

Divergent Reactivity of D-A Cyclopropanes under PTC Conditions, Ring-Opening vs. Decyanation Reaction

Department of Industrial Chemistry “Toso Montanari”, Center for Chemical Catalysis—C3, and INSTM RU Bologna, Alma Mater Studiorum—University of Bologna, V. Risorgimento 4, 40136 Bologna, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2023, 13(4), 760; https://doi.org/10.3390/catal13040760
Submission received: 27 March 2023
/
Revised: 13 April 2023
/
Accepted: 14 April 2023
/
Published: 16 April 2023
(This article belongs to the Special Issue Feature Papers in Catalysis in Organic and Polymer Chemistry)
Abstract
:The divergent reactivity of D-A cyclopropane, under PTC conditions, is herein reported. Thus, a ring-opening or a decyanation reaction can be achieved by reacting 2-arylcyclopropane-1,1-dicarbonitriles 1 with thioacetic acid in different reaction conditions. The use of solid Cs2CO3 leads unexpectedly to the synthesis of new D-A cyclopropane derivatives via a decyanation reaction, followed by diastereoselective acetylation, whereas the use of an aqueous solution of Cs2CO3 results in a typical ring-opening reaction with the formation of S-thiolate products. Therefore, the use of tailored reaction conditions allows one to obtain either cyclic or open-chain products in moderate to good yields.
1. Introduction
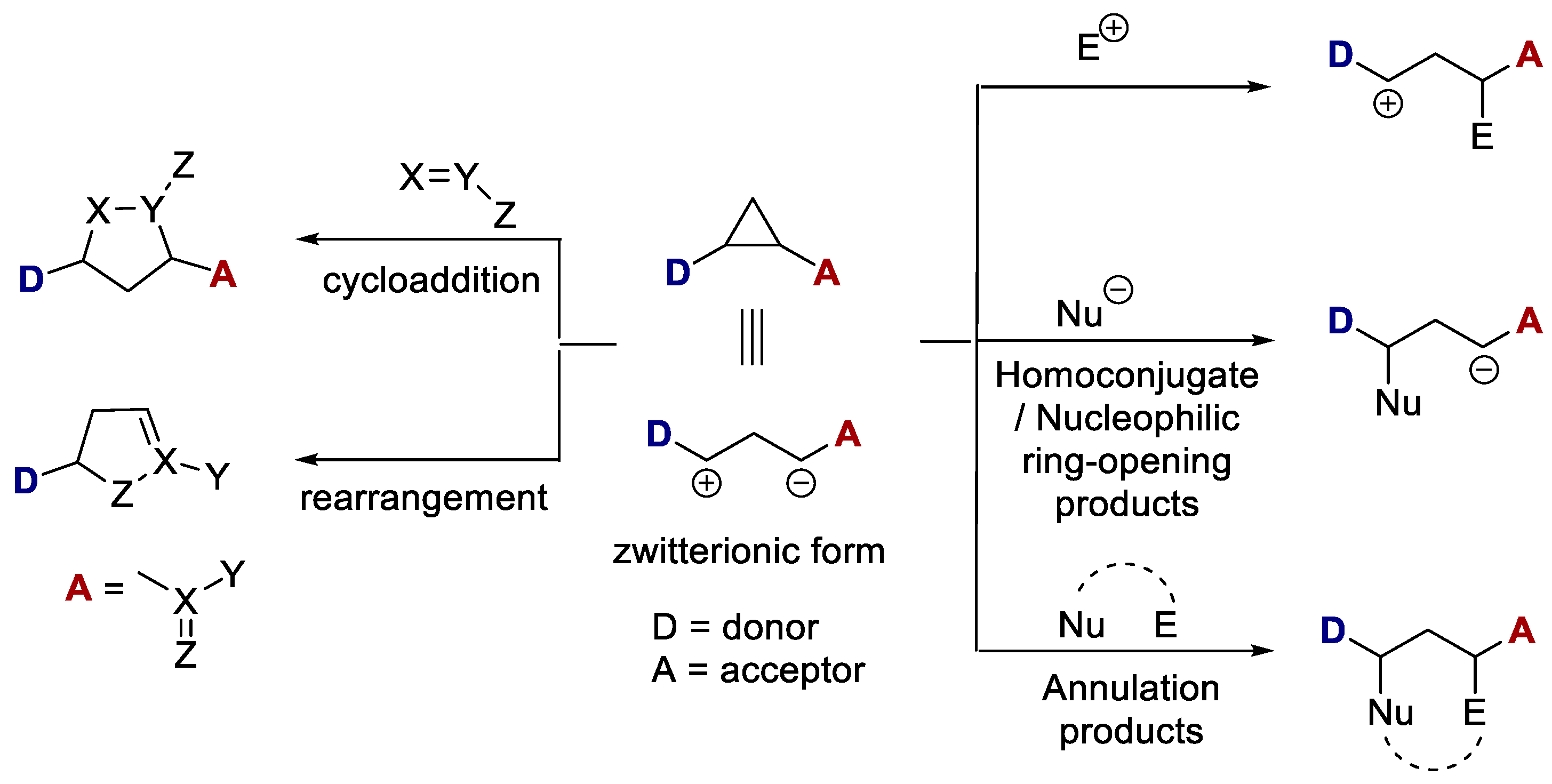
Cyclopropane is the smallest possible saturated cyclic structure with a ring strain (Baeyer strain energy) of about 110–115 kJ mol−1 [1]. At the same time, the C-C bonds of an unsubstituted cyclopropane are rather kinetically inert and, despite the strain, the molecule does not tend to give up on its cyclic structure. This energy barrier, however, descends significantly in activated cyclopropanes where donor (D) and acceptor (A) groups are installed vicinally in a three-membered ring system. The relatively weak chemical bond between the donor- and acceptor-substituted carbon atoms of the cyclopropane may be rationalized by a zwitterionic relationship (a 1,3-dipole) in which the negative and positive charges are stabilized by the acceptor and donor substituent(s), respectively (Scheme 1). The acceptor groups are often carbonyl derivatives, such as esters, ketones, and nitriles, whereas electron-rich aryls, alkenyl and heteroatoms are typically used as donor groups. Generally, two acceptor groups in a geminal position, which guarantee better activation, are employed. Reissig suggested referring to them as “donor–acceptor-substituted cyclopropanes” [2,3], which was later reduced to donor-acceptor D-A cyclopropanes.
The synergistic “push–pull” effect of vicinal charge-stabilizing groups boosts the high polarization of the C-C bond, allowing ring rupture under mild conditions. It also favours a multitude of different reactions with both nucleophiles and electrophiles, including moderately active ones, as well as diverse ambiphilic reagents. Nucleophilic attack occurs at the donor end, leading to homoconjugated products, while the electrophilic one occurs at the acceptor end to afford cation equivalents for further transformations. The ring-opening reaction of D-A cyclopropanes has evolved into an effective strategy to assemble functionalized carbon scaffolds. Moreover, with suitable reacting partners containing both nucleophilic and electrophilic sites, cascade reactions may also proceed through ring-opening and annulative ring-closure in what is a formal cycloaddition [3,4,5].
Cycloadditions of activated D-A cyclopropanes with dipolarophiles, 1,3-dipoles, or dienes represent a valuable tool for accessing highly functionalized five-, six-, or seven-membered-ring systems [6,7,8] (Scheme 1). Rearrangements that result in ring enlargement with the insertion of the acceptor in a cyclic structure are also possible [9].
Early synthetic applications of activated cyclopropanes were published in the 1960s and 1970s, and the first “golden age” for D-A cyclopropanes was entered in the 1980s, when all the fundamental reaction types were reported [10,11]. In 2014, Werz [12] and France [13] reviewed the 2000s as the second “golden age” of D–A cyclopropanes. Studies of their reactivity and catalytic asymmetric reactions of D–A cyclopropanes were next summarized in several reviews [5,14,15,16,17,18,19,20].
D-A cyclopropanes may be activated by (i) thermal activation [21,22], (ii) Lewis or Brønsted acid/base-mediated activation [23,24], and (iii) low-valent transition metal catalysis [25,26,27,28]. Recently, few reports regarding organocatalytic activation have been reported [23,29,30,31]. However, to the best of our knowledge, the reactivity of D-A cyclopropanes has never been studied under phase-transfer catalysis (PTC) [32,33,34,35]. Having maturated a broad expertise in the use of PTC in recent years [36,37,38,39,40,41,42,43], and inspired by the versatility of DA-cyclopropanes, we decided to study their reactivity with nucleophiles under PTC conditions.
2. Results
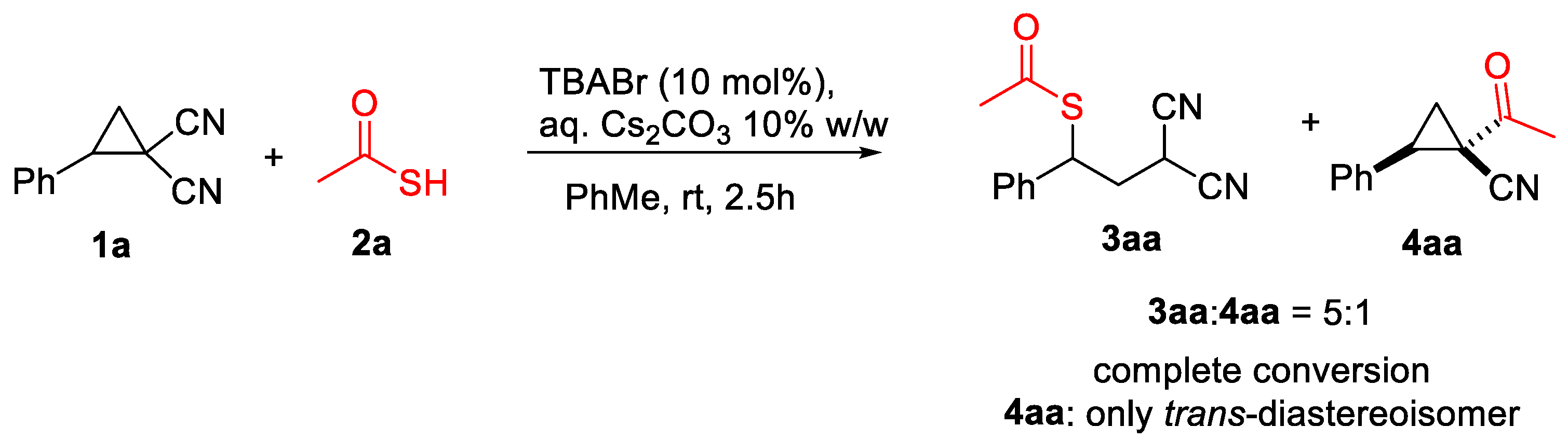
We started our investigation using 2-phenylcyclopropane-1,1-dicarbonitrile 1a as a model of D-A cyclopropane compounds, tetra-n-butylammonium bromide (TBABr) as a PTC catalyst, and a 10% w/w Cs2CO3 aqueous solution as the base. After some disappointing results using indole, diphenylphosphite, thiols, ene-carbamates, and sulfoxonium ylides as nucleophiles, which did not lead to the formation of the expected products, we observed reactivity when using thioacetic acid 2a as a reaction partner.
Surprisingly, besides product 3aa, derived from the expected ring-opening of the D-A cyclopropane, we observed the formation of compound 4aa as a single trans-diastereoisomer [44], obtained by the formal replacement of one of the cyano groups with an acetyl moiety, in a 5:1 ratio favouring 3aa (Scheme 2).
