
Isoselective Ring-Opening Polymerization of rac-Lactide Catalyzed by Simple Potassium Amidate Complexes Containing Polycyclic Aryl Group †
Abstract
:1. Introduction
2. Results and Discussion
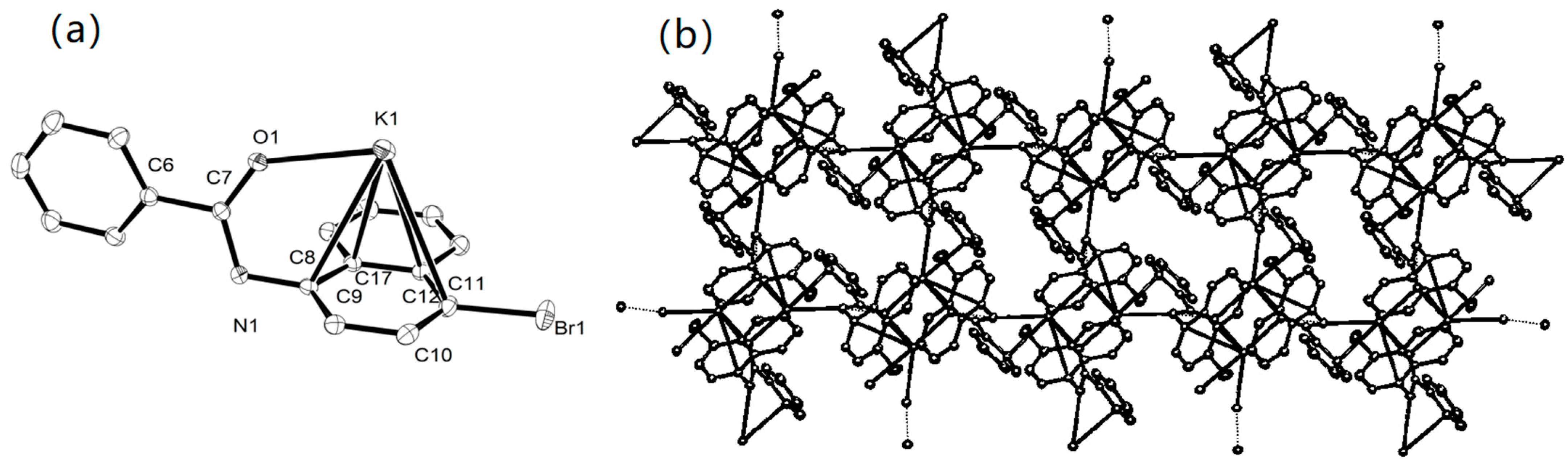
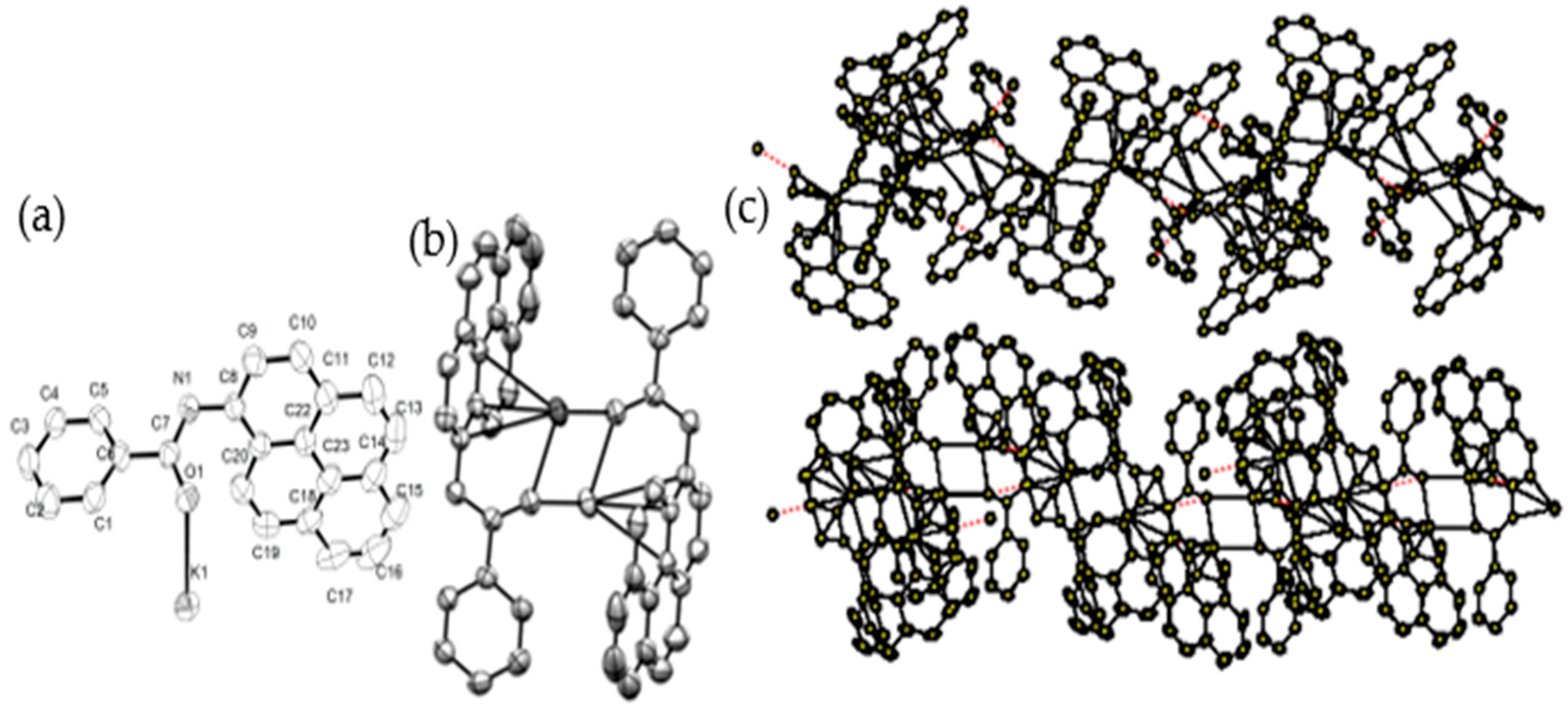
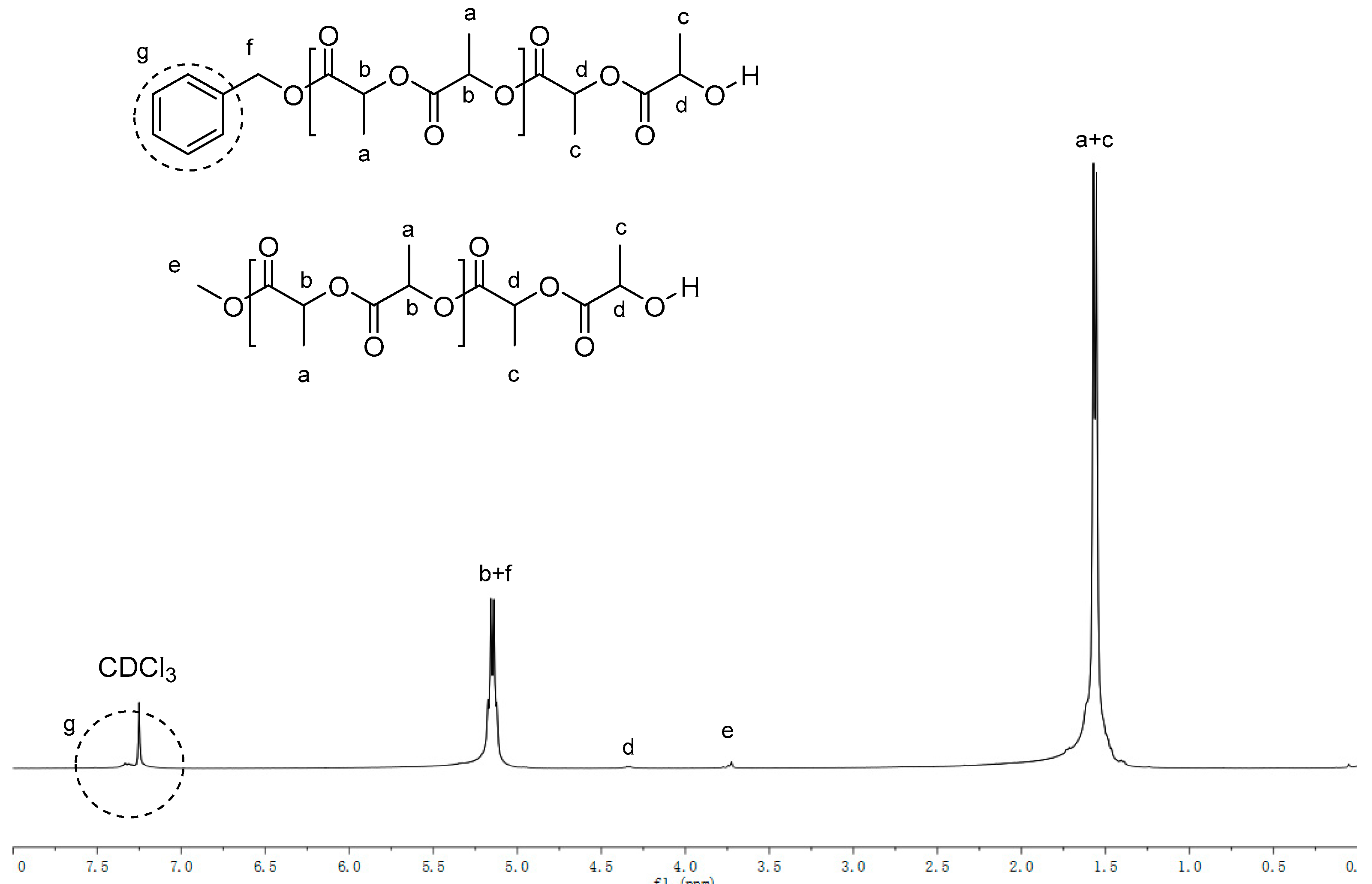
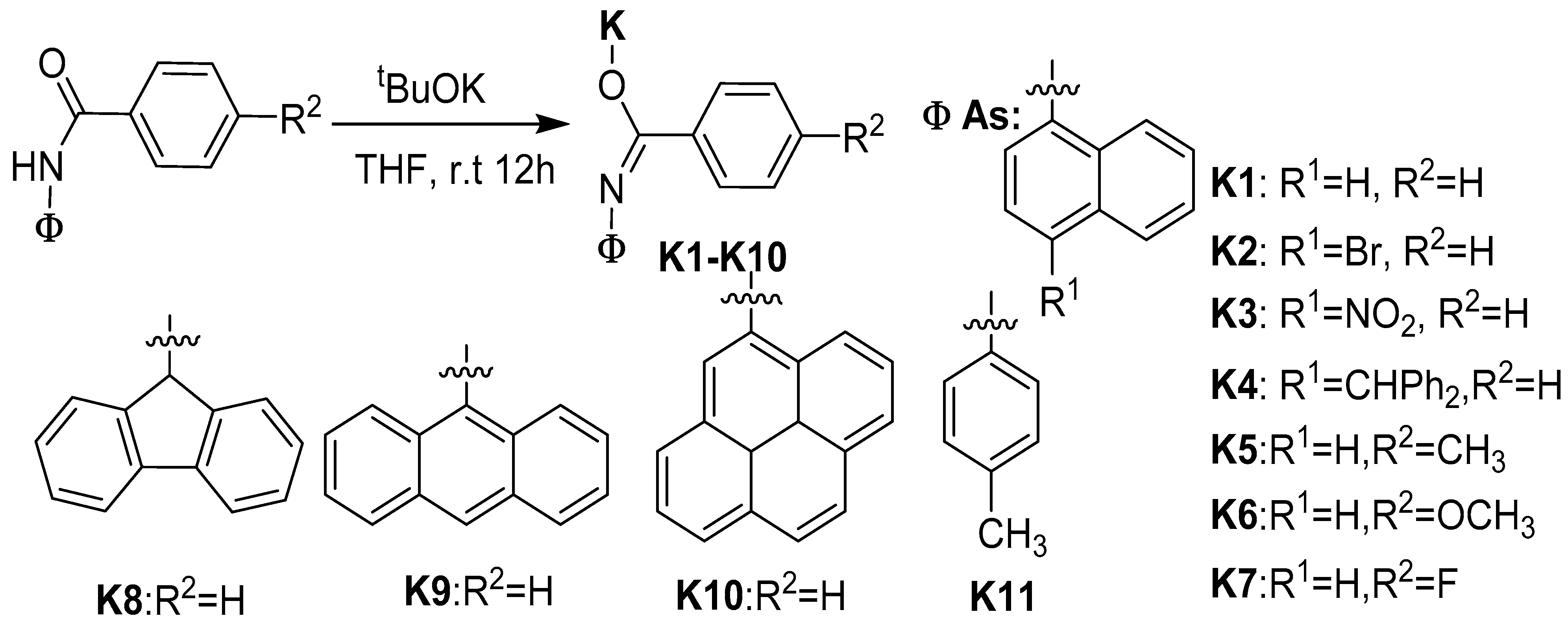
2.1. Synthesis and Characterization of K1–K10
2.2. Ring-Opening Polymerization of L-LA by K1–K10

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Cat | LA:K:BnOH | T/°C | t/min | Solvent | Conv. b (%) | Mncalcdc (×10 4 g mol−1) | Mnd (×10 4 g mol−1) | PDI d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | K2 | 500:1:1 | 30 | 15 | Toluene | 42 | 3.06 | 0.88 | 1.73 |
| 2 | K2 | 500:1:1 | 40 | 15 | Toluene | 88 | 6.33 | 1.50 | 1.77 |
| 3 | K2 | 500:1:1 | 50 | 15 | Toluene | 96 | 6.93 | 1.87 | 2.04 |
| 4 | K2 | 500:1:1 | 50 | 10 | Toluene | 87 | 6.27 | 1.28 | 1.93 |
| 5 | K2 | 500:1:1 | 50 | 5 | Toluene | 77 | 5.59 | 1.23 | 2.59 |
| 6 | K2 | 500:1:0 | 50 | 15 | Toluene | 94 | 6.79 | 3.85 | 1.49 |
| 7 | K2 | 500:1:2 | 50 | 15 | Toluene | 100 | 3.62 | 1.05 | 1.74 |
| 8 | K2 | 500:1:5 | 50 | 15 | Toluene | 100 | 1.49 | 0.96 | 1.34 |
| 9 | K2 | 500:1:10 | 50 | 15 | Toluene | 100 | 0.73 | 0.87 | 1.32 |
| 10 | K2 | 750:1:1 | 50 | 15 | Toluene | 68 | 7.31 | 3.38 | 1.93 |
| 11 | K2 | 1000:1:1 | 50 | 15 | Toluene | 55 | 7.93 | 4.18 | 1.65 |
| 12 | K2 | 1000:1:1 | 50 | 30 | Toluene | 59 | 8.48 | 4.52 | 1.79 |
| 13 | K2 | 1000:1:1 | 50 | 60 | Toluene | 61 | 8.76 | 4.78 | 1.75 |
| 14 | K2 | 1000:1:1 | 50 | 120 | Toluene | 68 | 9.86 | 6.27 | 1.64 |
| 15 | K2 | 500:1:1 | 50 | 15 | THF | 43 | 3.14 | 7.32 | 1.67 |
| 16 | K2 | 500:1:1 | 50 | 15 | Hexane | 0 | |||
| 17 | K2 | 500:1:1 | 50 | 15 | DCM | 41 | 2.97 | 5.02 | 1.87 |
| 18 | K1 | 500:1:1 | 50 | 10 | Toluene | 78 | 5.61 | 2.20 | 1.70 |
| 19 | K3 | 500:1:1 | 50 | 10 | Toluene | 92 | 6.61 | 1.88 | 1.90 |
| 20 | K4 | 500:1:1 | 50 | 10 | Toluene | 71 | 5.11 | 1.45 | 1.54 |
| 21 | K5 | 500:1:1 | 50 | 10 | Toluene | 85 | 6.13 | 2.30 | 1.68 |
| 22 | K6 | 500:1:1 | 50 | 10 | Toluene | 95 | 6.85 | 2.46 | 1.81 |
| 23 | K7 | 500:1:1 | 50 | 10 | Toluene | 78 | 5.63 | 1.38 | 1.67 |
| 24 | K8 | 500:1:1 | 50 | 10 | Toluene | 73 | 5.30 | 2.34 | 1.63 |
| 25 | K9 | 500:1:1 | 50 | 10 | Toluene | 97 | 7.00 | 1.87 | 1.93 |
| 26 | K10 | 500:1:1 | 50 | 10 | Toluene | 82 | 5.94 | 1.70 | 2.02 |
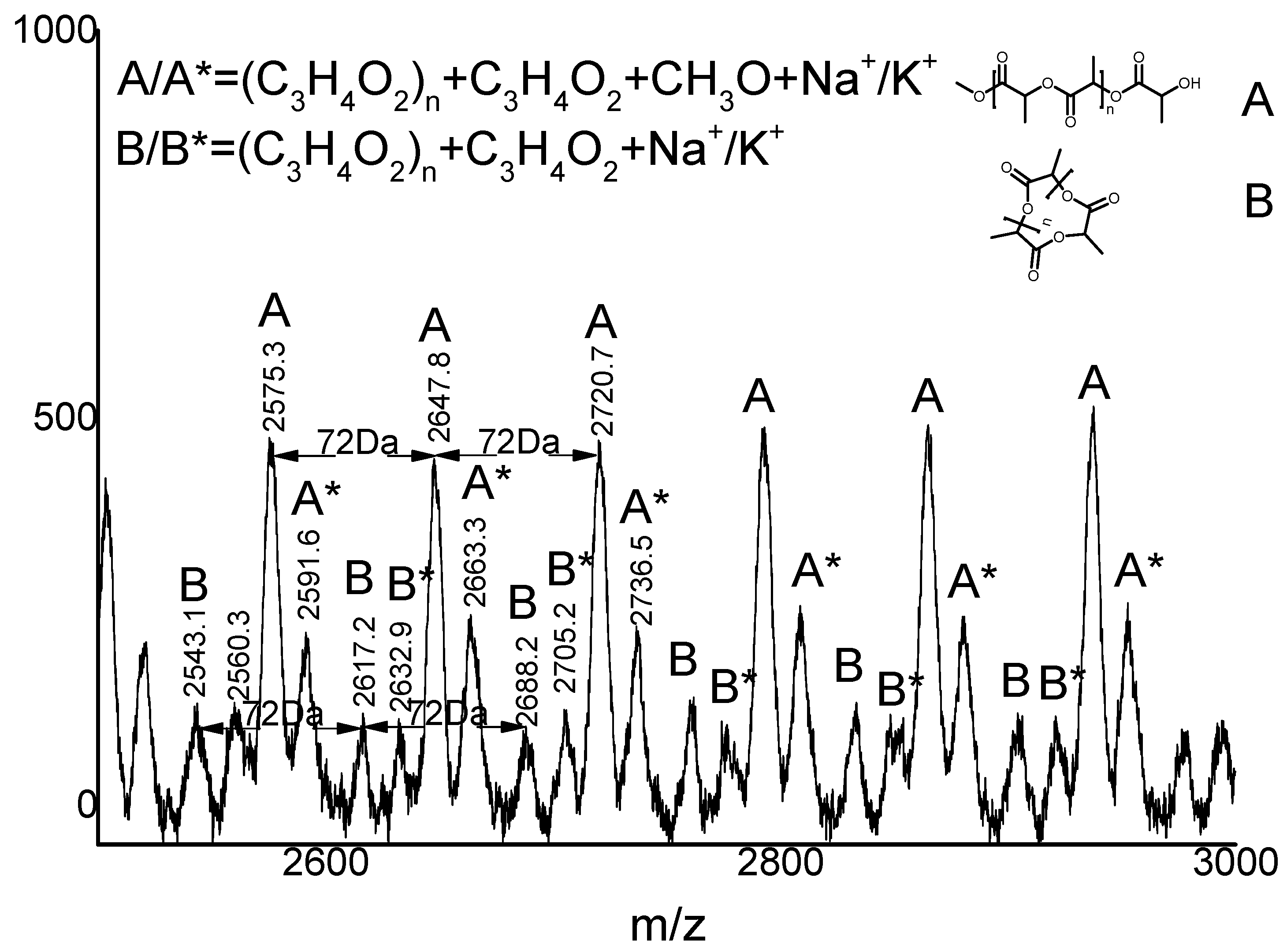
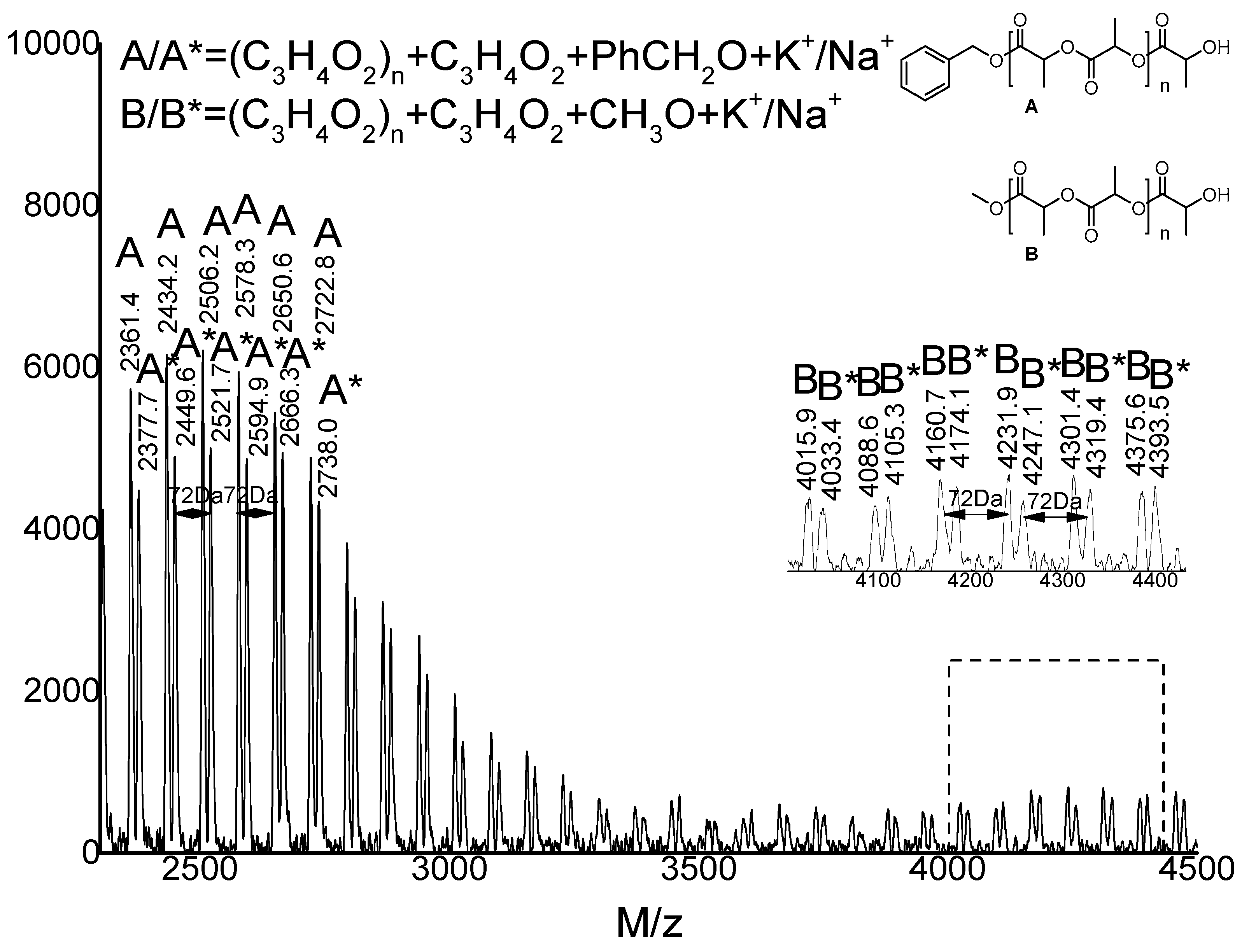
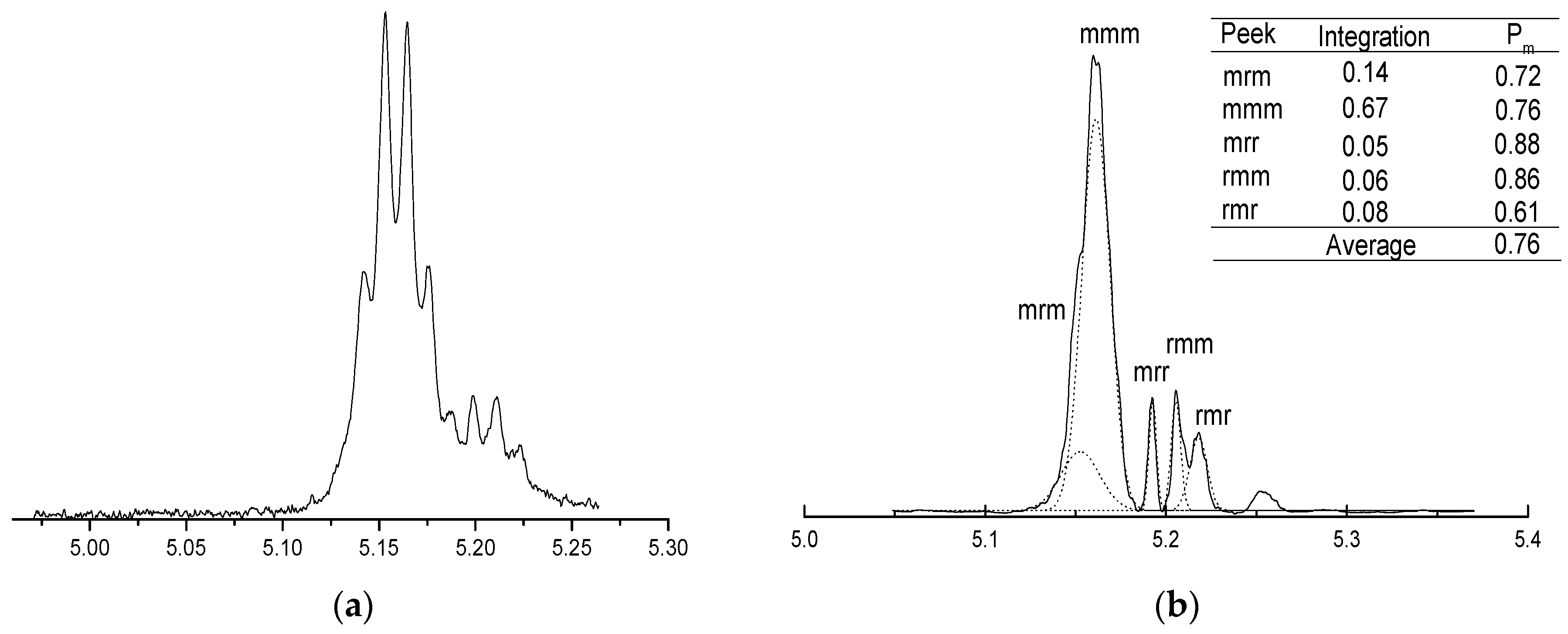
2.3. Microstructure Analysis of PLLA
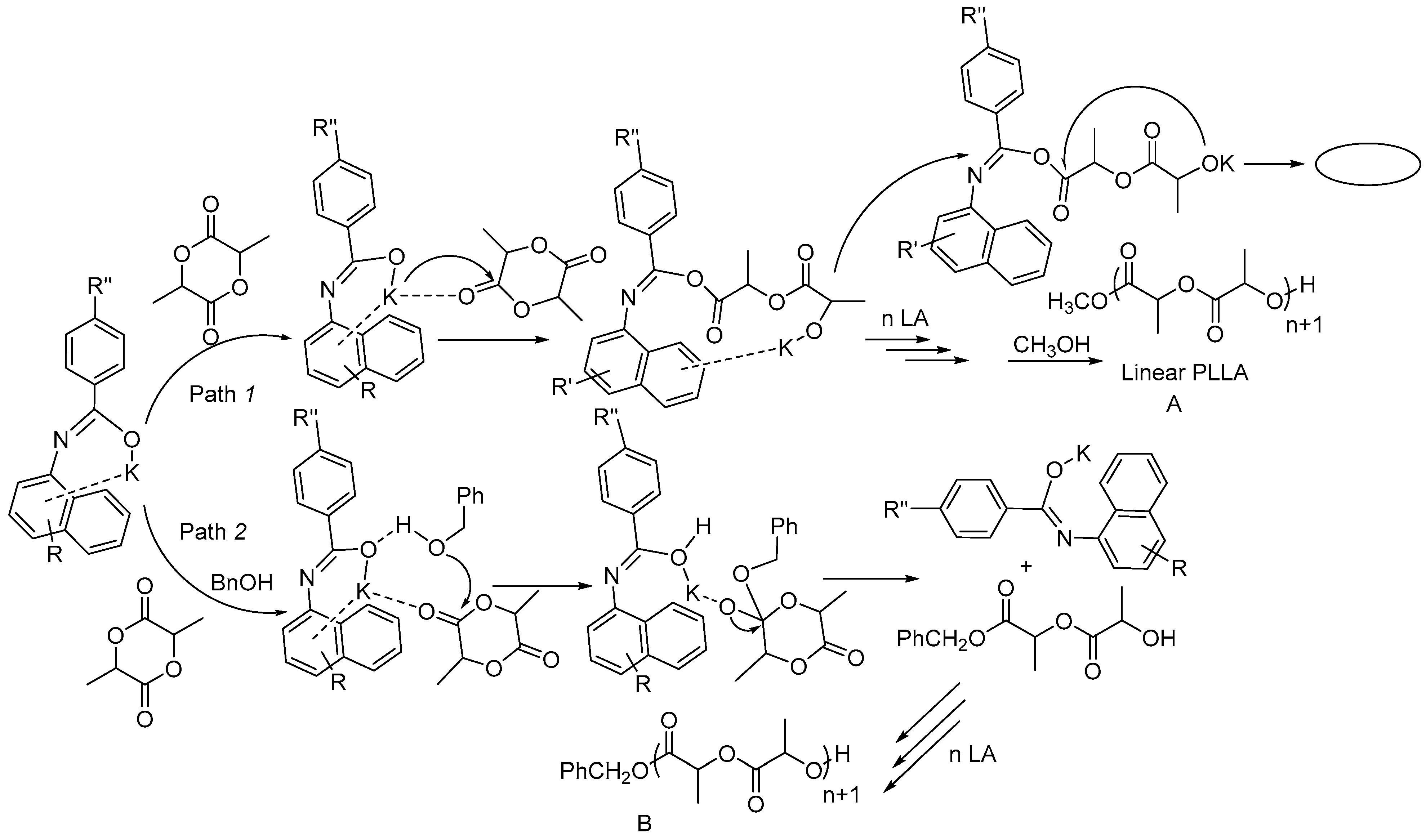
2.4. Ring-opening Polymerization of rac-Lactide by K1–K11
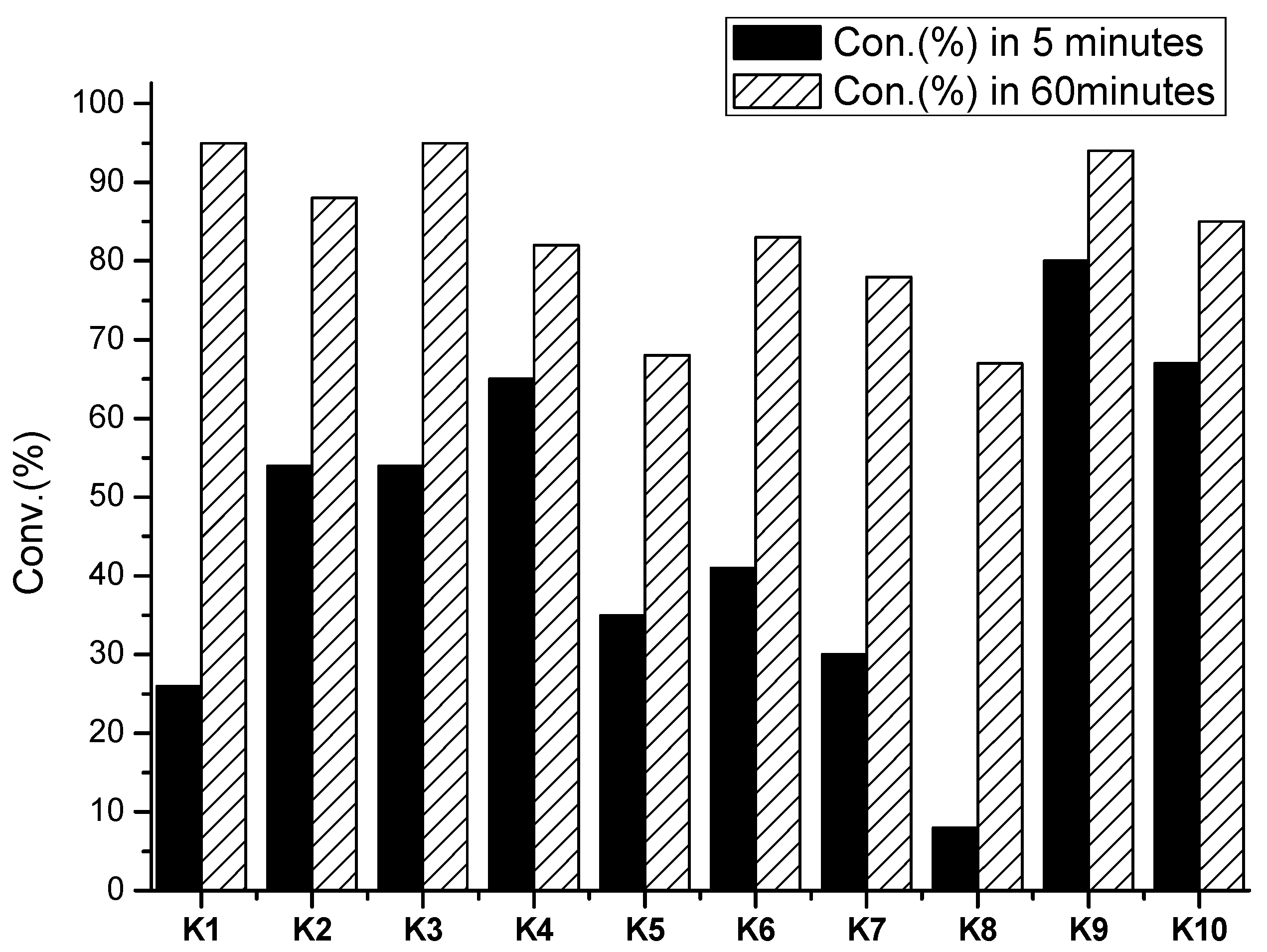
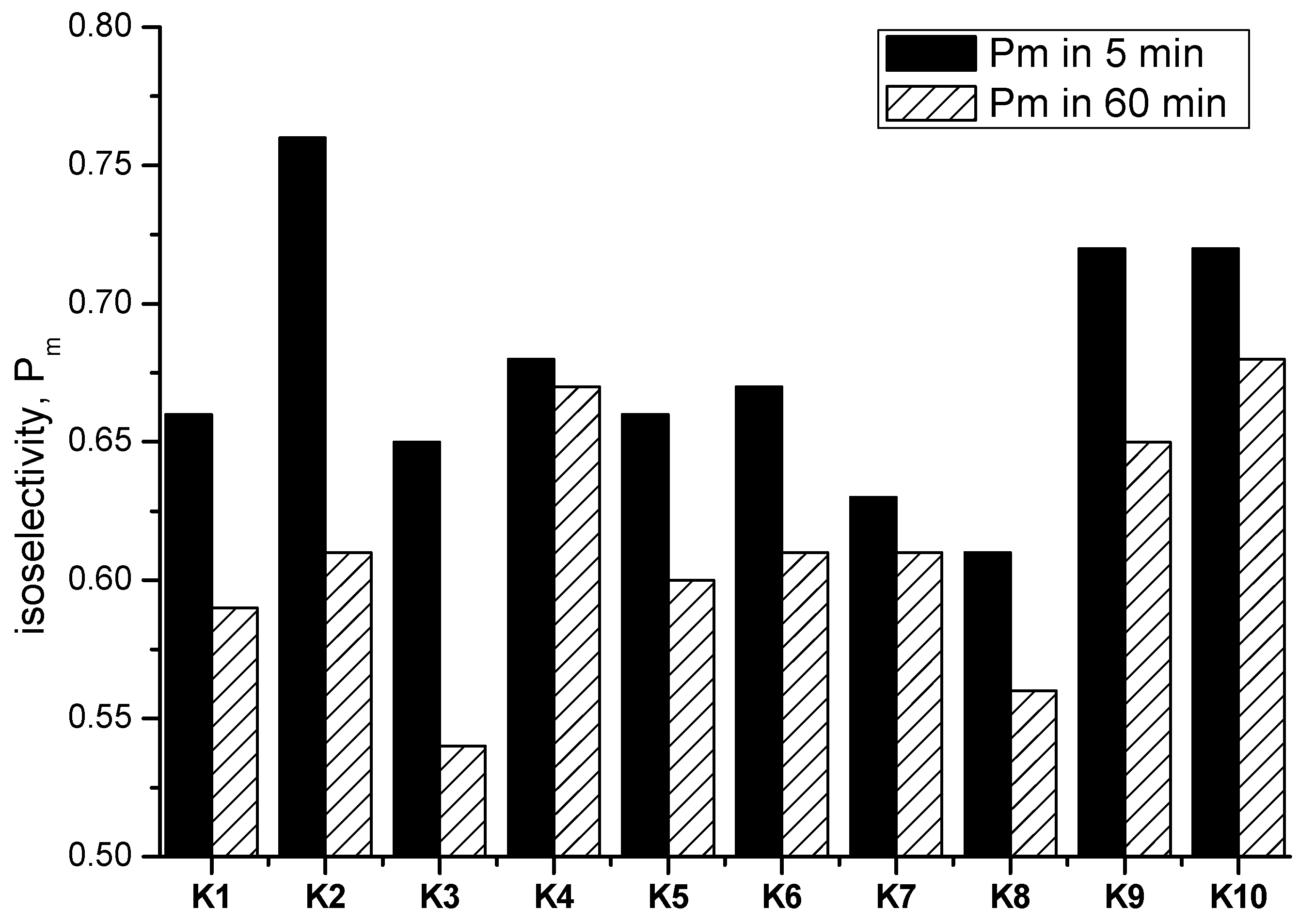
