2.1. The Reaction of (iprPCP)Ir with 2-Phenoxy-1-phenylethanol 1
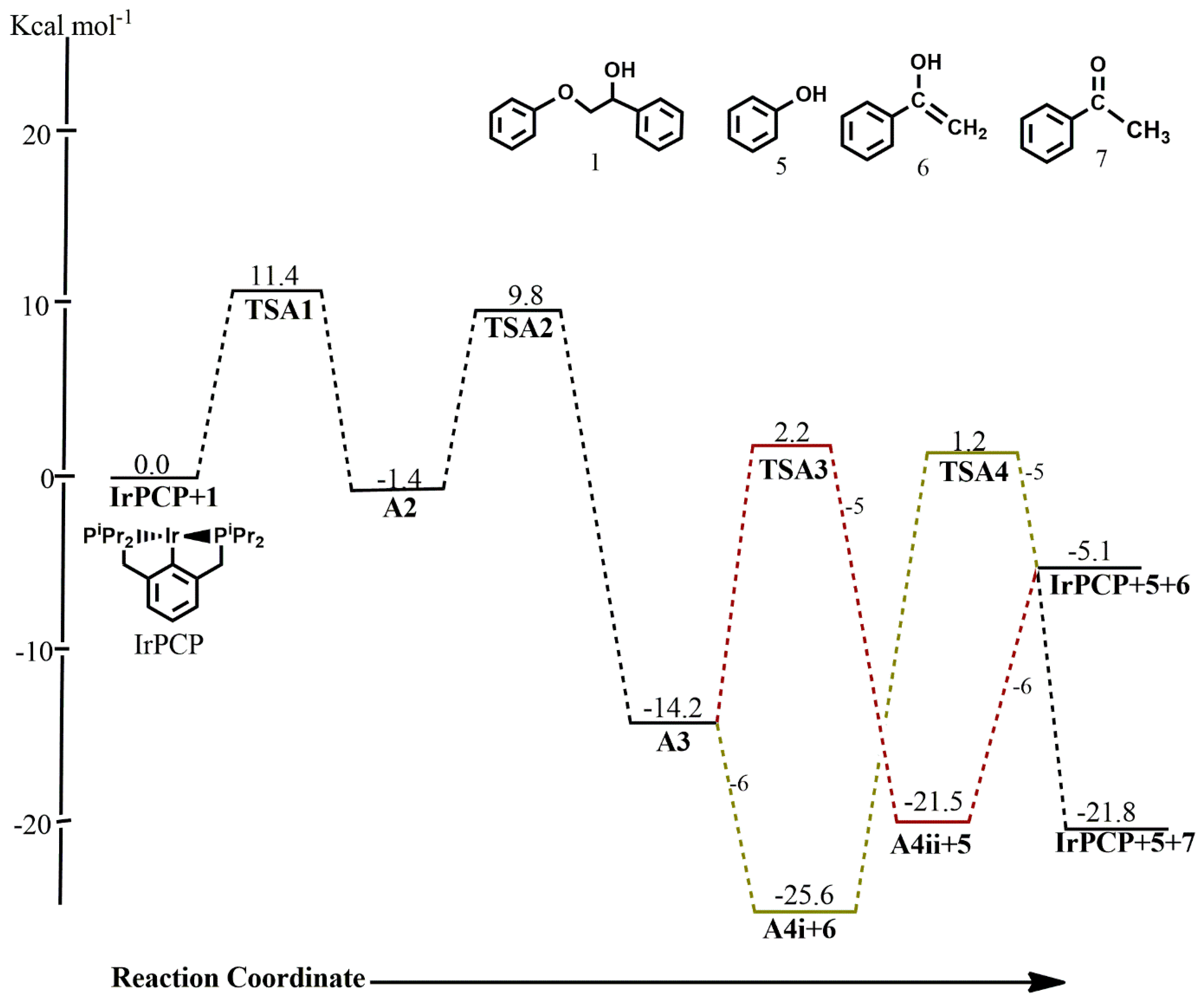
The optimized geometries of the stationary points, as well as the Gibbs free energy profile for the reaction of
(iprPCP)Ir with
2-phenoxy-
1-phenylethanol
1 along path
A are shown in
Figure 1 and
Figure 2. All stationary points in the catalytic cycle were sought on both the singlet and triplet state potential energy surface (PES), but triplet-state energies were found to be highly unstable, with the reactants being 104 kcal mol
−1 less stable on the triplet surface than on the singlet surface (
Table 1).
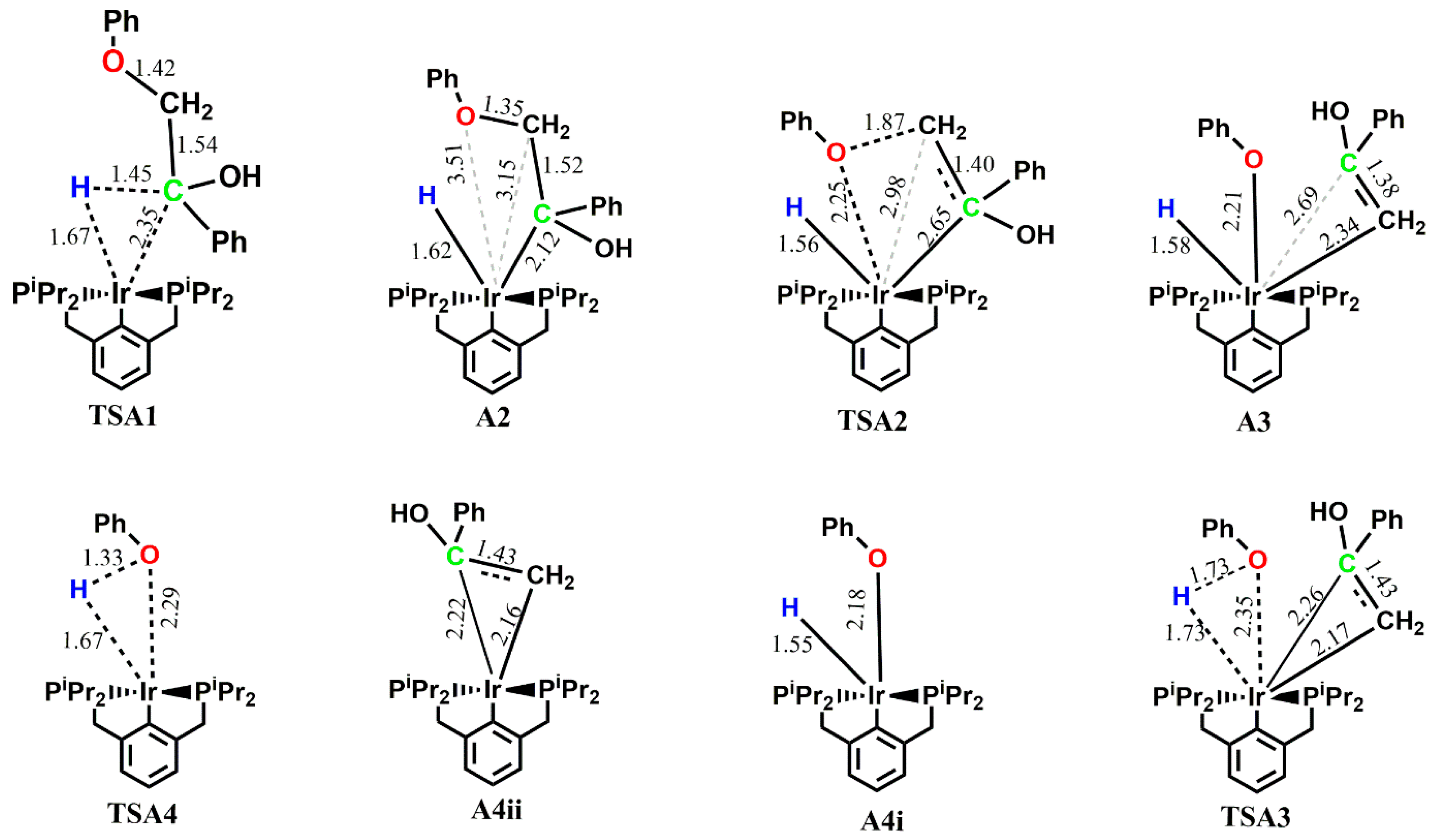
The first step in pathway A, which is the C-H addition, proceeds with an activation energy barrier of 11.4 kcal mol
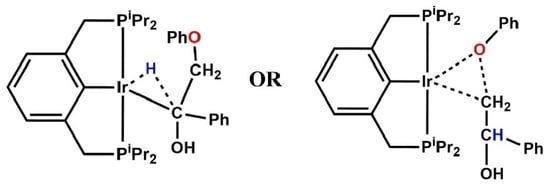
−1, leading to a five-coordinate complex
A2. Both the hydrogen being extracted by the metal and the β-O-4 group sit axially to the pincer backbone in
TSA1, and the transition state is a three-membered structure involving the α-carbon, the corresponding hydrogen and the metal. The Ir-C bond in the three-membered structure transitions from a bond length of 2.35 Å in
TSA1 to 2.12 Å in
A2 (
dIr-C = 2.12 Å) and both the abstracted hydrogen and the β-O-4 compound retain their axial position in
A2. On the triplet PES, the activation energy barrier for the C-H addition is 34.1 kcal mol
−1 and the resulting triplet intermediate is 21.1 kcal mol
−1 less stable than the singlet state intermediate. The next step, which is the insertion of the ether C-O bond for subsequent cleavage to the metal center, proceeds via a four-membered transition state
TSA2 involving the metal, the α-carbon, the ether oxygen and the ether carbon, with an activation energy barrier of 11.2 kcal mol
−1. The bond between the iridium and the ether carbon transitions from having no significant interaction (
dIr-C = 3.15 Å) in
A2 to 2.34 Å in
A3, with the O-Ir-C angle moving from 63.9° in
A2 to 92.2° in
TSA2 to 53.1° in
A3. Triplet state structures for
TSA2 and
A3 were found not to exist on the reaction surface. In the six-coordinate intermediate (
iprPCP)Ir(H)(OPh)(enolate)
A3, the enolate which was proposed to form a π-complex with the metal center [
14] preferably binds to the metal center via the methylene carbon in an equatorial fashion while the phenolate sits axially to the pincer backbone. The two possible orders of product release were considered, and it was revealed that the thermodynamic product is the enolate, which rearranges to the more stable ketone while the kinetic product is the phenol. On the singlet surface, the formation of
