

Asymmetric Synthesis of trans-3-Alkoxyamino-4-Oxygenated-2-Piperidones Mediated by Transition-Metal-Free Dual C-H Oxidation and Its CAL-B-Assisted Enzymatic Resolution
 , , ,
, , , 
Abstract
:
1. Introduction
2. Results and Discussion
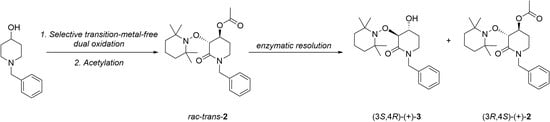
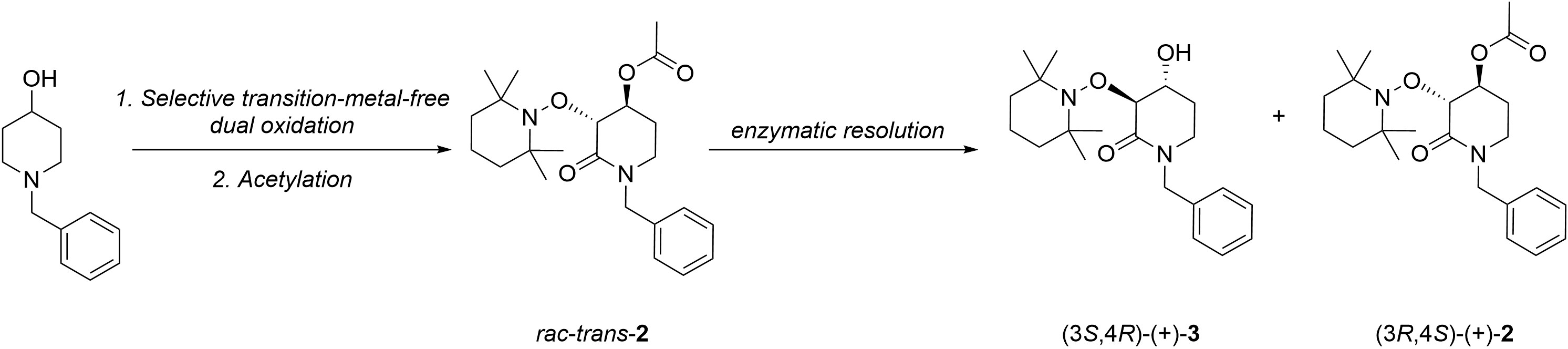
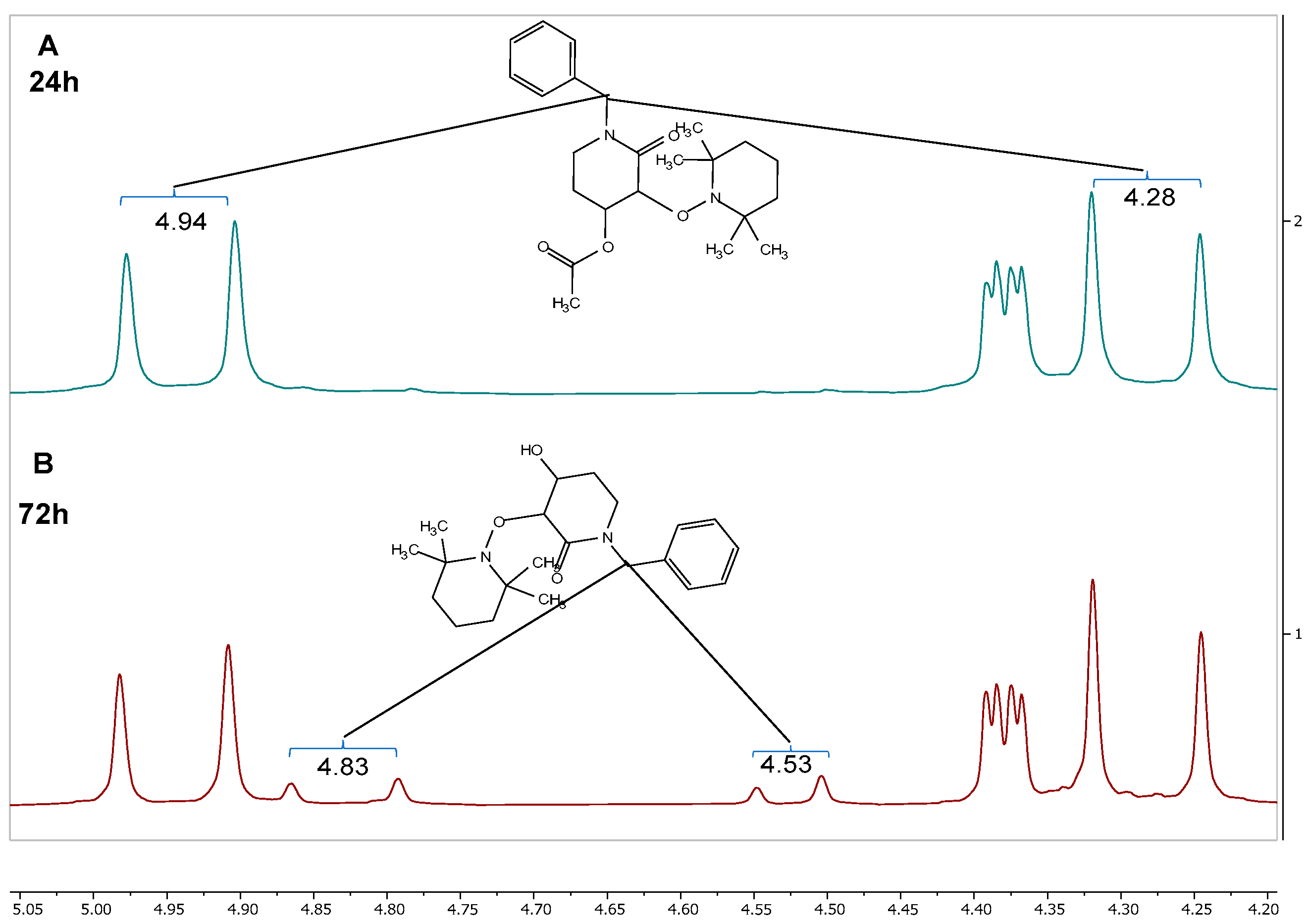
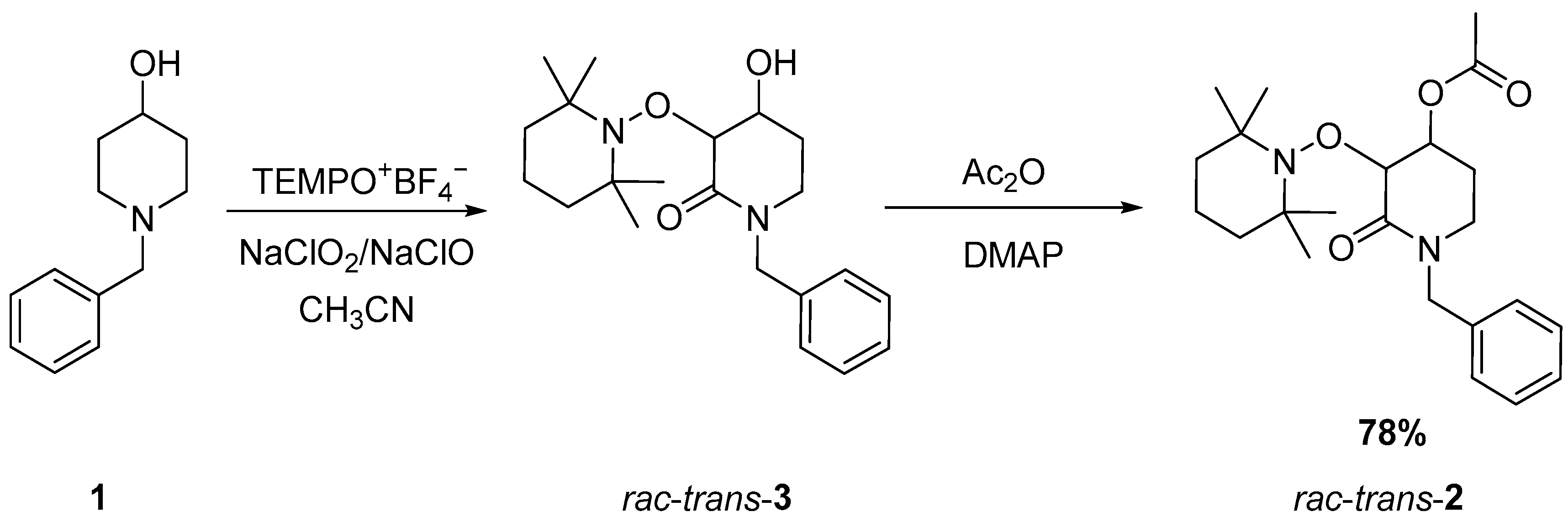
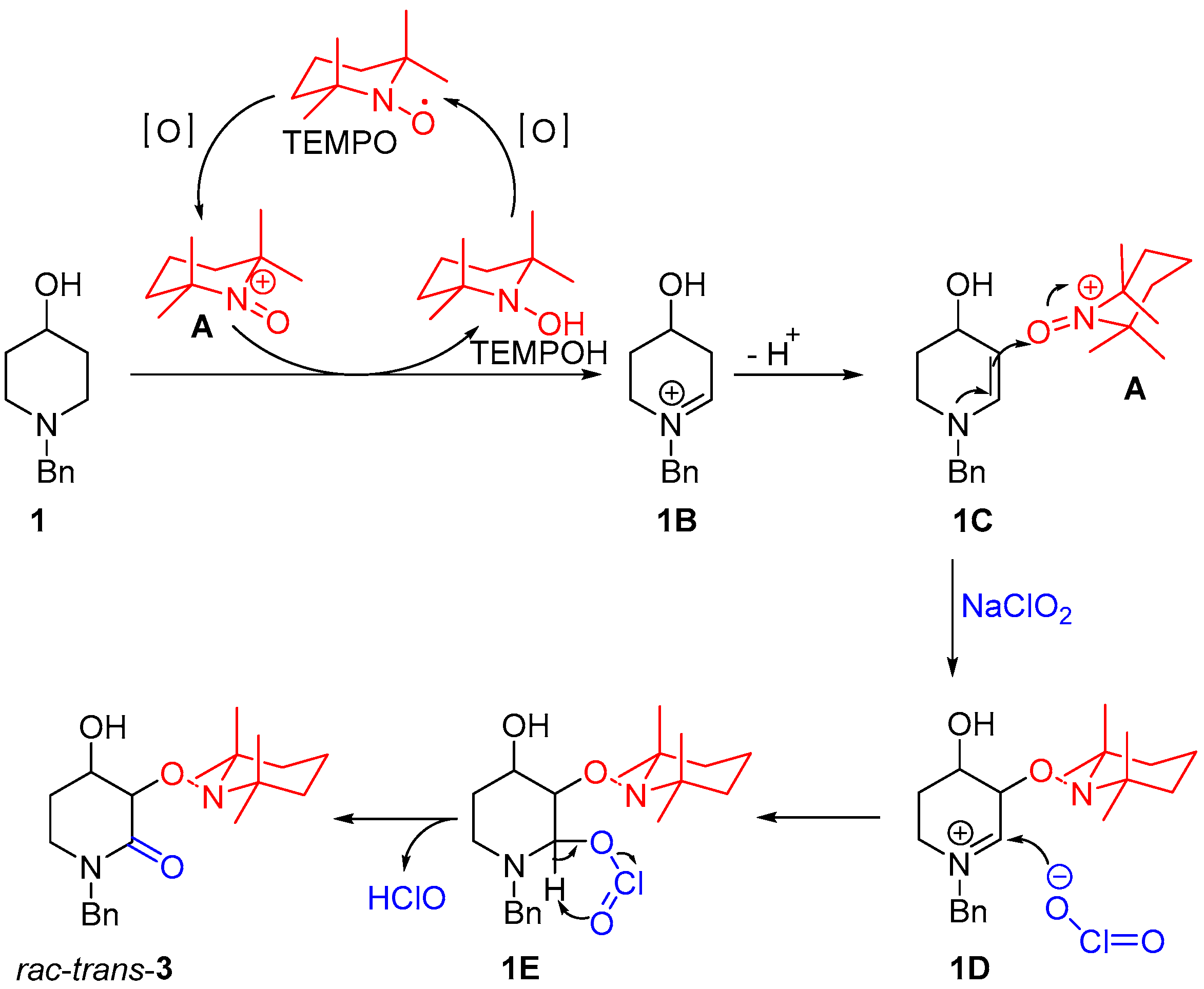
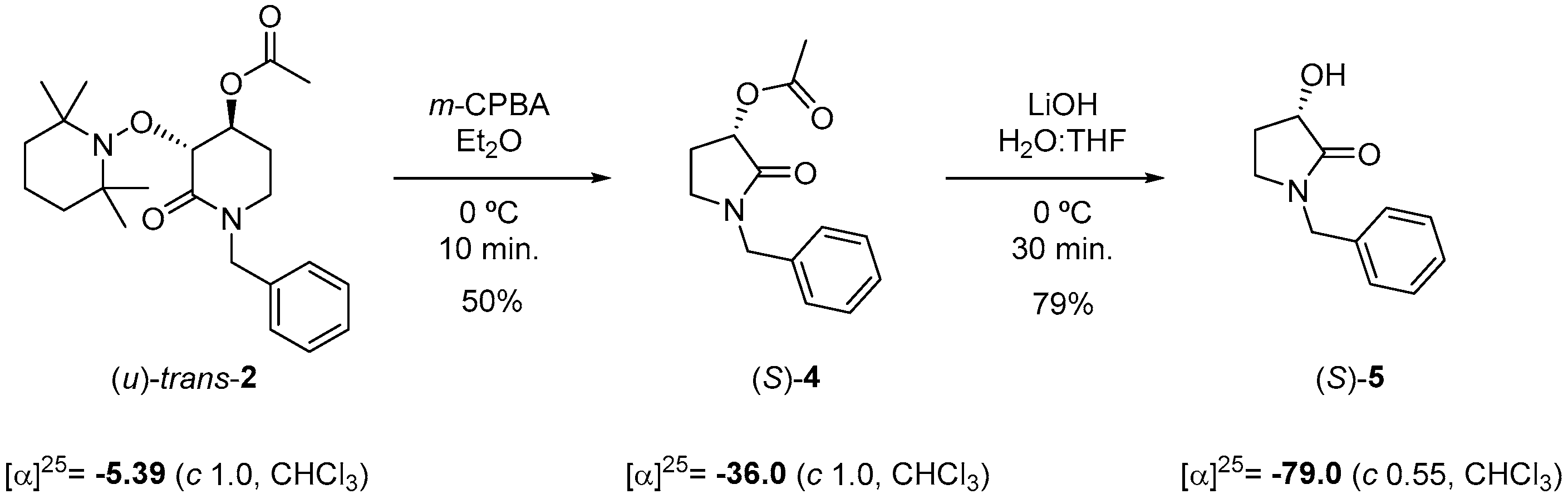
2.1. Synthesis of Rac-Trans-2
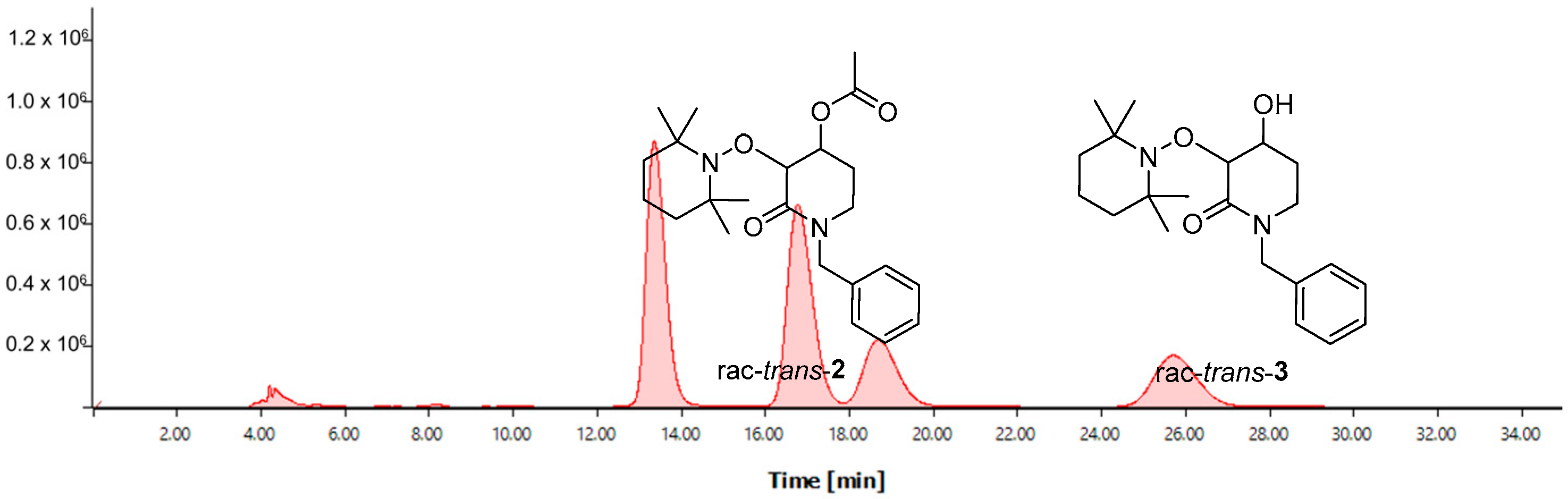
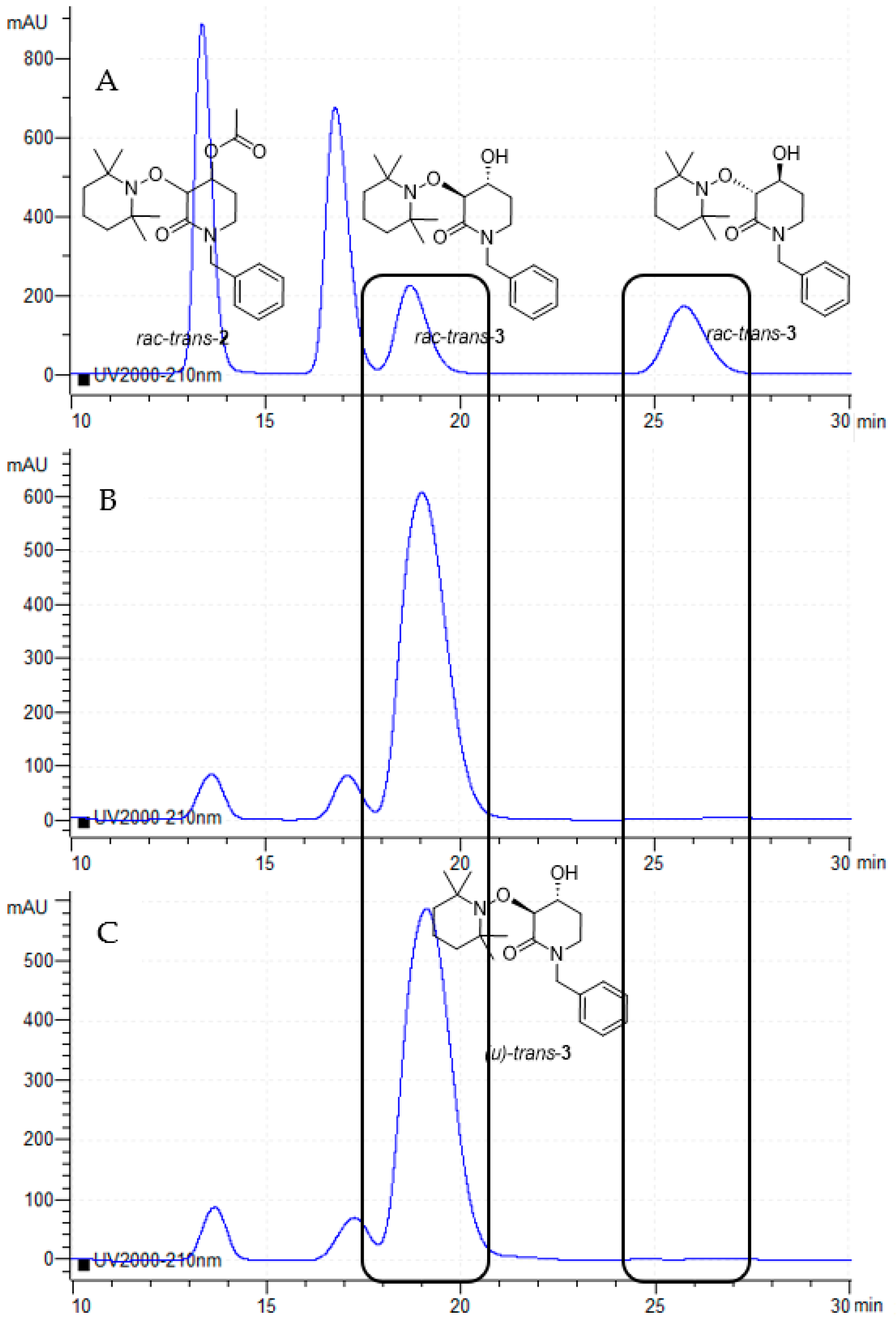
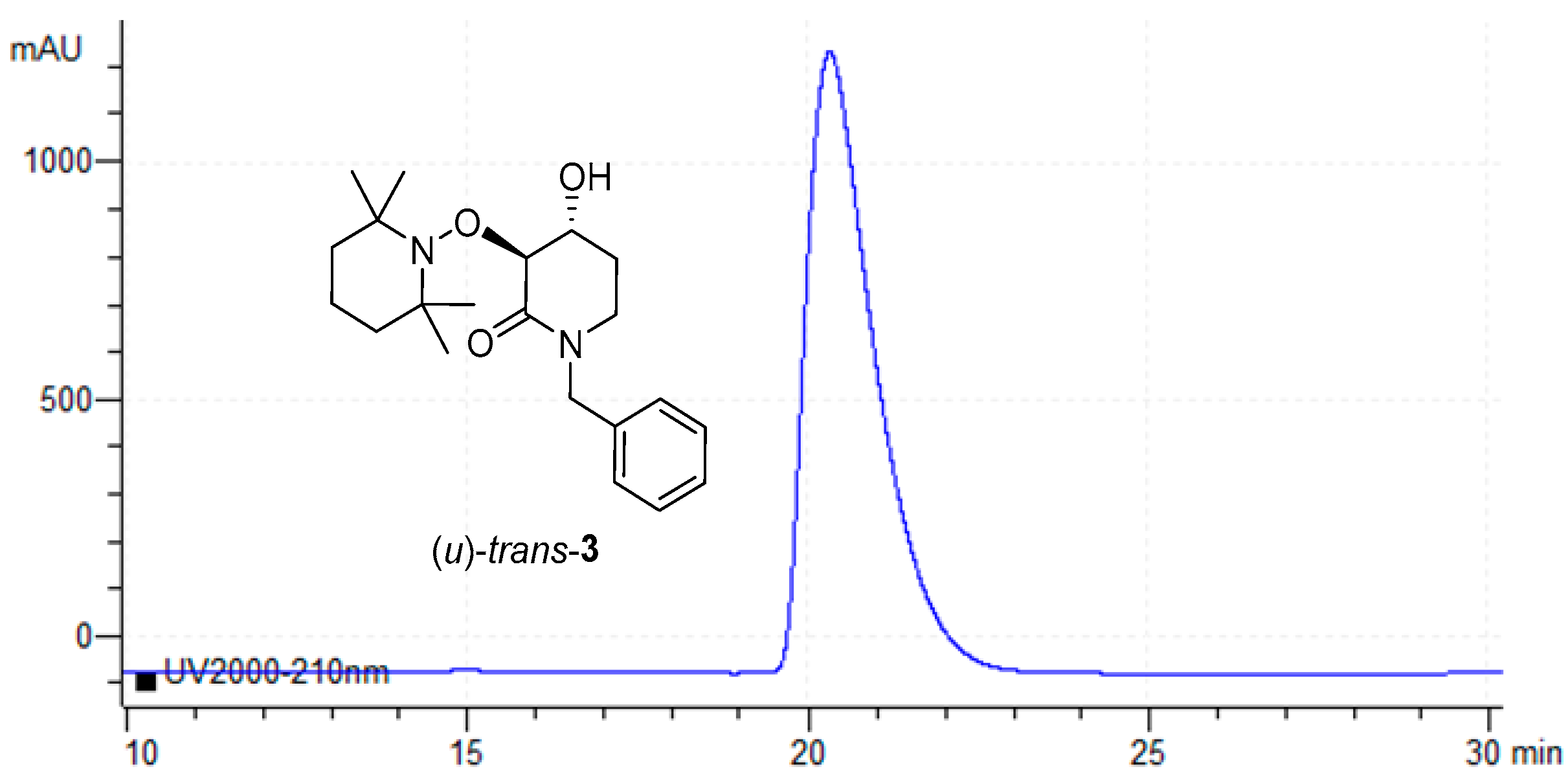
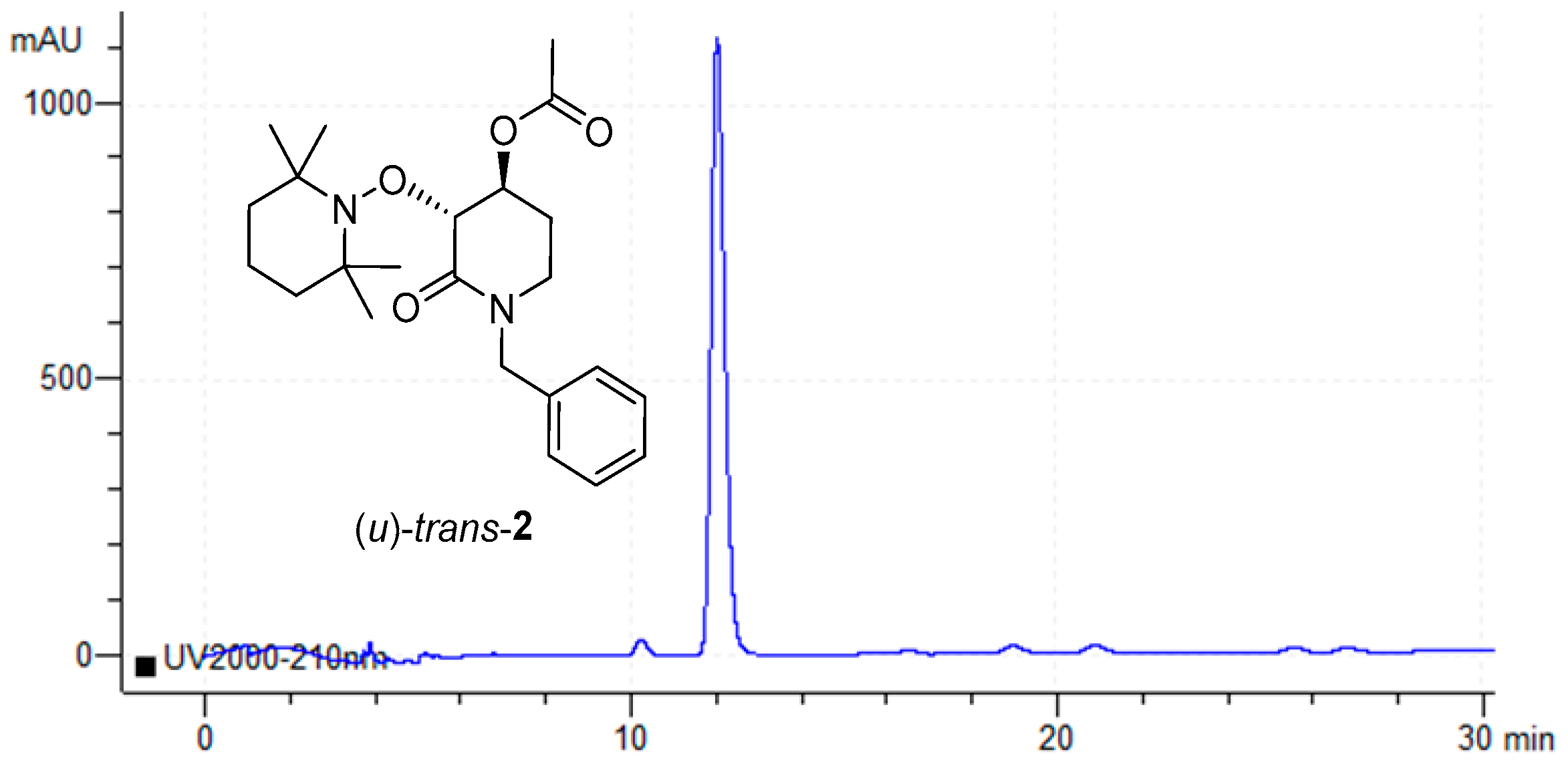
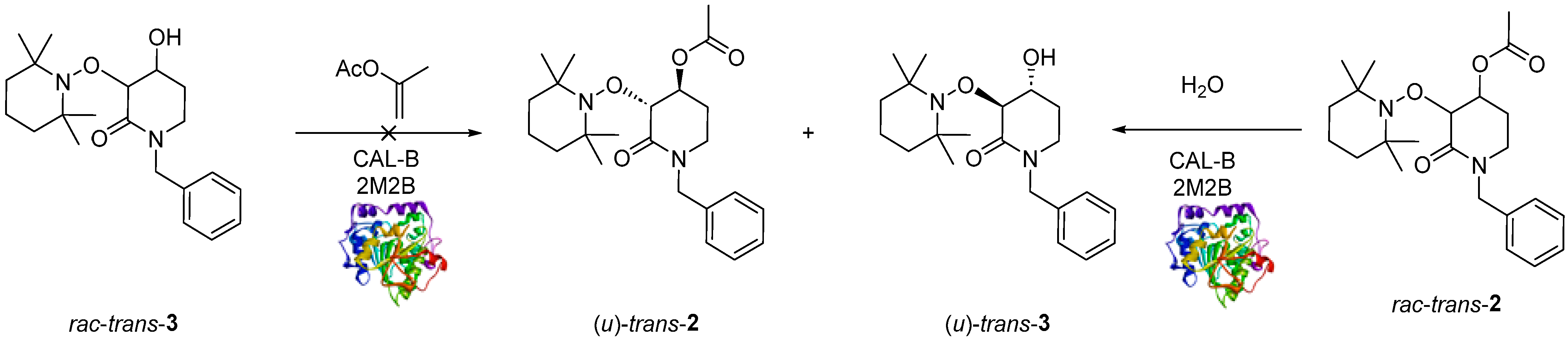
2.2. Enzymatic Resolution and Chiral-HPLC Enantiomeric Excess Evaluation
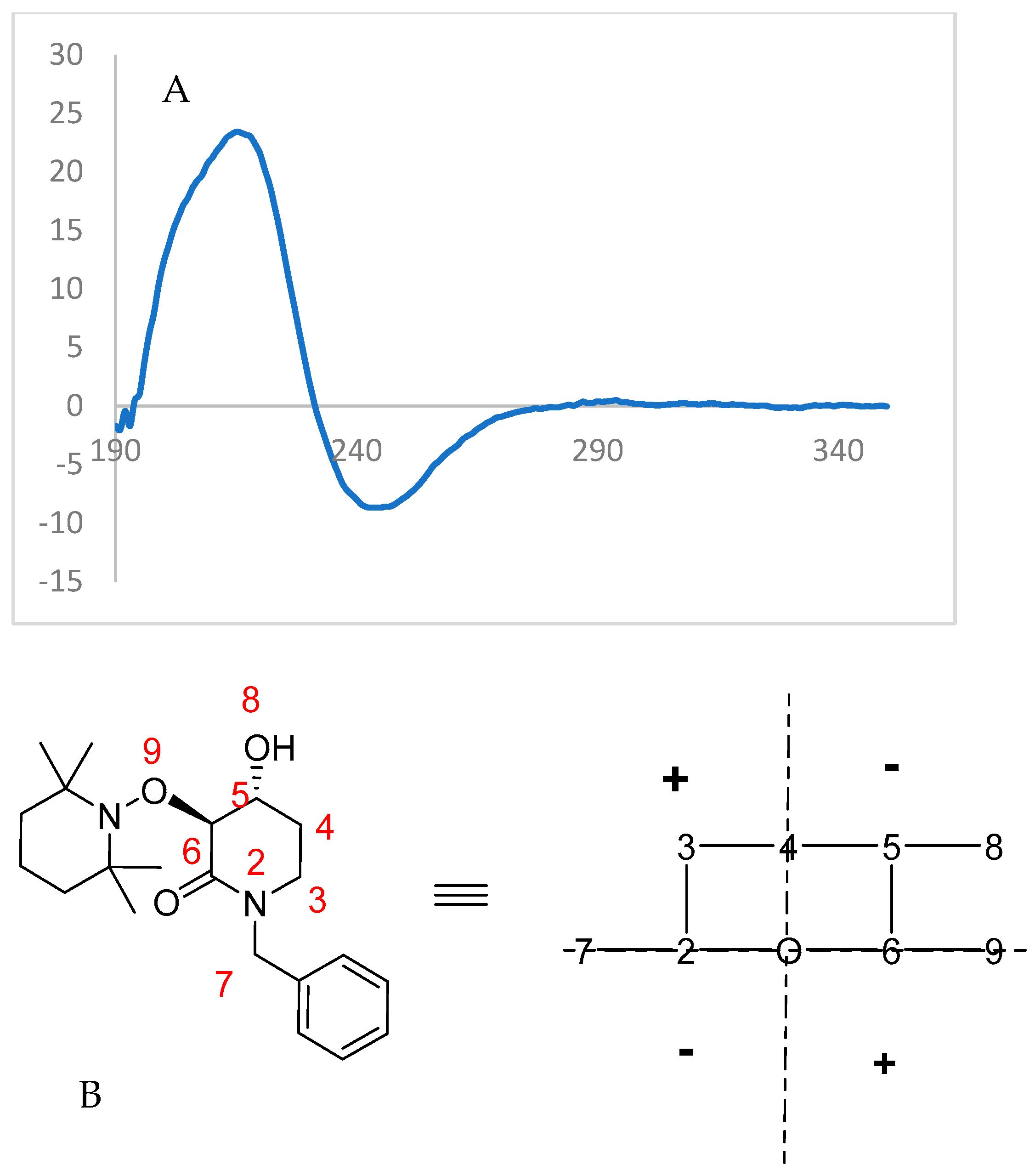
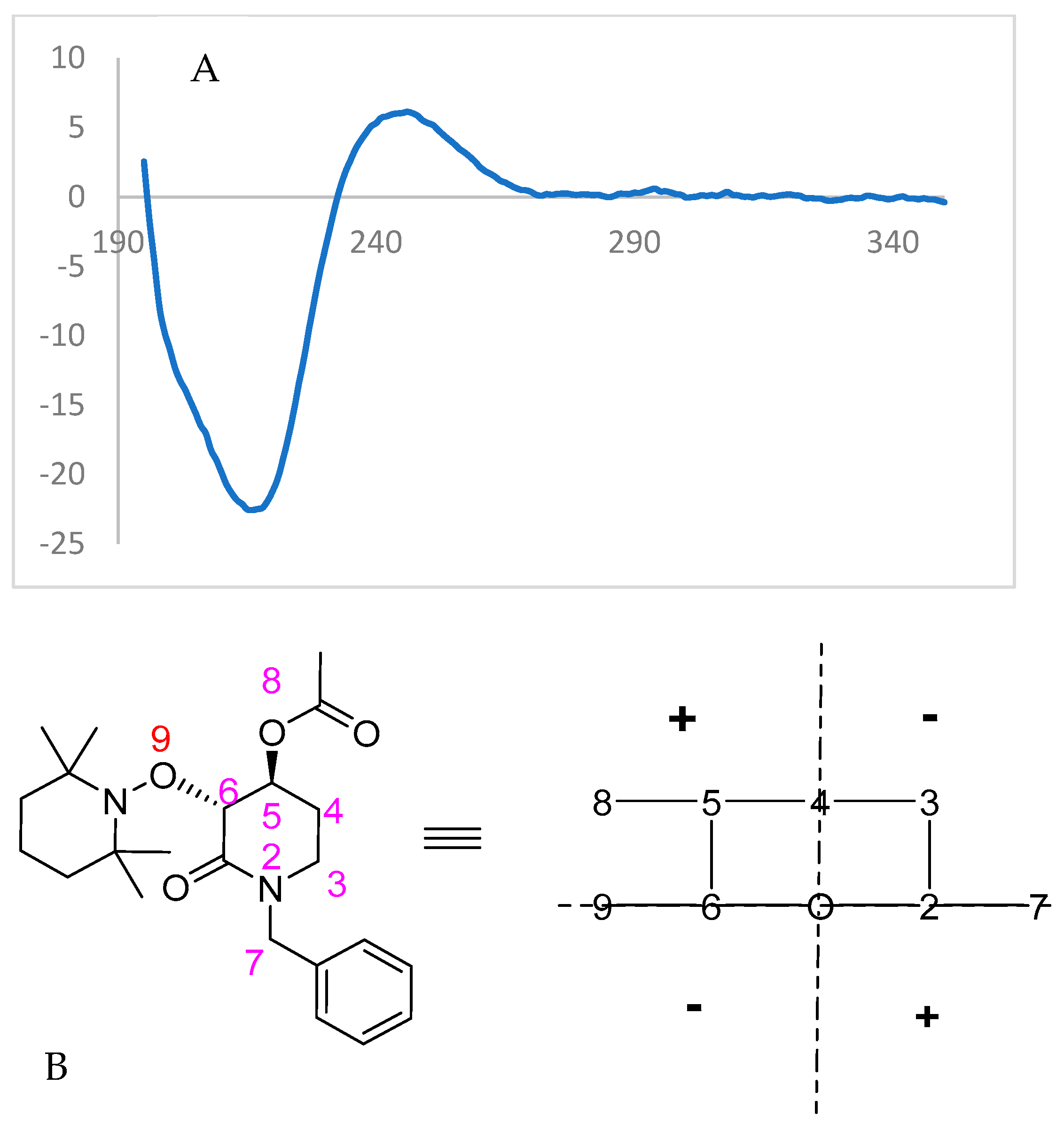
2.3. Chemical Correlation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kukula-Koch, W.A.; Widelski, J. Alkaloids. In Pharmacognosy Fundamentals, Applications and Strategy; Badal, S., Delgoda, R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 163–198. ISBN 9780128021040. [Google Scholar]
- Funayama, S.; Cordell, G.A. Alkaloids: A Treasury of Poisons and Medicines; Academic Press: London, UK, 2015; ISBN 9780124173026. [Google Scholar]