2.4.1. Polymerization Results of rac-LA in 60 min
2.4.2. Results of ROP toward rac-LA in 5 min
3. Experimental Section
3.1. General Procedures
3.2. Synthesis and Characterization of L1–L10
3.3. Synthesis of Potassium Complexes
3.4. General Procedure for the ROP of L- LA and rac-LA
3.5. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Iwata, T. Biodegradable and Bio-Based Polymers: Future Prospects of Eco-Friendly Plastics. Angew. Chem. Int. Ed. 2015, 54, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fevre, M.; Jones, G.O.; Waymouth, R.M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2018, 118, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Schwach, G.; Coudane, J.; Engel, R.; Vert, M. Zn lactate as initiator of DL-lactide ring opening polymerization and comparison with Sn octoate. Poly. Bull. 1996, 37, 771–776. [Google Scholar] [CrossRef]
- Wu, J.; Yu, T.L.; Chen, C.T.; Lin, C.C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Cabaret, O.D.; Vaca, B.M.; Bourissou, D. Controlled Ring-Opening Polymerization of Lactide and Glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef]
- Guillaume, S.M.; Kirillov, E.; Sarazin, Y.; Carpentier, J.F. Beyond Stereoselectivity, Switchable Catalysis: Some of the Last Frontier Challenges in Ring-Opening Polymerization of Cyclic Esters. Chem. Eur. J. 2015, 21, 7988–8003. [Google Scholar] [CrossRef]
- Sarazin, Y.; Carpentier, J.-F. Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides. Chem. Rev. 2015, 115, 3564–3614. [Google Scholar] [CrossRef] [PubMed]
- Santoro, O.; Zhang, X.; Redshaw, C. Synthesis of Biodegradable Polymers: A Review on the Use of Schiff-Base Metal Complexes as Catalysts for the Ring Opening Polymerization (ROP) of Cyclic Esters. Catalysts 2020, 10, 800. [Google Scholar] [CrossRef]
- Wu, L.-J.; Lee, W.; Ganta, P.K.; Chang, Y.-L.; Chang, Y.-C.; Chen, H.-Y. Multinuclear metal catalysts in ring-opening polymerization of ε-caprolactone and lactide: Cooperative and electronic effects between metal centers. Coord. Chem. Rev. 2023, 475, 214847. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Tolpygin, A.O.; Trifonov, A.A. Rare-earth metal complexes as catalysts for ring-opening polymerization of cyclic esters. Coord. Chem. Rev. 2019, 392, 83–145. [Google Scholar] [CrossRef]
- Strianese, M.; Pappalardo, D.; Mazzeo, M.; Lamberti, M.; Pellecchia, C. Salen-type aluminum and zinc complexes as two faced Janus compounds: Contribution to molecular sensing and polymerization catalysis. Dalton Trans. 2020, 49, 16533–16550. [Google Scholar] [CrossRef] [PubMed]
- Gadomska-Gajadhur, A.; Ruśkowski, P. Biocompatible Catalysts for Lactide Polymerization Catalyst Activity, Racemization Effect, and Optimization of the Polymerization Based on Design of Experiments. Org. Process Res. Dev. 2020, 24, 1435–1442. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C. Controlling the Ring-Opening Polymerization Process Using External Stimuli. Chin. J. Chem. 2020, 38, 282–286. [Google Scholar] [CrossRef]