A4i mixed with the free enolate
6, which can rearrange to the more stable ketone
7, is calculated to be the major resting state in the catalytic cycle with a free energy of −25.6 kcal mol
−1 relative to the reactants, whereas
A4i undergoes reductive elimination to give phenol with an activation barrier of 26.8 kcal mol
−1; this step is endergonic by 20.6 kcal mol
−1. In contrast, the release of phenol first proceeds with an activation barrier of 16.4 kcal mol
−1 and the subsequent release of the catalyst is endergonic by 16.4 kcal mol
−1. An exhaustive search for the transition state from
A3 and
A4ii that leads to the bond cleavage of the enolate and the release of the catalyst was not fruitful. The energies for both orders of product release show that subsequent removal of either the phenol or the enolate, which rearranges to the more stable ketone, to give back the catalyst is kinetically unfavorable. This means that the likely predominant products are
A4i+6+7 (the thermodynamic product) and
A4ii+5 (the kinetic product). These findings are consistent with reports from experimental work carried out by Kundu et al. where the intermediate prior to the release of phenol was isolated instead of the phenol and the catalyst [
14]. On the triplet PES, the formation of the
A4i is less stable than the singlet product by 83.5 kcal mol
−1 while formation of
A4ii is less stable by 118.1 kcal mol
−1. Although some of the minima (intermediates) and transition states in the catalytic cycle exist both in the singlet state and in the triplet state, the singlet state structures would deplete the triplet state reaction surface and no multiple-state reactivity is observed in the catalytic cycle.
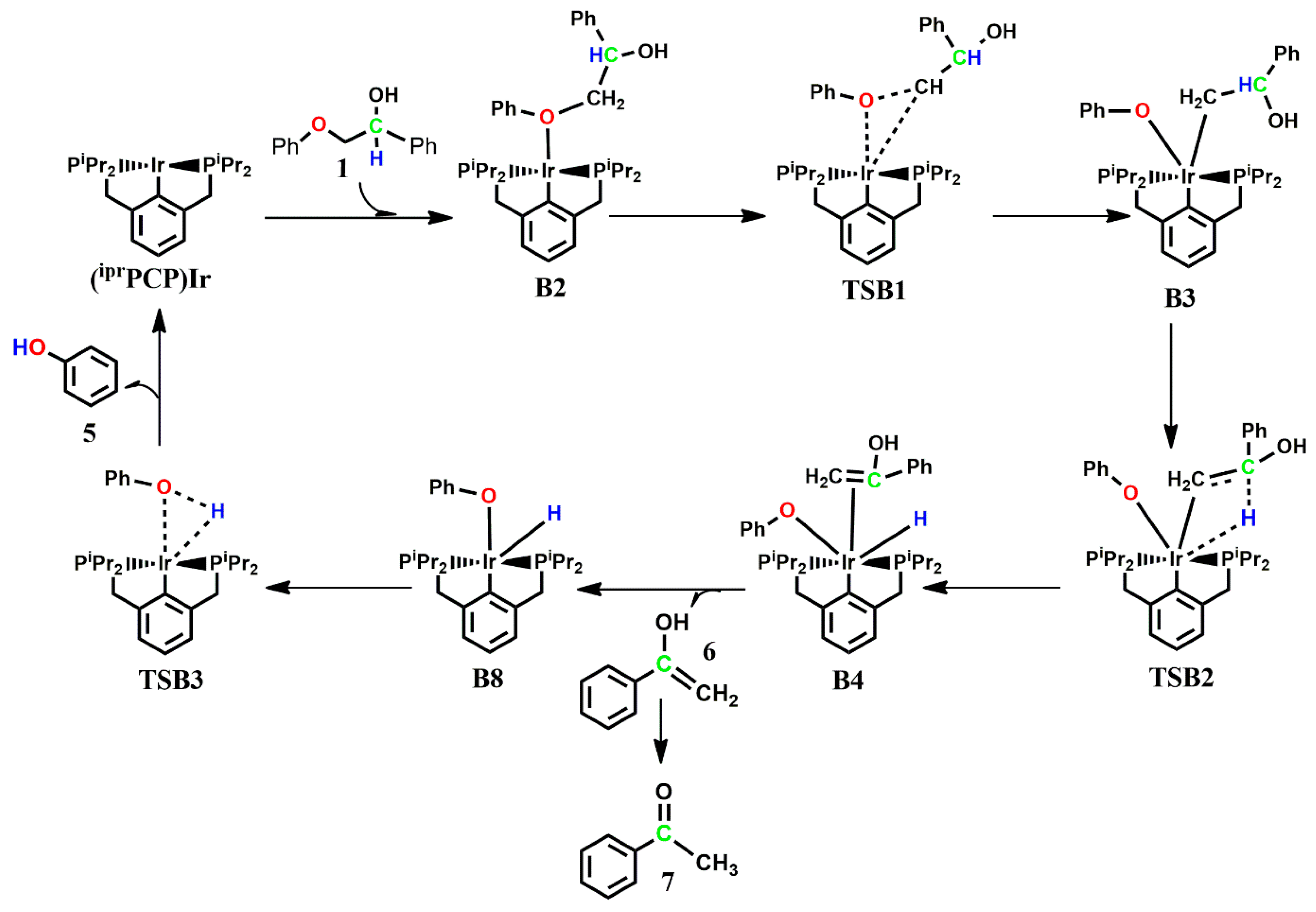
The optimized geometries of the stationary points, as well as the Gibbs free energy profile for the reaction of
(iprPCP)Ir with
2-phenoxy-
1-phenylethanol
1 along pathway
B are shown in
Figure 3 and
Figure 4. Triplet-state energies obtained along this pathway were also found to be less stable than the singlet-state energies (
Table 2). The reaction begins with the ether oxygen coordinating axially to the metal center, which is exergonic by −6.9 kcal mol
−1, and a bond is formed between the Ir and O to give
B2, which was not found to exist on the triplet surface. Subsequent addition of the
β-carbon to Ir and cleavage of the ether C-O bond takes place with an activation energy barrier of 34.1 kcal mol
−1.