We next started an optimization process of the reaction conditions in order to selectively direct the reaction to the formation of the new cyclopropane derivative 4aa, derived by a non-reductive decyanation reaction, or towards the open-chain product 3aa, indeed achieved by the conventional reactivity of D-A cyclopropanes.
It was immediately understood that performing the reaction in the same reaction conditions but using solid Cs2CO3 instead of the corresponding aqueous solution, the ratio between the two compounds 3aa and 4aa could be reversed in favour the new cyclopropane derivative 4aa (Table 1, entry 1). We next evaluated different ammonium salts, as reported in Table 1: tetramethylammonium hydroxide hydrate (TMAOH × 5H2O) afforded only traces of product 4aa and 3aa, whereas promising results were obtained by performing the reaction with tetra-n-butylammonium iodide (TBAI), trimethyloctadecylammonium bromide (TMODABr), or timethylbenzylammonium chloride (TMBACl), (entries 3–5). TMODABr gave a slightly lower degree of selection between products 3aa and 4aa (entry 4) but a higher yield value. No products were obtained in the absence of an ammonium salt (entry 6). An increase or decrease in the concentration of the reaction mixture resulted in lower yield values (entries 7 and 8). Interestingly, when performing the reaction with a slight excess of substrate 1a (entry 9) a yield increase and a better selectivity were obtained (compare entries 3 and 9). Lastly, a prolonged reaction time (entry 10) achieved slightly increased conversion, but meanwhile eroding the selectivity.
Subsequently, the screening of different bases (Table 2, entries 1–4), solvents (entries 5–10), and temperatures (entries 11, 12) was carried out. Cs2CO3 was confirmed as the best base, whereas better results were obtained using THF as a solvent (entry 9). Increasing the temperature to 60 °C (entry 11) resulted in a lower yield, while conducting the reaction at 0 °C (entry 12) for 18 h afforded product 4aa in a comparable yield.
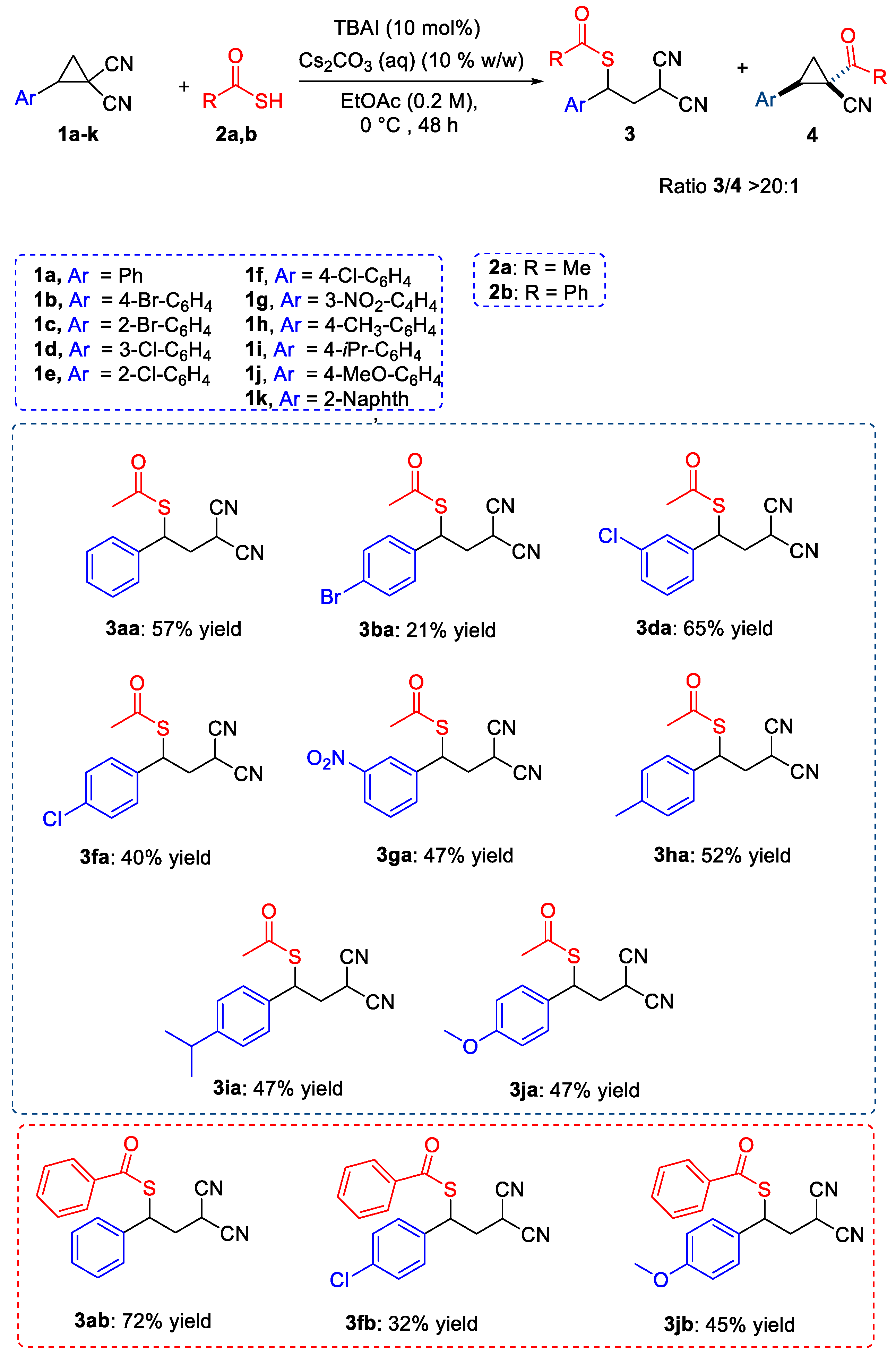
Having chosen the optimal reaction conditions for the selective obtainment of the decyaneted product 4 (Table 2, entry 9), we then moved to evaluate the generality of the reaction. As reported in Scheme 3, moderate to good yields and very good selectivity (ratio 4/3 > 20:1) could be obtained for all the D-A cyclopropane derivatives 1b–h employed regardless of the presence of electron-withdrawing or electron-donating substituents on the para-position of the aromatic ring (55–81%). The presence of orho-substituents on the aromatic ring was detrimental for the obtainable yields, while variable results were obtained with meta-substituted substrates. All new D-A cyclopropane derivatives 4 were obtained as single trans-diastereoisomers. Unfortunately, when thiobenzoic acid 2b was used in place of thioacetic acid, the corresponding decyanated cyclopropane derivatives were not obtained.
In addition, a screening of the reaction conditions for the selective obtainment of the ring-opening of D-A cyclopropanes 1 was performed. We restarted an optimization process of the reaction conditions in order to selectively direct the reaction towards the formation of the ring-opening product 3aa derived by a nucleophilic attack at the donor end of D-A cyclopropane 1a.
As previously mentioned, the use of an aqueous solution favoured the formation of product 3aa (Scheme 2). Moving from TBABr to TBAI (Table 3, entries 1 and 3), both selectivity and conversion improved. Better results were obtained working at 0 °C overnight (entries 4 and 5); a further improvement was also achieved using EtOAc as a solvent (entry 6). On the contrary, different ammonium salts besides TMODABr, different aqueous bases (K2CO3, Na2CO3, and NaHCO3), and other solvents (THF, CH2Cl2, Et2O, and TBME) tested were not conducive to any further improvements.
Having selected the optimal reaction conditions as the ones reported in Table 3 entry 6, we moved on to test the generality of the reaction.
As reported in Scheme 4, moderate to good yields and very good selectivity (ratio 3/4 >20:1) could be obtained for the D-A cyclopropane derivatives 1a, 1b, 1d, 1f, 1g–j, regardless of the presence of electron-withdrawing or electron-donating substituents on the aromatic ring. Thus, the presence of an EWG on para-position (1b, 1f) considerably lowered the yield, whereas the presence of an EDG on para-position (1h, 1i and 1j) led to comparable results with respect to 1a. No reactivity was observed in these reaction conditions, with D-A cyclopropanes 1c and 1e bearing a halogen in the ortho-position of the aromatic ring, probably due to a too-high steric constraint nearby the C2 of the cyclopropane ring where the nucleophilic attack had to occur.
The same reaction protocol was successfully employed with thiobenzoic acid 2b, obtaining products 3ab, 3fb, and 3jb in good or moderate yields.
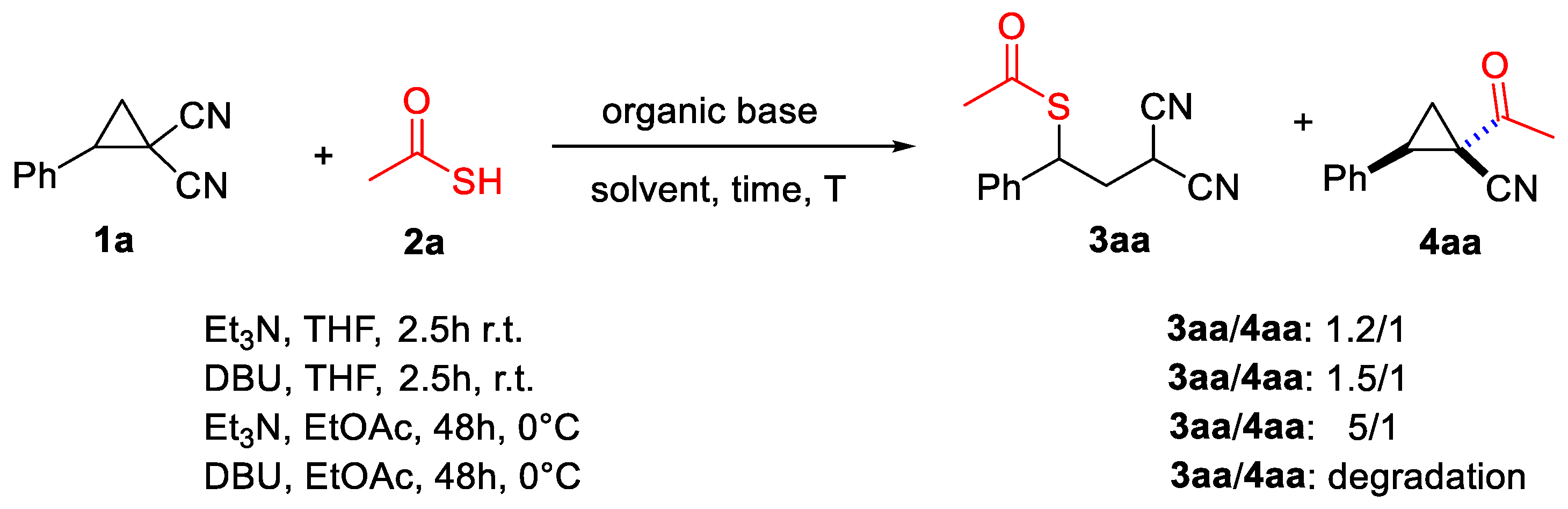
It is interesting to observe that the use of an organic base such as Et3N or DBU, although allowing the reaction, led to the formation of a mixture of product 3aa and 4aa without the selectivity obtainable in PTC conditions (Scheme 5).
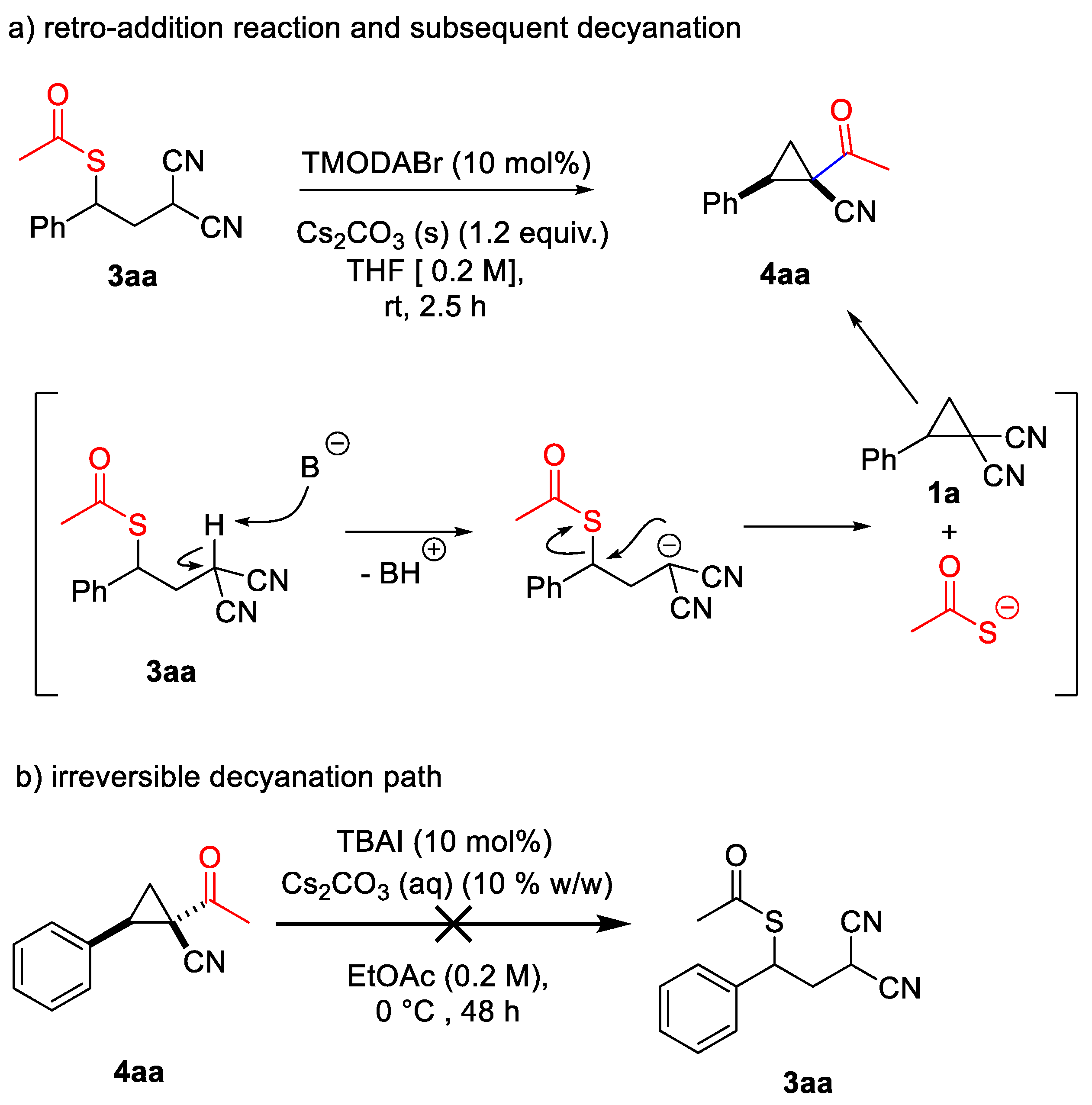
To shed some light on this intriguing and unusual divergent behaviour of D-A cyclopropanes 1, product 3aa was reacted with TMODABr in THF in the presence of solid Cs2CO3. This experiment afforded product 4aa via a retro-addition reaction and the subsequent decyanation of the restored D-A cyclopropane 1a (Scheme 6a). On the contrary, it was not possible to react 4aa in the standard reaction conditions to obtain the ring-opening product 3aa (Scheme 6b), indicating that the decyanation pathway is irreversible.