- Aniszewski, T. Alkaloids: Chemistry, Biology, Ecology, and Applications, 2nd ed.; Elsevier: Boston, MA, USA, 2015; ISBN 9780444594334. [Google Scholar]
- Buckingham, J.; Baggaley, K.H.; Roberts, A.D.; Szábo, L.F. (Eds.) Dictionary of Alkaloids with CDROM, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9781420077698. [Google Scholar]
- Mahajan, M.; Kumar, V.; Yadav, S.K. Alkaloids: Properties, Applications, and Pharmacological Effects; Cassiano, N.M., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 1–36. ISBN 13 9781612090962. [Google Scholar]
- von Nussbaum, F. In Alkaloids: Nature’s Curse or Blessing? By Manfred Hesse. Angew. Chem. Int. Ed. 2003, 42, 4852–4854. [Google Scholar] [CrossRef]
- Roberts, M.F.; Wink, M. (Eds.) Alkaloids: Biochemistry, Ecology, and Medicinal Applications; Plenum Press: New York, NY, USA, 1998; ISBN 0-306-45465-3. [Google Scholar]
- Cigan, E.; Eggbauer, B.; Schrittwieser, J.-H.; Kroutil, W. The role of biocatalysis in the asymmetric synthesis of alkaloids—An update. RCS Adv. 2021, 11, 28223–28270. [Google Scholar] [CrossRef]
- Lago, J.H.G.; Kato, M.J. 3α,4α-Epoxy-2-piperidone, a new minor derivative from leaves of Piper crassinervium Kunth (Piperaceae). Nat. Prod. Res. 2007, 21, 910–914. [Google Scholar] [CrossRef]
