Manganese in Biological and Synthetic Systems
5.1. Manganese-Containing Metalloenzymes
5.2. Synthetic Manganese Systems
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Arakawa, H.; Aresta, M.; Armor, J.N.; Barteau, M.A.; Beckman, E.J.; Bell, A.T.; Bercaw, J.E.; Creutz, C.; Dinjus, E.; Dixon, D.A.; et al. Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities. Chem. Rev. 2001, 101, 953–996. [Google Scholar] [CrossRef] [PubMed]
- Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem. Rev. 2005, 105, 2329–2363. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Zhao, J. Aliphatic C–H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Luo, T.; Lei, X. Late-Stage Diversification of Natural Products. ACS Cent. Sci. 2020, 6, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Quevedo, R.E.; Kohrt, J.T.; Oderinde, M.S.; Reilly, U.; White, M.C. Late-stage oxidative C(sp3)–H methylation. Nature 2020, 580, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Genovino, J.; Sames, D.; Hamann, L.G.; Touré, B.B. Accessing Drug Metabolites via Transition-Metal Catalyzed C–H Oxidation: The Liver as Synthetic Inspiration. Angew. Chem. Int. Ed. 2016, 55, 14218–14238. [Google Scholar] [CrossRef]
- Börgel, J.; Tanwar, L.; Berger, F.; Ritter, T. Late-stage aromatic C–H oxygenation. J. Am. Chem. Soc. 2018, 140, 16026–16031. [Google Scholar] [CrossRef]
- Haggin, J. Chemists seek greater recognition for catalysis. Chem. Eng. News 1993, 71, 22. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Concepts in homogeneous catalysis: The industrial view. J. Catal. 2003, 216, 23–31. [Google Scholar] [CrossRef]
- Weber, M.; Weber, M.; Kleine-Boymann, M. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2004; pp. 503–519. [Google Scholar]
- Fukuzumi, S.; Ohkubo, K. One-Step Selective Hydroxylation of Benzene to Phenol. Asian J. Org. Chem. 2015, 4, 836–845. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds; Academic Press: New York, NY, USA, 1981; pp. 315–339. [Google Scholar]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Lewis, J.C.; Coelho, P.S.; Arnold, F.H. Enzymatic functionalization of carbon–hydrogen bonds. Chem. Soc. Rev. 2011, 40, 2003–2021. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Sorokin, A.; Bernadou, J.; Meunier, B. Metalloporphyrin-catalyzed oxidation of 2-methylnaphthalene to vitamin K3 and 6-methyl-1,4-naphthoquinone by potassium monopersulfate in aqueous solution. J. Org. Chem. 1997, 62, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.; Meunier, B. Oxidation of polycyclic aromatic hydrocarbons catalyzed by iron tetrasulfophthalocyanine FePcS: Inverse isotope effects and oxygen labeling studies. Eur. J. Inorg. Chem. 1998, 1998, 1269–1281. [Google Scholar] [CrossRef]
- Higuchi, T.; Satake, C.; Hirobe, M. Selective quinone formation by oxidation of aromatics with heteroaromatic N-oxides catalyzed by ruthenium porphyrins. J. Am. Chem. Soc. 1995, 117, 8879–8880. [Google Scholar] [CrossRef]
- Khavasi, H.R.; Davarani, S.S.H.; Safari, N. Remarkable solvent effect on the yield and specificity of oxidation of naphthalene catalyzed by iron(III) porphyrins. J. Mol. Catal. A Chem. 2002, 188, 115–122. [Google Scholar] [CrossRef]
- Klein Gebbink, R.J.M.; Moret, M.-E. Non-Noble Metal Catalysis: Molecular Approaches and Reactions; John Wiley & Sons: Weinmar, Germany, 2019. [Google Scholar]
- Wallar, B.J.; Lipscomb, J.D. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 1996, 96, 2625–2658. [Google Scholar] [CrossRef]
- Que, L., Jr.; Ho, R.Y. Dioxygen activation by enzymes with mononuclear non-heme iron active sites. Chem. Rev. 1996, 96, 2607–2624. [Google Scholar] [CrossRef]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and functional aspects of metal sites in biology. Chem. Rev. 1996, 96, 2239–2314. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.; van Koten, G.; Klein Gebbink, R.J.M. Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: Recent developments in enzymology and modeling studies. Chem. Soc. Rev. 2008, 37, 2716–2744. [Google Scholar] [CrossRef]
- Que, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and nonheme high-valent iron and manganese oxo cores in biological and abiological oxidation reactions. ACS Cent. Sci. 2019, 5, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Chen, P.; Metz, M.; Lee, S.K.; Palmer, A.E. Oxygen binding, activation, and reduction to water by copper proteins. Angew. Chem. Int. Ed. 2001, 40, 4570–4590. [Google Scholar] [CrossRef]
- Pecoraro, V.L.; Baldwin, M.J.; Gelasco, A. Interaction of manganese with dioxygen and its reduced derivatives. Chem. Rev. 1994, 94, 807–826. [Google Scholar] [CrossRef]
- Law, N.A.; Caudle, M.T.; Pecoraro, V.L. Manganese redox enzymes and model systems: Properties, structures, and reactivity. Adv. Inorg. Chem. 1998, 46, 305–440. [Google Scholar]
- Youn, H.-D.; Kim, E.-J.; Roe, J.-H.; Hah, Y.C.; Kang, S.-O. A novel nickel-containing superoxide dismutase from Streptomyces spp. Biochem. J. 1996, 318, 889–896. [Google Scholar] [CrossRef]
- Barondeau, D.P.; Kassmann, C.J.; Bruns, C.K.; Tainer, J.A.; Getzoff, E.D. Nickel superoxide dismutase structure and mechanism. Biochemistry 2004, 43, 8038–8047. [Google Scholar] [CrossRef]
- Ragsdale, S.W. Nickel-based enzyme systems. J. Biol. Chem. 2009, 284, 18571–18575. [Google Scholar] [CrossRef]
- Kirby, A.J.; Hollfelder, F. From Enzyme Models to Model Enzymes; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.-K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.-S.; Zhou, J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000, 100, 235–350. [Google Scholar] [CrossRef]
- Neidig, M.L.; Solomon, E.I. Structure–function correlations in oxygen activating non-heme iron enzymes. Chem. Commun. 2005, 21, 5843–5863. [Google Scholar] [CrossRef]
- Matsumoto, K.; Tachikawa, S.; Hashimoto, N.; Nakano, R.; Yoshida, M.; Shindo, M. Aerobic C–H Oxidation of Arenes Using a Recyclable, Heterogeneous Rhodium Catalyst. J. Org. Chem. 2017, 82, 4305–4316. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Kozhushkov, S.I.; Ackermann, L.; Vaccaro, L. Heterogeneous catalytic approaches in C–H activation reactions. Green Chem. 2016, 18, 3471–3493. [Google Scholar] [CrossRef]
- Reay, A.J.; Fairlamb, I.J. Catalytic C–H bond functionalisation chemistry: The case for quasi-heterogeneous catalysis. Chem. Commun. 2015, 51, 16289–16307. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, N.; Amiri, A.; Ziarani, G.M.; Badiei, A. Review of some transition metal-based mesoporous catalysts for the direct hydroxylation of benzene to phenol (DHBP). Mol. Catal. 2021, 515, 111873. [Google Scholar] [CrossRef]
- Vicens, L.; Olivo, G.; Costas, M. Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar] [CrossRef]
- Bailey, C.L.; Drago, R.S. Utilization of O2 for the specific oxidation of organic substrates with cobalt(II) catalysts. Coord. Chem. Rev. 1987, 79, 321–332. [Google Scholar] [CrossRef]
- Carneiro, L.; Silva, A.R. Selective direct hydroxylation of benzene to phenol with hydrogen peroxide by iron and vanadyl based homogeneous and heterogeneous catalysts. Catal. Sci. Technol. 2016, 6, 8166–8176. [Google Scholar] [CrossRef]
- Han, J.W.; Jung, J.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Photocatalytic oxidation of benzene to phenol using dioxygen as an oxygen source and water as an electron source in the presence of a cobalt catalyst. Chem. Sci. 2017, 8, 7119–7125. [Google Scholar] [CrossRef]
- Anandababu, K.; Muthuramalingam, S.; Velusamy, M.; Mayilmurugan, R. Single-step benzene hydroxylation by cobalt(II) catalysts via a cobalt(III)-hydroperoxo intermediate. Catal. Sci. Technol. 2020, 10, 2540–2548. [Google Scholar] [CrossRef]
- Lemke, K.; Ehrich, H.; Lohse, U.; Berndt, H.; Jähnisch, K. Selective hydroxylation of benzene to phenol over supported vanadium oxide catalysts. Appl. Catal. A Gen. 2003, 243, 41–51. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, Y.; Li, G.; Hu, C. Room temperature direct oxidation of benzene to phenol using hydrogen peroxide in the presence of vanadium-substituted heteropolymolybdates. Appl. Catal. A Gen. 2005, 278, 251–261. [Google Scholar] [CrossRef]
- Masferrer-Rius, E. Manganese and Nickel Complexes as Catalysts for Aromatic Oxidation: Novel Methodologies for Non-Noble Metal Catalysis. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2022. [Google Scholar]