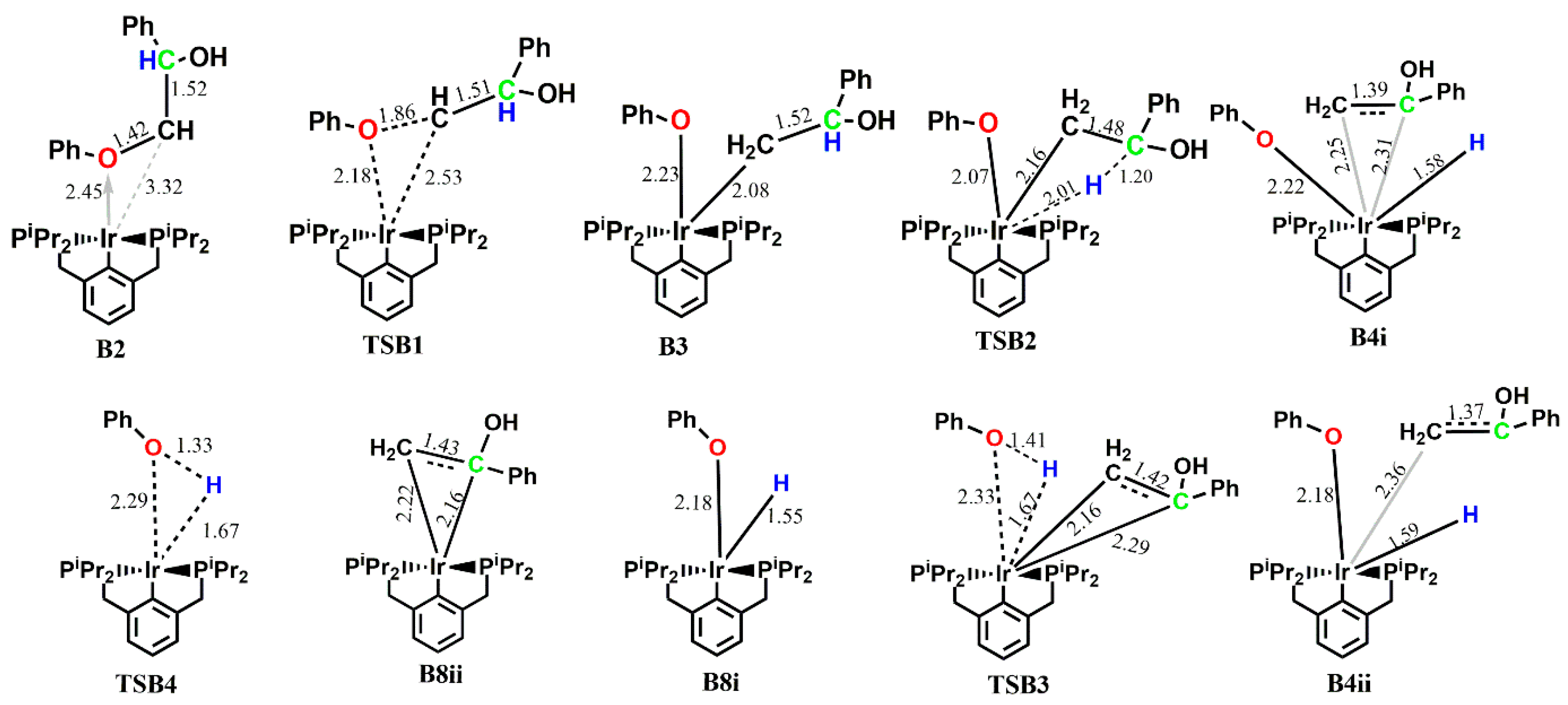
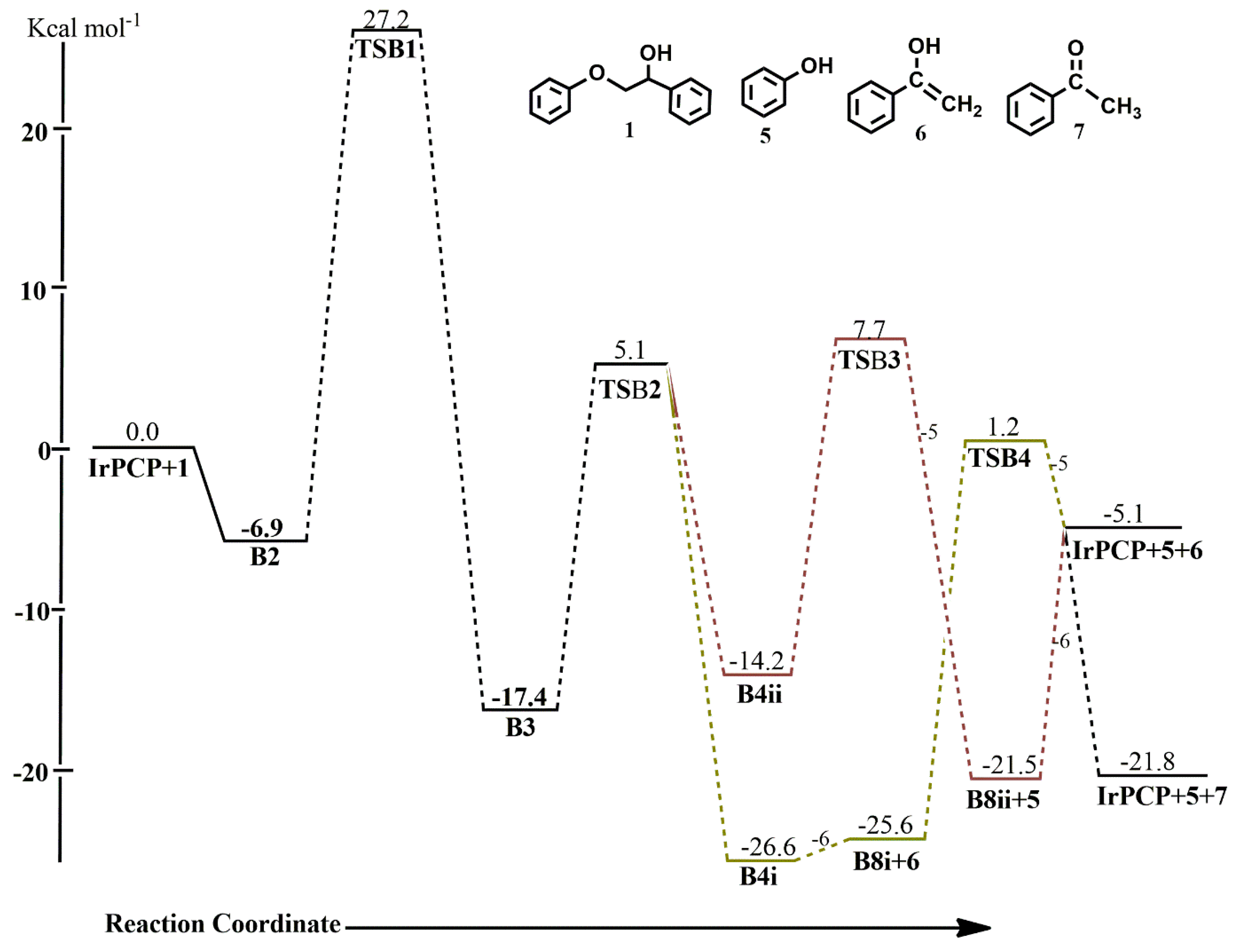
Here, the C-O bond insertion proceeds via a three-membered transition state TSB1, leading to the five-coordinate intermediate B3, which is exergonic by 10.5 kcal mol−1. The Ir-O bond length transitions from a bond length of 2.32 Å in B2 to 2.23 Å in B3, while the phenolate remains axial to the pincer backbone and the enolate sits in an equatorial fashion. The Ir-C(H2) bond also moves from having no significant interaction (dIr-C = 3.32 Å) in B2 to a covalent bond (dIr-C = 2.08 Å) in B3. The intermediate B3 was also located on the triplet PES, but was found to be less stable by 19.6 kcal mol−1, while the transition state leading to B3 was found not to exist on the triplet surface. Subsequent H abstraction by Ir from the α-carbon proceeds via a four-membered early transition state TSB2 with an activation energybarrier of 22.5 kcal mol−1, leading to a six-Ir-coordinated complex B4i. In the intermediate B4i, the enolate sits axially to the aromatic backbone of the pincer ligand, forming a π-complex with the iridium center and moving the phenolate and the abstracted hydrogen to the equatorial position. Neither TSB2 nor B4 could be located on the triplet potential energy surface. In order to determine the preferred order of product release, a different isomer of B4i which would allow for the release of the phenol first was located on the PES, but this isomer B4ii was found to be less stable by 12.4 kcal mol−1 than B4i.
The corresponding activation barrier for the initial release of the phenol from the predominant isomer of
B4i is 34.3 kcal mol
−1. Initial release of the enolate proceeds without barrier via the bond cleavage of the
η2-Ir-C bond in a slightly endergonic fashion (ΔG° = 1.0 kcal mol
−1), followed by the release of phenol which proceeds with an activation barrier of 26.8 kcal mol
−1 and is endergonic by 20.5 kcal mol
−1. This shows that the preferred order of product release is the initial release of the enolate followed by the release of the phenol. However, the energetics also show that the predominant product is
B8i+6+7, as subsequent release of the phenol and the catalyst is shown to be kinetically less favored. Similar to pathway A, the formation of
B8 and the transition state structure for the release of phenol are highly unstable on the triplet PES, and the major product formed is the enolate
6 which rearranges to the acetophenone 7 (essentially 7), as shown in
Figure 4. No multiple state reactivity is observed along path B.