- Kaler, S.; Jones, M.D. Recent advances in externally controlled ring-opening polymerisations. Dalton Trans. 2022, 51, 1241–1256. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Weidner, S.M. Syntheses of polylactides by means of tin catalysts. Polym. Chem. 2022, 13, 1618–1647. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications —A comprehensive review. Adv. Drug Deliver. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef]
- Lima, L.-T.; Aurasb, R.; Rubinob, M. Processing technologies for poly(lactic acid). Prog. Poly. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An Overview of Polylactides as Packaging Materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Oudega, M.; Gautier, S.E.; Chapon, P.; Fragoso, M.; Bates, M.L.; Parel, J.-M.; Bunge, M.B. Axonal regeneration into Schwann cell grafts within resorbable poly(α-hydroxyaciguidance channels in the adult rat spinal cord. Biomaterials 2001, 22, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Ma, P.X. Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering. Biomaterials 2004, 25, 4749–4757. [Google Scholar] [CrossRef] [PubMed]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyon, S.-H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Stanford, M.J.; Dove, A.P. Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 2010, 39, 486–494. [Google Scholar] [CrossRef]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef]
- Dijkstra, P.J.; Du, H.; Feijen, J. Single site catalysts for stereoselective ring-opening polymerization of lactides. Polym. Chem. 2011, 2, 520–527. [Google Scholar] [CrossRef]
- Kowalski, A.; Duda, A.; Penczek, S. Mechanism of Cyclic Ester Polymerization Initiated with Tin(II) Octoate. 2. Macromolecules Fitted with Tin(II) Alkoxide Species Observed Directly in MALDI−TOF Spectra. Macromolecules 2000, 33, 689–695. [Google Scholar] [CrossRef]
- Dove, A.P.; Gibson, V.C.; Marshall, E.L.; Rzepa, H.S.; White, A.J.P.; Williams, D.J. Synthetic, Structural, Mechanistic, and Computational Studies on Single-Site β-Diketiminate Tin(IInitiators for the Polymerization of rac-Lactide. J. Am. Chem. Soc. 2006, 128, 9834–9843. [Google Scholar] [CrossRef]
- Kremer, A.B.; Mehrkhodavandi, P. Dinuclear catalysts for the ring opening polymerization of lactide. Coord. Chem. Rev. 2019, 380, 35–37. [Google Scholar] [CrossRef]
- Carpentier, J.-F. Rare-Earth Complexes Supported by Tripodal Tetradentate Bis(phenolatLigands: A Privileged Class of Catalysts for Ring-Opening Polymerization of Cyclic Esters. Organometallics 2015, 34, 4175–4189. [Google Scholar] [CrossRef]