It is noteworthy that, using the first set of reaction conditions, namely solid Cs2CO3 and TMODABr, 4ka was obtained in 57% yield, in addition to product 5ka (12% yield) derived from the ring-opening of 4ka itself (Scheme 7); no selectivity could be obtained using aqueous Cs2CO3 and TBAI, since a mixture of products 3ka, 4ka, and 5ka was obtained.
The divergent behaviour of D-A cyclopropane 1 appeared to be strictly related to the typology of the base used, since the presence of solid Cs2CO3 allowed the unprecedented decyanation pathway to be activated in favour of the new cyclopropane derivative 4, whereas an aqueous solution of the same base pushed the reaction towards the ring-opening product 3. The extraction of water molecules into the organic phase in liquid–liquid systems probably decreases the reactivity of the thiolate by solvating it. On the other hand, in the solid–liquid mode, the anions are naked, and their reactivity is higher [45].
Moreover, the presence of a long aliphatic chain in the ammonium salt structure increased the selectivity between products 4 and 3, possibly due to the onset of considerable steric hindrance nearby the C2 of the cyclopropane ring, or to an increase in the reactivity of the thiolate.
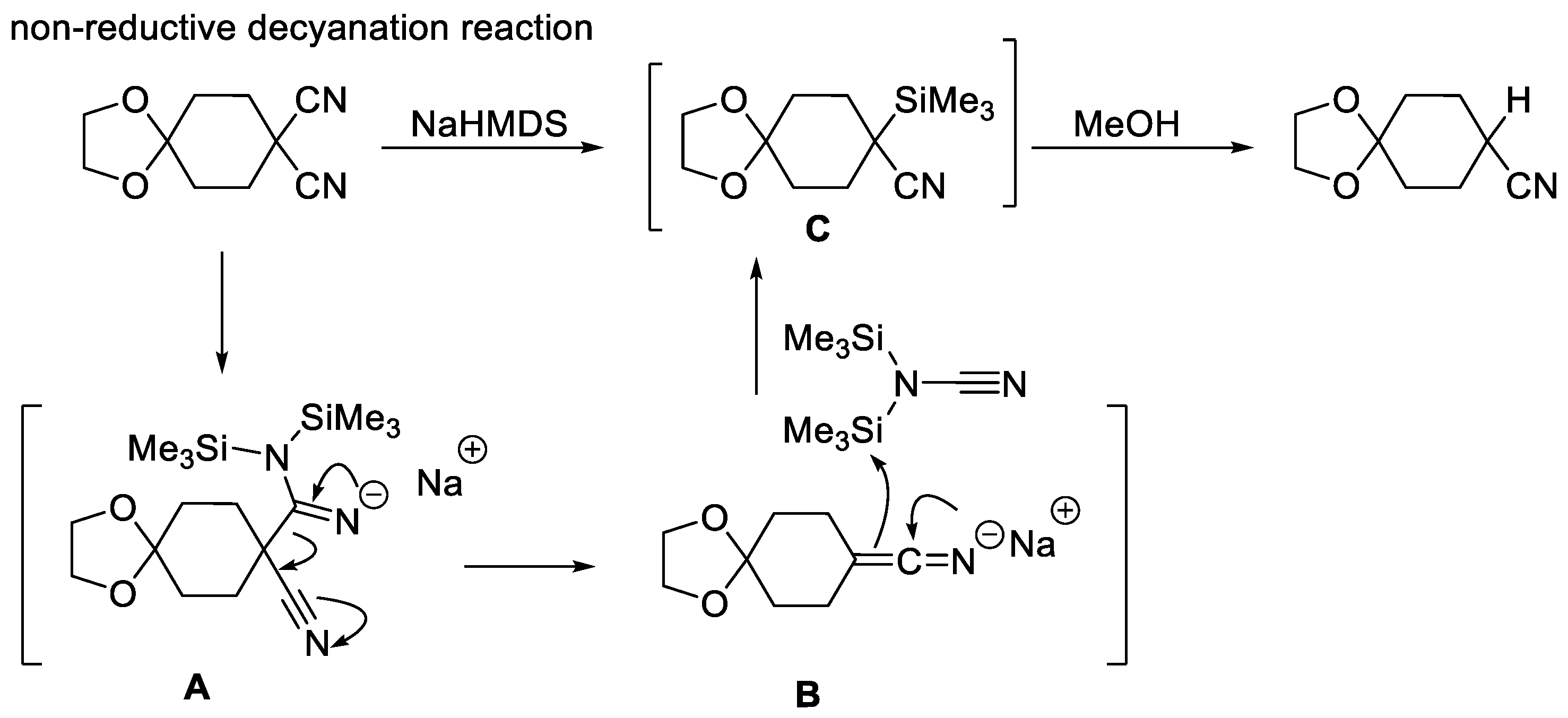
We then tried to devise a sound mechanistic hypothesis accounting for the formation of the unusual product 4aa in the reaction. In the literature, only one example of non-reductive decyanation reactions, of cyclic and acyclic disubstituted malononitriles, has been reported so far (Scheme 8) by Tanino [46] and co-workers, using sodium bis(trimethylsilyl)amide (NaHMDS) followed by methanol. The authors reported that the anionic intermediate A decomposes into α-cyano anion B and bis(trimethylsilyl)cyanamide, which readily undergoes an inter-molecular transfer of a silyl group. The reactive anion B is immediately captured by a silyl group to give C. The silyl group of C is then removed in the same pot simply by adding methanol to the reaction mixture.
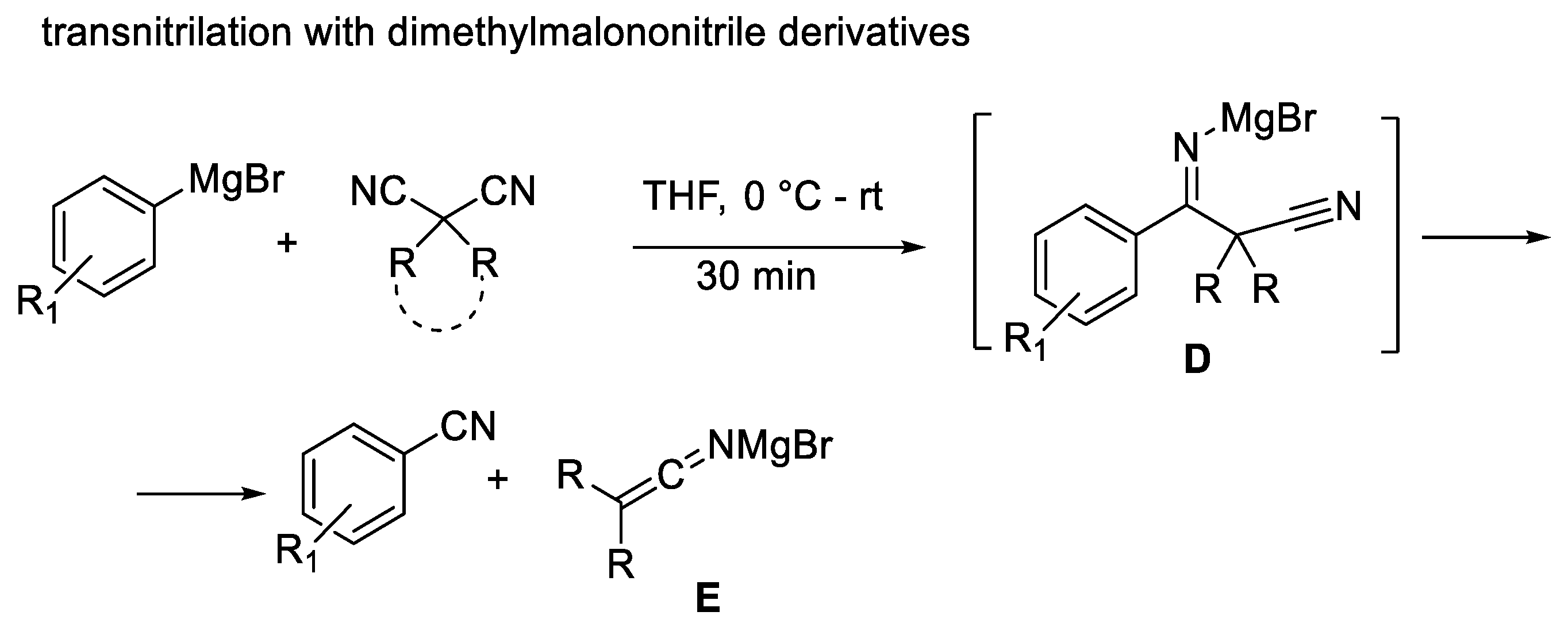
More recently, Reeves [47] and co-workers reported a transnitrilation of aryl Grignard and aryllithium reagents with dimethylmalononitrile derivatives via the intermediacy of imine D and the subsequent formation of ketimine E (Scheme 9).
On this basis, we envisioned that the thiolate, formed by the deprotonation of thioacetic acid by the inorganic base, attacks the electrophilic carbon of one of the two cyano groups, with the subsequent formation of a cyano anion I that evolves to ketimine anion II by the elimination of acetyl thiocyanate. The anion II is then captured by an acetyl group to form cyclopropane 4 with the concomitant formation of thiocyanate. The acetyl group enters at the less hindered side of the α-cyano carbanion, that is, trans to the phenyl ring (Scheme 10).
A few controlling experiments were performed in order to verify the proposed mechanism. First of all, an FeCl3 1M aqueous solution was added to the reaction mixture conducted in the reaction condition to obtain product 4, resulting in the development of an intense reddish-brown coloration, indicative of the formation of an iron complex with the thiocyanate ions present at the end of the reaction (Figure 1) [48].
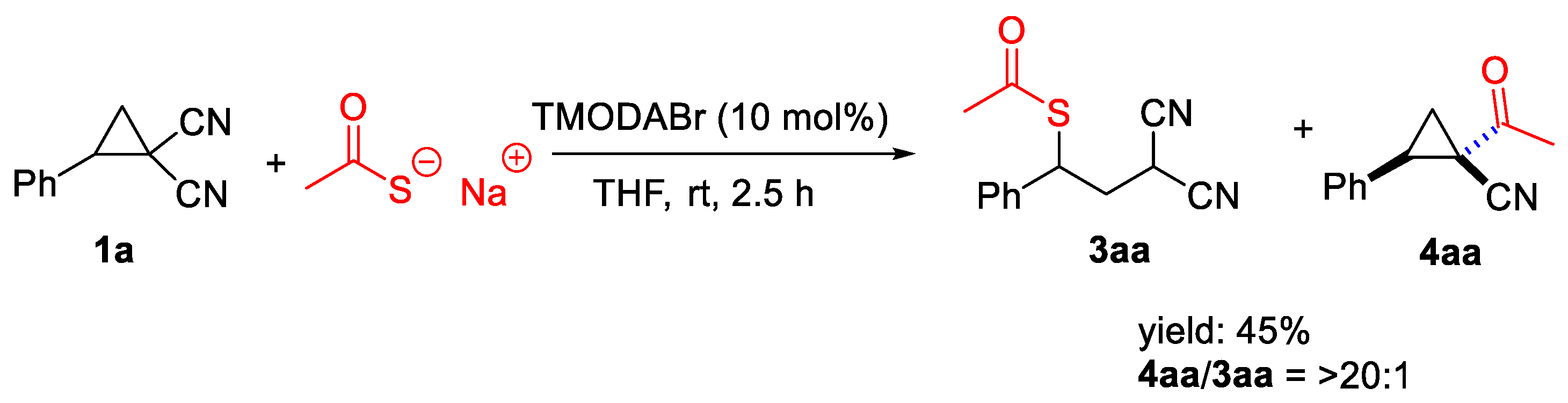
Next, D-A cyclopropane 1a was reacted with sodium thioacetate as a nucleophile, leading to the acquisition of product 4aa in a 45% yield (Scheme 11).
3. Materials and Methods
3.1. General Methods
The 1H and 13C NMR spectra were recorded on a Varian Mercury 400 spectrometer. Chemical shifts (δ) are reported in ppm relative to residual solvents signals [49] for 1H and 13C NMR. Signal patterns are indicated as follows: bs, broad singlet; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants (J) are given in Hertz (Hz). The 13C NMR were acquired with the 1H broad-band decoupled mode. Mass spectra were recorded using micromass LCT spectrometer using electrospray (ES) ionization techniques or FOCUS/DSQ using electron impact (EI) ionization techniques (relative intensities are given in brackets). The purification of reaction products was carried out by flash chromatography (FC) on silica gel (230–400 mesh) or by gravimetric chromatography using 70–230 mesh silica.
3.2. Materials
Analytical-grade solvents and commercially available reagents were used as received, unless otherwise noted.
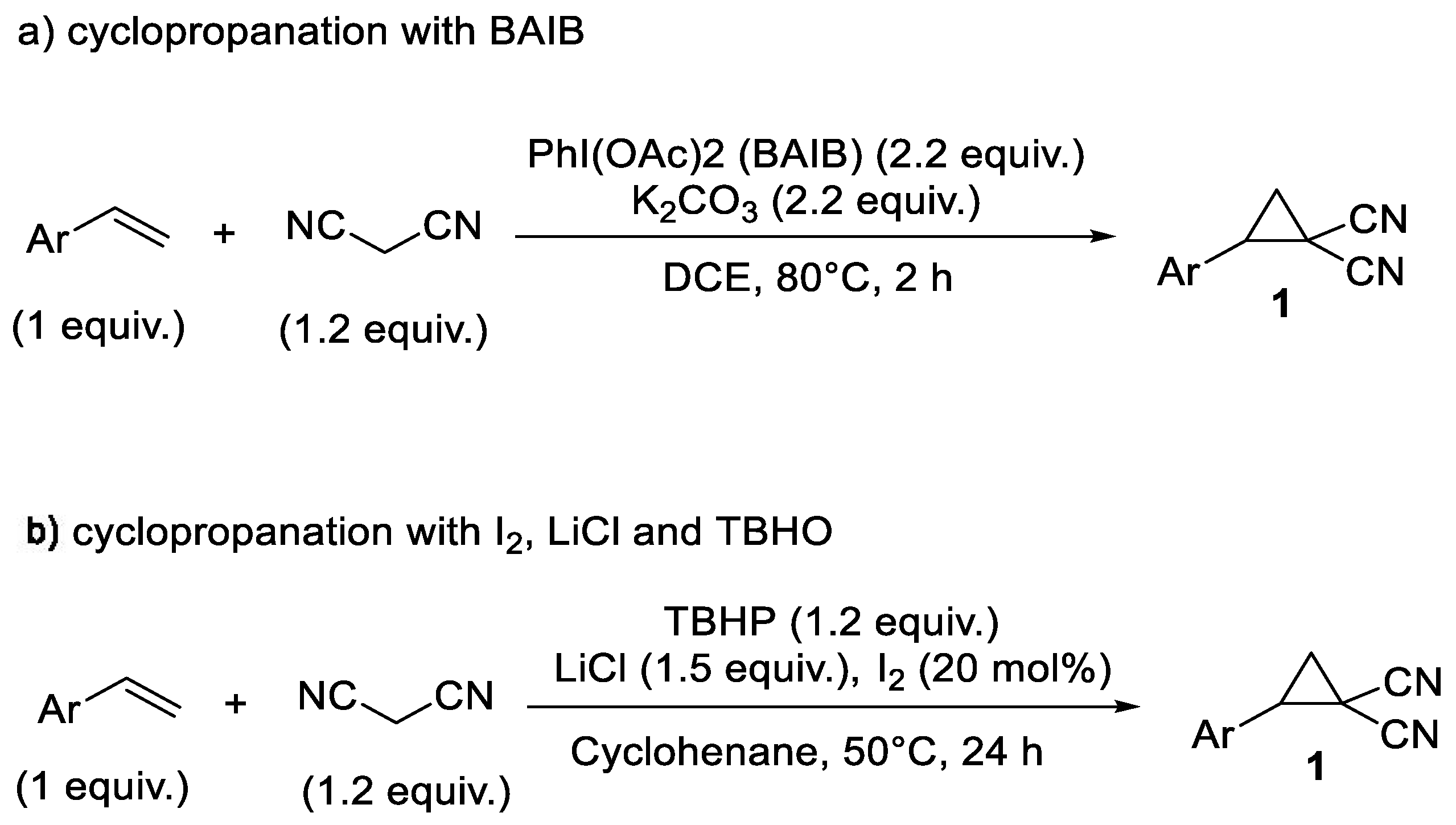
Cyclopropane 1 was obtained from the corresponding styrene derivatives and malononitrile following a literature procedure using bisacetoxyiodobenzene (BAIB) and K2CO3 [50], as reported in Scheme 12a, or using iodine, LiCl, and tert-butyl hydroperoxide (TBHP) [51], as reported in Scheme 12b.
The corresponding styrene derivatives, if not commercially available, were obtained by Wittig reactions from aldehydes.
3.3. General Procedure for the Synthesis of Products 4
In a 4 mL vial equipped with a magnetic stirring bar, D-A cyclopropane 1 (1.5 equiv., 0.3 mmol) was dissolved in 1000 µL of THF. TMODABr (10 mol% 0.02 mmol, 7.8 mg), thioacetic acid (1.0 equiv, 0.2 mmol, 14.3 μL), and Cs2CO3 (1.2 equiv., 0.24 mmol, 78.2 mg) were added in this order. The resulting suspension was stirred for 2.5 h at room temperature and then directly pre-purified by a short plug on silica gel using DCM and Et2O as eluents. After the evaporation of the solvent, the crude product was analysed by 1H-NMR and then purified through chromatography on silica gel to afford the desired compounds 4 as single diastereoisomers.
1-acetyl-2-phenylcyclopropane-1-carbonitrile 4aa