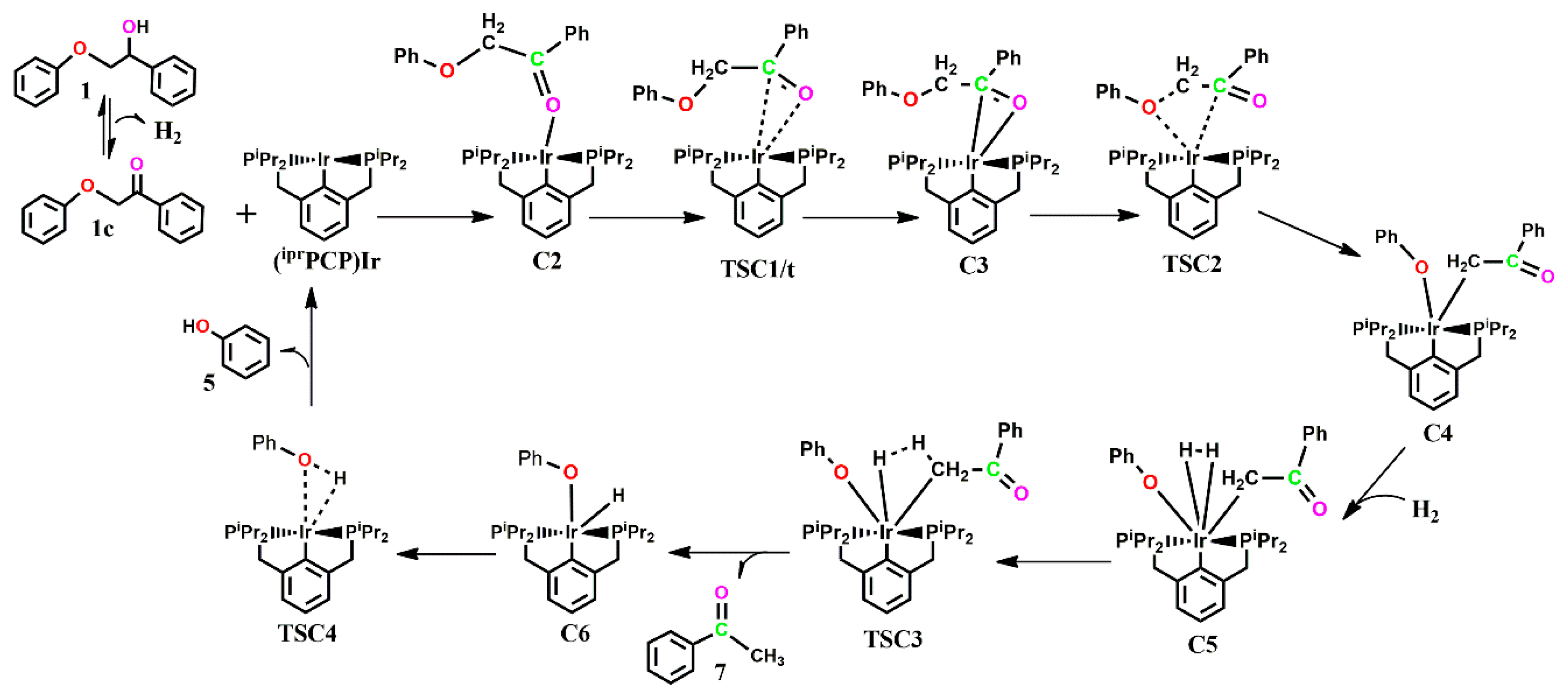
The optimized geometries of the stationary points, as well as the Gibbs free energy profile for the reaction of
(iprPCP)Ir with
2-phenoxy-
1-phenylethanol
1 along pathway
C are shown in
Figure 5 and
Figure 6. The reaction begins with the substrate binding to the catalyst via the ketone oxygen after undergoing dehydrogenative equilibrium in an exergonic fashion to give the adduct
C2.
C2 is located both on the singlet and triplet surface, with the triplet state structure being less stable by 12.9 kcal mol
−1 (
Table 3). The complex
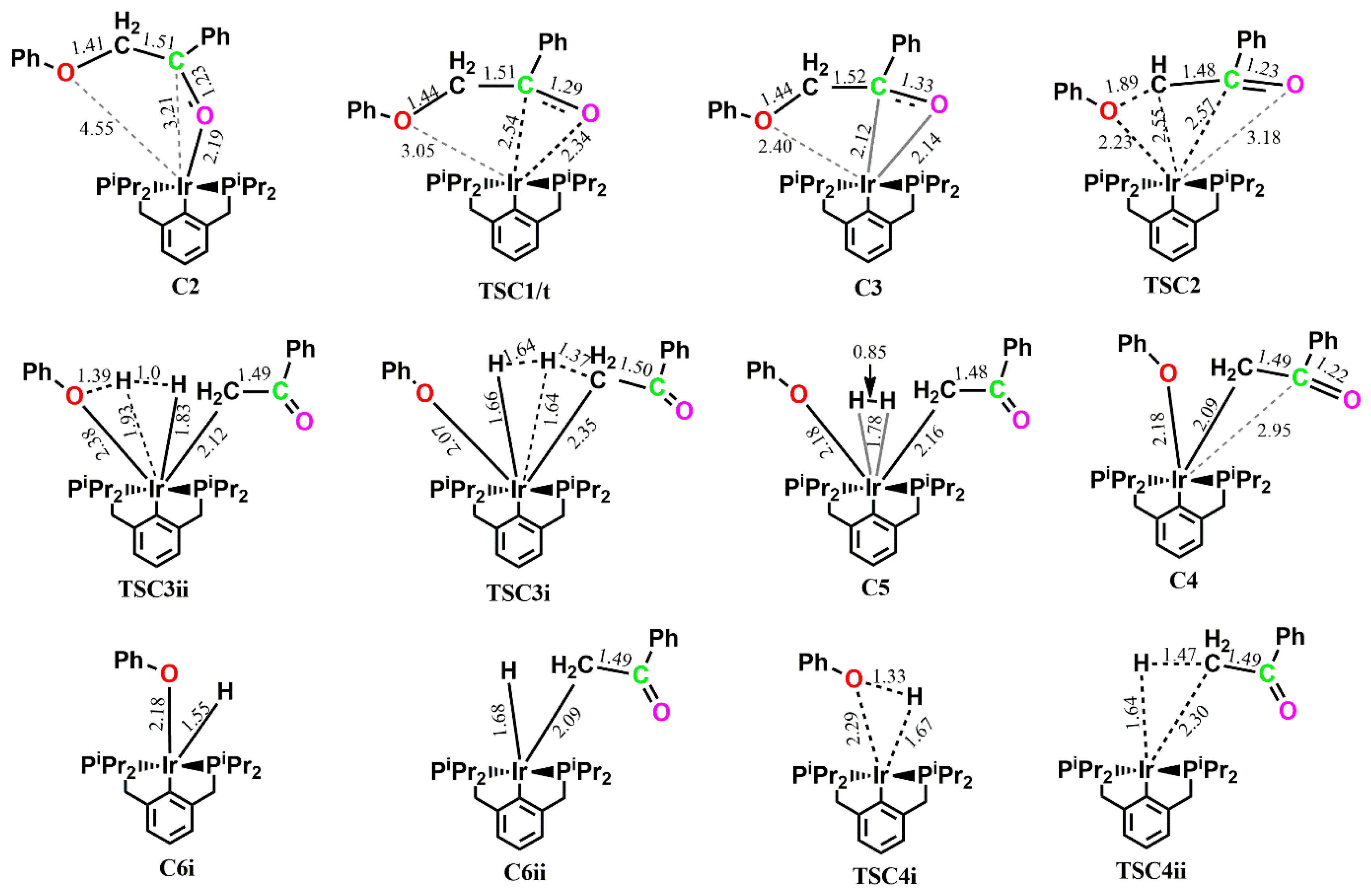
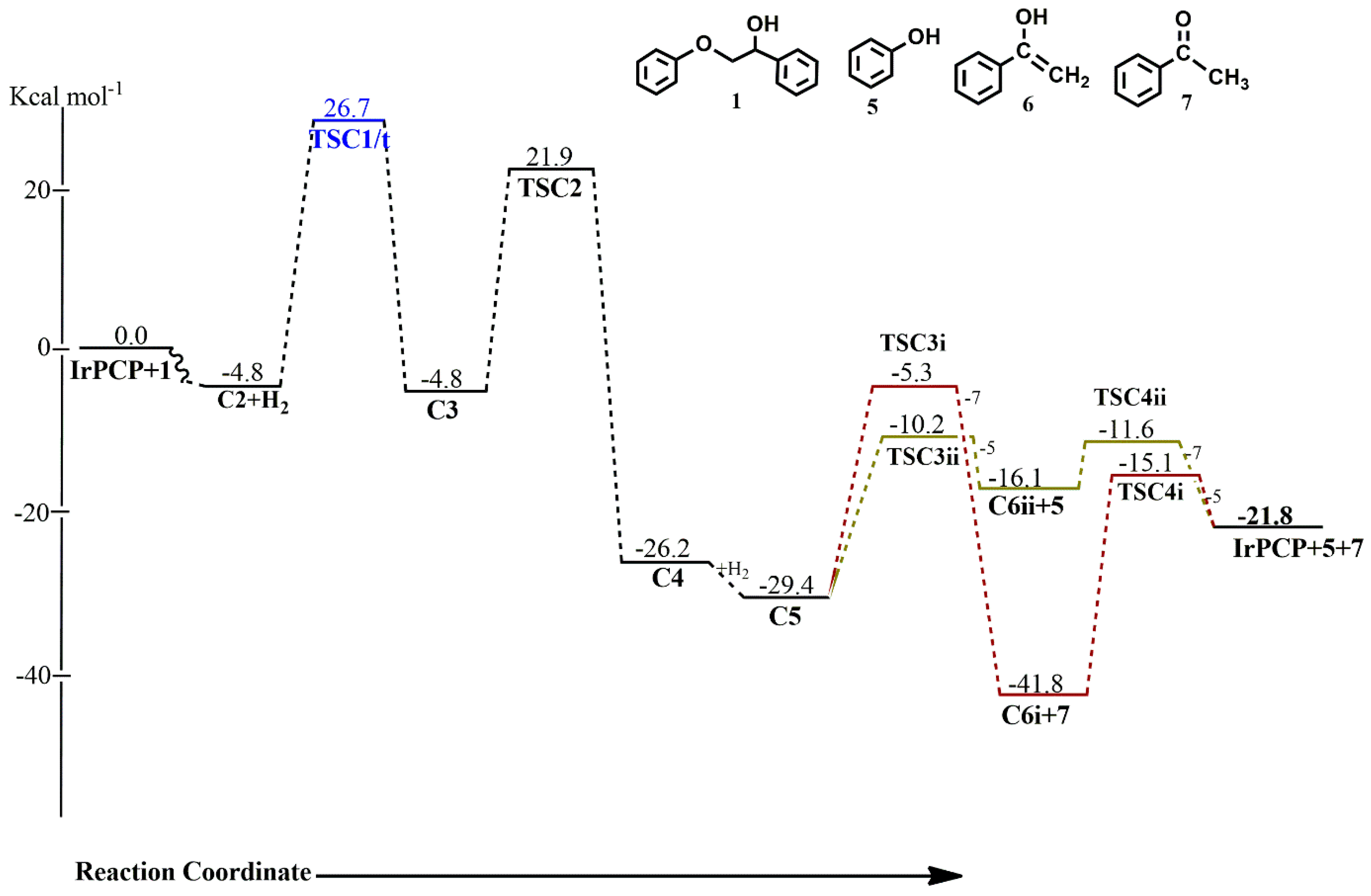
C2, which is proposed to undergo direct C-O bond cleavage, rather undergoes initial rearrangement to form a π-complex across the ketone C-O bond to the Ir center, allowing the ether oxygen of the substrate to sit in a much closer proximity to the metal center to allow for subsequent C-O addition. This rearrangement is thermoneutral and proceeds via
TSC1, which could only be located on the triplet PES with an activation barrier of 31.5 kcal mol
−1. Although the activation barrier for the formation of
C3 would be lower on the triplet surface, the more stable singlet-state
C2 would deplete the surface of the triplet-state
C2, making the formation of
C3 proceed from singlet-state
C2 via triplet-state
TSC1 to singlet-state
C3. The intermediate
C3 is also located on the triplet surface, but is found to be less stable by 30.4 kcal mol
−1 than the singlet-state
C3. Subsequent C-O bond cleavage follows via
TSC2, which involves the ether oxygen, the ether carbon, α-carbon and the Ir, with an activation barrier of 26.7 kcal mol
−1 to give a five-membered Ir-complex
C5, containing the phenoxide and what should have been a π-complex between the C-C bond and the Ir, but which is optimized as a C-bound ketone. The formation of
C4 is exergonic by 21.4 kcal mol
−1 on the singlet PES while the triplet state
C4 is less stable by 20.4 kcal mol
−1. Coordination of initially released H
2 is exergonic by 3.2 kcal mol
−1 leading to an
η2–H
2 complex
C5, shown to be the major resting state in the kinetically preferred catalytic cycle. In
C5, which is only located to the singlet PES, the H
2 ligand binds axially to the aromatic backbone of the pincer ligand while both the phenoxide and ketone are arranged equatorial to it, allowing for reductive elimination of either the phenol or the acetophenone. The release of the acetophenone first proceeds via
TSC3i with a barrier of 24.1 kcal mol
−1, followed by the release of the phenol via
TSC4i with a barrier of 26.7 kcal mol
−1, while the barriers for release of the phenol first, followed by subsequent release of the acetophenone, proceeding via
TSC3ii and
TSC4ii, are 19.2 kcal mol
−1 and 4.5 kcal mol
−1, respectively. The formation of
C6i+7 from the initial release of the acetophenone is seen as the major resting state (ΔG = −41.8 kcal mol