- Marin, P.; Venditto, V.; Thomas, C.M. Polymerization of rac-Lactide Using Achiral Iron Complexes: Access to Thermally Stable Stereocomplexes. Angew. Chem. Int. Ed. 2019, 58, 12585–12589. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Duan, R.; Li, X.; Hu, C.; Wang, X.; Chen, X. Breaking the Paradox between Catalytic Activity and Stereoselectivity: Rac-Lactide Polymerization by Trinuclear Salen–Al Complexes. Macromolecules 2018, 51, 906–913. [Google Scholar] [CrossRef]
- Ren, F.; Li, X.; Xian, J.; Han, X.; Cao, L.; Pan, X.; Wu, J. Bench-stable potassium complexes for living and isoselective ring-opening polymerization of rac-lactide. J. Polym. Sci. 2022, 60, 2847–2854. [Google Scholar] [CrossRef]
- Katalin, D.-P.; Oldenburg, F.J.; Menzel, J.P.; Springer, M.; Dawe, L.N.; Kozak, C.M. Lithium, sodium, potassium and calcium amine-bis(phenolatcomplexes in the ring-opening polymerization of rac-lactide. Dalton Trans. 2020, 49, 1531–1544. [Google Scholar]
- Spassky, N.; Wisniewski, M.; Pluta, C.; Borgne, A.L. Highly stereoelective polymerization of rac-(D, L)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile Copolymerization of Glycolide and rac-Lactide by Dimethyl(salicylaldiminato)aluminum Compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective Ring-Opening Polymerization of Racemic Lactide Using Aluminum-Achiral Ligand Complexes: Exploration of a Chain-End Control Mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef]
- Hormnirun, P.; Marshall, E.L.; Gibson, V.C.; White, A.J.P.; Williams, D.J. Remarkable Stereocontrol in the Polymerization of Racemic Lactide Using Aluminum Initiators Supported by Tetradentate Aminophenoxide Ligands. J. Am. Chem. Soc. 2004, 126, 2688–2689. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective Ring-Opening Polymerization of a Racemic Lactide by Using Achiral Salen–and Homosalen–Aluminum Complexes. Chem. Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef]
- Hormnirun, P.; Marshall, E.L.; Gibson, V.C.; Pugh, R.I.; White, A.J.P. Study of ligand substituent effects on the rate and stereoselectivity of lactide polymerization using aluminum salen-type initiators. Proc. Natl. Acad. Sci. USA 2006, 103, 15343–15348. [Google Scholar] [CrossRef]
- Tang, Z.; Chen, X.; Pang, X.; Yang, Y.; Zhang, X.; Jing, X. Stereoselective Polymerization of rac-Lactide Using a Monoethylaluminum Schiff Base Complex. Biomacromolecules 2004, 5, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, D.; Zhang, W.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent progress in the application of group 1, 2 & 13 metal complexes as catalysts for the ring opening polymerization of cyclic esters. Inorg. Chem. Front. 2019, 6, 2619–2652. [Google Scholar]