Following the general procedure and using cyclopropane 1a (50 mg), product 4aa was obtained in 61% yield (23 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.44–7.31 (m, 3H), 7.30–7.18 (m, 2H), 3.12 (t, J = 9.1 Hz, 1H), 2.58 (s, 3H), 2.21 (dd, J = 9.1, 4.9 Hz, 1H), 2.11 (dd, J = 8.4, 4.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ = 198.6, 133.1, 128.8 (2C), 128.6, 128.2 (2C), 118.3, 38.4, 30.3, 29.4, 24.7. MS (ESI) m/z: 208 [M + Na]+ The trans-relative configuration of compound 4aa was determined by a comparison with data in the literature [44] and by NOE experiments (see supplementary material). [44].
1-acetyl-2-(4-bromophenyl)cyclopropane-1-carbonitrile 4ba

Following the general procedure and using cyclopropane 1b (74 mg), product 4ba was obtained in 81% yield (43 mg) after chromatographic purification on silica gel (2:1 = DCM: n-hexane as eluent) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.50 (d, J = 8.3 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 3.05 (t, J = 8.7 Hz, 1H), 2.56 (s, 3H), 2.17 (dd, J = 9.1, 5.0 Hz, 1H), 2.03 (dd, J = 9.1, 5.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ = 198.2, 132.2, 131.9 (2C), 129.8 (2C), 122.7, 118.1, 37.4, 30.1, 29.4, 24.7. MS (ESI) m/z: 286, 288 [M + Na]+.
1-acetyl-2-(2-bromophenyl)cyclopropane-1-carbonitrile 4ca