−1) in the reaction cycle, also showing that the acetophenone
7 would be the predominant product obtained, while the phenol
5 is the kinetic product. The release of the catalyst is also seen to be kinetically feasible if the pathway that follows the initial release of the phenol is considered. The
η2–H
2 complex
C5, or the transition states
TS3i and
TS3ii, could not be located on the triplet PES, while the intermediates C6i and C6ii, as well as the transition states
TS4i and
TS4ii that were found on the triplet PES, were highly unstable. A two-state reactivity is observed along this pathway (path C) with the transition state for the rearrangement of
C2 to
C3 located only in the triplet state. However, the other stationary points on the catalytic cycle, which exist both on the singlet surface as well as on the triplet surface, show the singlet state structures to be the ground state structures of the system.
Table 4,
Table 5 and
Table 6 show comparisons in activation energies, energies of formation and apparent activation energies (
δE) for the entire cycles calculated for the three pathways and for the two different ligand combinations. For pathway A, where the ether bond cleavage proceeds via the initial C-H addition, the
δE was calculated to be 15.2 kcal mol
−1, while the
δE for pathway B and pathway C, where ether bond cleavage proceeds via direct C-O bond addition, were calculated to be 32.0 kcal mol
−1 and 27.0 kcal mol
−1, respectively. Taking into account preferred product outcomes, the
δE for pathway A reduces to 11.1 kcal mol
−1 when the phenol is the desired product, while the
δE for pathway C increases to 46.7 kcal mol
−1 when the acetophenone is the desired product. From these apparent activation energies, the selectivity for the initial C-H addition pathway for this type of cleavage reaction, as observed in studies carried out by Choi et al. [
14] and Haibach et al. [
13], has been demonstrated. The activation barrier for the ether bond cleavage is also lower in pathway A (
Ea = 16.4 kcal mol
−1) than in pathways B (
Ea = 34.1 kcal mol
−1) and C (
Ea = 26.7 kcal mol
−1). For all three pathways, the phenol is observed as the kinetic product, while the enolate/acetophenone is observed as the thermodynamic product, and release of the catalyst becomes more kinetically feasible when phenol is the desired product. It should also be noted that for pathways B and C, in which the reaction proceeds via direct C-O bond addition, C-O bond cleavage in pathway C, which involves the presence of the ketone functionality of the substrate, is kinetically favored by 7.4 kcal mol
−1 over pathway B. These results support both experimental [
12] and theoretical [
20] findings, which have reported that the presence of the ketone functionality facilitates the C-O bond cleavage and that the bond dissociation energy of the ketone is far less than its alcohol counterpart.