- Capron, M.A.; Wiemer, D.F. Piplaroxide, an Ant-Repellent Piperidine Epoxide from Piper tuberculatum. J. Nat. Prod. 1996, 59, 794. [Google Scholar] [CrossRef]
- Zhi-Yong, J.; Wen-Feng, L.; Chao-Guang, H.; Xiang-Zhong, H. New Amide Alkaloids from Piper longum. Fitoterapia 2013, 84, 222–226. [Google Scholar]
- Osorio-Nieto, U.; Chamorro-Arenas, D.; Quintero, L.; Höpfl, H.; Sartillo-Piscil, F. Transition Metal-Free Selective Double sp3 C-H Oxidation of Cyclic Amines to 3-Alkoxyamine Lactams. J. Org. Chem. 2016, 81, 8625–8632. [Google Scholar] [CrossRef]
- Griffiths, R.-J.; Burley, G.-A.; Talbot, E.-P.-A. Transition-Metal-Free Amine Oxidation: A Chemoselective Strategy for the Late-Stage Formation of Lactams. Org. Lett. 2017, 19, 870–873. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J.-P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. [Google Scholar] [CrossRef] [PubMed]
- Blanksby, S.-J.; Ellison, G.-B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.-K.; Youn, S.W. C-H Activation: A Complementary Tool in the Total Synthesis of Complex Natural Products. Chem. Eur. J. 2012, 18, 9452–9474. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef]
- Tan, P.-W.; Seayad, J. Advances in amide and thioamide assisted C(sp3)-H functionalization. Tetrahedron Lett. 2019, 60, 151338. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, U. Remote C-H bond activation/transformations: A continuous growing synthetic tool; Part II. Catal. Rev. 2018, 60, 497–565. [Google Scholar] [CrossRef]