Following the general procedure and using cyclopropane 1c (74 mg), product 4ca was obtained in 36% yield (19 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.65 (dd, J = 8.0, 1.2 Hz, 1H), 7.33 (td, J = 7.5, 1.2 Hz, 1H), 7.27–7.22 (m, 1H), 7.17 (dt, J = 7.6, 1.3 Hz, 1H), 3.08 (t, J = 8.6 Hz, 1H), 2.63 (s, 3H), 2.26 (dd, J = 8.8, 4.9 Hz, 1H), 2.10 (dd, J = 8.5, 5.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ = 198.6, 133.5, 133.0, 130.3, 129.2, 127.8, 126.9, 117.9, 39.4, 29.3, 29.3, 24.3. MS (ESI) m/z: 286, 288 [M + Na]+.
1-acetyl-2-(3-chlorophenyl)cyclopropane-1-carbonitrile 4da

Following the general procedure and using s cyclopropane 1d (60 mg), product 4da was obtained in 65% yield (28 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.47 (dd, J = 7.7, 1.5 Hz, 1H), 7.32 (td, J = 7.6, 1.8 Hz, 1H), 7.28 (td, J = 7.5,1.5, 1H), 7.18 (dd, J = 7.4, 1.8 Hz, 1H), 3.10 (t, J = 8.6 Hz, 1H), 2.62 (s, 3H), 2.26 (dd, J = 8.9, 4.9 Hz, 1H), 2.09 (dd, J = 8.4, 4.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 198.5, 136.4, 131.9, 130.0, 129.75, 129.0, 127.2, 118.0, 36.9, 29.2, 29.0, 23.8. MS (ESI) m/z: 242 [M + Na]+.
1-acetyl-2-(2-chlorophenyl)cyclopropane-1-carbonitrile 4ea

Following the general procedure and using cyclopropane 1e (60 mg), product 4ea was obtained in 32% yield (14 mg) after chromatographic purification on silica gel (3:1 = DCM:n-hexane as eluent) as a pale-yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.33–7.30 (m, 2H), 7.26–7.23 (bs, 1H), 7.13–7.10 (m, 1H), 3.07 (t, J = 8.7 Hz, 1H), 2.58 (s, 3H), 2.17 (dd, J = 9.1, 5.0 Hz, 1H), 2.06 (dd, J = 8.3, 5.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 198.2, 135.2, 134.7, 130.05, 128.8, 128.7, 126.2, 117.95, 37.2, 30.0, 29.5, 24.6. MS (ESI) m/z: 242 [M + Na]+.
1-acetyl-2-(4-chlorophenyl)cyclopropane-1-carbonitrile 4fa

Following the general procedure and using cyclopropane 1f (60 mg), product 4fa was obtained in 68% yield (30 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.38–7.33 (m, 2H), 7.21–7.17 (m, 2H), 3.09 (t, J = 8.75 Hz, 1H), 2.58 (s, 3H), 2.20 (dd, J = 9.2, 5.0 Hz, 1H), 2.06 (dd, J = 8.3, 5.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 198.3, 134.6, 131.7, 129.5, 129.05, 118.1, 37.3, 30.1, 29.5, 24.8. MS (ESI) m/z: 242 [M + Na]+.
1-acetyl-2-(3-nitrophenyl)cyclopropane-1-carbonitrile 4ga

Following the general procedure and using cyclopropane 1g (64 mg), product 4ga was obtained in 36% (17 mg) yield after chromatographic purification on silica gel (4:1 = n-hexane: EtOAc as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 8.26–8.20 (m, 1H), 8.17–8.13 (m, 1H), 7.62–7.57 (m, 2H), 3.22 (t, J = 8.7 Hz, 1H), 2.62 (s, 3H), 2.25 (dd, J = 9.1, 5.2 Hz, 1H), 2.16 (dd, J = 8.2, 5.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ = 197.8, 148.4, 135.5, 134.0, 129.9, 123.6, 123.5, 117.6, 36.4, 29.9, 29.5, 24.6. MS (ESI) m/z: 253 [M + Na]+.
1-acetyl-2-(p-tolyl)cyclopropane-1-carbonitrile 4ha

Following the general procedure and using cyclopropane 1h (55 mg), product 4ha was obtained in 62% (25 mg) yield after chromatographic purification on silica gel (4:1 = n-hexane: EtOAc as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.22–7.10 (m, 4H), 3.09 (t, J = 8.8 Hz, 1H), 2.57 (s, 3H), 2.35 (s, 3H), 2.19 (dd, J = 9.2, 4.9 Hz, 1H), 2.08 (dd, J = 8.4, 4.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ = 198.6, 138.5, 130.05, 129.5, 128.0, 118.5, 38.5, 30.4, 29.4, 24.7, 21.2. MS (ESI) m/z: 222 [M + Na]+.
1-acetyl-2-(4-isopropylphenyl)cyclopropane-1-carbonitrile 4ia

Following the general procedure and using cyclopropane 1i (63 mg), product 4ia was obtained in 55% yield (25 mg) after chromatographic purification on silica gel (4:1 = n-hexane: EtOAc as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.26–7.22 (m, 2H), 7.19–7.15 (m, 2H), 3.08 (t, J = 8.8 Hz, 1H), 2.91 (hept, J = 6.9 Hz, 1H), 2.57 (s, 3H), 2.20 (dd, J = 9.2, 4.8 Hz, 1H), 2.08 (dd, J = 8.4, 4.8 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ = 198.6, 149.4, 130.4, 128.1, 126.9, 118.5, 38.5, 33.8, 30.3, 29.4, 24.9, 23.8, MS (ESI) m/z: 250 [M + Na]+.
1-acetyl-2-(4-methoxyphenyl)cyclopropane-1-carbonitrile 4ja

Following the general procedure and using cyclopropane 1j (59 mg), product 4ja was obtained in 56% yield (24 mg) after chromatographic purification on silica gel (3:1 = n-hexane: EtOAc as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.21–7.15 (m, 2H), 6.94–6.87 (m, 2H), 3.81 (s, 3H), 3.08 (dd, J = 9.1, 8.4 Hz, 1H), 2.57 (s, 3H), 2.20 (dd, J = 9.2, 4.9 Hz, 1H), 2.09–2.02 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 198.6, 160.0, 129.4, 125.0, 118.6, 114.2, 55.3, 38.5, 30.4, 29.4, 24.9. MS (ESI) m/z: 238 [M + Na]+.
1-acetyl-2-(naphthalen-2-yl)cyclopropane-1-carbonitrile 4ka

Following the general procedure and using cyclopropane 1k (65 mg), product 4ka was obtained in 57% yield (27 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ = 7.93–7.76 (m, 3H), 7.76–7.69 (m, 1H), 7.57–7.44 (m, 2H), 7.36 (dd, J = 8.5, 1.9 Hz, 1H), 3.29 (t, J = 8.7 Hz, 1H), 2.61 (s, 3H), 2.33–2.22 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 198.55, 133.2, 133.1, 128.7, 127.9, 127.75, 127.5, 126.6, 126.55, 125.6, 118.4, 38.6, 30.3, 29.5, 24.8. MS (ESI) m/z: 258 [M + Na]+.
3.4. General Procedure for the Synthesis of Products 3
In a 4 mL vial equipped with a magnetic stirring bar, D-A cyclopropane 1 (1.0 equiv., 0.2 mmol) was dissolved in 1000 µL of EtOAc. TBAI (10 mol%, 0.02 mmol, 7.4 mg), thioacetic acid (1.5 equiv, 0.3 mmol, 21.4 µL), or thiobenzoic acid 2b (1.5 equiv, 0.3 mmol, 36 µL) and Cs2CO3 (aq, 10% w/w, 500 µL) were added in this order. The resulting suspension was stirred for 48 h at 0 °C and then directly pre-purified by a short plug on silica gel using DCM and Et2O as eluents. After the evaporation of the solvent, the crude product was analysed by 1H-NMR and then purified through chromatography on silica gel to afford the desired compounds 3 as oils.
S-(3,3-dicyano-1-phenylpropyl) ethanethioate 3aa