- Dai, Z.; Sun, Y.; Xiong, J.; Pan, X.; Wu, J. Alkali-Metal Monophenolates with a Sandwich-Type Catalytic Center as Catalysts for Highly Isoselective Polymerization of rac-Lactide. ACS Macro Lett. 2015, 4, 556–560. [Google Scholar] [CrossRef]
- Sun, Y.; Xiong, J.; Dai, Z.; Pan, X.; Tang, N.; Wu, J. Stereoselective Alkali-Metal Catalysts for Highly Isotactic Poly(rac-lactidSynthesis. Inorg. Chem. 2016, 55, 136–143. [Google Scholar] [CrossRef]
- Dai, Z.; Sun, Y.; Xiong, J.; Pan, X.; Tang, N.; Wu, J. Simple sodium and potassium phenolates as catalysts for highly isoselective polymerization of rac-lactide. Catal. Sci. Technol. 2016, 6, 515–520. [Google Scholar] [CrossRef]
- Wu, B.-B.; Tiana, L.-L.; Wang, Z.-X. Ring-opening polymerization of rac-lactide catalyzed by crown ether complexes of sodium and potassium iminophenoxides. RSC Adv. 2017, 7, 24055–24063. [Google Scholar] [CrossRef]
- Chen, C.; Cui, Y.; Mao, X.; Pan, X.; Wu, J. Suppressing Cyclic Polymerization for Isoselective Synthesis of High-Molecular-Weight Linear Polylactide Catalyzed by Sodium/Potassium Sulfonamidate Complexes. Macromolecules 2017, 50, 83–96. [Google Scholar] [CrossRef]
- Wu, B.-B.; Wang, Z.-X. Crown ether complexes of potassium quinolin-8-olates: Synthesis, characterization and catalysis toward the ring-opening polymerization of rac-lactide. RSC Adv. 2017, 7, 11657–11664. [Google Scholar] [CrossRef]
- Fernández-M, M.; Ortega, P.; Cuenca, T.; Cano, J.; Mosquera, M.E.G. Alkali-Metal Compounds with Bio-Based Ligands as Catalysts for Isoselective Lactide Polymerization: Influence of the Catalyst Aggregation on the Polymerization Control. Organometallics 2020, 39, 2278–2286. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, J.; Sun, Y.; Tang, N.; Wu, J. Highly Iso-Selective and Active Catalysts of Sodium and Potassium Monophenoxides Capped by a Crown Ether for the Ring-Opening Polymerization of rac-Lactide. Macromolecules 2014, 47, 7789–7796. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, J.; Mao, X.; Cong, Y.; Cui, Y.; Pan, X.; Wu, J. Isoselective Polymerization of rac-Lactide Catalyzed by Ion-Paired Potassium Amidinate Complexes. Inorg. Chem. 2018, 57, 3158–3168. [Google Scholar] [CrossRef]
- Yao, C.; Yang, Y.; Ma, H. Potassium complexes supported by monoanionic tetradentate amino-phenolate ligands: Synthesis, structure and catalysis in the ring-opening polymerization of rac-lactide. Dalton Trans. 2017, 46, 6087–6097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Wang, S.; Solan, G.A.; Liang, T.; Rajendrana, N.M.; Sun, W.-H. Sodium iminoquinolates with cubic and hexagonal prismatic motifs: Synthesis, characterization and their catalytic behavior toward the ROP of rac-lactide. Inorg. Chem. Front. 2016, 3, 1178–1189. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, W.; Cao, F.; Solan, G.A.; Zhang, X.; Jiang, Y.; Hao, X.; Sun, W.-H. Potassium N-arylbenzimidates as readily accessible and benign (pre)catalysts for the ring opening polymerization of ε-CL and L-LA. Mol. Catal. 2020, 498, 111280. [Google Scholar] [CrossRef]