- Li, B.; Ali, A.-I.-M.; Ge, H. Recent Advances in Using Transition-Metal-Catalyzed C–H Functionalization to Build Fluorescent Materials. Chem 2020, 6, 2591–2657. [Google Scholar] [CrossRef]
- Romero-Ibañez, J.; Cruz-Gregorio, S.; Sandoval-Lira, J.; Hernández-Pérez, J.M.; Quintero, L.; Sartillo-Piscil, F. Transition-metal-free deconstructive lactamization of piperidines. Angew. Chem. Int. Ed. 2019, 58, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ibañez, J.; Fuentes, L.; Sartillo-Piscil, F. Transition-Metal-Free Functionalization of Saturated and Unsaturated Amines to Bioactive Alkaloids Mediated by Sodium Chlorite. Synlett 2021, 32, 1385–1396. [Google Scholar] [CrossRef]
- Recoba-Torres, A.; Cruz-Gregorio, A.; Quintero, L.; Sandoval-Lira, J.; Romero-Ibañez, J.; Sartillo-Piscil, F. Selective deconstructive lactamization of the indolo [2,3-α]quinolizine skeleton for the total synthesis of (+) and (−)-cuscutamine. Eur. J. Org. Chem. 2022, 2022, e202200292. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Hydrolases: Catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, 39, 4504–4523. [Google Scholar] [CrossRef] [PubMed]