- Rajeev, A.; Balamurugan, M.; Sankaralingam, M. Rational Design of First-Row Transition Metal Complexes as the Catalysts for Oxidation of Arenes: A Homogeneous Approach. ACS Catal. 2022, 12, 9953–9982. [Google Scholar] [CrossRef]
- Fenton, H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. Üeber die Katalyse des Hydroperoxydes. Naturwissenschaften 1932, 20, 948–950. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. A Math. Phys. Sci. 1934, 147, 332–351. [Google Scholar]
- Walling, C. Intermediates in the reactions of Fenton type reagents. Acc. Chem. Res. 1998, 31, 155–157. [Google Scholar] [CrossRef]
- MacFaul, P.A.; Wayner, D.; Ingold, K. A radical account of “Oxygenated Fenton chemistry”. Acc. Chem. Res. 1998, 31, 159–162. [Google Scholar] [CrossRef]
- Goldstein, S.; Meyerstein, D. Comments on the mechanism of the “Fenton-like” reaction. Acc. Chem. Res. 1999, 32, 547–550. [Google Scholar] [CrossRef]
- Merz, J.; Waters, W. 511. The oxidation of aromatic compounds by means of the free hydroxyl radical. J. Chem. Soc. 1949, 2427–2433. [Google Scholar] [CrossRef]
- Smith, J.L.; Norman, R. 539. Hydroxylation. Part I. The oxidation of benzene and toluene by Fenton’s reagent. J. Chem. Soc. 1963, 2897–2905. [Google Scholar] [CrossRef]
- Walling, C.; Johnson, R.A. Fenton’s reagent. V. Hydroxylation and side-chain cleavage of aromatics. J. Am. Chem. Soc. 1975, 97, 363–367. [Google Scholar] [CrossRef]
- Kurata, T.; Watanabe, Y.; Katoh, M.; Sawaki, Y. Mechanism of aromatic hydroxylation in the Fenton and related reactions. One-electron oxidation and the NIH shift. J. Am. Chem. Soc. 1988, 110, 7472–7478. [Google Scholar] [CrossRef]
- Cussó, O.; Garcia-Bosch, I.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Asymmetric epoxidation with H2O2 by manipulating the electronic properties of non-heme iron catalysts. J. Am. Chem. Soc. 2013, 135, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- MacFaul, P.A.; Ingold, K.; Wayner, D.; Que, L. A Putative Monooxygenase Mimic Which Functions via Well-Disguised Free Radical Chemistry1. J. Am. Chem. Soc. 1997, 119, 10594–10598. [Google Scholar] [CrossRef]
- Ingold, K.U.; MacFaul, P.A. Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier, B., Ed.; Imperial College Press: London, UK, 2000; p. 45. [Google Scholar]
- Hiatt, R.; Clipsham, J.; Visser, T. The induced decomposition of tert-butyl hydroperoxide. Can. J. Chem. 1964, 42, 2754–2757. [Google Scholar] [CrossRef]
- Udenfriend, S.; Clark, C.T.; Axelrod, J.; Brodie, B.B. Ascorbic acid in aromatic hydroxylation. I. A model system for aromatic hydroxylation. J. Biol. Chem. 1954, 208, 731–738. [Google Scholar] [CrossRef]
- Brodie, B.B.; Axelrod, J.; Shore, P.A.; Udenfriend, S. Ascorbic acid in aromatic hydroxylation II. Products formed by reaction of substrates with ascorbic acid, ferrous ion, and oxygen. J. Biol. Chem. 1954, 208, 741–750. [Google Scholar] [CrossRef]
- Slavik, R.; Peters, J.-U.; Giger, R.; Bürkler, M.; Bald, E. Synthesis of potential drug metabolites by a modified Udenfriend reaction. Tetrahedron Lett. 2011, 52, 749–752. [Google Scholar] [CrossRef]
- Mathieu, D.; Bartoli, J.F.; Battioni, P.; Mansuy, D. Monooxygenation of aromatic compounds by dioxygen with bioinspired systems using non-heme iron catalysts and tetrahydropterins: Comparison with other reducing agents and interesting regioselectivity favouring meta-hydroxylation. Tetrahedron 2004, 60, 3855–3862. [Google Scholar] [CrossRef]
- Metelitsa, D.I. Mechanisms of the hydroxylation of aromatic compounds. Russ. Chem. Rev. 1971, 40, 563. [Google Scholar] [CrossRef]
- Kunai, A.; Hata, S.; Ito, S.; Sasaki, K. The role of oxygen in the hydroxylation reaction of benzene with Fenton’s reagent. Oxygen 18 tracer study. J. Am. Chem. Soc. 1986, 108, 6012–6016. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; De Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef] [PubMed]
- Cussó, O.; Ribas, X.; Costas, M. Biologically inspired non-heme iron-catalysts for asymmetric epoxidation; design principles and perspectives. Chem. Commun. 2015, 51, 14285–14298. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic; Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- De Montellano, P.R.O.; Kunze, K.L.; Beilan, H.S. Chiral orientation of prosthetic heme in the cytochrome P-450 active site. J. Biol. Chem. 1983, 258, 45–47. [Google Scholar] [CrossRef]
- Tshuva, E.Y.; Lippard, S.J. Synthetic models for non-heme carboxylate-bridged diiron metalloproteins: Strategies and tactics. Chem. Rev. 2004, 104, 987–1012. [Google Scholar] [CrossRef]
- Hegg, E.L.; Que, L., Jr. The 2-His-1-carboxylate facial triad—An emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 1997, 250, 625–629. [Google Scholar] [CrossRef]
- Que, L. One motif—Many different reactions. Nat. Struct. Biol. 2000, 7, 182–184. [Google Scholar] [CrossRef]
- Koehntop, K.D.; Emerson, J.P.; Que, L. The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem. 2005, 10, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kryatov, S.V.; Rybak-Akimova, E.V.; Schindler, S. Kinetics and mechanisms of formation and reactivity of non-heme iron oxygen intermediates. Chem. Rev. 2005, 105, 2175–2226. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, E.G.; Lipscomb, J.D. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 2008, 4, 186–193. [Google Scholar] [CrossRef]
- Gibson, D.; Resnick, S.; Lee, K.; Brand, J.; Torok, D.; Wackett, L.; Schocken, M.; Haigler, B. Desaturation, dioxygenation, and monooxygenation reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp. strain 9816-4. J. Bacteriol. 1995, 177, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.D.; Parales, J.V.; Gibson, D.T.; Lipscomb, J.D. Single Turnover Chemistry and Regulation of O2 Activation by the Oxygenase Component of Naphthalene 1,2-Dioxygenase. J. Biol. Chem. 2001, 276, 1945–1953. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- Meunier, B.; Bernadou, J. Active iron-oxo and iron-peroxo species in cytochromes P450 and peroxidases; oxo-hydroxo tautomerism with water-soluble metalloporphyrins. In Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; Springer: New York, NY, USA, 2000; pp. 1–35. [Google Scholar]
- Cooper, H.L.R.; Groves, J.T. Molecular probes of the mechanism of cytochrome P450. Oxygen traps a substrate radical intermediate. Arch. Biochem. Biophys. 2011, 507, 111–118. [Google Scholar] [CrossRef]
- Kille, S.; Zilly, F.E.; Acevedo, J.P.; Reetz, M.T. Regio-and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. [Google Scholar] [CrossRef]
- Narayan, A.R.; Jiménez-Osés, G.; Liu, P.; Negretti, S.; Zhao, W.; Gilbert, M.M.; Ramabhadran, R.O.; Yang, Y.-F.; Furan, L.R.; Li, Z.; et al. Enzymatic hydroxylation of an unactivated methylene C–H bond guided by molecular dynamics simulations. Nat. Chem. 2015, 7, 653–660. [Google Scholar] [CrossRef]
- Roiban, G.D.; Agudo, R.; Reetz, M.T. Cytochrome P450 Catalyzed Oxidative Hydroxylation of Achiral Organic Compounds with Simultaneous Creation of Two Chirality Centers in a Single C-H Activation Step. Angew. Chem. Int. Ed. 2014, 53, 8659–8663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shafer, B.M.; Demars, M.D.; Stern, H.A.; Fasan, R. Controlled oxidation of remote sp3 C–H bonds in artemisinin via P450 catalysts with fine-tuned regio-and stereoselectivity. J. Am. Chem. Soc. 2012, 134, 18695–18704. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Finzel, B.; Gunsalus, I.; Wagner, G.C.; Kraut, J. The 2.6-Å crystal structure of Pseudomonas putida cytochrome P-450. J. Biol. Chem. 1985, 260, 16122–16130. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Narasimhulu, S.; Havran, L.M.; Winkler, J.D.; Poulos, T.L. Crystal structure of cytochrome P450cam complexed with its catalytic product, 5-exo-hydroxycamphor. J. Am. Chem. Soc. 1995, 117, 6297–6299. [Google Scholar] [CrossRef]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Kells, P.M.; Ouellet, H.; Santos-Aberturas, J.; Aparicio, J.F.; Podust, L.M. Structure of cytochrome P450 PimD suggests epoxidation of the polyene macrolide pimaricin occurs via a hydroperoxoferric intermediate. Chem. Biol. 2010, 17, 841–851. [Google Scholar] [CrossRef]
- Shah, M.B.; Jang, H.-H.; Zhang, Q.; Stout, C.D.; Halpert, J.R. X-ray crystal structure of the cytochrome P450 2B4 active site mutant F297A in complex with clopidogrel: Insights into compensatory rearrangements of the binding pocket. Arch. Biochem. Biophys. 2013, 530, 64–72. [Google Scholar] [CrossRef]