Following the general procedure and using cyclopropane 1a (33.6 mg) and thioacetic acid 2a (21.4 µL), product 3aa was obtained in 57% yield (28 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (600 MHz, CDCl3) δ = 7.42–7.32 (m, 3H), 7.31–7.27 (m, 2H), 4.73 (dd, J = 9.3, 6.6 Hz, 1H), 3.53 (dd, J = 9.0, 6.4 Hz, 1H), 2.71 (ddd, J = 13.8, 9.0, 6.6 Hz, 1H), 2.58 (ddd, J = 13.8, 9.4, 6.4 Hz, 1H), 2.35 (s, 3H). 13C NMR (151 MHz, CDCl3) δ = 193.5, 136.9, 129.5, 128.9, 127.6, 111.9, 111.6, 44.8, 37.1, 30.4, 20.9. MS (ESI) m/z: 267 [M + Na]+.
S-(1-(4-bromophenyl)-3,3-dicyanopropyl) ethanethioate 3ba

Following the general procedure and using cyclopropane 1b (49.4 mg) and thioacetic acid 2a (21.4 µL), product 3ba was obtained in 21% yield (14 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.52 (d, J = 8.5 Hz, 2H), 7.19 (d, J = 8.5 Hz, 2H), 4.70 (dd, J = 8.9, 7.1 Hz, 1H), 3.59 (dd, J = 8.6, 6.9 Hz, 1H), 2.69 (ddd, J = 13.9, 8.6, 7.1 Hz, 1H), 2.56 (ddd, J = 13.9, 8.9, 6.9 Hz, 1H), 2.36 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 193.0, 136.4, 132.6, 129.3, 122.9, 111.7, 111.4, 44.2, 36.7, 30.4, 20.9. MS (ESI) m/z: 345, 347 [M + Na]+.
S-(1-(3-chlorophenyl)-3,3-dicyanopropyl) ethanethioate 3da

Following the general procedure and using cyclopropane 1d (40 mg) and thioacetic acid 2a (21.4 µL), product 3da was obtained in 65% yield (36 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.35–7.28 (m, 3H), 7.24–7.18 (m, 1H), 4.72 (dd, J = 8.69, 7.24 Hz, 1H) 3.61 (dd, J = 8.43, 6.98 Hz, 1H), 2.69 (ddd, J = 13.9, 8.4, 7.3 Hz, 1H), 2.59 (ddd, J = 13.5, 8.7, 7.0 Hz, 1H), 2.37 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 193.0, 139.45, 135.4, 130.8, 129.2, 127.7, 125.9, 111.7, 111.5, 44.32, 36.85, 30.46, 20.99. MS (ESI) m/z: 301 [M + Na]+.
S-(1-(4-chlorophenyl)-3,3-dicyanopropyl) ethanethioate 3fa

Following the general procedure and using cyclopropane 1f (40 mg) and thioacetic acid 2a (21.4 µL), product 3fa was obtained in 40% yield (22 mg) after chromatographic purification on silica gel (3:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.38–7.33 (m, 2H), 7.27–7.23 (m, 2H), 4.72 (dd, J = 8.9, 7.1 Hz, 1H), 3.58 (dd, J = 8.5, 6.85 Hz, 1H), 2.69 (ddd, J = 13.9, 8.6, 7.1 Hz, 1H), 2.56 (ddd, J = 13.9, 8.7, 6.8 Hz, 1H), 2.36 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 193.1, 135.9, 134.9, 129.7, 129.0, 111.7, 111.4, 44.2, 36.85, 30.5, 21.0. MS (ESI) m/z: 301 [M + Na]+.
S-(3,3-dicyano-1-(3-nitrophenyl)propyl) ethanethioate 3ga

Following the general procedure using substrate 1g (43 mg) and thioacetic acid 2a (21.4 µL), product 3ga was obtained in 47% yield (25 mg) after chromatographic purification on silica gel (4:1 = n-hexane: EtOAc as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 8.26–8.18 (m, 2H), 7.73–7.68 (m, 1H), 7.63–7.57 (m, 1H), 4.87 (t, J = 8.0 Hz, 1H), 3.76 (t, J = 7.5 Hz, 1H), 2.82–2.64 (m, 2H), 2.39 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 194.0, 161.2, 142.5, 133.1, 130.05, 123.3, 122.3, 116.9, 69.7, 51.8, 42.1, 29,4. MS (ESI) m/z: 312 [M + Na]+.
S-(3,3-dicyano-1-(p-tolyl)propyl) ethanethioate 3ha

Following the general procedure and using cyclopropane 1h (36 mg) and thioacetic acid 2a (21.4 µL), product 3ha was obtained in 52% yield (27 mg) after chromatographic purification on silica gel (4:1 = n-hexane: Et2O as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.17 (br s, 4H), 4.70 (dd, J = 9.6, 6.5 Hz, 1H), 3.52 (dd, J = 9.2, 6.3 Hz, 1H) 2.71 (ddd, J = 13.6, 9.3, 6.5 Hz, 1H), 2.56 (ddd, J = 13.8, 9.6, 6.4 Hz, 1H), 2.34 (s, 3H), 2.33 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 193.6, 139.0, 133.8, 130.2, 127.5, 112.0, 111.6, 44.5, 37.15, 35.1, 30.4, 20.9. MS (ESI) m/z: 312 [M + Na]+.
S-(3,3-dicyano-1-(4-isopropylphenyl)propyl) ethanethioate 3ia

Following the general procedure using substrate 1i (42 mg) and thioacetic acid 2a (21.4 µL), product 3ia was obtained in 47% yield (27 mg) after chromatographic purification on silica gel (6:1 = n-hexane: EtOAc as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.25–7.19 (m, 4H), 4.72 (dd, J = 9.6, 6.5 Hz, 1H), 3.53 (dd, J = 9.3, 6.25 Hz, 1H), 2.90 (hept, J = 6.9, 1H) 2.73 (ddd, J = 13.7, 9.3, 6.5 Hz, 1H), 2.60–2.51 (m, 1H) 2.36 (s, 3H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ = 193.6, 149.9, 134.05, 127.6, 127.5, 112.0, 111.6, 44.5, 37.2, 33.8, 30.4, 23.8, 20.9. MS (ESI) m/z: 309 [M + Na]+.
S-(3,3-dicyano-1-(4-methoxyphenyl)propyl) ethanethioate 3ja

Following the general procedure using substrate 1j (40 mg) and thioacetic acid 2a (21.4 µL), product 3ja was obtained in 47% yield (51 mg) after chromatographic purification on silica gel (4:1 = n-hexane: EtOAc as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.24–7.20 (m, 2H), 6.92–6.87 (m, 2H), 4.72 (dd, J = 9.7, 6.4 Hz, 1H), 3.81 (s, 3H) 3.53 (dd, J = 9.2, 6.2 Hz, 1H), 2.72 (ddd, J = 13.7, 9.2, 6.4 Hz, 1H), 2.54 (ddd, J = 13.7, 9.7, 6.25 Hz, 1H) 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 193.7, 159.9, 128.8, 128.6, 114.9, 112.0, 111.6, 55.35, 44.3, 37.2, 30.4, 20.9. MS (ESI) m/z: 297 [M + Na]+.
S-(3,3-dicyano-1-phenylpropyl) benzothioate 3ab

Following the general procedure using cyclopropane 1a (33.6 mg) and thiobenzoic acid 2b (36 µL), product 3ab was obtained in 72% yield (44 mg) after chromatographic purification on silica gel (2:1 = DCM: n-hexane as eluent) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.97–7.90 (m, 2H), 7.64–7.56 (m, 1H), 7.53–7.31 (m, 7H), 4.97 (dd, J = 9.4, 6.5 Hz, 1H), 3.62 (dd, J = 9.0, 6.5 Hz, 1H), 2.86 (ddd, J = 13.8, 8.9, 6.5 Hz, 1H), 2.69 (ddd, J = 13.8, 9.4, 6.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ = 189.6, 137.0, 135.9, 134.1, 129.64, 129.61, 128.8, 127.8, 127.5, 112.0, 111.6, 44.8, 37.4, 21.0. MS (ESI)m/z: 329 [M + Na]+.
S-(3,3-dicyano-1-(4-chlorophenyl)propyl) benzothioate 6fa

Following the general procedure using cyclopropane 1f (40 mg) and thiobenzoic acid 2b (36 µL), product 3fb was obtained in 32% yield (22 mg) after chromatographic purification on silica gel (1:1 = n-hexane: EtOAc as eluent) a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.95–7.90 (m, 2H), 7.64–7.58 (m, 1H), 7.52–7.42 (m, 2H), 7.42–7.32 (m, 4H), 4.95 (dd, J = 8.9, 7.0 Hz, 1H), 3.67 (dd, J = 8.5, 6.9 Hz, 1H), 2.83 (ddd, J = 13.9, 8.5, 7.0 Hz, 1H), 2.67 (ddd, J = 13.9, 9.0, 6.9 Hz, 1H). MS (ESI)m/z: 363 [M + Na]+.
S-(3,3-dicyano-1-(4-methoxyphenyl)propyl) benzothioate 3jb