- Bordes, I.; Recatalá, J.; Świderek, K.; Moliner, V. Is Promiscuous CALB a Good Scaffold for Designing New Epoxidases? Molecules 2015, 20, 17789–17806. [Google Scholar] [CrossRef] [Green Version]
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Nat. Acad. Sci. USA 1985, 82, 3192–3196. [Google Scholar] [CrossRef] [Green Version]
- Flores-Sánchez, P.; Escalante, J.; Castillo, E. Enzymatic Resolution of N-Protected-β3-Amino Methyl Esters, Using Lipase B from Candida Antarctica. Tetrahedron Assym. 2005, 16, 629–634. [Google Scholar] [CrossRef]
- Torres-Gavilán, A.; Escalante, J.; Regla, I.; López-Munguía, A.; Castillo, E. “Easy-On, Easy-Off” resolution of Chiral 1-Phenylethylamine Catalyzed by Candida Antarctica Lipase, B. Tetrahedron Asym. 2007, 18, 2621–2624. [Google Scholar] [CrossRef]
- Priego, J.; Ortíz-Nava, C.; Carrillo-Morales, M.; López-Munguía, A.; Escalante, J.; Castillo, E. Solvent Engineering: An Effective Tool to Direct Chemoselectivity in a Lipase-Catalyzed Michael Addition. Tetrahedron 2009, 65, 536–539. [Google Scholar] [CrossRef]
- Rivera-Ramírez, J.D.; Escalante, J.; López-Munguía, A.; Marty, A.; Castillo, E. Thermodynamically Controlled Chemoselectivity in Lipase-Catalyzed Aza-Michael Additions. J. Mol. Catal. B Enzym. 2015, 112, 76–82. [Google Scholar] [CrossRef]