- Dawson, J.H.; Sono, M. Cytochrome P-450 and chloroperoxidase: Thiolate-ligated heme enzymes. Spectroscopic determination of their active-site structures and mechanistic implications of thiolate ligation. Chem. Rev. 1987, 87, 1255–1276. [Google Scholar] [CrossRef]
- Mueller, E.J.; Loida, P.J.; Sligar, S.G. Twenty-five Years of P450 cam Research. In Cytochrome P450; Springer: New York, NY, USA, 1995; pp. 83–124. [Google Scholar]
- King, N.K.; Winfield, M. Oxygen uptake and evolution by iron porphyrin enzymes. Aust. J. Chem. 1959, 12, 47–64. [Google Scholar]
- Dunford, H.; Stillman, J. Structure and functional properties of peroxidases and catalases. Coord. Chem. Rev. 1976, 19, 187–251. [Google Scholar] [CrossRef]
- Meunier, B.; Bernadou, J. Metal-oxo species in P450 enzymes and biomimetic models. Oxo-hydroxo tautomerism with water-soluble metalloporphyrins. Top. Catal. 2002, 21, 47–54. [Google Scholar] [CrossRef]
- Rittle, J.; Green, M.T. Cytochrome P450 compound I: Capture, characterization, and C–H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; McClusky, G.A. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976, 98, 859–861. [Google Scholar] [CrossRef]
- Groves, J.T.; McClusky, G.A.; White, R.E.; Coon, M.J. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. [Google Scholar] [CrossRef]
- Ullrich, R.; Hofrichter, M. Enzymatic hydroxylation of aromatic compounds. Cell. Mol. Life Sci. 2007, 64, 271–293. [Google Scholar] [CrossRef]
- de Montellano, P.R.O.; de Voss, J.J. Substrate oxidation by cytochrome P450 enzymes. In Cytochrome P450—Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic; Plenum Publishers: New York, NY, USA, 2005; pp. 183–245. [Google Scholar]
- Groves, J.T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006, 100, 434–447. [Google Scholar] [CrossRef]
- Guroff, G.; Daly, J.W.; Jerina, D.M.; Renson, J.; Witkop, B.; Udenfriend, S. Hydroxylation-induced migration: The NIH shift. Science 1967, 157, 1524–1530. [Google Scholar] [CrossRef]
- Daly, J.; Jerina, D.; Witkop, B. Arene oxides and the NIH shift: The metabolism, toxicity and carcinogenicity of aromatic compounds. Experientia 1972, 28, 1129–1149. [Google Scholar] [CrossRef]
- Barry, S.M.; Challis, G.L. Mechanism and catalytic diversity of Rieske non-heme iron-dependent oxygenases. ACS Catal. 2013, 3, 2362–2370. [Google Scholar] [CrossRef]
- Karlsson, A.; Parales, J.V.; Parales, R.E.; Gibson, D.T.; Eklund, H.; Ramaswamy, S. Crystal structure of naphthalene dioxygenase: Side-on binding of dioxygen to iron. Science 2003, 299, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Kauppi, B.; Lee, K.; Carredano, E.; Parales, R.E.; Gibson, D.T.; Eklund, H.; Ramaswamy, S. Structure of an aromatic-ring-hydroxylating dioxygenase–naphthalene 1,2-dioxygenase. Structure 1998, 6, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Parales, R.E.; Parales, J.V.; Gibson, D.T. Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity. J. Bacteriol. 1999, 181, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; De Los Santos, E.L.; Alkhalaf, L.M.; Challis, G.L. Rieske non-heme iron-dependent oxygenases catalyse diverse reactions in natural product biosynthesis. Nat. Prod. Rep. 2018, 35, 622–632. [Google Scholar] [CrossRef]
- Martins, B.M.; Svetlitchnaia, T.; Dobbek, H. 2-Oxoquinoline 8-monooxygenase oxygenase component: Active site modulation by Rieske-[2Fe-2S] center oxidation/reduction. Structure 2005, 13, 817–824. [Google Scholar] [CrossRef]
- Hsueh, K.-L.; Westler, W.M.; Markley, J.L. NMR investigations of the Rieske protein from Thermus thermophilus support a coupled proton and electron transfer mechanism. J. Am. Chem. Soc. 2010, 132, 7908–7918. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Altier, D.J.; Stubna, A.; Popescu, C.V.; Münck, E.; Lipscomb, J.D. Benzoate 1,2-dioxygenase from Pseudomonas putida: Single turnover kinetics and regulation of a two-component Rieske dioxygenase. Biochemistry 2002, 41, 9611–9626. [Google Scholar] [CrossRef]
- Bugg, T.D.; Ramaswamy, S. Non-heme iron-dependent dioxygenases: Unravelling catalytic mechanisms for complex enzymatic oxidations. Curr. Op. Chem. Biol. 2008, 12, 134–140. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Lipscomb, J.D. Hydrogen peroxide-coupled cis-diol formation catalyzed by naphthalene 1,2-dioxygenase. J. Biol. Chem. 2003, 278, 829–835. [Google Scholar] [CrossRef]
- Lippard, S.J. Hydroxylation of C–H bonds at carboxylate-bridged diiron centres. Philos. Trans. R. Soc. Lond. Ser. A 2005, 363, 861–877. [Google Scholar] [CrossRef]
- Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Que, L., Jr.; True, A.E. Dinuclear iron-and manganese-oxo sites in biology. Prog. Inorg. Chem. 1990, 38, 97–200. [Google Scholar]
- Merkx, M.; Kopp, D.A.; Sazinsky, M.H.; Blazyk, J.L.; Müller, J.; Lippard, S.J. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew. Chem. Int. Ed. 2001, 40, 2782–2807. [Google Scholar] [CrossRef]
- Notomista, E.; Lahm, A.; Di Donato, A.; Tramontano, A. Evolution of bacterial and archaeal multicomponent monooxygenases. J. Mol. Evol. 2003, 56, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.G.; Batchelor, P.J.; Morcomb, S.M. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 2003, 27, 449–479. [Google Scholar] [CrossRef]
- Green, J.; Dalton, H. Substrate specificity of soluble methane monooxygenase. Mechanistic implications. J. Biol. Chem. 1989, 264, 17698–17703. [Google Scholar] [CrossRef]
- Siewert, I.; Limberg, C. Low-Molecular-Weight Analogues of the Soluble Methane Monooxygenase (sMMO): From the Structural Mimicking of Resting States and Intermediates to Functional Models. Chem. Eur. J. 2009, 15, 10316–10328. [Google Scholar] [CrossRef]
- Sazinsky, M.H.; Bard, J.; Di Donato, A.; Lippard, S.J. Crystal Structure of the Toluene/o-Xylene Monooxygenase Hydroxylase from Pseudomonas stutzeri OX1. J. Biol. Chem. 2004, 279, 30600–30610. [Google Scholar] [CrossRef]
- Sazinsky, M.H.; Dunten, P.W.; McCormick, M.S.; DiDonato, A.; Lippard, S.J. X-ray Structure of a Hydroxylase Regulatory Protein Complex from a Hydrocarbon-Oxidizing Multicomponent Monooxygenase, Pseudomonas sp. OX1 Phenol Hydroxylase. Biochemistry 2006, 45, 15392–15404. [Google Scholar] [CrossRef]
- Chauhan, S.; Barbieri, P.; Wood, T.K. Oxidation of trichloroethylene, 1,1-dichloroethylene, and chloroform by toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 1998, 64, 3023–3024. [Google Scholar] [CrossRef]
- Mitchell, K.H.; Rogge, C.E.; Gierahn, T.; Fox, B.G. Insight into the mechanism of aromatic hydroxylation by toluene 4-monooxygenase by use of specifically deuterated toluene and p-xylene. Proc. Natl. Acad. Sci. USA 2003, 100, 3784–3789. [Google Scholar] [CrossRef] [PubMed]
- Friedle, S.; Reisner, E.; Lippard, S.J. Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes. Chem. Soc. Rev. 2010, 39, 2768–2779. [Google Scholar] [CrossRef]
- Colby, J.; Stirling, D.I.; Dalton, H. The soluble methane mono-oxygenase of Methylococcus capsulatus (Bath). Its ability to oxygenate n-alkanes, n-alkenes, ethers, and alicyclic, aromatic and heterocyclic compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef]
- Kovaleva, E.; Neibergall, M.; Chakrabarty, S.; Lipscomb, J.D. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc. Chem. Res. 2007, 40, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Tinberg, C.E.; Lippard, S.J. Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): Evidence for a multi-step, proton-dependent reaction pathway. Biochemistry 2009, 48, 12145–12158. [Google Scholar] [CrossRef]
- Valentine, A.M.; Stahl, S.S.; Lippard, S.J. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J. Am. Chem. Soc. 1999, 121, 3876–3887. [Google Scholar] [CrossRef]
- Beauvais, L.G.; Lippard, S.J. Reactions of the peroxo intermediate of soluble methane monooxygenase hydroxylase with ethers. J. Am. Chem. Soc. 2005, 127, 7370–7378. [Google Scholar] [CrossRef]
- Song, W.J.; Lippard, S.J. Mechanistic studies of reactions of peroxodiiron(III) intermediates in T201 variants of toluene/o-xylene monooxygenase hydroxylase. Biochemistry 2011, 50, 5391–5399. [Google Scholar] [CrossRef]
- Murray, L.J.; Naik, S.G.; Ortillo, D.O.; García-Serres, R.; Lee, J.K.; Huynh, B.H.; Lippard, S.J. Characterization of the arene-oxidizing intermediate in ToMOH as a diiron(III) species. J. Am. Chem. Soc. 2007, 129, 14500–14510. [Google Scholar] [CrossRef]
- Kappock, T.J.; Caradonna, J.P. Pterin-dependent amino acid hydroxylases. Chem. Rev. 1996, 96, 2659–2756. [Google Scholar] [CrossRef]