Following the general procedure using 1j (40 mg) and thiobenzoic acid 2b (36 µL), product 3jb was obtained in 45% yield (30 mg) after chromatographic purification on silica gel (2:1 = DCM: n-hexane as eluent) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.95–7.90 (m, 2H), 7.63–7.57 (m, 1H), 7.50–7.42 (m, 2H), 7.35–7.29 (m, 2H), 6.97–6.90 (m, 2H), 4.92 (dd, J = 9.7, 6.2 Hz, 1H), 3.82 (s, 3H), 3.61 (dd, J = 9.2, 6.2 Hz, 1H), 2.86 (ddd, J = 13.7, 9.2, 6.2 Hz, 1H), 2.64 (ddd, J = 13.7, 9.7, 6.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ = 189.8, 160.0, 136.1, 134.0, 129.0, 128.8, 128.6, 127.4, 114.9, 112.0, 111.6, 55.4, 44.3, 37.4, 21.0. MS (ESI)m/z: 359 [M + Na]+.
4. Conclusions
In summary, the reactivity of some D-A cyclopropanes with thioacetic (and thiobenzoic) acid under PTC conditions was explored. This study, which constitutes a rare example of PTC reactions with cyclopropane substrates, led to the discovery of an unprecedented decyanation–acetylation reaction, affording 1-acetyl-1-cyano cyclopropanes 4. This process was found to compete with a typical cyclopropane ring-opening reaction leading to adducts 3. An investigation of the parameters affecting the two divergent pathways pointed to the nature of the inorganic base (solid vs. aqueous) as the key factor. With this insight, the screening of PT catalysts, solvents, and temperatures led to the creation of two complementary conditions, enabling excellent control over the product produced by the reaction. Thus, a series of cyclopropanes 4 were selectivity obtained in moderate to good yields using the first set of conditions, while the second set led to their ring-opened counterparts 3 with comparable results. Conversely, the low selectivity observed with common homogeneous organic bases in this reaction highlights the unique possibilities offered by the combination of PTC with D-A cyclopropanes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13040760/s1, NMR spectra of selected compounds.
Author Contributions
Conceptualization: G.D.B., L.B. and M.F.; methodology: G.D.B., P.V., L.B. and M.F.; writing—original draft preparation: M.F.; writing—review and editing: G.D.B., L.B. and M.F.; project administration: M.F. and L.B.; funding acquisition: M.F. and L.B. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge financial support by the University of Bologna (RFO program). This work was supported by the Italian Ministry for University and Research (MUR, PRIN 2020, 2020AEX4TA project).
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
We thank the bachelor students of our department who, in the past few years, have developed part of this project: Cosimo Zaccaria, Alessandro Coatti, Alice Mammi, and Magdalena Medrzycka. We thank Luca Zuppiroli for performing the mass spectrometry measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de Meijere, A. Bonding properties of cyclopropane and their chemical consequences. Angew. Chem. Int. Ed. Engl. 1979, 18, 809–826. [Google Scholar] [CrossRef]
- Reissig, H.-U.; Hirsch, E. Donor-Acceptor substituted cyclopropanes: Synthesis and ring opening to 1,4-dicarbonyl compounds. Angew. Chem. Int. Ed. 1980, 19, 813–814. [Google Scholar] [CrossRef]
- Reissig, H.-U.; Zimmer, R. Donor−Acceptor-substituted cyclopropane derivatives and their application in organic synthesis. Chem. Rev. 2003, 103, 1151–1196. [Google Scholar] [CrossRef] [PubMed]
- Reissig, H.-U. Donor-Acceptor-Substituted Cyclopropanes: Versatile Building Blocks in Organic Synthesis. Top. Curr. Chem. 1988, 144, 73–135. [Google Scholar] [CrossRef]
- Yu, M.; Pagenkopf, B.L. Recent advances in donor–acceptor (DA) cyclopropanes. Tetrahedron 2005, 61, 321–347. [Google Scholar] [CrossRef]
- Carson, C.A.; Kerr, M.A. Heterocycles from cyclopropanes: Applications in natural product synthesis. Chem. Soc. Rev. 2009, 38, 3051–3060. [Google Scholar] [CrossRef]
- Agrawal, D.; Yadav, V. Silylmethyl-substituted cyclopropyl and other strained ring systems: Cycloaddition with dipolarophiles. Chem. Commun. 2008, 6471–6488. [Google Scholar] [CrossRef]
- Lebold, T.P.; Kerr, M.A. Intramolecular annulations of donor–acceptor cyclopropanes. Pure Appl. Chem. 2010, 82, 1797–1812. [Google Scholar] [CrossRef]
- Meazza, M.; Guo, H.; Rios, R. Synthetic applications of vinyl cyclopropane opening. Org. Biomol. Chem. 2017, 15, 2479–2490. [Google Scholar] [CrossRef]
- Danishefsky, S. Electrophilic cyclopropanes in organic synthesis. Acc. Chem. Res. 1979, 12, 66–72. [Google Scholar] [CrossRef]
- Wenkert, E. Oxycyclopropanes in organochemical synthesis. Acc. Chem. Res. 1980, 13, 27–31. [Google Scholar] [CrossRef]
- Schneider, T.F.; Kaschel, J.; Werz, D.B. A New Golden Age for Donor–Acceptor Cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. [Google Scholar] [CrossRef] [PubMed]
- Cavitt, M.A.; Phun, L.H.; France, S. Intramolecular donor–acceptor cyclopropane ring-opening cyclizations. Chem. Soc. Rev. 2014, 43, 804–818. [Google Scholar] [CrossRef] [PubMed]
- De Nanteuil, F.; De Simone, F.; Frei, R.; Benfatti, F.; Serrano, E.; Waser, J. Cyclization and annulation reactions of nitrogen-substituted cyclopropanes and cyclobutanes. Chem. Commun. 2014, 50, 10912–10928. [Google Scholar] [CrossRef]
- Grover, H.K.; Emmett, M.R.; Kerr, M.A. Carbocycles from donor–acceptor cyclopropanes. Org. Biomol. Chem. 2015, 13, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Budynina, E.M.; Ivanov, K.L.; Sorokin, I.D.; Melnikov, M.Y. Ring opening of Donor–Acceptor cyclopropanes with N-Nucleophiles. Synthesis 2017, 49, 3035–3068. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, X.; Feng, X. Asymmetric catalytic reactions of Donor–Acceptor cyclopropanes. Angew. Chem. Int. Ed. 2021, 60, 9192–9204. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Trushkov, I.V. Donor-Acceptor cyclopropanes in the synthesis of carbocycles. Chem. Rec. 2019, 19, 2189–2208. [Google Scholar] [CrossRef]
- Ghosh, K.; Das, S. Recent advances in ring-opening of donor acceptor cyclopropanes using C-nucleophiles. Org. Biomol. Chem. 2021, 19, 965–982. [Google Scholar] [CrossRef]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropane. Chem. Rev. 2021, 121, 227–263. [Google Scholar] [CrossRef]
- Dolfini, J.E.; Menich, K.; Corliss, P.; Cavanaugh, K.; Danishefsky, S.; Chakrabartty, S. The reaction of enamines with activated cyclopropanes. Tetrahedron Lett. 1966, 37, 4421–4426. [Google Scholar] [CrossRef]
- England, D.B.; Kuss, T.D.O.; Keddy, R.G.; Kerr, M.A. Cyclopentannulation of 3-alkylindoles: A synthesis of a tetracyclic subunit of the kopsane alkaloids. J. Org. Chem. 2001, 66, 4704–4709. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Vidal-Albalat, A.; Jørgensen, J.; Barløse, C.L.; Jessen, K.S.; Iversen, M.V.; Jørgensen, K.A. Directing the activation of Donor-Acceptor cyclopropanes towards stereoselective 1,3-dipolar cycloaddition reactions by brønsted base catalysis. Angew. Chem. Int. Ed. 2017, 56, 11831–11835. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.; Sánchez-Díez, E.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J.L. Catalytic generation of Donor-Acceptor cyclopropanes under N-heterocyclic carbene activation and their stereoselective reaction with alkylideneoxindoles. Adv. Synth. Catal. 2017, 359, 1678–1683. [Google Scholar] [CrossRef]
- Saya, L.; Fernández, I.; López, F.; Mascareñas, J.L. Nickel-catalyzed intramolecular [3 + 2 + 2] cycloadditions of alkylidenecyclopropanes. A straightforward entry to fused 6,7,5-tricyclic systems. Org. Lett. 2014, 16, 5008–5011. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.A.; Burnie, A.J.; Negru, D.E. Rhodium-Catalyzed [(3 + 2) + 1] carbocyclization reactions of alkynylidenecyclopropanes with carbon monoxide: Regiospecific construction of polysubstituted phenols. Org. Lett. 2014, 16, 4356–4359. [Google Scholar] [CrossRef]
- Kuila, B.; Mahajan, D.; Singh, P.; Bhargava, G. Nickel catalyzed [3 + 2] cycloaddition reaction of bis(methylenecyclopropane) with cyclic and acyclic dienophiles. Enantioselective (8 + 3) Cycloadditions by Activation of Donor–Acceptor Cyclopropanes Employing Chiral Brønsted Base Catalysis. Tetrahedron Lett. 2015, 56, 1307–1311. [Google Scholar] [CrossRef]
- Corti, V.; Marcantonio, E.; Mamone, M.; Giungi, A.; Fochi, M.; Bernardi, L. Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines. Catalysts 2020, 10, 150. [Google Scholar] [CrossRef]
- McLeod, D.A.; Thøgersen, M.K.; Barløse, C.L.; Skipper, M.L.; Obregón, E.B.; Jørgensen, K.A. Enantioselective (8+3) Cycloadditions by Activation of Donor–Acceptor Cyclopropanes Employing Chiral Brønsted Base Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202206096. [Google Scholar] [CrossRef]
- Halskov, K.S.; Kniep, F.; Lauridsen, V.H.; Iversen, E.H.; Donslund, B.S.; Jørgensen, K.A. Organocatalytic Enamine-Activation of Cyclopropanes for Highly Stereoselective Formation of Cyclobutanes. J. Am. Chem. Soc. 2015, 137, 1685–1691. [Google Scholar] [CrossRef]
- Ortega, A.; Manzano, R.; Uria, U.; Carrillo, L.; Reyes, E.; Tejero, T.; Merino, P.; Vicario, J.L. Catalytic Enantioselective Cloke–Wilson Rearrangement. Angew. Chem. Int. Ed. 2018, 57, 8225–8229. [Google Scholar] [CrossRef] [PubMed]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 3rd ed.; VCH: Weinheim, Germany, 1993. [Google Scholar] [CrossRef]
- Starks, C.; Liotta, C.; Halpern, M. Phase-Transfer Catalysis: Fundamentals, Applications and Industrial Perspectives; Chapman & Hall: New York, NY, USA, 1994; ISBN 978-94-011-0687-0. [Google Scholar]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Marianacci, O.; Micheletti, G.; Bernardi, L.; Fini, F.; Fochi, F.; Pettersen, D.; Sgarzani, V.; Ricci, A. Organocatalytic asymmetric mannich reactions with N-Boc and N-Cbz protected α-amido sulfones. Chem. Eur. J. 2007, 13, 8338–8351. [Google Scholar] [CrossRef]
- Fini, F.; Micheletti, G.; Bernardi, L.; Pettersen, D.; Fochi, M.; Ricci, A. An easy entry to optically active α-amino phosphonic acid derivatives using phase-transfer catalysis (PTC). Chem. Commun. 2008, 4345–4347. [Google Scholar] [CrossRef]
- Gioia, C.; Fini, F.; Mazzanti, A.; Bernardi, L.; Ricci, A. Organocatalytic asymmetric formal [3 + 2] cycloaddition with in situ-generated N-carbamoyl nitrones. J. Am. Chem. Soc. 2009, 131, 9614–9615. [Google Scholar] [CrossRef]
- Bernardi, L.; Fini, F.; Gochi, M.; Ricci, A. Organocatalyzed Enantioselective Synthesis of Nitroalkanes Bearing All-Carbon Quaternary Stereogenic Centers through Conjugate Addition of Acetone Cyanohydrin. Synlett 2008, 1857–1861. [Google Scholar] [CrossRef]
- Cassani, C.; Bernardi, L.; Fini, F.; Ricci, A. Catalytic asymmetric mannich reactions of sulfonylacetates. Angew. Chem. Int. Ed. 2009, 48, 5694–5697. [Google Scholar] [CrossRef]
- Mazzotta, S.; Gramigna, L.; Bernardi, L.; Ricci, A. One-Pot synthesis of optically active β-amino-α-methylene carbonyl derivatives from α-amidosulfones using quinine-based Phase-Transfer Catalysts. Org. Process Res. Dev. 2010, 14, 687–691. [Google Scholar] [CrossRef]
- Bernardi, L.; Fochi, M.; Carbone, R.; Martinelli, A.; Fox, M.E.; Cobley, C.J.; Kandagatla, B.; Oruganti, S.; Dahanukar, V.H.; Carlone, A. Organocatalytic Asymmetric Conjugate Additions to Cyclopent-1-enecarbaldehyde: A Critical Assessment of Organocatalytic Approaches towards the Telaprevir Bicyclic Core. Chem. Eur. J. 2015, 21, 19208–19222. [Google Scholar] [CrossRef]
- Bertuzzi, G.; Silvestrini, F.; Moimare, P.; Pecorari, D.; Mazzanti, A.; Bernardi, L.; Fochi, M. Chemodivergent Preparation of Various Heterocycles via Phase-Transfer Catalysis: Enantioselective Synthesis of Functionalized Piperidines. Adv. Synth. Catal. 2020, 362, 1167–1175. [Google Scholar] [CrossRef]
- Luo, Y.-C.; Ma, H.; Hu, X.-Q.; Xu, P.-F. Sc(OTf)3 Catalyzed [4 + 2]-Annulation Reaction between Electron-Rich Phenols and Donor—Acceptor Cyclopropanes: Synthesis of Polysubstituted Dihydronaphthols. J. Org. Chem. 2017, 82, 1013–1023. [Google Scholar] [CrossRef]
- Naik, S.D.; Doraiswamy, L.K. Mathematical modeling of solid-liquid phase-transfer catalysis. Chem. Eng. Sci. 1997, 52, 4533–4546. [Google Scholar] [CrossRef]
- Domon, D.; Iwakura, M.; Tanino, K. Non-reductive decyanation reactions of disubstituted malononitrile derivatives promoted by NaHMDS. Tetrahedron Lett. 2017, 58, 1957–1960. [Google Scholar] [CrossRef]
- Reeves, J.T.; Malapit, C.A.; Buono, F.G.; Sidhu, K.P.; Marsini, M.A.; Avery Sader, C.; Fandrick, K.R.; Busacca, C.A.; Senanayake, C.H. Transnitrilation from Dimethylmalononitrile to Aryl Grignard and Lithium Reagents: A Practical Method for Aryl Nitrile Synthesis. J. Am. Chem. Soc. 2015, 137, 9481–9488. [Google Scholar] [CrossRef]
- Jeffery, G.H.; Bassett, J.; Mendham, J.; Denney, R.C. Vogel’s Textbook of Quantitative Chemical Analysis, 5th ed.; Longman Scientific and Technical: Harlow, UK, 1989. [Google Scholar]
- Gottlieb, H.E.; Kottlyar, V.; Nudelman, A.J. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, M.; Dong, Z.; Liang, F.; Zhang, J. Hypervalent iodine(iii)-mediated cyclopropa(e)nation of alkenes/alkynes under mild conditions. Org. Biomol. Chem. 2014, 12, 1341–1350. [Google Scholar] [CrossRef]
- Yoshimura, A.; Jones, T.N.; Yusubov, M.S.; Zhdankin, V.V. Hypoiodite-Mediated Catalytic Cyclopropanation of Alkenes with Malononitrile. Adv. Synth. Catal. 2014, 356, 3336–3340. [Google Scholar] [CrossRef]
Scheme 1.
Reactivity of D-A cyclopropanes.