- Montaudo, G.; Montaudo, M.S.; Puglisi, C.; Samperi, F.; Spassky, N.; LeBorgne, A.; Wisniewski, M. Evidence for Ester-Exchange Reactions and Cyclic Oligomer Formation in the Ring-Opening Polymerization of Lactide with Aluminum Complex Initiators. Macromolecules 1996, 29, 6461–6465. [Google Scholar] [CrossRef]
- Spassky, N.; Simic, V.; Montaudo, M.S.; Hubert-Pfalzgraf, L.G. Inter- and intramolecular ester exchange reactions in the ring-opening polymerization of (D,L)-lactide using lanthanide alkoxide initiators. Macromol. Chem. Phys. 2000, 201, 2432–2440. [Google Scholar] [CrossRef]
- Save, M.; Schappacher, M.; Soum, A. Controlled Ring-Opening Polymerization of Lactones and Lactides Initiated by Lanthanum Isopropoxide, 1. General Aspects and Kinetics. Macromol. Chem. Phys. 2002, 203, 889–899. [Google Scholar] [CrossRef]
- Iwasa, N.; Katao, S.; Liu, J.; Fujiki, M.; Furukawa, Y.; Nomura, K. Notable Effect of Fluoro Substituents in the Imino Group in Ring-Opening Polymerization of ε-Caprolactone by Al Complexes Containing Phenoxyimine Ligands. Organometallics 2009, 28, 2179–2187. [Google Scholar] [CrossRef]
- Cayuela, J.; Bounor-Legaré, V.; Cassagnau, P.; Michel, A. Ring-Opening Polymerization of ε-Caprolactone Initiated with Titanium n-Propoxide or Titanium Phenoxide. Macromolecules 2006, 39, 1338–1346. [Google Scholar] [CrossRef]
- Baran, J.; Duda, A.; Kowalski, A.; Szymanski, R.; Penczek, S. Intermolecular chain transfer to polymer with chain scission: General treatment and determination of kp/ktr in L, L-lactide polymerization. Macromol. Rapid Commun. 1997, 18, 325–333. [Google Scholar] [CrossRef]
- Huang, B.-H.; Ko, B.-T.; Athar, T.; Lin, C.-C. Synthesis, Characterization, and Structural Determination of Polynuclear Lithium Aggregates and Factors Affecting Their Aggregation. Inorg. Chem. 2006, 45, 7348–7356. [Google Scholar] [CrossRef]
- Maudoux, N.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. Aluminum, Indium, and Mixed Yttrium−Lithium Complexes Supported by a Chiral Binap-Based Fluorinated Dialkoxide: Structural Features and Heteroselective ROP of Lactide. Organometallics 2014, 33, 5740–5748. [Google Scholar] [CrossRef]
- García-Valle, F.M.; Estivill, R.; Gallegos, C.; Cuenca, T.; Mosquera, M.E.G.; Tabernero, V.; Cano., J. Metal and Ligand-Substituent Effects in the Immortal Polymerization of rac-Lactide with Li, Na, and K Phenoxo-imine ComplexesVanessa Tabernero. Organometallics 2015, 34, 477–487. [Google Scholar] [CrossRef]