- Flatmark, T.; Stevens, R.C. Structural insight into the aromatic amino acid hydroxylases and their disease-related mutant forms. Chem. Rev. 1999, 99, 2137–2160. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, P.F. Tetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999, 68, 355–381. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P. Life as aerobes: Are there simple rules for activation of dioxygen by enzymes? J. Biol. Inorg. Chem. 2001, 6, 1–13. [Google Scholar] [CrossRef]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine hydroxylase the initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [CrossRef]
- Davis, M.D.; Kaufman, S. Evidence for the formation of the 4a-carbinolamine during the tyrosine-dependent oxidation of tetrahydrobiopterin by rat liver phenylalanine hydroxylase. J. Biol. Chem. 1989, 264, 8585–8596. [Google Scholar] [CrossRef]
- Francisco, W.A.; Tian, G.; Fitzpatrick, P.F.; Klinman, J.P. Oxygen-18 kinetic isotope effect studies of the tyrosine hydroxylase reaction: Evidence of rate limiting oxygen activation. J. Am. Chem. Soc. 1998, 120, 4057–4062. [Google Scholar] [CrossRef]
- Bassan, A.; Blomberg, M.R.; Siegbahn, P.E. Mechanism of Dioxygen Cleavage in Tetrahydrobiopterin-Dependent Amino Acid Hydroxylases. Chem. Eur. J. 2003, 9, 106–115. [Google Scholar] [CrossRef]
- Kemsley, J.N.; Wasinger, E.C.; Datta, S.; Mitić, N.; Acharya, T.; Hedman, B.; Caradonna, J.P.; Hodgson, K.O.; Solomon, E.I. Spectroscopic and Kinetic Studies of PKU Inducing Mutants of Phenylalanine Hydroxylase: Arg158Gln and Glu280Lys. J. Am. Chem. Soc. 2003, 125, 5677–5686. [Google Scholar] [CrossRef]
- Dix, T.A.; Bollag, G.E.; Domanico, P.; Benkovic, S.J. Phenylalanine hydroxylase: Absolute configuration and source of oxygen of the 4a-hydroxytetrahydropterin species. Biochemistry 1985, 24, 2955–2958. [Google Scholar] [CrossRef]
- Daly, J.; Levitt, M.; Guroff, G.; Udenfriend, S. Isotope studies on the mechanism of action of adrenal tyrosine hydroxylase. Arch. Biochem. Biophys. 1968, 126, 593–598. [Google Scholar] [CrossRef]
- Siegmund, H.-U.; Kaufman, S. Hydroxylation of 4-methylphenylalanine by rat liver phenylalanine hydroxylase. J. Biol. Chem. 1991, 266, 2903–2910. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Martinez, A.; Teigen, K.; Jensen, V.R. Formation of the Iron–Oxo Hydroxylating Species in the Catalytic Cycle of Aromatic Amino Acid Hydroxylases. Chem. Eur. J. 2011, 17, 3746–3758. [Google Scholar] [CrossRef]
- Olsson, E.; Martinez, A.; Teigen, K.; Jensen, V.R. Substrate Hydroxylation by the Oxido–Iron Intermediate in Aromatic Amino Acid Hydroxylases: A DFT Mechanistic Study. Eur. J. Inorg. Chem. 2011, 2011, 2720–2732. [Google Scholar] [CrossRef]
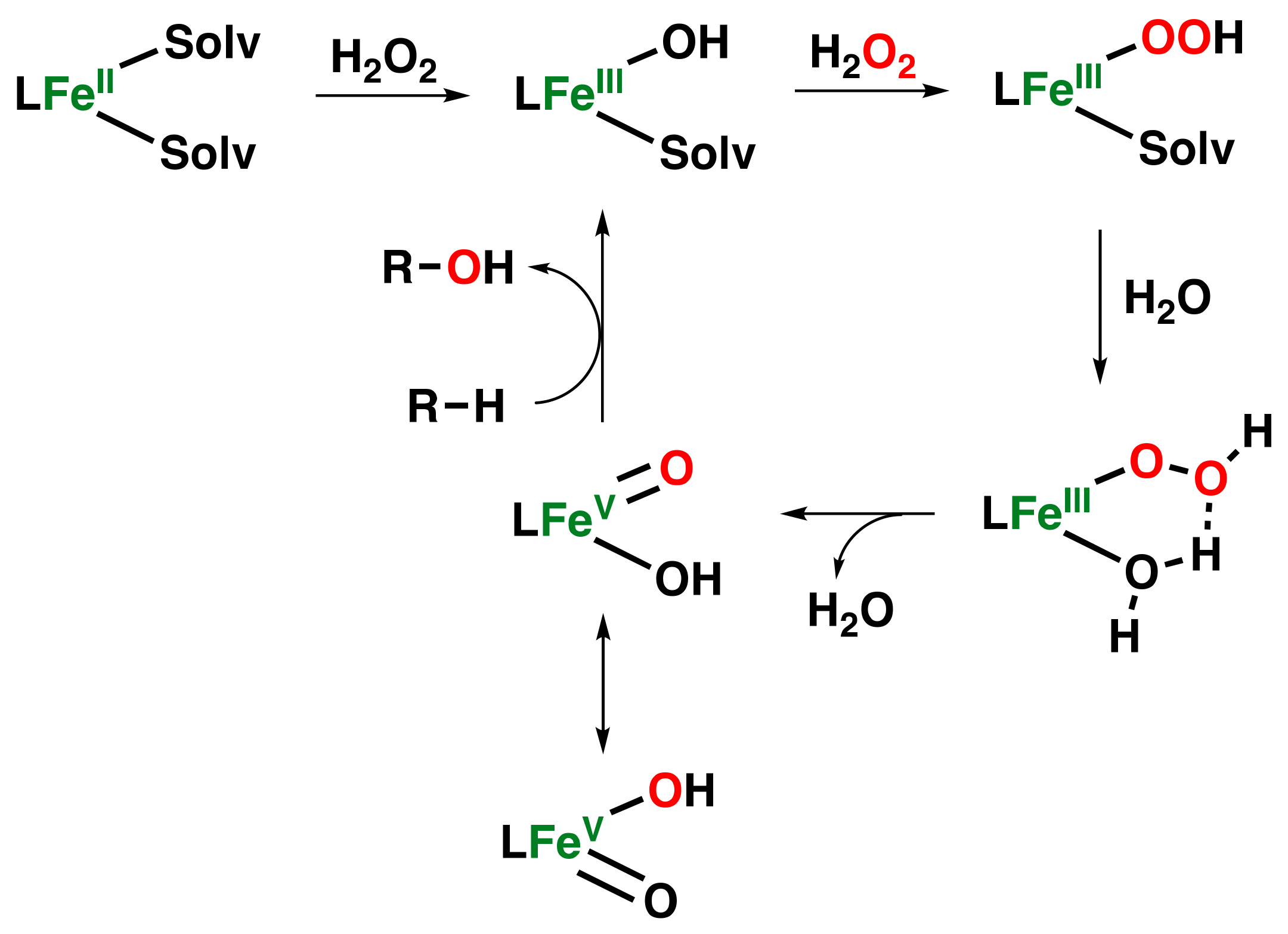
- Kal, S.; Xu, S.; Que, L., Jr. Bio-inspired Nonheme Iron Oxidation Catalysis: Involvement of Oxoiron(V) Oxidants in Cleaving Strong C–H Bonds. Angew. Chem. Int. Ed. 2020, 59, 7332–7349. [Google Scholar] [CrossRef] [PubMed]
- Bryliakov, K.P.; Talsi, E.P. Active sites and mechanisms of bioinspired oxidation with H2O2, catalyzed by non-heme Fe and related Mn complexes. Coord. Chem. Rev. 2014, 276, 73–96. [Google Scholar] [CrossRef]
- Oloo, W.N.; Que, L., Jr. Bioinspired Nonheme Iron Catalysts for C–H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Cussó, O.; Costas, M. Biologically inspired C–H and C=C oxidations with hydrogen peroxide catalyzed by iron coordination complexes. Chem. Asian J. 2016, 11, 3148–3158. [Google Scholar] [CrossRef]
- Olivo, G.; Cussó, O.; Borrell, M.; Costas, M. Oxidation of alkane and alkene moieties with biologically inspired nonheme iron catalysts and hydrogen peroxide: From free radicals to stereoselective transformations. J. Biol. Inorg. Chem. 2017, 22, 425–452. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. Non-heme oxoiron(V) intermediates in chemo-, regio- and stereoselective oxidation of organic substrates. Coord. Chem. Rev. 2019, 384, 126–139. [Google Scholar] [CrossRef]
- Kim, C.; Chen, K.; Kim, J.; Que, L. Stereospecific alkane hydroxylation with H2O2 catalyzed by an iron(II) –tris(2-pyridylmethyl)amine complex. J. Am. Chem. Soc. 1997, 119, 5964–5965. [Google Scholar] [CrossRef]
- Chen, K.; Que, L. Stereospecific alkane hydroxylation by non-heme iron catalysts: Mechanistic evidence for an FeVO active species. J. Am. Chem. Soc. 2001, 123, 6327–6337. [Google Scholar] [CrossRef] [PubMed]
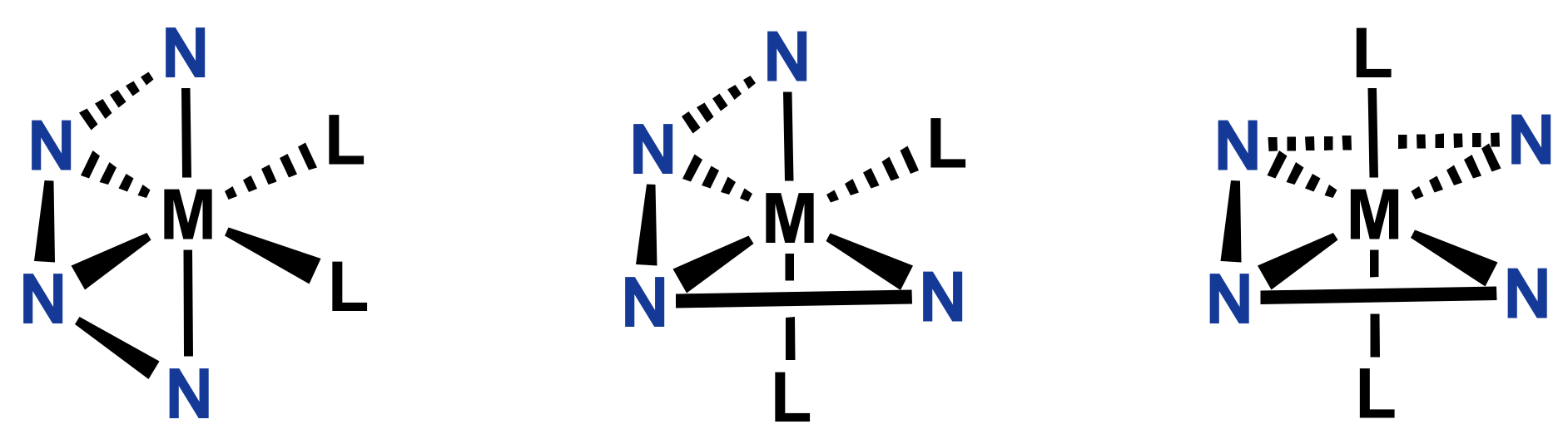
- Aldrich-Wright, J.R.; Vagg, R.S.; Williams, P.A. Design of chiral picen-based metal complexes for molecular recognition of α-aminoacids and nucleic acids. Coord. Chem. Rev. 1997, 166, 361–389. [Google Scholar] [CrossRef]
- Knof, U.; von Zelewsky, A. Predetermined chirality at metal centers. Angew. Chem. Int. Ed. 1999, 38, 302–322. [Google Scholar] [CrossRef]
- Ng, C.; Sabat, M.; Fraser, C.L. Metal complexes with cis α topology from stereoselective quadridentate ligands with amine, pyridine, and quinoline donor groups. Inorg. Chem. 1999, 38, 5545–5556. [Google Scholar] [CrossRef]
- Costas, M.; Que, J. Lawrence, Ligand topology tuning of iron-catalyzed hydrocarbon oxidations. Angew. Chem. Int. Ed. 2002, 41, 2179–2181. [Google Scholar] [CrossRef]
- Lee, D.; Park, H. Ligand Taxonomy for Bioinorganic Modeling of Dioxygen-Activating Non-Heme Iron Enzymes. Chem. Eur. J. 2020, 26, 5916–5926. [Google Scholar]
- Gamba, I.; Codolà, Z.; Lloret-Fillol, J.; Costas, M. Making and breaking of the O–O bond at iron complexes. Coord. Chem. Rev. 2017, 334, 2–24. [Google Scholar] [CrossRef]
- Dantignana, V.; Company, A.; Costas, M. Oxoiron(V) Complexes of Relevance in Oxidation Catalysis of Organic Substrates. Isr. J. Chem. 2020, 60, 1004–1018. [Google Scholar] [CrossRef]
- Borrell, M.; Costas, M. Mechanistically driven development of an iron catalyst for selective syn-dihydroxylation of alkenes with aqueous hydrogen peroxide. J. Am. Chem. Soc. 2017, 139, 12821–12829. [Google Scholar] [CrossRef]
- Chen, K.; Costas, M.; Kim, J.; Tipton, A.K.; Que, L. Olefin cis-dihydroxylation versus epoxidation by non-heme iron catalysts: Two faces of an FeIII–OOH coin. J. Am. Chem. Soc. 2002, 124, 3026–3035. [Google Scholar] [CrossRef]
- Borrell, M.; Costas, M. Greening oxidation catalysis: Iron catalyzed alkene syn-dihydroxylation with aqueous hydrogen peroxide in green solvents. ACS Sustain. Chem. Eng. 2018, 6, 8410–8416. [Google Scholar] [CrossRef]
- Prat, I.; Mathieson, J.S.; Güell, M.; Ribas, X.; Luis, J.M.; Cronin, L.; Costas, M. Observation of Fe(V)=O using variable-temperature mass spectrometry and its enzyme-like C–H and C=C oxidation reactions. Nat. Chem. 2011, 3, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Arakawa, K.; Funabiki, T.; Kodera, M. An Iron(III)–Monoamidate Complex Catalyst for Selective Hydroxylation of Alkane C–H Bonds with Hydrogen Peroxide. Angew. Chem. Int. Ed. 2012, 51, 3448–3452. [Google Scholar] [CrossRef]
- Xu, S.; Veach, J.J.; Oloo, W.N.; Peters, K.C.; Wang, J.; Perry, R.H.; Que, L. Detection of a transient FeV(O)(OH) species involved in olefin oxidation by a bio-inspired non-haem iron catalyst. Chem. Commun. 2018, 54, 8701–8704. [Google Scholar] [CrossRef] [PubMed]
- Borrell, M.; Andris, E.; Navrátil, R.; Roithová, J.; Costas, M. Characterized cis-FeV(O)(OH) intermediate mimics enzymatic oxidations in the gas phase. Nat. Commun. 2019, 10, 901. [Google Scholar] [CrossRef]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A synthetically useful, self-assembling MMO mimic system for catalytic alkene epoxidation with aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194–7195. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science 2010, 327, 566–571. [Google Scholar] [CrossRef]
- White, M.C. Adding aliphatic C–H bond oxidations to synthesis. Science 2012, 335, 807–809. [Google Scholar] [CrossRef]
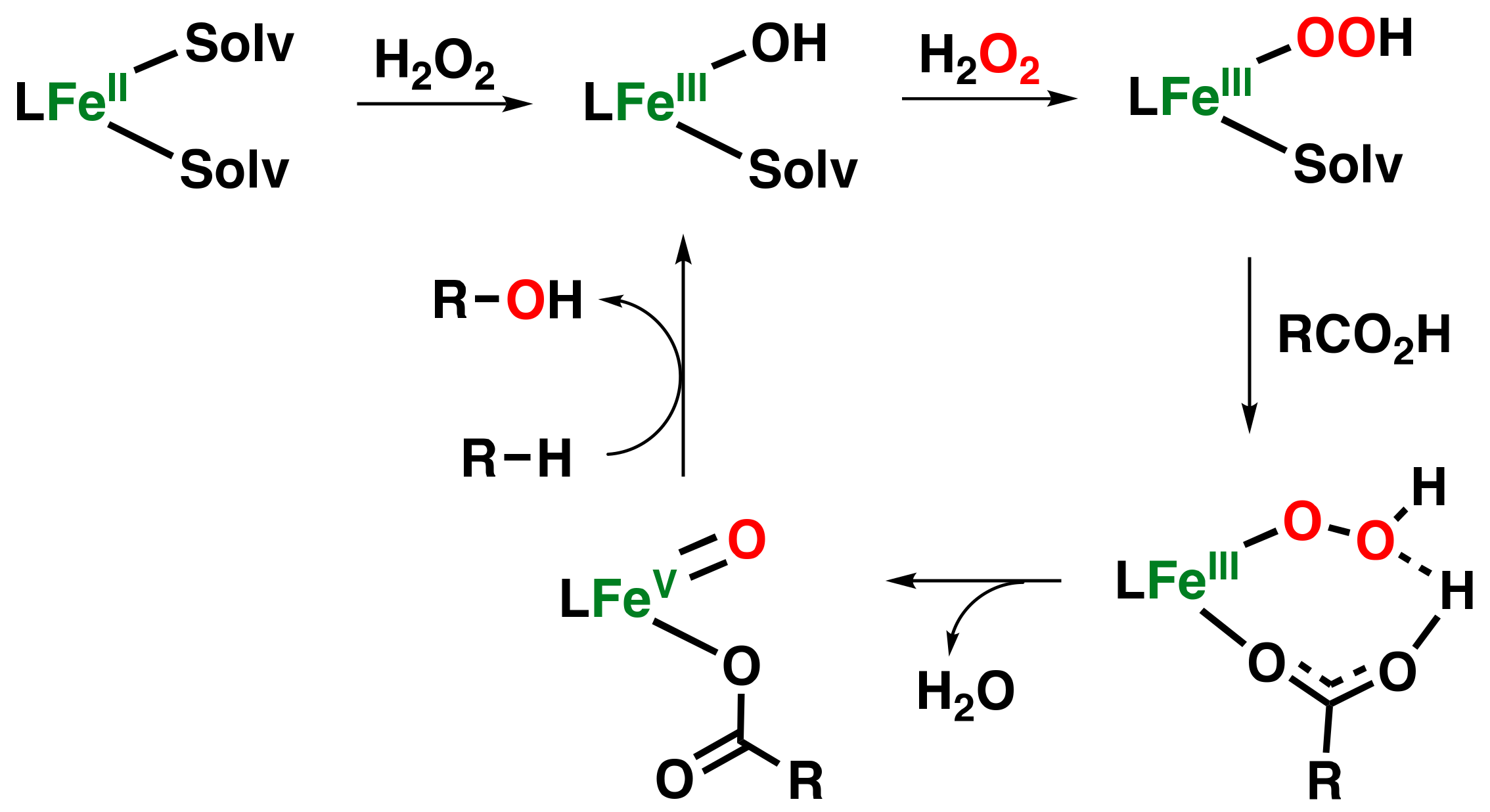
- Mas-Ballesté, R.; Que, L. Iron-catalyzed olefin epoxidation in the presence of acetic acid: Insights into the nature of the metal-based oxidant. J. Am. Chem. Soc. 2007, 129, 15964–15972. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Talsi, E.P. Direct C–H Oxidation of Aromatic Substrates in the Presence of Biomimetic Iron Complexes. In Frontiers of Green Catalytic Selective Oxidations; Springer: Singapore, 2019; pp. 253–276. [Google Scholar]
- Kitajima, N.; Ito, M.; Fukui, H.; Morooka, Y. A reaction mimic of tyrosine hydroxylase: Hydroxylation of a phenoxo ferric complex to a catecholato complex with mCPBA. J. Am. Chem. Soc. 1993, 115, 9335–9336. [Google Scholar] [CrossRef]