Scheme 2.
Reaction conditions: 1a 0.1 mmol, thioacetic acid; 2a 0.15 mmol, 1 mL 10% w/w Cs2CO3, TBABr (10 mol%), PhMe 1 mL (0.1 M).
Scheme 2.
Reaction conditions: 1a 0.1 mmol, thioacetic acid; 2a 0.15 mmol, 1 mL 10% w/w Cs2CO3, TBABr (10 mol%), PhMe 1 mL (0.1 M).

Scheme 3.
Substrate scope of product 4.

Scheme 4.
Substrate scope of products 3.

Scheme 5.
Use of organic bases.

Scheme 6.
(a) Conversion of product 3aa in 4aa; (b) stability of 4aa under the reaction conditions.

Scheme 7.
Reactivity of 4k.

Scheme 8.
Non-reductive decyanation reaction.

Scheme 9.
Transnitrilation reaction.

Scheme 10.
Mechanistic hypothesis.

Figure 1.
Visualization of thiocyanate by complexation with FeCl3.

Scheme 11.
Use of sodium thioacetate.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Ammonium salt screening 1.
 | ||||||
| Entry | Ammonium Salt (10 mol%) | Solvent (M) | T (° C) | t (h) | NMR Yield of 4aa (%) 2 | Ratio 4aa/3aa 3 |
| 1 | TBABr | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 32 | 11/1 |
| 2 | TMAOH × 5H2O | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | trace | 14/1 |
| 3 | TBAI | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 34 | 17/1 |
| 4 | TMODABr | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 46 | 14/1 |
| 5 | TMBACl | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 10 | >20/1 |
| 6 | // | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | ||
| 7 | TMODABr | PhMe (250 µL, 0.4 M) | r.t. | 2.5 | 19 | 10/1 |
| 8 | TMODABr | PhMe (1000 µL, 0.1 M) | r.t. | 2.5 | 31 | >20/1 |
| 9 4 | TMODABr | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 52 | >20/1 |
| 10 4 | TMODABr | PhMe (500 µL, 0.2 M) | r.t. | 18 | 45 | 7/1 |
1 Reaction conditions: 1a (0.1 mmol), thioacetic acid (0.15 mmol), cat. (10 mol%), solid Cs2CO3 (0.12 mmol) in PhMe, rt, 2.5 h; 2 determined by 1H-NMR using m-dinitrobenzene as internal standard; 3 determined by 1 H NMR on the crude reaction mixture; 4 1a (0.15 mmol), thioacetic acid (0.1 mmol), cat. (10 mol%), Cs2CO3 (0.12 mmol) in PhMe (500 µL), rt, 2.5 h.
Table 2.
Reaction condition screening 1.
 | ||||||
| Entry | Base | Solvent | T (° C) | t (h) | NMR Yield (%) 2 | Ratio 4aa/3aa 3 |
| 1 | Cs2CO3(s) | PhMe | r.t. | 2.5 | 52 | >20/1 |
| 2 | K2CO3(s) | PhMe | r.t. | 2.5 | 19 | >20/1 |
| 3 | KHCO3(s) | PhMe | r.t. | 2.5 | 7 | >20/1 |
| 4 | K3PO4(s) | PhMe | r.t. | 2.5 | 29 | >20/1 |
| 5 | Cs2CO3(s) | CH2Cl2 | r.t. | 2.5 | 50 | >20/1 |
| 6 | Cs2CO3(s) | EtOAc | r.t. | 2.5 | 39 | >20/1 |
| 7 | Cs2CO3(s) | Et2O | r.t. | 2.5 | 28 | >20/1 |
| 8 | Cs2CO3(s) | MTBE | r.t. | 2.5 | 43 | >20/1 |
| 9 | Cs2CO3(s) | THF | r.t. | 2.5 | 69 | >20/1 |
| 10 | Cs2CO3(s) | 2-Me-THF | r.t. | 2.5 | 46 | >20/1 |
| 11 | Cs2CO3(s) | THF | 60 | 2.5 | 23 | >20/1 |
| 12 | Cs2CO3(s) | THF | 0 | 18 | 60 | >20/1 |
1 1a (0.15 mmol), thioacetic acid (0.1 mmol), TMODABr (10 mol%), base (0.12 mmol) in solvent (500 µL), 2.5 h; 2 determined by 1H NMR using m-dinitrobenzene as internal standard; 3 determined by 1H NMR on the crude reaction mixture.
Table 3.
Reaction condition screening 1.
 | ||||||
| Entry | Ammonium Salt | Solvent (M) | T (° C) | t (h) | NMR Yield 3aa (%) 2 | Ratio 3aa/4aa 3 |
| 1 | TBABr | PhMe (1000 µL, 0.1 M) | r.t. | 2.5 | 20 | 5/1 |
| 2 | TBABr | PhMe (500 µL, 0.1 M) | r.t. | 2.5 | 20 | 11/1 |
| 3 | TBAI | PhMe (500 µL, 0.2 M) | r.t. | 2.5 | 23 | >20:1 |
| 4 | TBABr | PhMe (500 µL, 0.2 M) | 0 | 48 | 43 | >20:1 |
| 5 | TBAI | PhMe (500 µL, 0.2 M) | 0 | 48 | 46 | >20:1 |
| 6 | TBAI | EtOAc (500 µL, 0.2 M) | 0 | 48 | 64 | >20:1 |
1 1a (0.1 mmol), thioacetic acid (0.1.5 mmol), ammonium salt (10 mol%), Cs2CO3 (1 mL 10% w/w) in solvent (x µL), 2.5 h; 2 determined by 1H NMR using m-dinitrobenzene as internal standard; 3 determined by 1H NMR on the crude reaction mixture.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bisag, G.D.; Viola, P.; Bernardi, L.; Fochi, M. Divergent Reactivity of D-A Cyclopropanes under PTC Conditions, Ring-Opening vs. Decyanation Reaction. Catalysts 2023, 13, 760. https://doi.org/10.3390/catal13040760
AMA Style
Bisag GD, Viola P, Bernardi L, Fochi M. Divergent Reactivity of D-A Cyclopropanes under PTC Conditions, Ring-Opening vs. Decyanation Reaction. Catalysts. 2023; 13(4):760. https://doi.org/10.3390/catal13040760
Chicago/Turabian StyleBisag, Giorgiana Denisa, Pietro Viola, Luca Bernardi, and Mariafrancesca Fochi. 2023. "Divergent Reactivity of D-A Cyclopropanes under PTC Conditions, Ring-Opening vs. Decyanation Reaction" Catalysts 13, no. 4: 760. https://doi.org/10.3390/catal13040760
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.