2.3. The Reaction of (iprPCP)Co with 2-Phenoxy-1-phenylehtanol
Figure 7,
Figure 8,
Figure 9,
Figure 10,
Figure 11 and
Figure 12 show the optimized geometries as well as the Gibbs free energy profiles for the catalytic dehydroaryloxylation of
2-phenoxy-
1-phenylehtanol
1 using (
iprPCP)Co along pathways A, B and C, respectively. All stationary points have been sought on both the singlet and triplet potential energy surface as shown in
Table 7,
Table 8 and
Table 9, with the triplet state reactants being 77.2 kcal mol
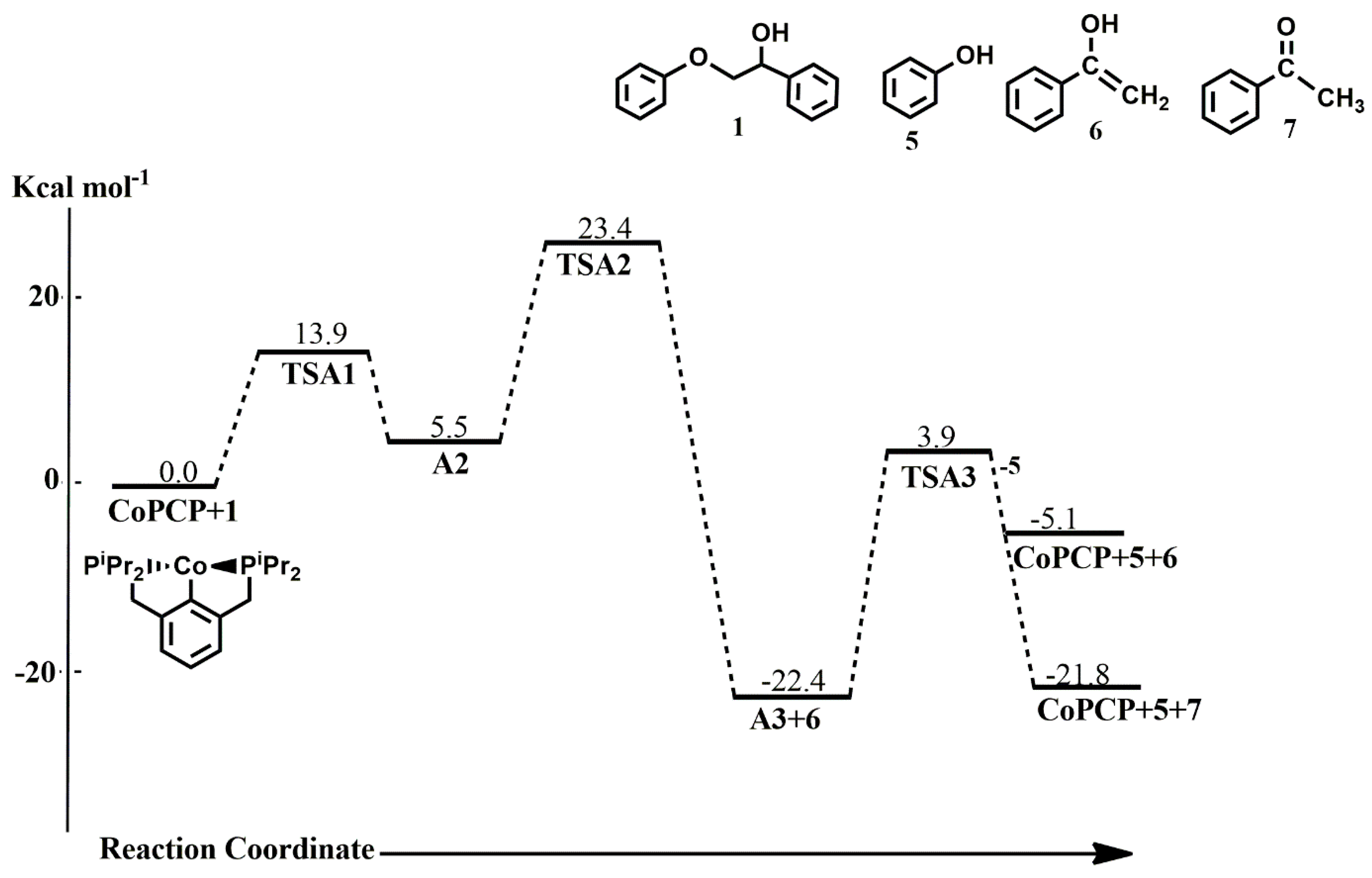
−1 less stable than the singlet state reactants. Along pathway A, the (
iprPCP)Co-catalyzed cleavage of
1 proceeds initially via C-H addition through
TSA1 with an activation barrier of 13.9 kcal mol
−1 to form
A2, whereas C-O bond insertion follows via
TSA2 with an activation barrier of 17.9 kcal mol
−1. The abstracted hydrogen sits axially to the aromatic backbone of the pincer ligand in
TSA1 but shifts to an equatorial position during the C-O bond cleavage, while the cleaved phenolate replaces it in the axial position in
TSA2. It is found that the six-membered intermediate is not formed after the C-O bond cleavage occurs, as the enolate is released in this step with a reaction energy of 27.9 kcal mol
−1 to form
A3, containing only the phenoxide and the hydrogen ligand. The transition states for the C-H addition and C-O insertion, as well as the intermediate
A2, were found not to exist on the triplet surface. Finally, reductive elimination of phenol
5 proceeds via
TSA3 with an activation barrier of 26.3 kcal mol
−1 leading to the regeneration of the catalyst. The formation of
A3+[6+7] at −22.4 kcal mol
−1 is the major resting state in the catalytic cycle, being only lower than the final products
CoPCP +[5+7] by 0.6 kcal mol
−1. On the triplet surface, the formation of
A3 is endergonic by 24.1 kcal mol
−1, making it less stable than the singlet state
A3 by 46.5 kcal mol
−1. These energies show that the major product formed from the
iprPCPCo-catalyzed cleavage of a lignin β-O-4 compound is acetophenone and a two-state reactivity was not observed in the catalytic cycle.
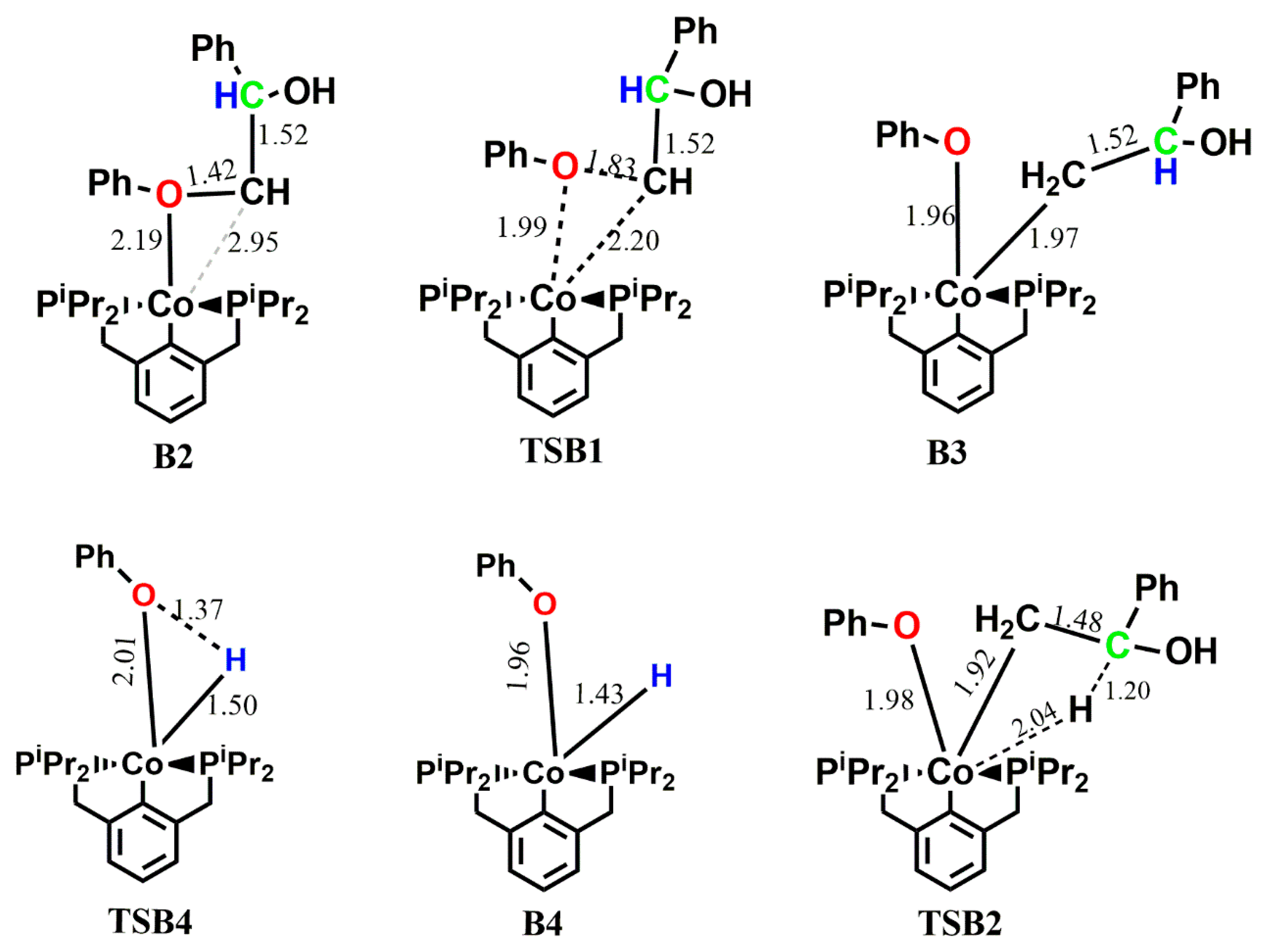
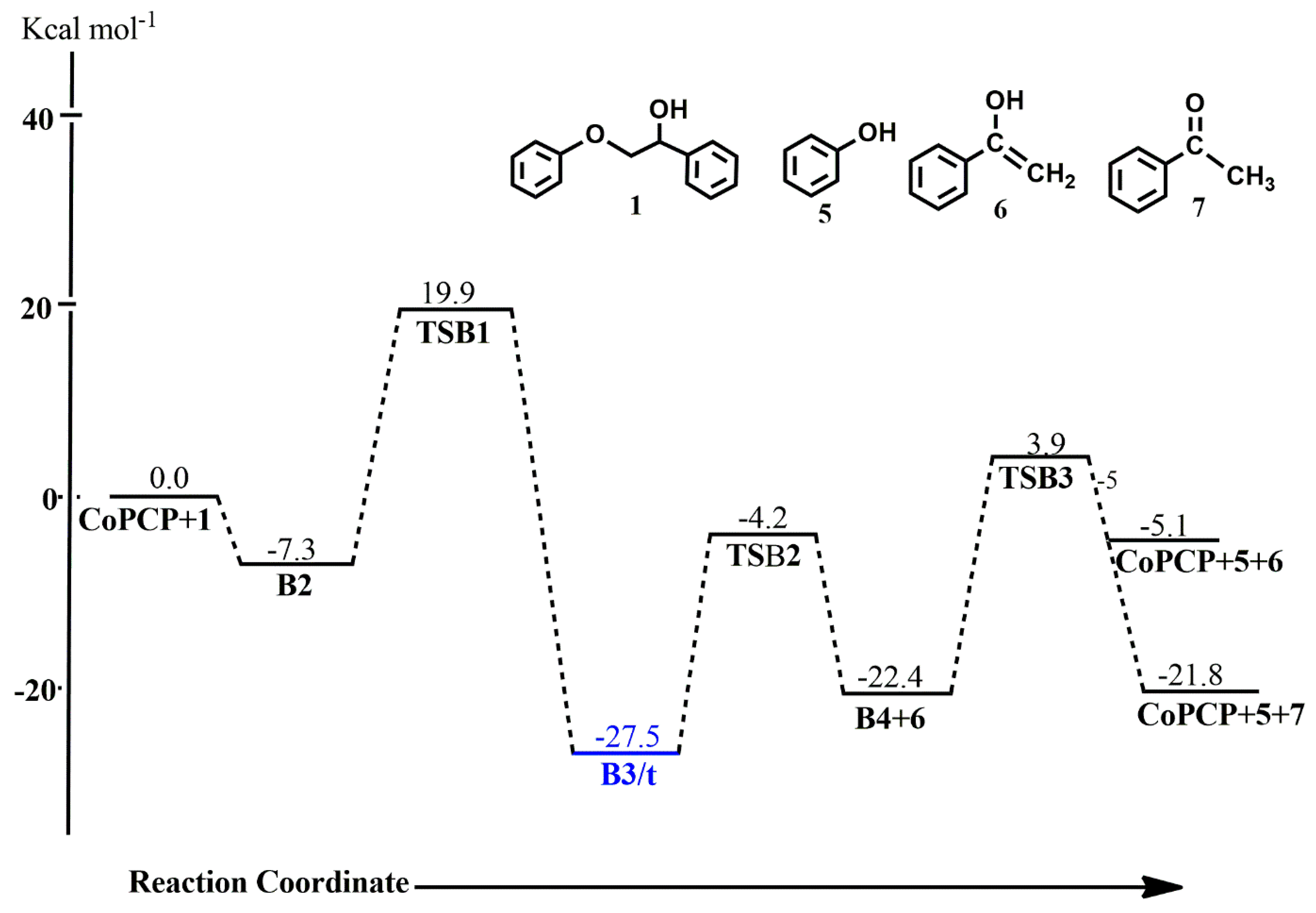
In the (iprPCP)Co-catalyzed reaction of 1 along pathway B, the ether oxygen forms a covalent bond with the cobalt complex to give an adduct B2 in an exergonic reaction (ΔG° = −7.3 kcal mol−1). Direct ether cleavage via C-O bond addition follows with an activation energy barrier of 27.2 kcal mol−1, leading to the O-bound and C-bound complex B3. Here, a two-state reactivity is observed, as the triplet state B3 is found to be more stable than the singlet state B3 by 9.8 kcal mol−1. The ether-carbon transitions