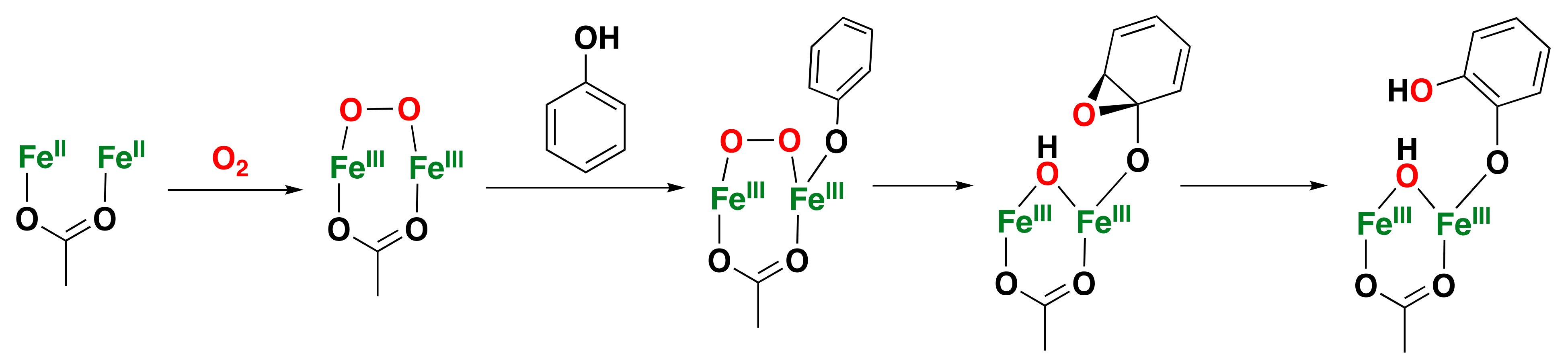
- Ménage, S.; Galey, J.B.; Hussler, G.; Seité, M.; Fontecave, M. Aromatic Hydroxylation by H2O2 and O2 Catalyzed by a μ-Oxo Diiron(III) Complex. Angew. Chem. Int. Ed. 1996, 35, 2353–2355. [Google Scholar] [CrossRef]
- Ménage, S.; Galey, J.-B.; Dumats, J.; Hussler, G.; Seité, M.; Luneau, I.G.; Chottard, G.; Fontecave, M. O2 activation and aromatic hydroxylation performed by diiron complexes. J. Am. Chem. Soc. 1998, 120, 13370–13382. [Google Scholar] [CrossRef]
- Mekmouche, Y.; Ménage, S.; Toia-Duboc, C.; Fontecave, M.; Galey, J.B.; Lebrun, C.; Pécaut, J. H2O2-Dependent Fe-Catalyzed Oxidations: Control of the Active Species. Angew. Chem. Int. Ed. 2001, 40, 949–952. [Google Scholar] [CrossRef]
- Lange, S.J.; Miyake, H.; Que, L. Evidence for a nonheme Fe(IV)=O species in the intramolecular hydroxylation of a phenyl moiety. J. Am. Chem. Soc. 1999, 121, 6330–6331. [Google Scholar] [CrossRef]
- Jensen, M.P.; Mehn, M.P.; Que, L., Jr. Intramolecular Aromatic Amination through Iron-Mediated Nitrene Transfer. Angew. Chem. Int. Ed. 2003, 42, 4357–4360. [Google Scholar] [CrossRef]
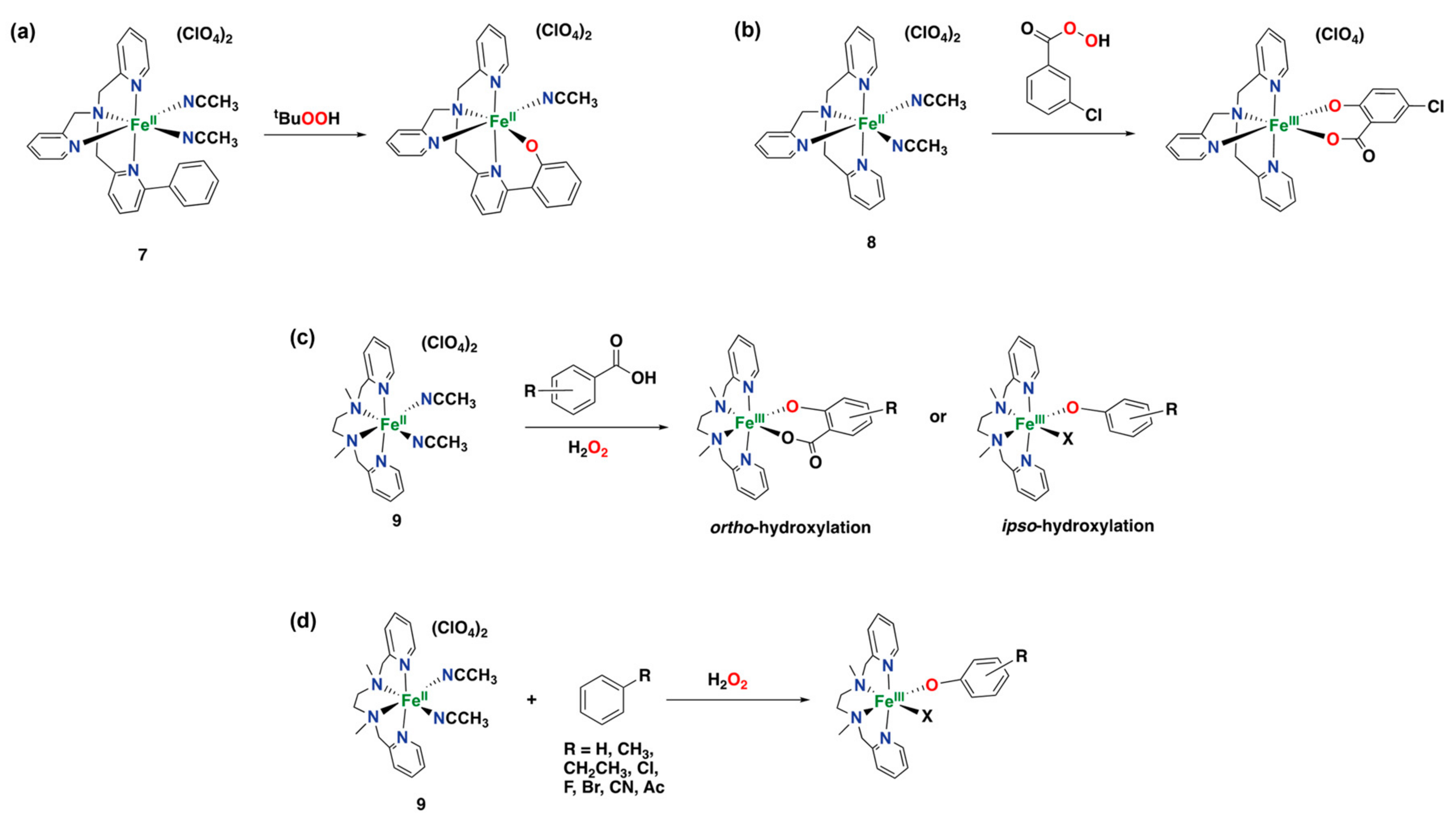
- Jensen, M.P.; Lange, S.J.; Mehn, M.P.; Que, E.L.; Que, L. Biomimetic Aryl Hydroxylation Derived from Alkyl Hydroperoxide at a Nonheme Iron Center. Evidence for an FeIV=O Oxidant. J. Am. Chem. Soc. 2003, 125, 2113–2128. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.Y.; Seo, M.S.; Lim, M.H.; Consugar, M.B.; Park, M.J.; Rohde, J.-U.; Han, J.; Kim, K.M.; Kim, J.; Que, L., Jr. Self-hydroxylation of perbenzoic acids at a nonheme iron(II) center. Chem. Commun. 2005, 5644–5646. [Google Scholar] [CrossRef]
- Taktak, S.; Flook, M.; Foxman, B.M.; Que, L., Jr.; Rybak-Akimova, E.V. ortho-Hydroxylation of benzoic acids with hydrogen peroxide at a non-heme iron center. Chem. Commun. 2005, 5301–5303. [Google Scholar] [CrossRef]
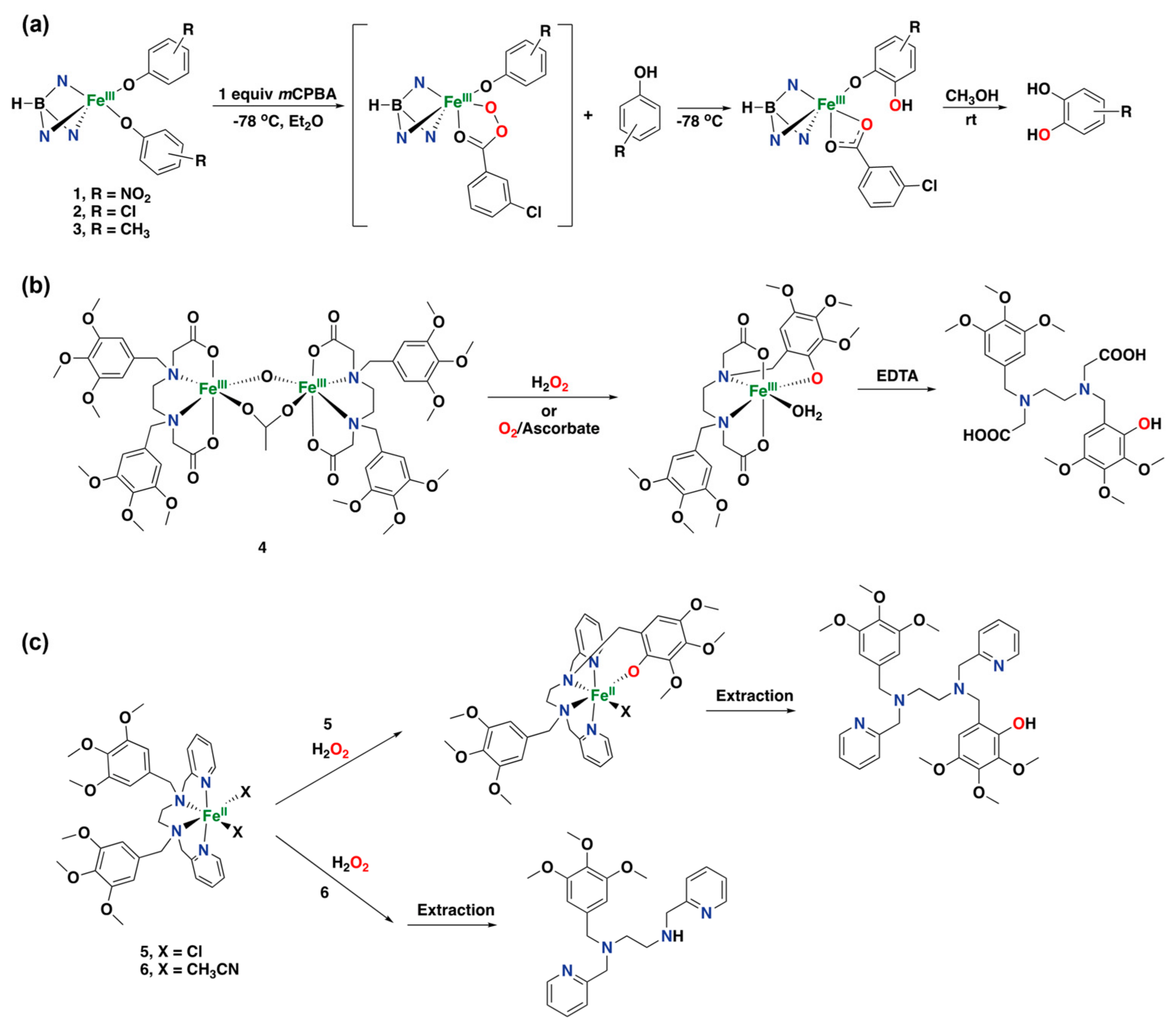
- Makhlynets, O.V.; Das, P.; Taktak, S.; Flook, M.; Mas-Ballesté, R.; Rybak-Akimova, E.V.; Que, L., Jr. Iron-Promoted ortho-and/or ipso-Hydroxylation of Benzoic Acids with H2O2. Chem. Eur. J. 2009, 15, 13171–13180. [Google Scholar] [CrossRef]
- Makhlynets, O.V.; Rybak-Akimova, E.V. Aromatic Hydroxylation at a Non-Heme Iron Center: Observed Intermediates and Insights into the Nature of the Active Species. Chem. Eur. J. 2010, 16, 13995–14006. [Google Scholar] [CrossRef]
- Makhlynets, O.V.; Oloo, W.N.; Moroz, Y.S.; Belaya, I.G.; Palluccio, T.D.; Filatov, A.S.; Müller, P.; Cranswick, M.A.; Que, L.; Rybak-Akimova, E.V. H2O2 activation with biomimetic non-haem iron complexes and AcOH: Connecting the g= 2.7 EPR signal with a visible chromophore. Chem. Commun. 2014, 50, 645–648. [Google Scholar] [CrossRef]
- Ansari, A.; Kaushik, A.; Rajaraman, G. Mechanistic Insights on the ortho-Hydroxylation of Aromatic Compounds by Non-heme Iron Complex: A Computational Case Study on the Comparative Oxidative Ability of Ferric-Hydroperoxo and High-Valent FeIV=O and FeV=O Intermediates. J. Am. Chem. Soc. 2013, 135, 4235–4249. [Google Scholar] [CrossRef]
- Ségaud, N.; Rebilly, J.-N.; Sénéchal-David, K.; Guillot, R.; Billon, L.; Baltaze, J.-P.; Farjon, J.; Reinaud, O.; Banse, F. Iron Coordination Chemistry with New Ligands Containing Triazole and Pyridine Moieties. Comparison of the Coordination Ability of the N-Donors. Inorg. Chem. 2013, 52, 691–700. [Google Scholar] [CrossRef]
- Martinho, M.; Banse, F.; Bartoli, J.-F.; Mattioli, T.A.; Battioni, P.; Horner, O.; Bourcier, S.; Girerd, J.-J. New example of a non-heme mononuclear iron(IV) oxo complex. Spectroscopic data and oxidation activity. Inorg. Chem. 2005, 44, 9592–9596. [Google Scholar] [CrossRef]
- Rebilly, J.N.; Zhang, W.; Herrero, C.; Dridi, H.; Sénéchal-David, K.; Guillot, R.; Banse, F. Hydroxylation of aromatics by H2O2 catalyzed by mononuclear non-heme iron complexes: Role of triazole hemilability in substrate-induced bifurcation of the H2O2 activation mechanism. Chem. Eur. J. 2020, 26, 659–668. [Google Scholar] [CrossRef]
- de Visser, S.P.; Oh, K.; Han, A.-R.; Nam, W. Combined Experimental and Theoretical Study on Aromatic Hydroxylation by Mononuclear Nonheme Iron(IV)–Oxo Complexes. Inorg. Chem. 2007, 46, 4632–4641. [Google Scholar] [CrossRef]
- Bartoli, J.-F.; Lambert, F.; Morgenstern-Badarau, I.; Battioni, P.; Mansuy, D. Unusual efficiency of a non-heme iron complex as catalyst for the hydroxylation of aromatic compounds by hydrogen peroxide: Comparison with iron porphyrins. Comptes Rendus Chim. 2002, 5, 263–266. [Google Scholar] [CrossRef]
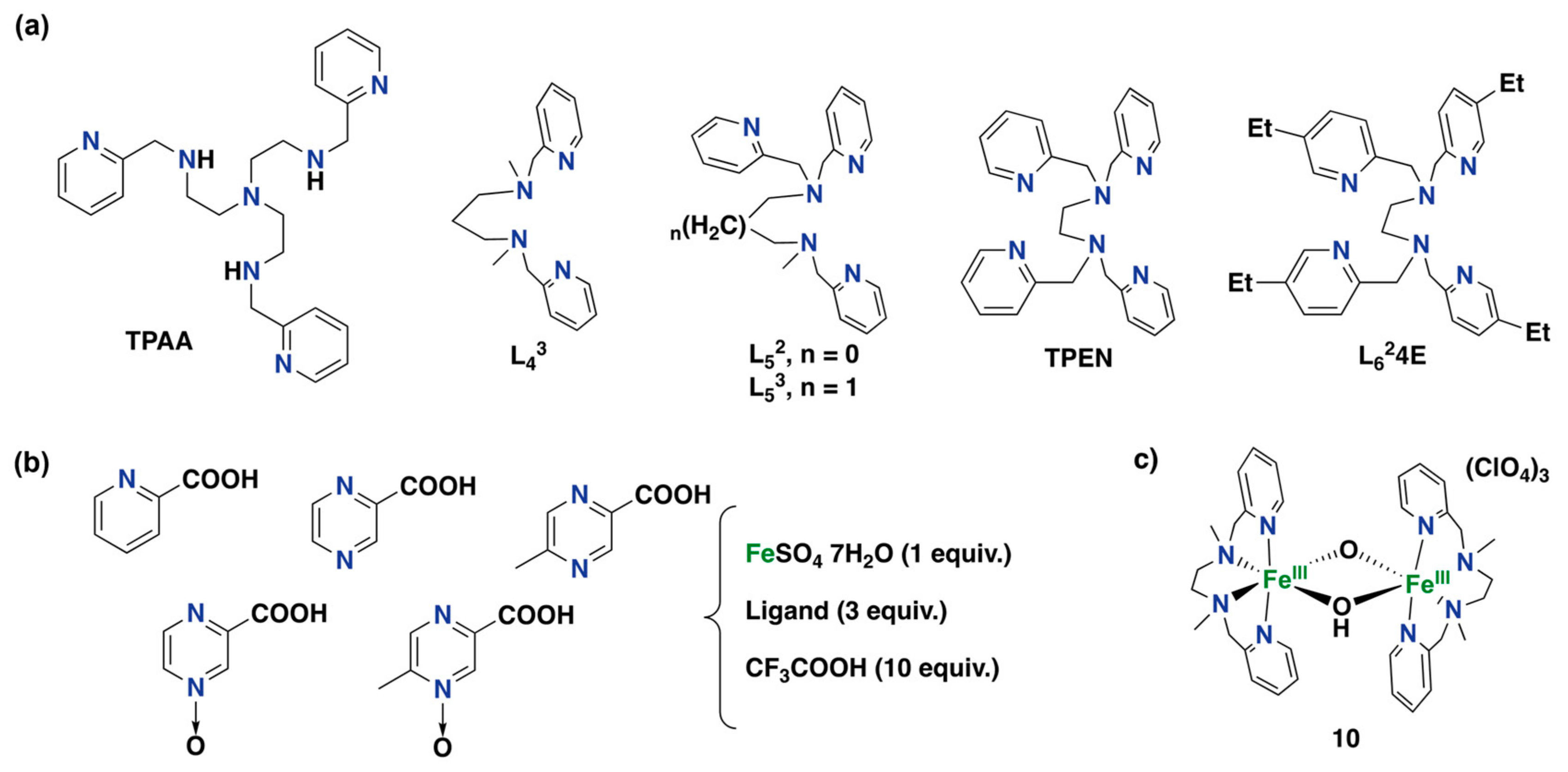
- Balland, V.; Mathieu, D.; Pons-Y-Moll, N.; Bartoli, J.F.; Banse, F.; Battioni, P.; Girerd, J.-J.; Mansuy, D. Non-heme iron polyazadentate complexes as catalysts for oxidations by H2O2: Particular efficiency in aromatic hydroxylations and beneficial effects of a reducing agent. J. Mol. Catal. A Chem. 2004, 215, 81–87. [Google Scholar] [CrossRef]
- Thibon, A.; Bartoli, J.-F.; Guillot, R.; Sainton, J.; Martinho, M.; Mansuy, D.; Banse, F. Non-heme iron polyazadentate complexes as catalysts for aromatic hydroxylation by H2O2: Particular efficiency of tetrakis(2-pyridylmethyl)ethylenediamine–iron(II) complexes. J. Mol. Catal. A 2008, 287, 115–120. [Google Scholar] [CrossRef]
- Bianchi, D.; Bortolo, R.; Tassinari, R.; Ricci, M.; Vignola, R. A novel iron-based catalyst for the biphasic oxidation of benzene to phenol with hydrogen peroxide. Angew. Chem. Int. Ed. 2000, 39, 4321–4323. [Google Scholar] [CrossRef]
- Bianchi, D.; Bertoli, M.; Tassinari, R.; Ricci, M.; Vignola, R. Direct synthesis of phenols by iron-catalyzed biphasic oxidation of aromatic hydrocarbons with hydrogen peroxide. J. Mol. Catal. A Chem. 2003, 200, 111–116. [Google Scholar] [CrossRef]
- Bianchi, D.; Bertoli, M.; Tassinari, R.; Ricci, M.; Vignola, R. Ligand effect on the iron-catalysed biphasic oxidation of aromatic hydrocarbons by hydrogen peroxide. J. Mol. Catal. A Chem. 2003, 204, 419–424. [Google Scholar] [CrossRef]
- Kejriwal, A.; Bandyopadhyay, P.; Biswas, A.N. Aromatic hydroxylation using an oxo-bridged diiron(III) complex: A bio-inspired functional model of toluene monooxygenases. Dalton Trans. 2015, 44, 17261–17267. [Google Scholar] [CrossRef] [PubMed]
- Raba, A.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. Catalytic hydroxylation of benzene and toluene by an iron complex bearing a chelating di-pyridyl-di-NHC ligand. Chem. Commun. 2014, 50, 11454–11457. [Google Scholar] [CrossRef]
- Raba, A.; Cokoja, M.; Ewald, S.; Riener, K.; Herdtweck, E.; Pöthig, A.; Herrmann, W.A.; Kühn, F.E. Synthesis and Characterization of Novel Iron(II) Complexes with Tetradentate Bis(N-heterocyclic carbene)–Bis (pyridine)(NCCN) Ligands. Organometallics 2012, 31, 2793–2800. [Google Scholar] [CrossRef]
- Rogers, M.M.; Stahl, S.S. N-Heterocyclic carbenes as ligands for high-oxidation-state metal complexes and oxidation catalysis. In N-Heterocyclic Carbenes in Transition Metal Catalysis; Springer: New York, NY, USA, 2006; pp. 21–46. [Google Scholar]
- Strassner, T. The role of NHC ligands in oxidation catalysis. In Organometallic Oxidation Catalysis; Springer: Berlin/Heidelberg, Germany, 2006; pp. 125–148. [Google Scholar]
- Lindhorst, A.C.; Schütz, J.; Netscher, T.; Bonrath, W.; Kühn, F.E. Catalytic oxidation of aromatic hydrocarbons by a molecular iron–NHC complex. Catal. Sci. Technol. 2017, 7, 1902–1911. [Google Scholar] [CrossRef]
- Silva, G.C.; Carvalho, N.M.; Horn, A., Jr.; Lachter, E.R.; Antunes, O.A. Oxidation of aromatic compounds by hydrogen peroxide catalyzed by mononuclear iron(III) complexes. J. Mol. Catal. A Chem. 2017, 426, 564–571. [Google Scholar] [CrossRef]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Capocasa, G.; Olivo, G.; Barbieri, A.; Lanzalunga, O.; Di Stefano, S. Direct hydroxylation of benzene and aromatics with H2O2 catalyzed by a self-assembled iron complex: Evidence for a metal-based mechanism. Catal. Sci. Technol. 2017, 7, 5677–5686. [Google Scholar] [CrossRef]
- Ticconi, B.; Colcerasa, A.; Di Stefano, S.; Lanzalunga, O.; Lapi, A.; Mazzonna, M.; Olivo, G. Oxidative functionalization of aliphatic and aromatic amino acid derivatives with H2O2 catalyzed by a nonheme imine based iron complex. RSC Adv. 2018, 8, 19144–19151. [Google Scholar] [CrossRef]
- Capocasa, G.; Di Berto Mancini, M.; Frateloreto, F.; Lanzalunga, O.; Olivo, G.; Di Stefano, S. Easy Synthesis of a Self-Assembled Imine-Based Iron(II) Complex Endowed with Crown-Ether Receptors. Eur. J. Org. Chem. 2020, 23, 3390–3397. [Google Scholar] [CrossRef]