- Roşca, S.-C.; Roşca, D.-A.; Dorcet, V.; Kozak, C.M.; Kerton, F.M.; Carpentier, J.-F.; Sarazin, Y. Alkali aminoether-phenolate complexes: Synthesis, structural characterization and evidence for an activated monomer ROP mechanism. Dalton Trans. 2013, 42, 9361–9375. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, C.; Tabernero, V.; Mosquera, M.E.G.; Cuenca, T.; Cano, J. Comparative Study of Lactide Polymerization with Lithium, Sodium, Potassium, Magnesium, Calcium, and Zinc Azo naphthoxide Complexes. Eur. J. Inorg. Chem. 2015, 30, 5124–5132. [Google Scholar] [CrossRef]
- Cui, Y.; Chen., C.; Sun, Y.; Wu, J.; Pan., X. Isoselective mechanism of the ring-opening polymerization of rac-lactide catalyzed by chiral potassium binolates. Inorg. Chem. Front. 2017, 4, 261–269. [Google Scholar] [CrossRef]
- Stirling, E.; Champouret, Y.; Visseaux, M. Catalytic metal-based systems for controlled statistical copolymerisation of lactide with a lactone. Polym. Chem. 2018, 9, 2517–2531. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howardd, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]












| Bond Lengths of K2 (Å) | Bond Lengths of K10 (Å) | ||||||
|---|---|---|---|---|---|---|---|
| K1-O1 | 2.598(2) | K1-C11 | 3.413(3) | K1-O1 | 2.593(4) | K1-C21 3 | 3.058(5) |
| K1-O1 1 | 2.598(2) | K1-C12 | 3.232(3) | K1-N1 1 | 2.693(4) | K1-C22 3 | 3.239(5) |
| K1-C8 | 3.249(3) | K1-C17 | 3.149(3) | K1-C8 3 | 3.180(5) | K1-O1 3 | 2.633(4) |
| K1-C9 | 3.496(3) | K1-N1 1 | 2.733(2) | K1-C9 3 | 3.511(5) | K1-N1 4 | 2.693(4) |
| Bond angles of K2 (°) | Bond angles of K10 (°) | ||||||
| O1-K1-O1 1 | 80.39(7) | O1-K1-K1 3 | 44.94(8) | ||||
| O1 1-K1-N1 1 | 139.25(8) | O1-K1-O1 3 | 89.01(11) | ||||
| O1-K1-C8 | 53.33(7) | O1-K1-N1 4 | 132.82(13) | ||||
| O1-K1-K17 | 64.14(7) | O1 3-K1-N1 4 | 111.91(12) | ||||
| O1-K1-C9 | 70.39(7) | O1-K1-C8 3 | 119.49(13) | ||||
| O1-K1-C11 | 102.57(7) | O1-K1-C9 3 | 141.63(13) | ||||
| O1-K1-C12 | 89.92(7) | O1-K1-C21 3 | 99.66(13) | ||||
| O1-K1-C22 3 | 101.49(13) | ||||||
| Run | Cat | LA:K:BnOH | T/°C | t/min | Solvent | Conv. b (%) | Mncalcdc (×10 4gmol−1) | Mnd (×10 4 g mol−1) | PDI d | Pme |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | K2 | 100:1:1 | 20 | 60 | Toluene | 100 | 1.45 | 0.64 | 2.10 | |
| 2 | K2 | 250:1:1 | 20 | 60 | Toluene | 77 | 2.78 | 1.27 | 1.81 | |
| 3 | K2 | 100:1:1 | 0 | 60 | Toluene | 85 | 1.24 | 0.43 | 2.23 | |
| 4 | K2 | 100:1:1 | 0 | 120 | Toluene | 100 | 1.45 | 0.61 | 1.85 | |
| 5 | K2 | 100:1:1 | −30 | 60 | Toluene | 85 | 1.23 | 0.45 | 2.03 | |
| 6 | K2 | 100:1:1 | −78 | 5 | Toluene | 54 | 0.78 | 0.26 | 1.79 | 0.76 |
| 7 | K2 | 100:1:1 | −78 | 10 | Toluene | 67 | 0.97 | 0.43 | 1.35 | 0.75 |
| 8 | K2 | 100:1:1 | −78 | 60 | Toluene | 83 | 1.21 | 0.58 | 1.72 | 0.61 |