- Pérez-Venegas, M.; Reyes-Rangel, G.; Neri, A.; Escalante, J.; Juaristi, E. Mechanochemical Enzymatic Resolution of N -Benzylated-β 3 -Amino Esters. Beilstein J. Org. Chem. 2017, 13, 1728–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Rojas, M.; Rivera-Ramírez, J.; Ávila-Ortiz, C.; Juaristi, E.; González-Muñoz, F.; Castillo, E.; Escalante, J. One-Pot Lipase-Catalyzed Enantioselective Synthesis of (R)-(−)-N-Benzyl-3-(Benzylamino)butanamide: The Effect of Solvent Polarity on Enantioselectivity. Molecules 2017, 22, 2189. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Rojas, M.A.; Castillo, E.; Razo-Hernández, R.S.; Pastor, N.; Juaristi, E.; Escalante, J. Effect of the Substituent and Amino Group Position on the Lipase-Catalyzed Resolution of γ-Amino Esters: A Molecular Docking Study Shedding Light on Candida Antarctica Lipase B Enantioselectivity. Eur. J. Org. Chem. 2021, 2021, 4790–4802. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, G.K.; Ortega-Rojas, M.A.; Chavelas-Hernández, L.; Razo-Hernández, R.S.; Valdéz-Camacho, J.R.; Escalante, J. Solvent-Free Lipase-Catalyzed Transesterification of Alcohols with Methyl Esters Under Vacuum-Assisted Conditions. ChemistrySelect 2022, 7, e202202643. [Google Scholar] [CrossRef]
- Otera, J.; Nishikido, J. Kinetic Resolution. In Esterification- Methods, Reactions, and Applications; Wiley-VCH: Hoboken, NJ, USA, 2010; pp. 197–205. ISBN 978-3-527-32289-3. [Google Scholar]