- Olivo, G.; Arancio, G.; Mandolini, L.; Lanzalunga, O.; Di Stefano, S. Hydrocarbon oxidation catalyzed by a cheap nonheme imine-based iron(II) complex. Catal. Sci. Technol. 2014, 4, 2900–2903. [Google Scholar] [CrossRef]
- Olivo, G.; Giosia, S.; Barbieri, A.; Lanzalunga, O.; Di Stefano, S. Alcohol oxidation with H2O2 catalyzed by a cheap and promptly available imine based iron complex. Org. Biomol. Chem. 2016, 14, 10630–10635. [Google Scholar] [CrossRef]
- Ticconi, B.; Capocasa, G.; Cerrato, A.; Di Stefano, S.; Lapi, A.; Marincioni, B.; Olivo, G.; Lanzalunga, O. Insight into the chemoselective aromatic vs. side-chain hydroxylation of alkylaromatics with H2O2 catalyzed by a non-heme imine-based iron complex. Catal. Sci. Technol. 2021, 11, 171–178. [Google Scholar] [CrossRef]
- Olivo, G.; Nardi, M.; Vìdal, D.; Barbieri, A.; Lapi, A.; Gómez, L.; Lanzalunga, O.; Costas, M.; Di Stefano, S. C–H Bond Oxidation Catalyzed by an Imine-Based Iron Complex: A Mechanistic Insight. Inorg. Chem. 2015, 54, 10141–10152. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Zima, A.M.; Tkachenko, N.V.; Bryliakov, K.P.; Talsi, E.P. Direct Evaluation of the Reactivity of Nonheme Iron(V)–Oxo Intermediates toward Arenes. ACS Catal. 2018, 8, 5255–5260. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Bryliakov, K.P.; Talsi, E.P. EPR Spectroscopic Detection of the Elusive FeV=O Intermediates in Selective Catalytic Oxofunctionalizations of Hydrocarbons Mediated by Biomimetic Ferric Complexes. ACS Catal. 2015, 5, 2702–2707. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Dramatic effect of carboxylic acid on the electronic structure of the active species in Fe(PDP)-catalyzed asymmetric epoxidation. ACS Catal. 2016, 6, 5399–5404. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Iron-catalyzed enantioselective epoxidations with various oxidants: Evidence for different active species and epoxidation mechanisms. ACS Catal. 2017, 7, 60–69. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Lubov, D.P.; Bryliakov, K.P.; Talsi, E.P. Aromatic C–H oxidation by non-heme iron(V)-oxo intermediates bearing aminopyridine ligands. J. Mol. Catal. 2020, 483, 110708. [Google Scholar] [CrossRef]
- Van Heuvelen, K.M.; Fiedler, A.T.; Shan, X.; De Hont, R.F.; Meier, K.K.; Bominaar, E.L.; Münck, E.; Que, L. One-electron oxidation of an oxoiron(IV) complex to form an [O=FeV=NR]+ center. Proc. Natl. Acad. Sci. USA 2012, 109, 11933–11938. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; García-España, E.; Basallote, M.G.; Münck, E.; Que, L., Jr.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C–H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef] [PubMed]
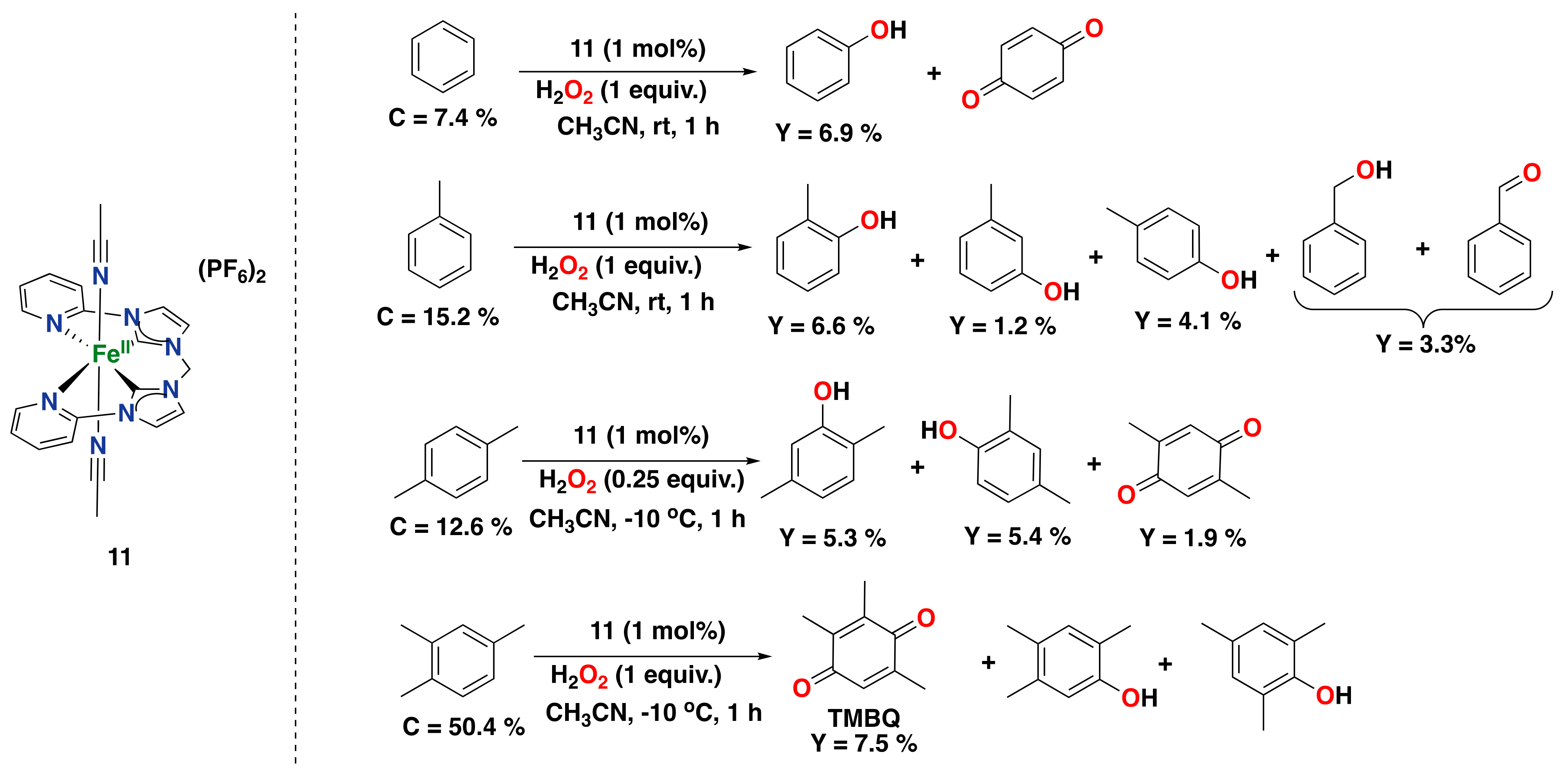
- Tkachenko, N.V.; Ottenbacher, R.V.; Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Efficient Aromatic C–H Oxidation with H2O2 in the Presence of Iron Complexes of the PDP Family. ChemCatChem 2018, 10, 4052–4057. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Lyakin, O.Y.; Zima, A.M.; Talsi, E.P.; Bryliakov, K.P. Effect of different carboxylic acids on the aromatic hydroxylation with H2O2 in the presence of an iron aminopyridine complex. J. Organomet. Chem. 2018, 871, 130–134. [Google Scholar] [CrossRef]
- Kal, S.; Draksharapu, A.; Que, L., Jr. Sc3+ (or HClO4) Activation of a Nonheme FeIII–OOH Intermediate for the Rapid Hydroxylation of Cyclohexane and Benzene. J. Am. Chem. Soc. 2018, 140, 5798–5804. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Van Heuvelen, K.M.; Meier, K.K.; Münck, E.; Que, L., Jr. Sc3+-triggered oxoiron(IV) formation from O2 and its non-heme iron(II) precursor via a Sc3+–peroxo–Fe3+ intermediate. J. Am. Chem. Soc. 2013, 135, 10198–10201. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-M.; Bang, S.; Kim, Y.M.; Cho, J.; Hong, S.; Nomura, T.; Ogura, T.; Troeppner, O.; Ivanović-Burmazović, I.; Sarangi, R. A mononuclear nonheme iron(III)–peroxo complex binding redox-inactive metal ions. Chem. Sci. 2013, 4, 3917–3923. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, W.-J.; Lu, X.; Yang, H.; Chen, Z.; Liao, R.-Z.; Yin, G. Nonredox metal ions promoted olefin epoxidation by iron(II) complexes with H2O2: DFT calculations reveal multiple channels for oxygen transfer. Inorg. Chem. 2017, 56, 15138–15149. [Google Scholar] [CrossRef]
- Nodzewska, A.; Watkinson, M. Remarkable increase in the rate of the catalytic epoxidation of electron deficient styrenes through the addition of Sc(OTf)3 to the MnTMTACN catalyst. Chem. Commun. 2018, 54, 1461–1464. [Google Scholar] [CrossRef]
- Chatterjee, S.; Paine, T.K. Olefin cis-Dihydroxylation and Aliphatic C–H Bond Oxygenation by a Dioxygen-Derived Electrophilic Iron–Oxygen Oxidant. Angew. Chem. Int. Ed. 2015, 54, 9338–9342. [Google Scholar] [CrossRef] [PubMed]
- Kal, S.; Que, L., Jr. Activation of a Non-Heme FeIII-OOH by a Second FeIII to Hydroxylate Strong C–H Bonds: Possible Implications for Soluble Methane Monooxygenase. Angew. Chem. Int. Ed. 2019, 58, 8484–8488. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, H.; Cai, H.; Zhang, J.; Gong, X.; Han, W. Iron-catalyzed arene C–H hydroxylation. Science 2021, 374, 77–81. [Google Scholar] [CrossRef] [PubMed]
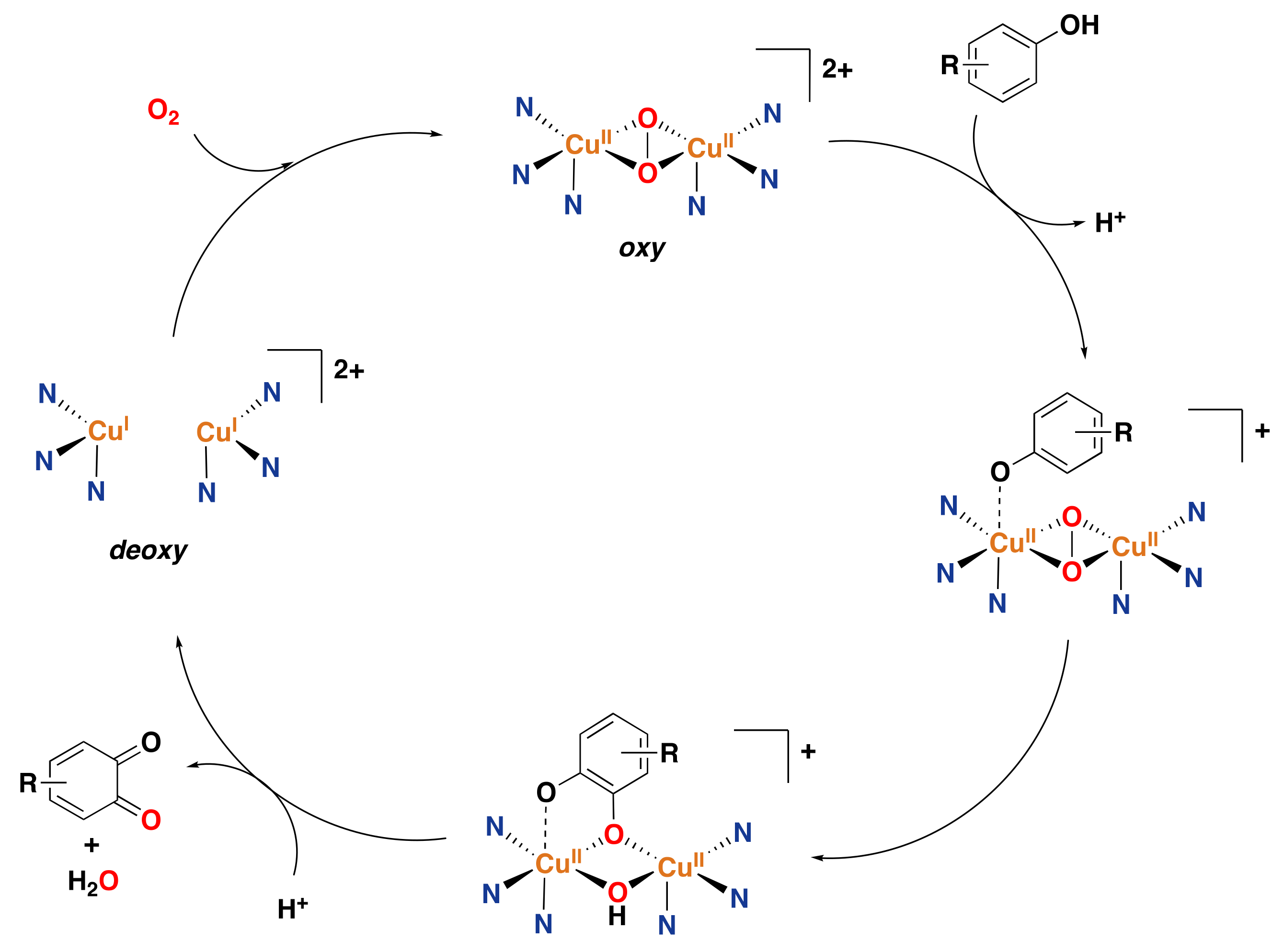
- Klinman, J.P. Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem. Rev. 1996, 96, 2541–2562. [Google Scholar] [CrossRef]
- Fontecave, M.; Pierre, J.-L. Oxidations by copper metalloenzymes and some biomimetic approaches. Coord. Chem. Rev. 1998, 170, 125–140. [Google Scholar] [CrossRef]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Mol. Biol. 1998, 5, 1084–1090. [Google Scholar] [CrossRef]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper oxidases and oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef]
- Rolff, M.; Schottenheim, J.; Decker, H.; Tuczek, F. Copper–O2 reactivity of tyrosinase models towards external monophenolic substrates: Molecular mechanism and comparison with the enzyme. Chem. Soc. Rev. 2011, 40, 4077–4098. [Google Scholar] [CrossRef]
- Decker, H.; Schweikardt, T.; Tuczek, F. The first crystal structure of tyrosinase: All questions answered? Angew. Chem. Int. Ed. 2006, 45, 4546–4550. [Google Scholar] [CrossRef]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Garcia-Bosch, I.; Company, A.; Costas, M. Structural and reactivity models for copper oxygenases: Cooperative effects and novel reactivities. Acc. Chem. Res. 2015, 48, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.D.; Hayes, J.C.; Gultneh, Y.; Cruse, R.W.; McKown, J.W.; Hutchinson, J.P.; Zubieta, J. Copper-mediated hydroxylation of an arene: Model system for the action of copper monooxygenases. Structures of a binuclear copper(I) complex and its oxygenated product. J. Am. Chem. Soc. 1984, 106, 2121–2128. [Google Scholar] [CrossRef]
- Karlin, K.D.; Nasir, M.S.; Cohen, B.I.; Cruse, R.W.; Kaderli, S.; Zuberbuehler, A.D. Reversible dioxygen binding and aromatic hydroxylation in O2-reactions with substituted xylyl dinuclear copper(I) complexes: Syntheses and low-temperature kinetic/thermodynamic and spectroscopic investigations of a copper monooxygenase model system. J. Am. Chem. Soc. 1994, 116, 1324–1336. [Google Scholar] [CrossRef]
- Pidcock, E.; Obias, H.V.; Zhang, C.X.; Karlin, K.D.; Solomon, E.I. Investigation of the reactive oxygen intermediate in an arene hydroxylation reaction performed by xylyl-bridged binuclear copper complexes. J. Am. Chem. Soc. 1998, 120, 7841–7847. [Google Scholar] [CrossRef]
- Becker, M.; Schindler, S.; Karlin, K.D.; Kaden, T.A.; Kaderli, S.; Palanché, T.; Zuberbühler, A.D. Intramolecular ligand hydroxylation: Mechanistic high-pressure studies on the reaction of a dinuclear copper(I) complex with dioxygen. Inorg. Chem. 1999, 38, 1989–1995. [Google Scholar] [CrossRef]
- Maiti, D.; Fry, H.C.; Woertink, J.S.; Vance, M.A.; Solomon, E.I.; Karlin, K.D. A 1: 1 copper–dioxygen adduct is an end-on bound superoxo copper(II) complex which undergoes oxygenation reactions with phenols. J. Am. Chem. Soc. 2007, 129, 264–265. [Google Scholar] [CrossRef]
- Maiti, D.; Lee, D.H.; Gaoutchenova, K.; Würtele, C.; Holthausen, M.C.; Narducci Sarjeant, A.A.; Sundermeyer, J.; Schindler, S.; Karlin, K.D. Reactions of a Copper(II) Superoxo Complex Lead to C–H and O–H Substrate Oxygenation: Modeling Copper-Monooxygenase C–H Hydroxylation. Angew. Chem. Int. Ed. 2008, 47, 82–85. [Google Scholar] [CrossRef]