from a position axial to the aromatic backbone in TSB1 to an equatorial position in B3, while the ether-oxygen sits axially to the aromatic backbone in both TSB1 and B3. Subsequent hydrogen abstraction via C-H addition to the metal center proceeds from the more stable triplet-state B3/t via singlet-state TSB2 with an activation energy barrier of 23.3 kcal mol−1 to form a singlet-state B4 which is endergonic by 5.1 kcal mol−1. Similar to pathway A, cleavage of the enolate occurs in this step leaving only the phenolate and the hydride bound to the metal center, as seen in intermediate B4 which then undergoes reductive elimination of the phenol and regeneration of the catalyst with an activation energy barrier of 26.3 kcal mol−1. The enolate 6, and the more stable acetophenone 7, to which it rearranges, are also shown to be the major products formed in this reaction. Apart from the intermediate B3, all other stationary points along the reaction cycle are found to be unstable on the triplet PES.
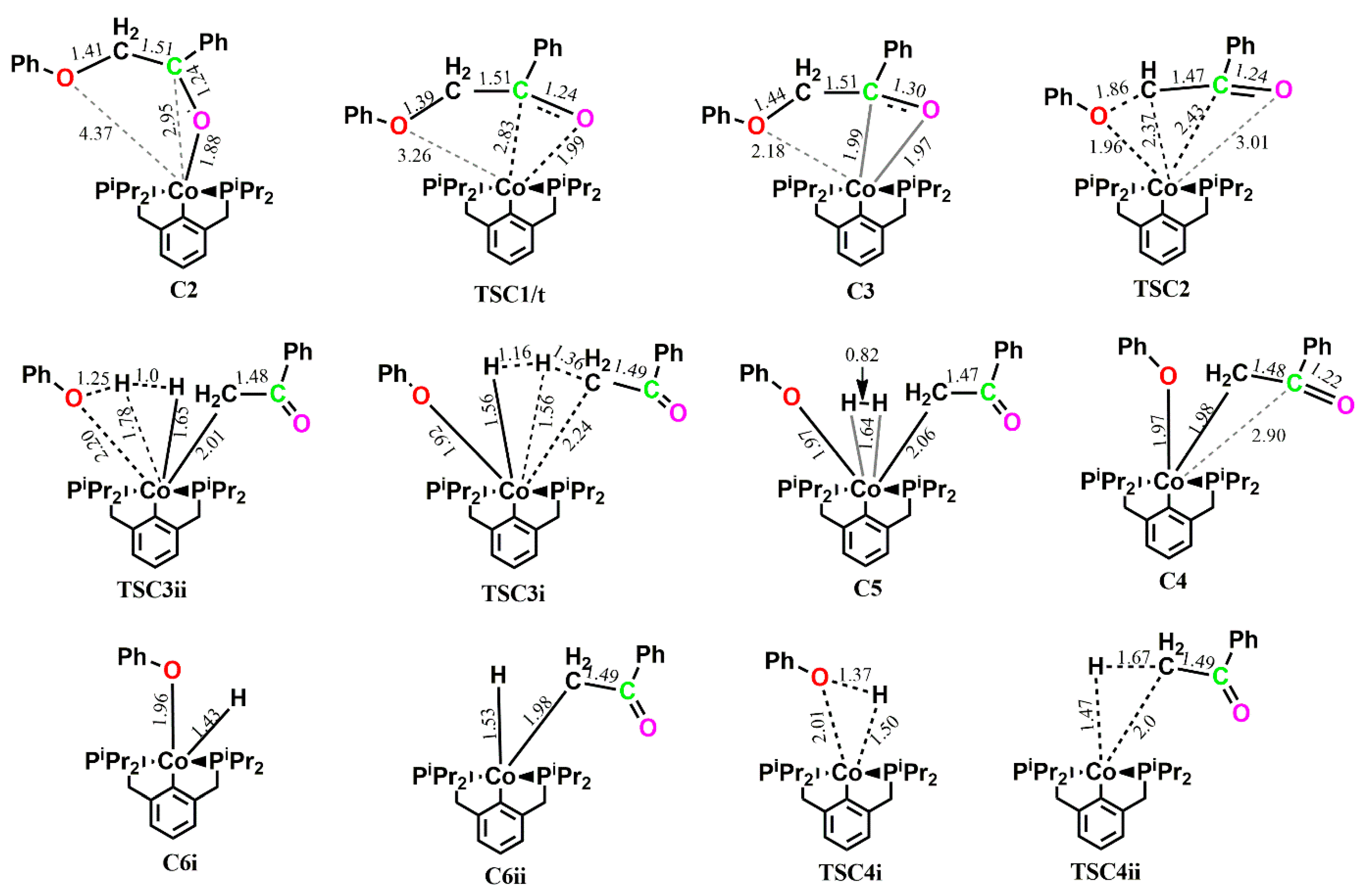
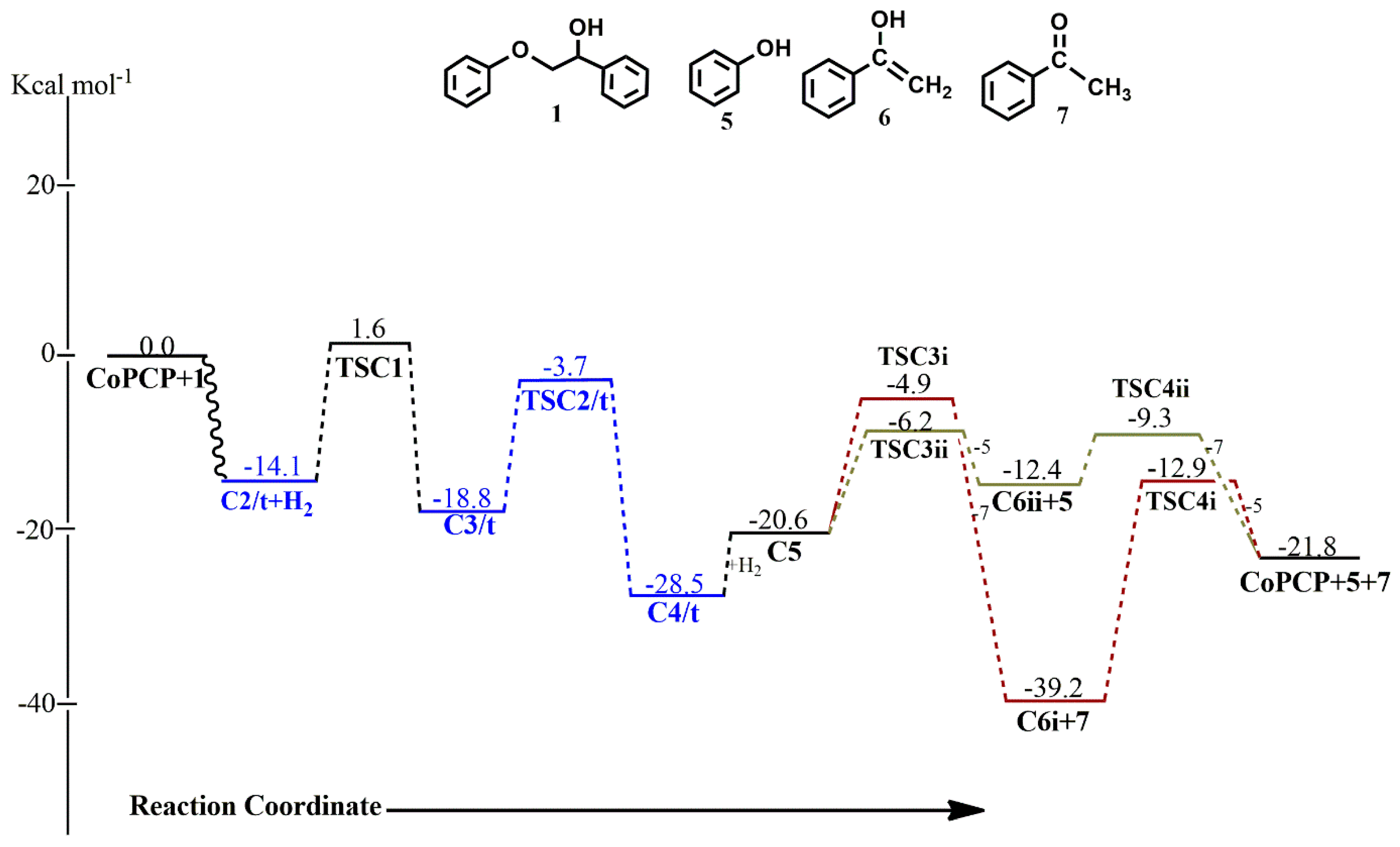
In the (
iprPCP)Co-catalyzed reaction of
1 along pathway C, all but one of the stationary points in the catalytic cycle exhibit multiple spin ground states, where more than one of those stationary points with lower energies belong to the higher spin state and a two-state reactivity is observed as shown in
Figure 12. The coordination of the ketone substrate to the metal center to form the adduct
C2 is exergonic by −14.1 kcal mol
−1 on the triplet surface and −9.9 kcal mol
−1 on the singlet surface. This causes subsequent rearrangement of the adduct to form
C3 containing the
η2-bound ketone to proceed via
TSC1 with an activation energy barrier of 15.7 kcal mol
−1 instead of a lower barrier of 11.5 kcal mol
−1. Similar to what was observed in the (
iprPCP)Ir-catalyzed reaction, the ketone-carbon transitions from having no significant interaction with the cobalt center in C2 (
dIr-C = 2.95 Å) to forming a covalent bond with the metal center in
C3 (
dIr-C = 1.99 Å), and moves from occupying an axial position relative to the aromatic backbone of the pincer ligand in
C2 to an equatorial position in
C3. The bond distance between the ether-oxygen and the metal center also reduces from 4.37 Å to 2.18 Å after the rearrangement of
C2 to
C3, bringing the ether oxygen in close proximity for subsequent C-O bond cleavage. Ether bond cleavage via
TSC2/t is kinetically more favored on the triplet surface with an activation energy barrier of 15.1 kcal mol
−1 (
Ea[singlet state] = 33.2 kcal mol
−1), leading to the formation of the triplet state O-bound and C-bound ketone
C4/t, which is exergonic by 28.5 kcal mol
−1 and more stable than the singlet-state