- Ohtani, T.; Nakatsukasa, H.; Kamezaw, M.; Tachibana, H.; Naoshima, Y. Enantioselectivity of Candida antarctica lipase for some synthetic substrates including aliphatic secondary alcohols. J. Mol. Catal. B Enzym. 1998, 4, 53–60. [Google Scholar] [CrossRef]
- Rocha, L.-C.; Rosset, I.-G.; Luiz, R.-F.; Raminelli, C.; Porto, A.-L.-M. Kinetic resolution of iodophenylethanols by Candida antarctica lipase and their application for the synthesis of chiral biphenyl compounds. Tetrahedron Asym. 2010, 18, 926–929. [Google Scholar] [CrossRef]
- Ferraz, H.-M.-C.; Bianco, G.-G.; Teixeira, C.-C.; Andrade, L.-H.; Porto, A.-L.-M. Enzymatic resolution of α-tetralols by CALB-catalyzed acetylation. Tetrahedron Asym. 2007, 18, 1070–1076. [Google Scholar] [CrossRef]
- Raminelli, C.; Comasseto, J.-V.; Andrade, L.-H.; Porto, A.-L.-M. Kinetic resolution of propargylic and allylic alcohols by Candida antarctica lipase (Novozyme 435). Tetrahedron Asym. 2004, 15, 3117–3122. [Google Scholar] [CrossRef]
- Ferreira, H.-V.; Rocha, L.-C.; Severino, R.-P.; Viana, R.-B.; Da Silva, A.-B.-F.; Porto, A.-L.-M. Enzymatic Resolution of Racemic Sulcatol by Lipase from Candida antarctica in a Large Scale. J. Iran. Chem. Soc. 2010, 7, 883–889. [Google Scholar] [CrossRef]