- Karlin, K.D.; Zhang, C.X.; Rheingold, A.L.; Galliker, B.; Kaderli, S.; Zuberbühler, A.D. Reversible dioxygen binding and arene hydroxylation reactions: Kinetic and thermodynamic studies involving ligand electronic and structural variations. Inorg. Chim. Acta 2012, 389, 138–150. [Google Scholar] [CrossRef]
- Lee, J.Y.; Peterson, R.L.; Ohkubo, K.; Garcia-Bosch, I.; Himes, R.A.; Woertink, J.; Moore, C.D.; Solomon, E.I.; Fukuzumi, S.; Karlin, K.D. Mechanistic insights into the oxidation of substituted phenols via hydrogen atom abstraction by a cupric–superoxo complex. J. Am. Chem. Soc. 2014, 136, 9925–9937. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.Y.; Cowley, R.E.; Ginsbach, J.W.; Siegler, M.A.; Solomon, E.I.; Karlin, K.D. A N3S(thioether)-ligated CuII-superoxo with enhanced reactivity. J. Am. Chem. Soc. 2015, 137, 2796–2799. [Google Scholar] [CrossRef]
- Mahapatra, S.; Kaderli, S.; Llobet, A.; Neuhold, Y.-M.; Palanché, T.; Halfen, J.A.; Young, V.G.; Kaden, T.A.; Que, L.; Zuberbühler, A.D.; et al. Binucleating ligand structural effects on (μ-peroxo)-and bis(μ-oxo) dicopper complex formation and decay: Competition between arene hydroxylation and aliphatic C–H bond activation. Inorg. Chem. 1997, 36, 6343–6356. [Google Scholar] [CrossRef]
- Sander, O.; Henß, A.; Näther, C.; Würtele, C.; Holthausen, M.C.; Schindler, S.; Tuczek, F. Aromatic Hydroxylation in a Copper Bis(imine) Complex Mediated by a μ-η2:η2 Peroxo Dicopper Core: A Mechanistic Scenario. Chem. Eur. J. 2008, 14, 9714–9729. [Google Scholar] [CrossRef] [PubMed]
- Menif, R.; Martell, A.E.; Squattrito, P.J.; Clearfield, A. New hexaaza macrocyclic binucleating ligands. Oxygen insertion with a dicopper(I) Schiff base macrocyclic complex. Inorg. Chem. 1990, 29, 4723–4729. [Google Scholar] [CrossRef]
- Santagostini, L.; Gullotti, M.; Monzani, E.; Casella, L.; Dillinger, R.; Tuczek, F. Reversible dioxygen binding and phenol oxygenation in a tyrosinase model system. Chem. Eur. J. 2000, 6, 519–522. [Google Scholar] [CrossRef]
- Palavicini, S.; Granata, A.; Monzani, E.; Casella, L. Hydroxylation of phenolic compounds by a peroxodicopper(II) complex: Further insight into the mechanism of tyrosinase. J. Am. Chem. Soc. 2005, 127, 18031–18036. [Google Scholar] [CrossRef]
- Itoh, S.; Kumei, H.; Taki, M.; Nagatomo, S.; Kitagawa, T.; Fukuzumi, S. Oxygenation of phenols to catechols by a (μ-η2:η2-peroxo) dicopper(II) complex: Mechanistic insight into the phenolase activity of tyrosinase. J. Am. Chem. Soc. 2001, 123, 6708–6709. [Google Scholar] [CrossRef]
- Battaini, G.; De Carolis, M.; Monzani, E.; Tuczek, F.; Casella, L. The phenol ortho-oxygenation by mononuclear copper(I) complexes requires a dinuclear μ-η2:η2-peroxodicopper(II) complex rather than mononuclear CuO2 species. Chem. Commun. 2003, 726–727. [Google Scholar] [CrossRef]
- Citek, C.; Lyons, C.T.; Wasinger, E.C.; Stack, T.D.P. Self-assembly of the oxy-tyrosinase core and the fundamental components of phenolic hydroxylation. Nat. Chem. 2012, 4, 317–322. [Google Scholar] [CrossRef]
- Company, A.; Palavicini, S.; Garcia-Bosch, I.; Mas-Ballesté, R.; Que, L., Jr.; Rybak-Akimova, E.V.; Casella, L.; Ribas, X.; Costas, M. Tyrosinase-Like Reactivity in a CuIII2(μ-O)2 Species. Chem. Eur. J. 2008, 14, 3535–3538. [Google Scholar] [CrossRef]
- Tachi, Y.; Aita, K.; Teramae, S.; Tani, F.; Naruta, Y.; Fukuzumi, S.; Itoh, S. Dicopper–Dioxygen Complex Supported by Asymmetric Pentapyridine Dinucleating Ligand. Inorg. Chem. 2004, 43, 4558–4560. [Google Scholar] [CrossRef]
- Garcia-Bosch, I.; Company, A.; Frisch, J.R.; Torrent-Sucarrat, M.; Cardellach, M.; Gamba, I.; Güell, M.; Casella, L.; Que, L., Jr.; Ribas, X.; et al. O2 Activation and Selective Phenolate ortho Hydroxylation by an Unsymmetric Dicopper μ-η1:η1-Peroxido Complex. Angew. Chem. Int. Ed. 2010, 49, 2406–2409. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Furutachi, H.; Kobino, M.; Tomii, M.; Nagatomo, S.; Tosha, T.; Osako, T.; Fujinami, S.; Itoh, S.; Kitagawa, T.; et al. Intramolecular arene hydroxylation versus intermolecular olefin epoxidation by (μ-η2:η2-peroxo)dicopper(II) complex supported by dinucleating ligand. J. Am. Chem. Soc. 2006, 128, 3874–3875. [Google Scholar] [CrossRef] [PubMed]
- Mirica, L.M.; Vance, M.; Rudd, D.J.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; Stack, T. A Stabilized μ-η2:η2 Peroxodicopper(II) Complex with a Secondary Diamine Ligand and Its Tyrosinase-like Reactivity. J. Am. Chem. Soc. 2002, 124, 9332–9333. [Google Scholar] [CrossRef] [PubMed]
- Mirica, L.M.; Rudd, D.J.; Vance, M.A.; Solomon, E.I.; Hodgson, K.O.; Hedman, B.; Stack, T.D.P. μ-η2:η2-Peroxodicopper(II) complex with a secondary diamine ligand: A functional model of tyrosinase. J. Am. Chem. Soc. 2006, 128, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- Mirica, L.M.; Vance, M.; Rudd, D.J.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; Stack, T.D.P. Tyrosinase reactivity in a model complex: An alternative hydroxylation mechanism. Science 2005, 308, 1890–1892. [Google Scholar] [CrossRef]
- Hamann, J.N.; Tuczek, F. New catalytic model systems of tyrosinase: Fine tuning of the reactivity with pyrazole-based N-donor ligands. Chem. Commun. 2014, 50, 2298–2300. [Google Scholar] [CrossRef]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D.P. Structure and spectroscopy of copper–dioxygen complexes. Chem. Rev. 2004, 104, 1013–1046. [Google Scholar] [CrossRef]
- Lewis, E.A.; Tolman, W.B. Reactivity of dioxygen–copper systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef]
- Halfen, J.A.; Mahapatra, S.; Wilkinson, E.C.; Kaderli, S.; Young, V.G., Jr.; Que, L., Jr.; Zuberbühler, A.D.; Tolman, W.B. Reversible cleavage and formation of the dioxygen O–O bond within a dicopper complex. Science 1996, 271, 1397–1400. [Google Scholar] [CrossRef]
- Tolman, W.B. Making and breaking the dioxygen O–O bond: New insights from studies of synthetic copper complexes. Acc. Chem. Res. 1997, 30, 227–237. [Google Scholar] [CrossRef]
- Op’t Holt, B.T.; Vance, M.A.; Mirica, L.M.; Heppner, D.E.; Stack, T.D.P.; Solomon, E.I. Reaction coordinate of a functional model of tyrosinase: Spectroscopic and computational characterization. J. Am. Chem. Soc. 2009, 131, 6421–6438. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.L.; Rodgers, K.R.; Tolman, W.B. Is the Bis(μ-oxo)dicopper Core Capable of Hydroxylating an Arene? Angew. Chem. Int. Ed. 1999, 38, 1139–1142. [Google Scholar] [CrossRef]
- Herres-Pawlis, S.; Verma, P.; Haase, R.; Kang, P.; Lyons, C.T.; Wasinger, E.C.; Flörke, U.; Henkel, G.; Stack, T.D.P. Phenolate hydroxylation in a bis(μ-oxo)dicopper(III) complex: Lessons from the guanidine/amine series. J. Am. Chem. Soc. 2009, 131, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S. Developing mononuclear copper–active-oxygen complexes relevant to reactive intermediates of biological oxidation reactions. Acc. Chem. Res. 2015, 48, 2066–2074. [Google Scholar] [CrossRef]
- Kunishita, A.; Teraoka, J.; Scanlon, J.D.; Matsumoto, T.; Suzuki, M.; Cramer, C.J.; Itoh, S. Aromatic Hydroxylation Reactivity of a Mononuclear Cu(II) –Alkylperoxo Complex. J. Am. Chem. Soc. 2007, 129, 7248–7249. [Google Scholar] [CrossRef]
- Kunishita, A.; Scanlon, J.D.; Ishimaru, H.; Honda, K.; Ogura, T.; Suzuki, M.; Cramer, C.J.; Itoh, S. Reactions of copper(II)-H2O2 adducts supported by tridentate bis(2-pyridylmethyl)amine ligands: Sensitivity to solvent and variations in ligand substitution. Inorg. Chem. 2008, 47, 8222–8232. [Google Scholar] [CrossRef]
- Würtele, C.; Gaoutchenova, E.; Harms, K.; Holthausen, M.C.; Sundermeyer, J.; Schindler, S. Crystallographic characterization of a synthetic 1:1 end-on copper dioxygen adduct complex. Angew. Chem. Int. Ed. 2006, 45, 3867–3869. [Google Scholar] [CrossRef]
- Conde, A.; Diaz-Requejo, M.M.; Pérez, P.J. Direct, copper-catalyzed oxidation of aromatic C–H bonds with hydrogen peroxide under acid-free conditions. Chem. Commun. 2011, 47, 8154–8156. [Google Scholar] [CrossRef]
- Vilella, L.; Conde, A.; Balcells, D.; Díaz-Requejo, M.M.; Lledós, A.; Pérez, P.J. A competing, dual mechanism for catalytic direct benzene hydroxylation from combined experimental-DFT studies. Chem. Sci. 2017, 8, 8373–8383. [Google Scholar] [CrossRef]
- Wu, L.; Zhong, W.; Xu, B.; Wei, Z.; Liu, X. Synthesis and characterization of copper(II) complexes with multidentate ligands as catalysts for the direct hydroxylation of benzene to phenol. Dalton Trans. 2015, 44, 8013–8020. [Google Scholar] [CrossRef]