C4 by 4.2 kcal mol
−1. The phenolate occupies the axial position relative to the aromatic backbone of the pincer ligand, while the ketone occupies the equatorial position. However, both the phenolate and the ketone move equatorially to the pincer ligand after hydrogenation occurs, putting the
η2–H
2 in the axial position in
C5. The hydrogenation step is endergonic by 18.2 kcal mol
−1 on the triplet surface and endergonic by 3.7 kcal mol
−1 on the singlet surface. Similar to the observed trend in the (
iprPCP)Ir-catalyzed reaction, the phenol is the kinetic product while the acetophenone is the thermodynamic product. Reductive elimination of the phenol first proceeds with an activation barrier of 14.4 kcal mol
−1 with the released product being formed at −12.4 kcal mol
−1, while initial release of the acetophenone proceeds with activation barrier of 15.7 kcal mol
−1 and the released product is formed at −39.2 kcal mol
−1. The intermediate
C5, and the transition states
TSC3i and
TSC3ii are all found to be more stable on the singlet surface. Subsequent release of the acetophenone from
C6ii, which proceeds with an activation energy barrier of 3.1 kcal mol
−1 on the singlet surface, and is lower than the triplet state activation energy barrier by 26.4 kcal mol
−1, leads to the release of the catalyst, which is exergonic by 9.4 kcal mol
−1, while subsequent release of the phenol from
C6i proceeds with an activation energy barrier of 26.3 kcal mol
−1 which is less kinetically favored for the release of the catalyst. Hence, the likely observed product from this sequence of product release would be
C6i+7.
Table 10,
Table 11 and
Table 12 show the comparison in activation energies, energies of formation and apparent activation energies (
δE) for the entire cycles, calculated for the three pathways and two different ligand combinations. The trend observed here in the activation energies and apparent activation energies (
δE) for the entire cycle is different from the (
iprPCP)Ir-catalyzed process. The activation barrier for the C-O bond addition is seen to be lower in pathway C (
Ea = 15.1 kcal mol
−1) than pathways A (
Ea = 17.9 kcal mol
−1) and B (
Ea = 27.2 kcal mol
−1), whereas, ultimately, the calculated apparent activation energies (
δE) for pathway C are shown to be kinetically favored by 15.7 kcal mol
−1 over pathway A and by 17.3 kcal mol
−1 over pathway B. These energetics show that the kinetically favored pathway for the (
iprPCP)Co-catalyzed cleavage of a β-O-4 compound is pathway C, which involves the ketone motif of the substrate and begins with the C-O bond addition, even when the desired product is acetophenone (
δE = 16.7 kcal mol
−1).






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}