| 9 | K2 | 100:1:1 | −78 | 120 | Toluene | 88 | 1.28 | 0.65 | 2.12 | |
| 10 | K2 | 100:1:0 | −78 | 60 | Toluene | 26 | 0.38 | 0.38 | 4.52 | |
| 11 | K2 | 100:1:0 | −78 | 120 | Toluene | 50 | 0.72 | 0.48 | 3.89 | |
| 12 | K2 | 100:1:2 | −78 | 60 | Toluene | 54 | 0.79 | 0.25 | 1.33 | |
| 13 | K2 | 100:1:1 | −78 | 60 | hexane | 54 | 0.79 | 0.56 | 1.41 | 0.58 |
| 14 | K2 | 100:1:1 | −78 | 60 | THF | 56 | 0.82 | 0.86 | 1.58 | 0.67 |
| 15 | K2 | 100:1:1 | −78 | 60 | DCM | 64 | 0.93 | 0.79 | 1.67 | 0.64 |
| 16 | K1 | 100:1:1 | −78 | 60 | Toluene | 95 | 1.38 | 1.21 | 1.91 | 0.59 |
| 17 | K3 | 100:1:1 | −78 | 60 | Toluene | 95 | 1.38 | 0.87 | 1.24 | 0.54 |
| 18 | K4 | 100:1:1 | −78 | 60 | Toluene | 82 | 1.19 | 1.23 | 1.17 | 0.67 |
| 19 | K5 | 100:1:1 | −78 | 60 | Toluene | 68 | 0.99 | 0.81 | 1.99 | 0.60 |
| 20 | K6 | 100:1:1 | −78 | 60 | Toluene | 83 | 1.21 | 0.75 | 1.83 | 0.61 |
| 21 | K7 | 100:1:1 | −78 | 60 | Toluene | 78 | 1.13 | 0.93 | 1.65 | 0.61 |
| 22 | K8 | 100:1:1 | −78 | 60 | Toluene | 67 | 0.97 | 0.66 | 1.21 | 0.56 |
| 23 | K9 | 100:1:1 | −78 | 60 | Toluene | 94 | 1.36 | 0.61 | 1.42 | 0.65 |
| 24 | K10 | 100:1:1 | −78 | 60 | Toluene | 85 | 1.23 | 0.60 | 1.56 | 0.68 |
| 25 | K11 f | 100:1:1 | −78 | 60 | Toluene | 63 | 0.92 | 0.63 | 1.69 | 0.54 |
| Run | Cat | LA:K:BnOH | Conv(%) b | MnCalcd c | Mn d | PDI d | Pme |
|---|---|---|---|---|---|---|---|
| 1 | K1 | 100:1:1 | 26 | 0.38 | 0.19 | 1.13 | 0.66 |
| 2 | K2 | 100:1:1 | 54 | 0.78 | 0.26 | 1.79 | 0.76 |
| 3 | K3 | 100:1:1 | 54 | 0.78 | 0.89 | 1.44 | 0.65 |
| 4 | K4 | 100:1:1 | 65 | 0.94 | 0.61 | 1.83 | 0.68 |
| 5 | K5 | 100:1:1 | 35 | 0.51 | 0.39 | 1.77 | 0.66 |
| 6 | K6 | 100:1:1 | 41 | 0.60 | 0.29 | 1.34 | 0.67 |
| 7 | K7 | 100:1:1 | 30 | 0.44 | 0.46 | 1.82 | 0.63 |
| 8 | K8 | 100:1:1 | 8 | 0.12 | 0.29 | 1.32 | 0.61 |
| 9 | K9 | 100:1:1 | 80 | 1.16 | 0.37 | 1.45 | 0.72 |
| 10 | K10 | 100:1:1 | 67 | 0.97 | 0.29 | 1.07 | 0.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Zhang, W.; Wang, X.; Wang, R.; Han, M.; Cao, F.; Hao, X. Isoselective Ring-Opening Polymerization of rac-Lactide Catalyzed by Simple Potassium Amidate Complexes Containing Polycyclic Aryl Group. Catalysts 2023, 13, 770. https://doi.org/10.3390/catal13040770
Gao J, Zhang W, Wang X, Wang R, Han M, Cao F, Hao X. Isoselective Ring-Opening Polymerization of rac-Lactide Catalyzed by Simple Potassium Amidate Complexes Containing Polycyclic Aryl Group. Catalysts. 2023; 13(4):770. https://doi.org/10.3390/catal13040770
Chicago/Turabian StyleGao, Jiahao, Wenjuan Zhang, Xing Wang, Rui Wang, Mingyang Han, Furong Cao, and Xiang Hao. 2023. "Isoselective Ring-Opening Polymerization of rac-Lactide Catalyzed by Simple Potassium Amidate Complexes Containing Polycyclic Aryl Group" Catalysts 13, no. 4: 770. https://doi.org/10.3390/catal13040770