- Tsuji, T.; Zaoputra, A.A.; Hitomi, Y.; Mieda, K.; Ogura, T.; Shiota, Y.; Yoshizawa, K.; Sato, H.; Kodera, M. Specific enhancement of catalytic activity by a dicopper core: Selective hydroxylation of benzene to phenol with hydrogen peroxide. Angew. Chem. Int. Ed. 2017, 56, 7779–7782. [Google Scholar] [CrossRef] [PubMed]
- Muthuramalingam, S.; Anandababu, K.; Velusamy, M.; Mayilmurugan, R. Benzene Hydroxylation by Bioinspired Copper(II) Complexes: Coordination Geometry versus Reactivity. Inorg. Chem. 2020, 59, 5918–5928. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Karlin, K.D.; Fukuzumi, S. One-step selective hydroxylation of benzene to phenol with hydrogen peroxide catalysed by copper complexes incorporated into mesoporous silica–alumina. Chem. Sci. 2016, 7, 2856–2863. [Google Scholar] [CrossRef] [PubMed]
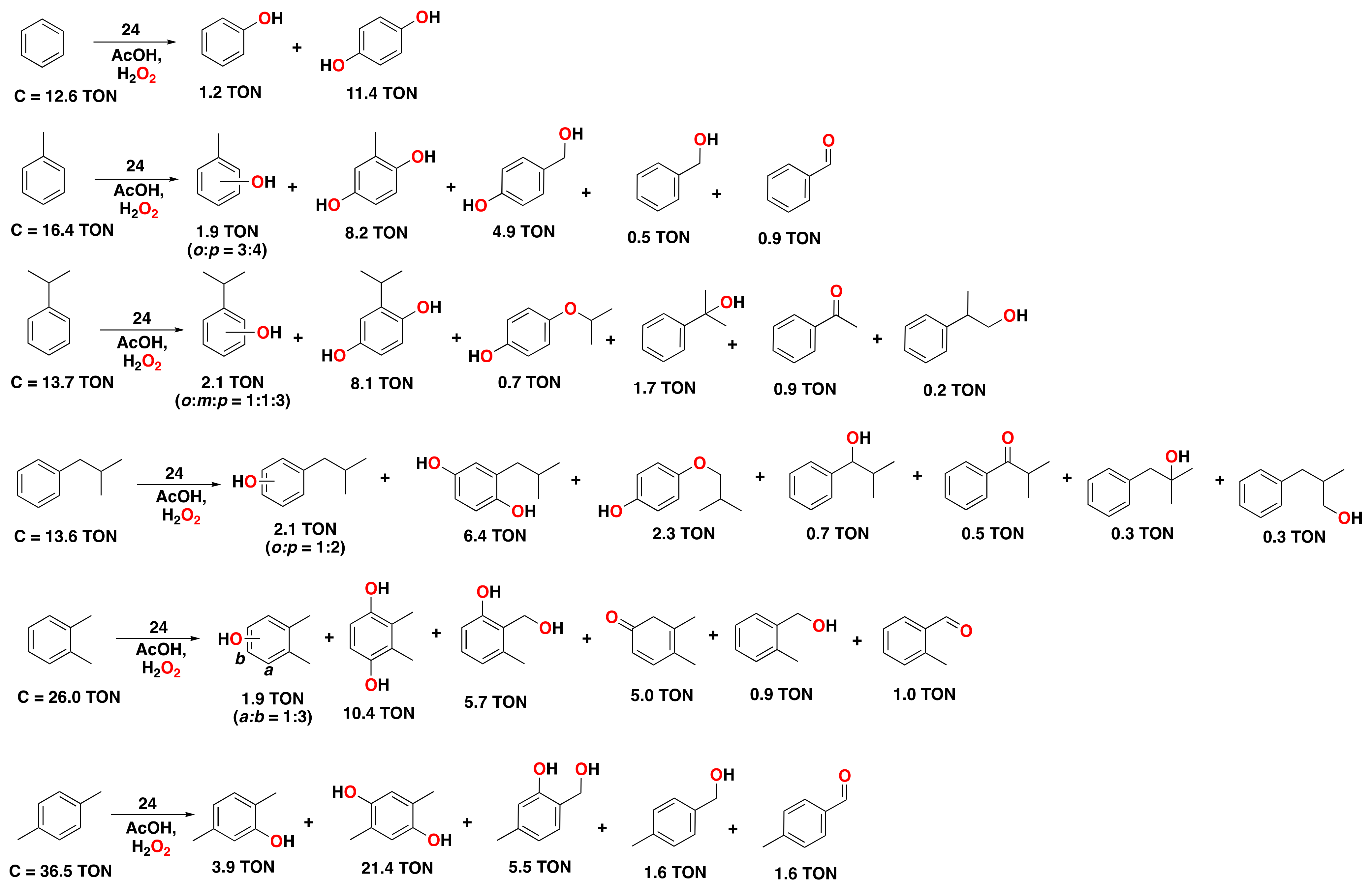
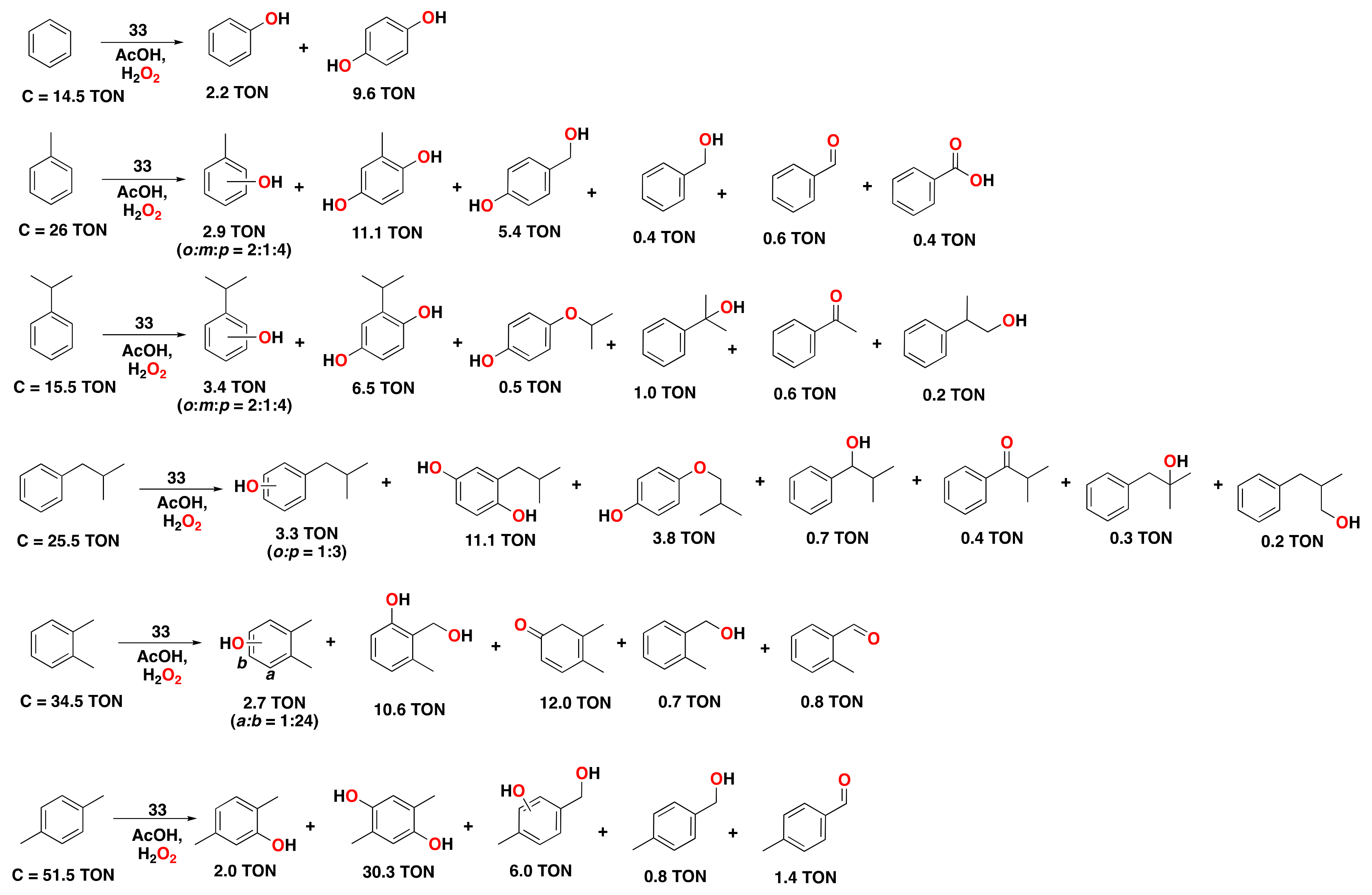
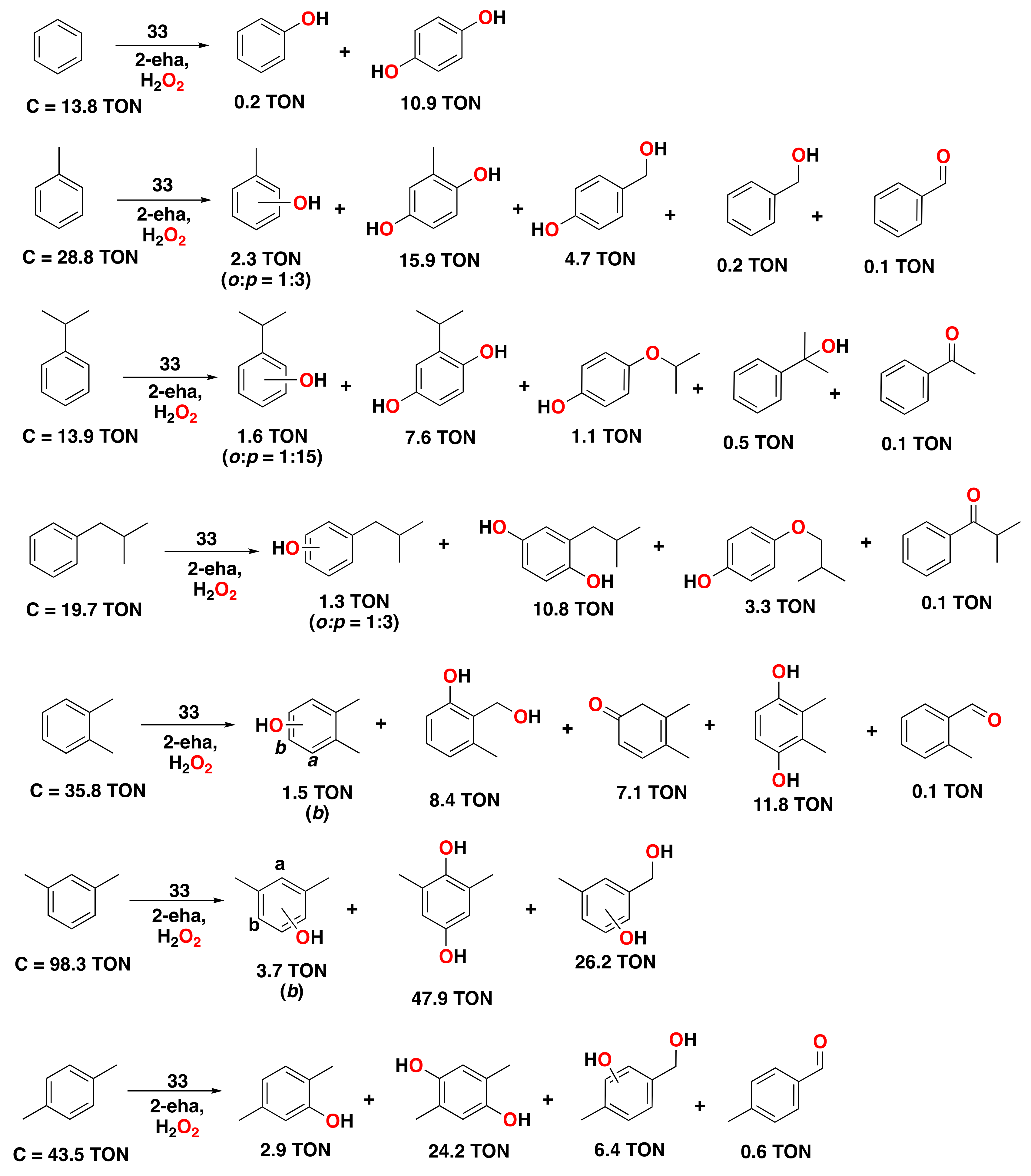
- Kumari, S.; Muthuramalingam, S.; Dhara, A.K.; Singh, U.; Mayilmurugan, R.; Ghosh, K. Cu(I) complexes obtained via spontaneous reduction of Cu(II) complexes supported by designed bidentate ligands: Bioinspired Cu(I) based catalysts for aromatic hydroxylation. Dalton Trans. 2020, 49, 13829–13839. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef]
- Corona, T.; Company, A. Spectroscopically characterized synthetic mononuclear nickel–oxygen species. Chem. Eur. J. 2016, 22, 13422–13429. [Google Scholar] [CrossRef]
- Kimura, E.; Machida, R. A mono-ozygenase model for selective aromatic hydroxylation with nickel(II)-macrocyclic polyamines. J. Chem. Soc. Chem. Commun. 1984, 499–500. [Google Scholar] [CrossRef]
- Honda, K.; Cho, J.; Matsumoto, T.; Roh, J.; Furutachi, H.; Tosha, T.; Kubo, M.; Fujinami, S.; Ogura, T.; Kitagawa, T.; et al. Oxidation Reactivity of Bis(μ-oxo) Dinickel(III) Complexes: Arene Hydroxylation of the Supporting Ligand. Angew. Chem. Int. Ed. 2009, 48, 3304–3307. [Google Scholar] [CrossRef]
- Kunishita, A.; Doi, Y.; Kubo, M.; Ogura, T.; Sugimoto, H.; Itoh, S. Ni(II)/H2O2 reactivity in bis[(pyridin-2-yl)methyl]amine tridentate ligand system. Aromatic hydroxylation reaction by bis(μ-oxo)dinickel(III) complex. Inorg. Chem. 2009, 48, 4997–5004. [Google Scholar] [CrossRef]
- Tano, T.; Doi, Y.; Inosako, M.; Kunishita, A.; Kubo, M.; Ishimaru, H.; Ogura, T.; Sugimoto, H.; Itoh, S. Nickel(II) Complexes of tpa Ligands with 6-Phenyl Substituents (Phntpa). Structure and H2O2-Reactivity. Bull. Chem. Soc. Jpn. 2010, 83, 530–538. [Google Scholar] [CrossRef]
- Hikichi, S.; Yoshizawa, M.; Sasakura, Y.; Akita, M.; Moro-oka, Y. First Synthesis and Structural Characterization of Dinuclear M(III) Bis(μ-oxo) Complexes of Nickel and Cobalt with Hydrotris(pyrazolyl)borate Ligand. J. Am. Chem. Soc. 1998, 120, 10567–10568. [Google Scholar] [CrossRef]
- Itoh, S.; Bandoh, H.; Nagatomo, S.; Kitagawa, T.; Fukuzumi, S. Aliphatic hydroxylation by a bis(μ-oxo)dinickel(III) complex. J. Am. Chem. Soc. 1999, 121, 8945–8946. [Google Scholar] [CrossRef]
- Shiren, K.; Ogo, S.; Fujinami, S.; Hayashi, H.; Suzuki, M.; Uehara, A.; Watanabe, Y.; Moro-oka, Y. Synthesis, Structures, and Properties of Bis(μ-oxo)nickel(III) and Bis(μ-superoxo)nickel(II) Complexes: An Unusual Conversion of a NiIII2(μ-O)2 Core into a NiII2(μ-OO)2 Core by H2O2 and Oxygenation of Ligand. J. Am. Chem. Soc. 2000, 122, 254–262. [Google Scholar] [CrossRef]
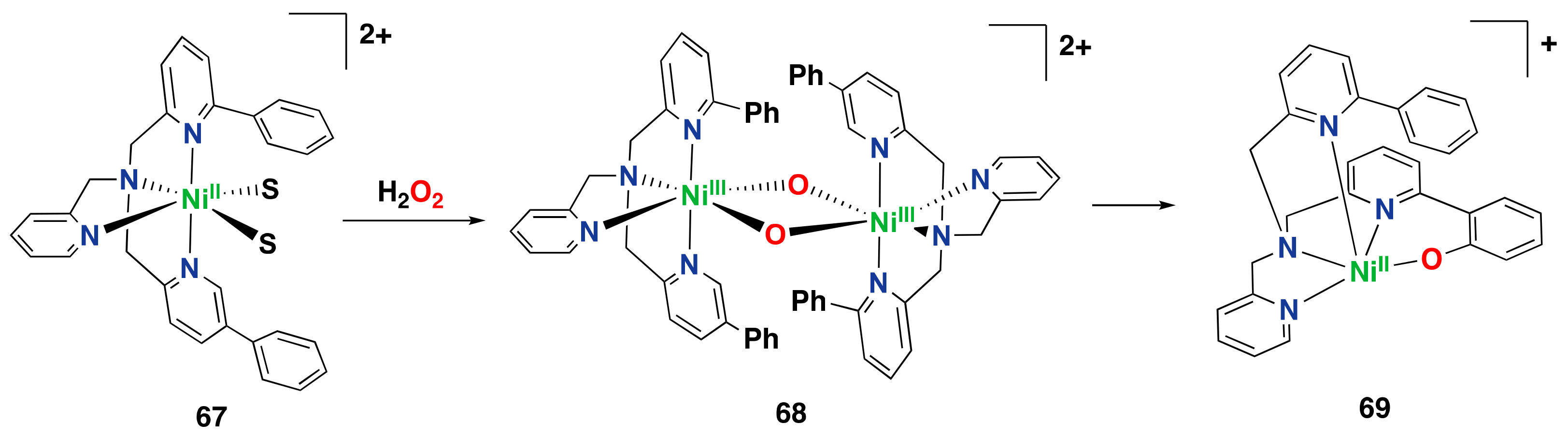
- Mandimutsira, B.S.; Yamarik, J.L.; Brunold, T.C.; Gu, W.; Cramer, S.P.; Riordan, C.G. Dioxygen activation by a nickel thioether complex: Characterization of a NiIII2(μ-O)2 Core. J. Am. Chem. Soc. 2001, 123, 9194–9195. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Bandoh, H.; Nakagawa, M.; Nagatomo, S.; Kitagawa, T.; Karlin, K.D.; Fukuzumi, S. Formation, Characterization, and Reactivity of Bis(μ-oxo)dinickel(III) Complexes Supported by A Series of Bis[2-(2-pyridyl)ethyl]amine Ligands. J. Am. Chem. Soc. 2001, 123, 11168–11178. [Google Scholar] [CrossRef]
- Schenker, R.; Mandimutsira, B.S.; Riordan, C.G.; Brunold, T.C. Spectroscopic and Computational Studies on [(PhTttBu)2Ni2(μ-O)2]: Nature of the Bis-μ-oxo (Ni3+)2 “Diamond” Core. J. Am. Chem. Soc. 2002, 124, 13842–13855. [Google Scholar] [CrossRef]
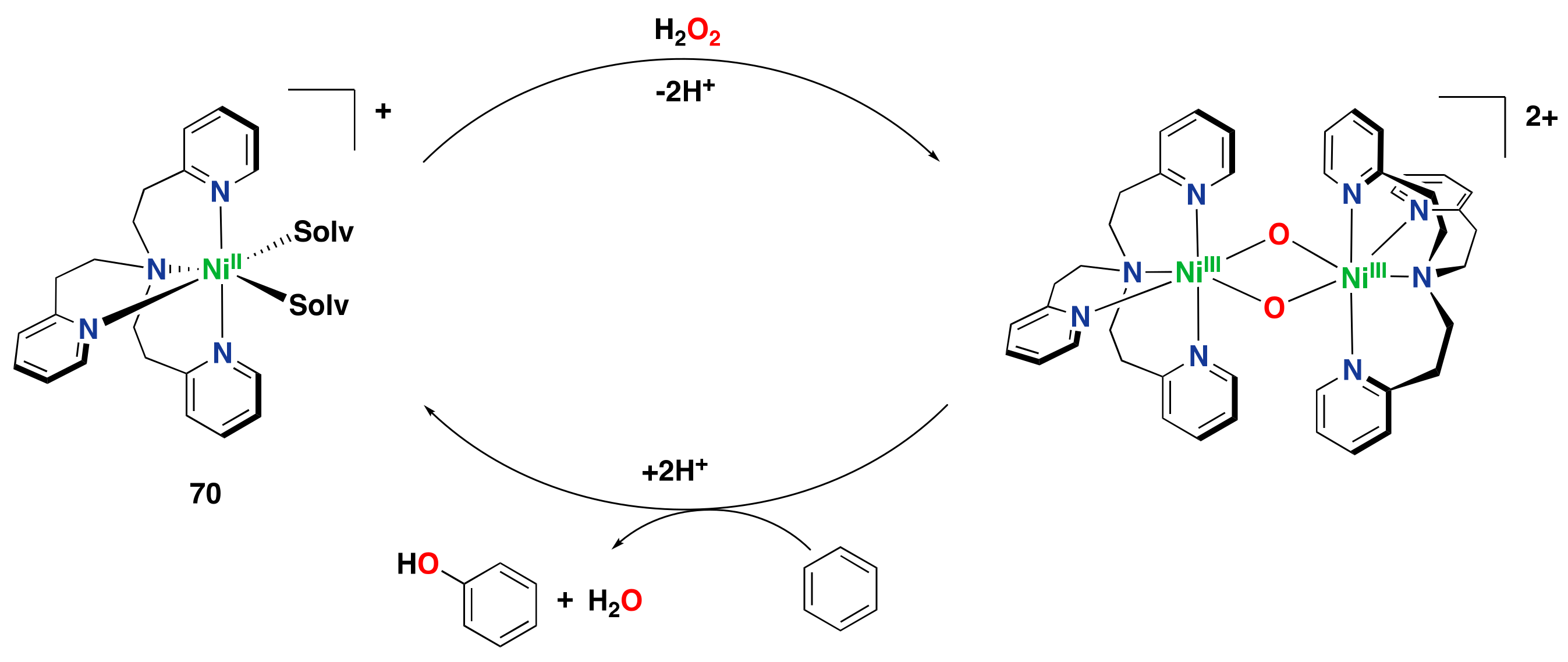
- Morimoto, Y.; Bunno, S.; Fujieda, N.; Sugimoto, H.; Itoh, S. Direct hydroxylation of benzene to phenol using hydrogen peroxide catalyzed by nickel complexes supported by pyridylalkylamine ligands. J. Am. Chem. Soc. 2015, 137, 5867–5870. [Google Scholar] [CrossRef]
- Morimoto, Y.; Takagi, Y.; Saito, T.; Ohta, T.; Ogura, T.; Tohnai, N.; Nakano, M.; Itoh, S. A Bis(μ-oxido)dinickel(III) Complex with a Triplet Ground State. Angew. Chem. Int. Ed. 2018, 57, 7640–7643. [Google Scholar] [CrossRef]
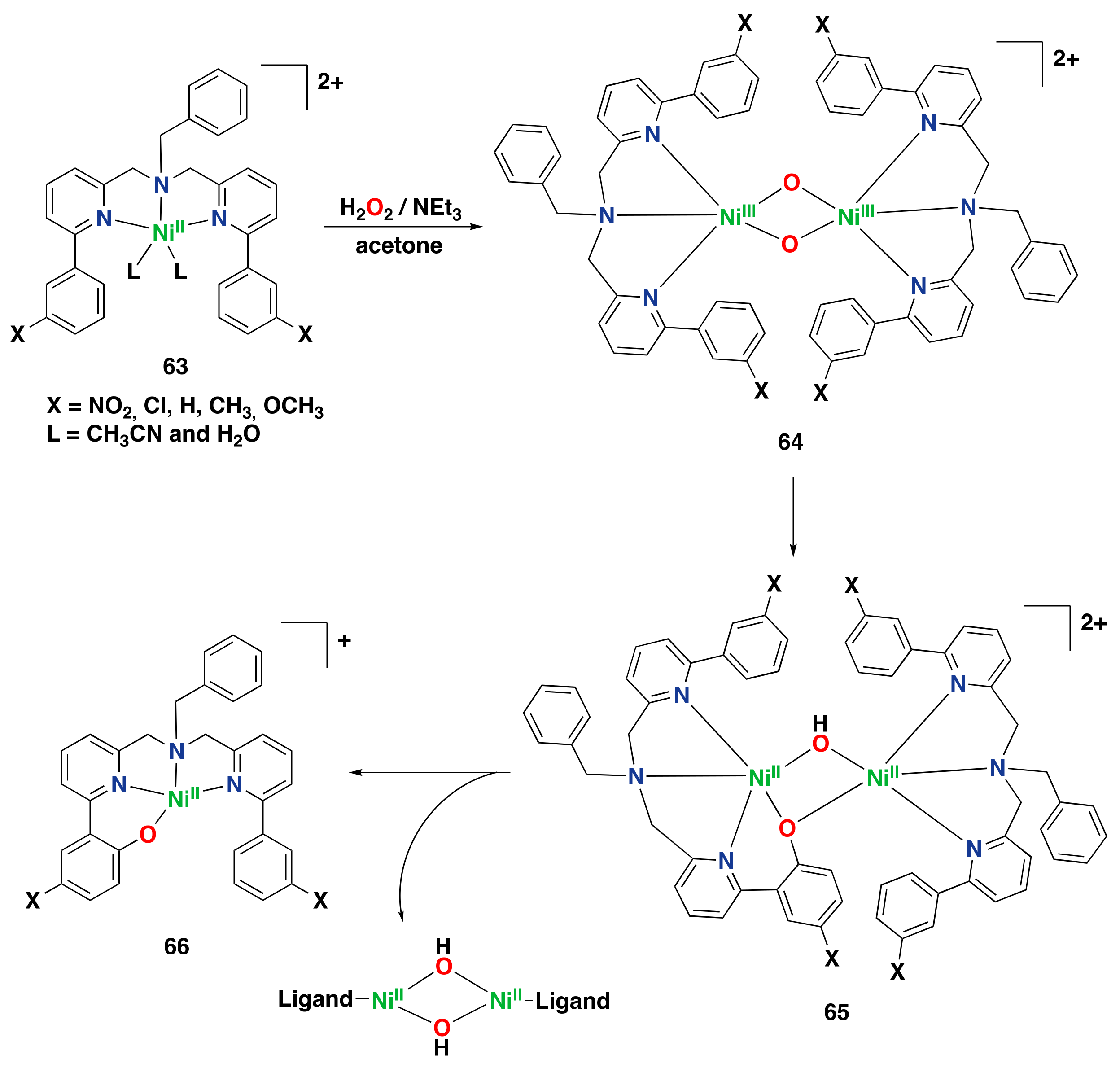
- Masferrer-Rius, E.; Hopman, R.M.; van der Kleij, J.; Lutz, M.; Klein Gebbink, R.J.M. On the Ability of Nickel Complexes Derived from Tripodal Aminopyridine Ligands to Catalyze Arene Hydroxylations. Chimia 2020, 74, 489–494. [Google Scholar] [CrossRef]
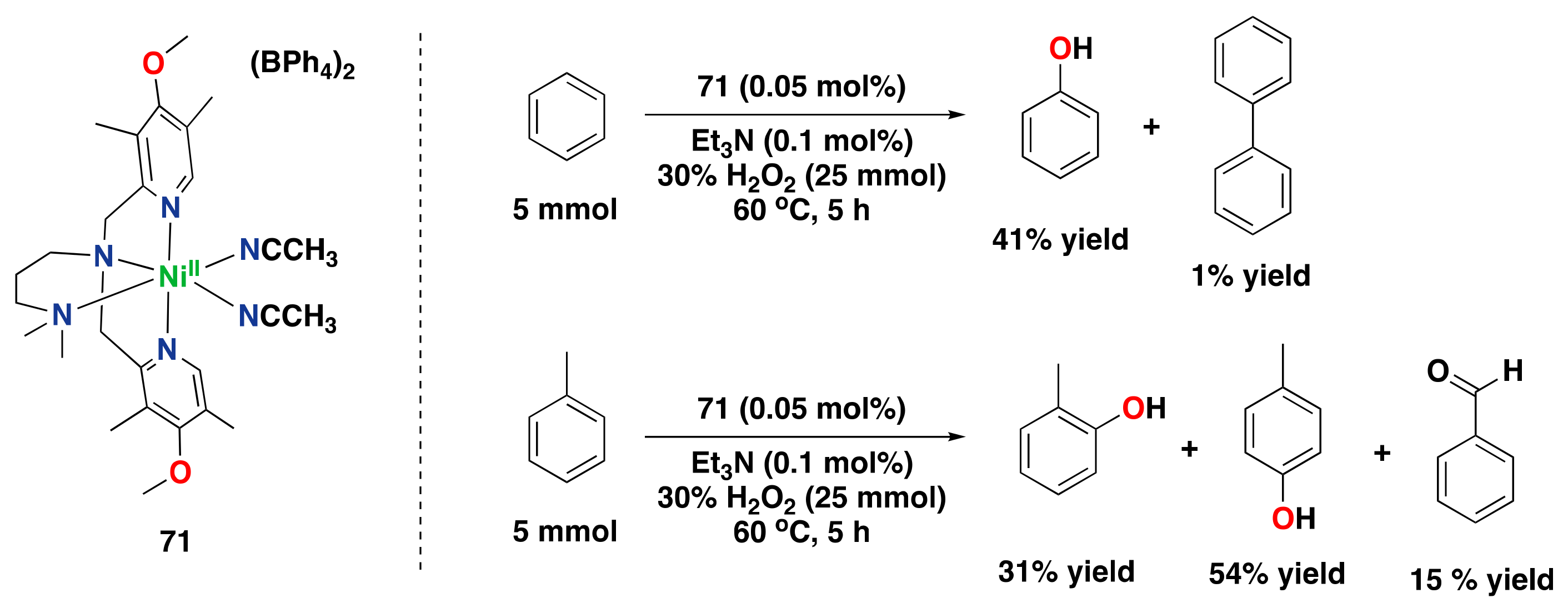
- Muthuramalingam, S.; Anandababu, K.; Velusamy, M.; Mayilmurugan, R. One step phenol synthesis from benzene catalysed by nickel(II) complexes. Catal. Sci. Technol. 2019, 9, 5991–6001. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef] [PubMed]
- Cady, C.W.; Crabtree, R.H.; Brudvig, G.W. Functional models for the oxygen-evolving complex of photosystem II. Coord. Chem. Rev. 2008, 252, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Cinco, R.M.; McFarlane Holman, K.L.; Robblee, J.H.; Yano, J.; Pizarro, S.A.; Bellacchio, E.; Sauer, K.; Yachandra, V.K. Calcium EXAFS establishes the Mn-Ca cluster in the oxygen-evolving complex of photosystem II. Biochemistry 2002, 41, 12928–12933. [Google Scholar] [CrossRef] [PubMed]
- Cahiez, G.; Duplais, C.; Buendia, J. Chemistry of organomanganese(II) compounds. Chem. Rev. 2009, 109, 1434–1476. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.R.; Dillon, B.R.; Thomas, S.P. Recent advances of manganese catalysis for organic synthesis. Eur. J. Org. Chem. 2016, 2016, 3912–3929. [Google Scholar] [CrossRef]
- Philip, R.M.; Radhika, S.; Abdulla, C.A.; Anilkumar, G. Recent Trends and Prospects in Homogeneous Manganese-Catalysed Epoxidation. Adv. Synth. Catal. 2021, 363, 1272–1289. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Z.; Fukuzumi, S.; Nam, W.; Wang, B. Artificial nonheme iron and manganese oxygenases for enantioselective olefin epoxidation and alkane hydroxylation reactions. Coord. Chem. Rev. 2020, 421, 213443. [Google Scholar] [CrossRef]
- Sun, W.; Sun, Q. Bioinspired manganese and iron complexes for enantioselective oxidation reactions: Ligand design, catalytic activity, and beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [CrossRef]
- Wu, X.; Seo, M.S.; Davis, K.M.; Lee, Y.-M.; Chen, J.; Cho, K.-B.; Pushkar, Y.N.; Nam, W. A highly reactive mononuclear non-heme manganese(IV)–Oxo complex that can activate the strong C–H bonds of alkanes. J. Am. Chem. Soc. 2011, 133, 20088–20091. [Google Scholar] [CrossRef]
- Aratani, Y.; Yamada, Y.; Fukuzumi, S. Selective hydroxylation of benzene derivatives and alkanes with hydrogen peroxide catalysed by a manganese complex incorporated into mesoporous silica–alumina. Chem. Commun. 2015, 51, 4662–4665. [Google Scholar] [CrossRef] [PubMed]
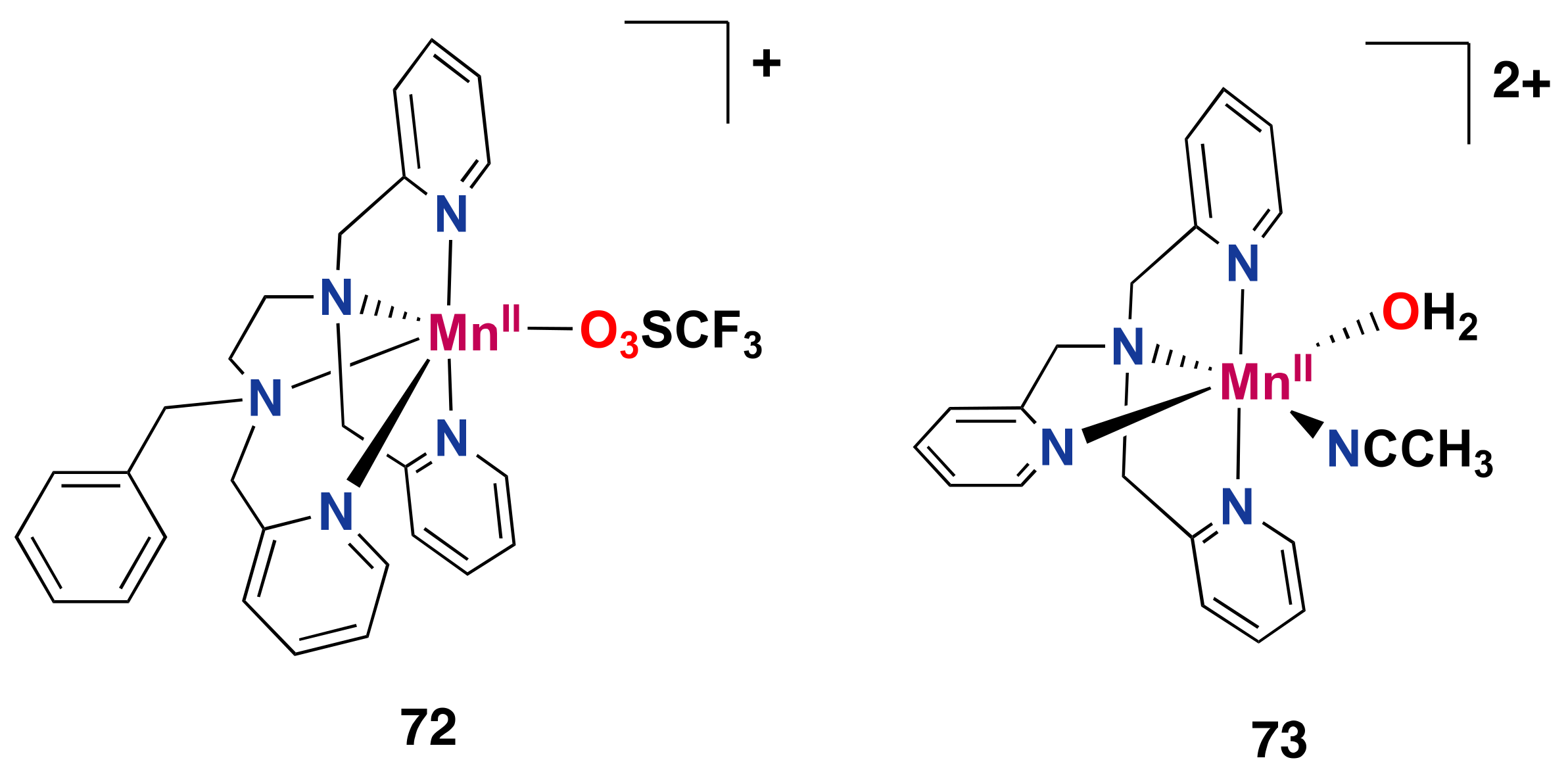
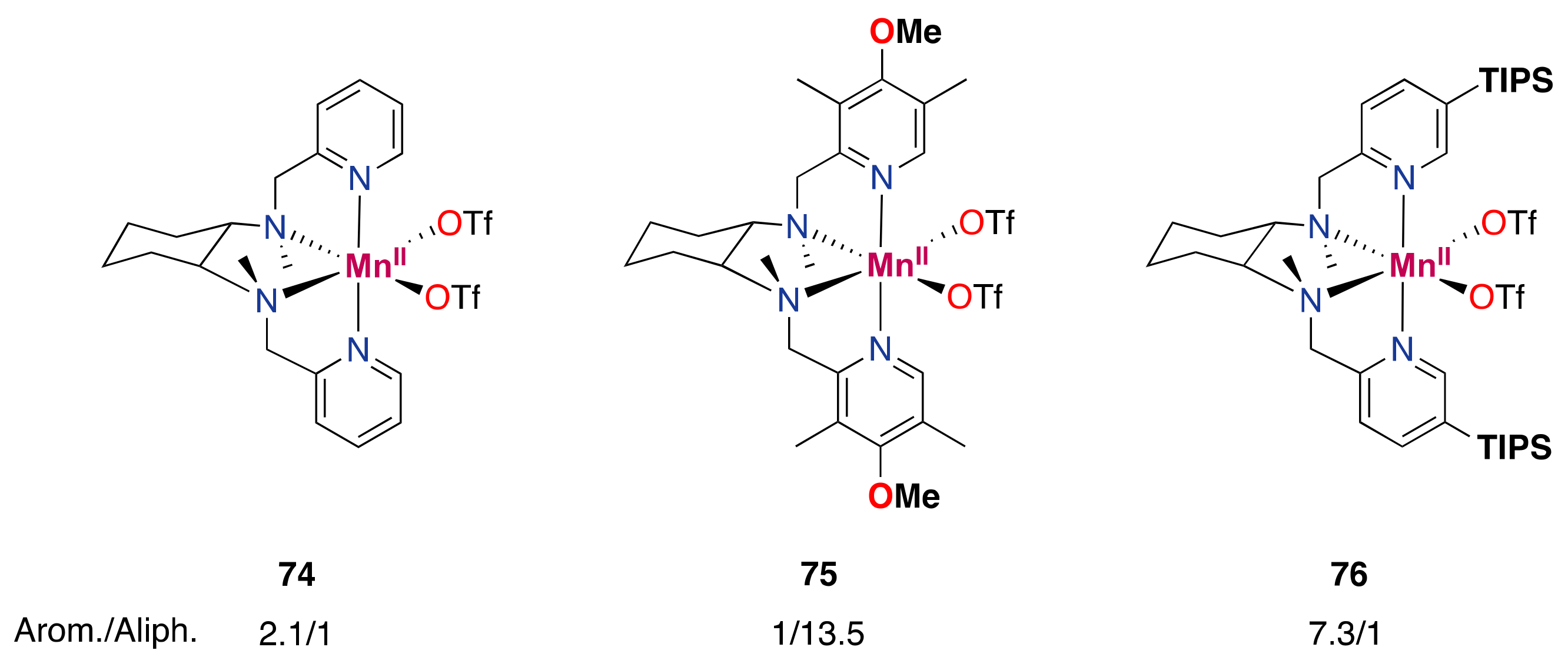
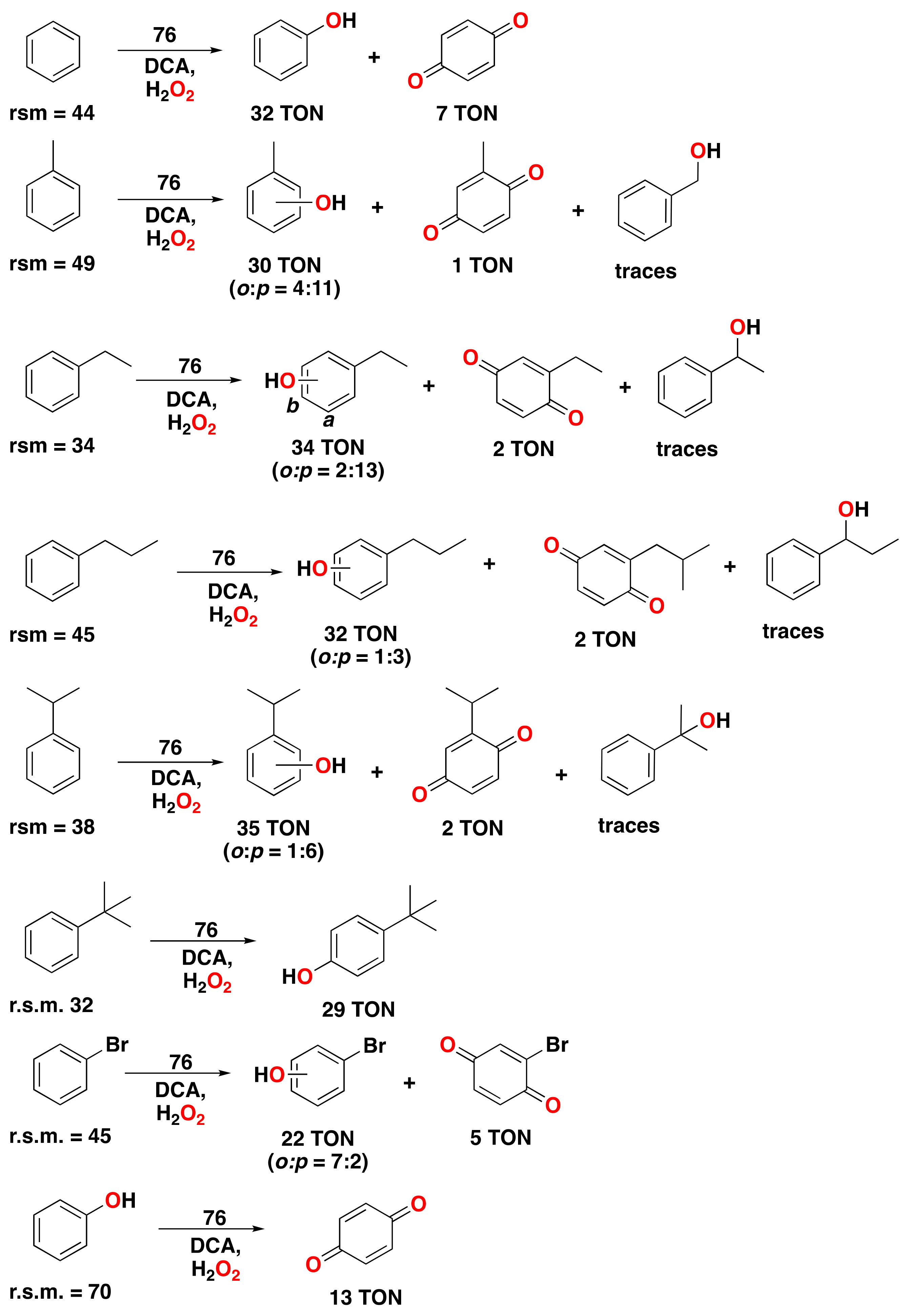
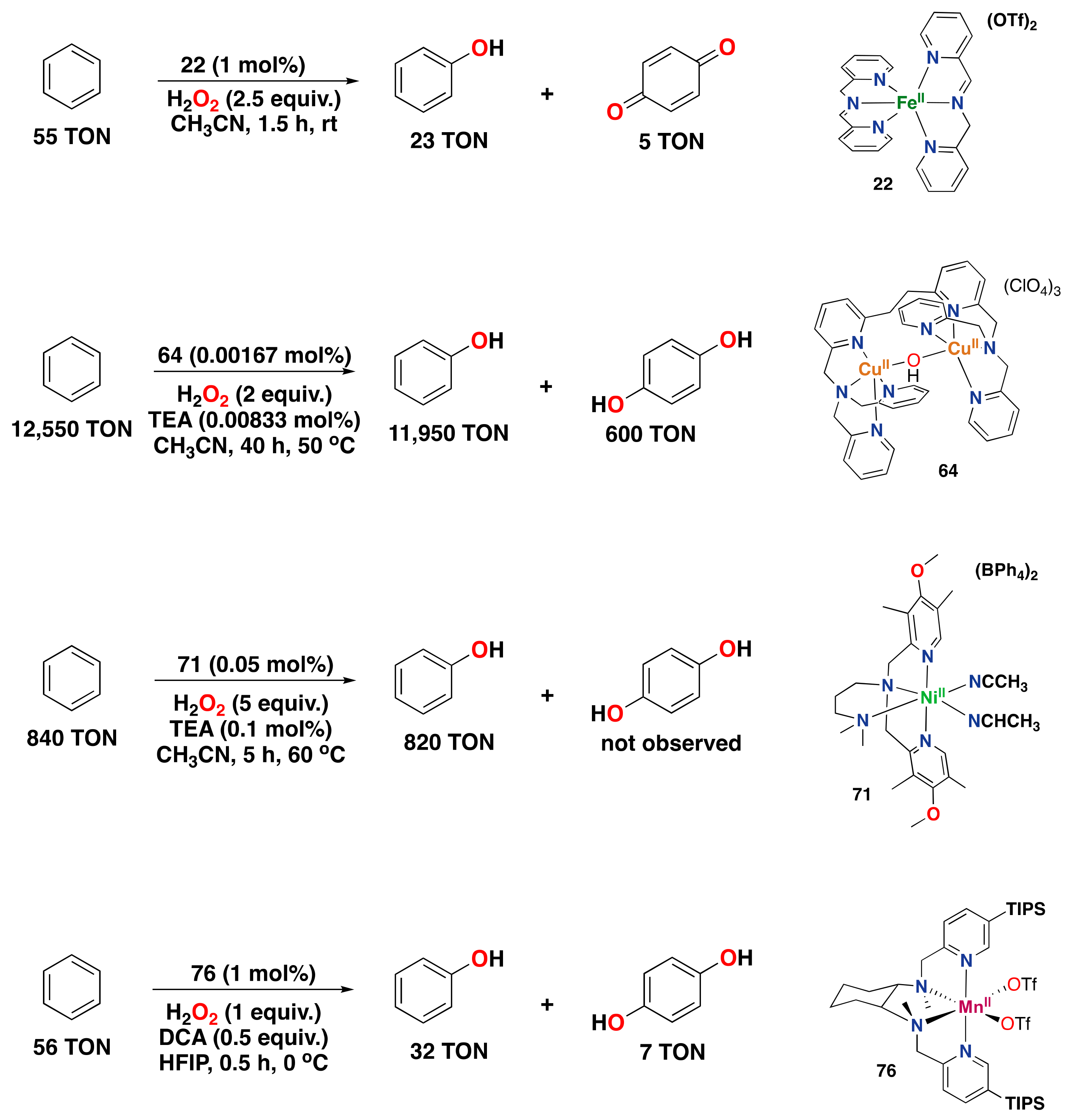
- Masferrer-Rius, E.; Borrell, M.; Lutz, M.; Costas, M.; Klein Gebbink, R.J.M. Aromatic C H Hydroxylation Reactions with Hydrogen Peroxide Catalyzed by Bulky Manganese Complexes. Adv. Synth. Catal. 2021, 363, 3785–3795. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masferrer-Rius, E.; Klein Gebbink, R.J.M. Non-Noble Metal Aromatic Oxidation Catalysis: From Metalloenzymes to Synthetic Complexes. Catalysts 2023, 13, 773. https://doi.org/10.3390/catal13040773
Masferrer-Rius E, Klein Gebbink RJM. Non-Noble Metal Aromatic Oxidation Catalysis: From Metalloenzymes to Synthetic Complexes. Catalysts. 2023; 13(4):773. https://doi.org/10.3390/catal13040773
Chicago/Turabian StyleMasferrer-Rius, Eduard, and Robertus J. M. Klein Gebbink. 2023. "Non-Noble Metal Aromatic Oxidation Catalysis: From Metalloenzymes to Synthetic Complexes" Catalysts 13, no. 4: 773. https://doi.org/10.3390/catal13040773