- Ferreira, H.-V.; Rocha, L.-C.; Severino, R.-P.; Porto, A.-L.-M. Syntheses of Enantiopure Aliphatic Secondary Alcohols and Acetates by Bioresolution with Lipase B from Candida antarctica. Molecules 2012, 17, 8955–8967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henández, J.G.; Frings, M.; Bolm, C. Mechanochemical enzymatic resolution of secondary alcohols under ball-milling conditions. Chem. Cat. Chem. 2016, 8, 1769–1772. [Google Scholar] [CrossRef]
- Bocquin, L.; Egholm-Jacobsen, E. Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol. Catalysts 2023, 13, 54. [Google Scholar] [CrossRef]
- Escorcia, A.-M.; Daza, M.-C.; Doerr, M. Computational study of the enantioselectivity of the O-acetylation of (R,S)-propranolol catalyzed by Candida antarctica lipase B. J. Mol. Catal. B Enzym. 2014, 108, 21–31. [Google Scholar] [CrossRef]
- Shen, J.-W.; Qi, J.-M.; Zhang, X.-J.; Liu, Z.-Q.; Zheng, Y.-G. Significantly increased catalytic activity of Candida antarctica lipase B for the resolution of cis-(±)-dimethyl 1-acetylpiperidine-2,3-dicarboxylate. Catal. Sci. Technol. 2018, 8, 4718–4725. [Google Scholar] [CrossRef]
- Husson, E.; Garcia-Matilla, V.; Humeau, C.; Chevalot, I.; Fournier, F.; Marc, I. Enzymatic acylation of a bifunctional molecule in 2-methyl-2-butanol: Kinetic modelling. Enzyme Microb. Technol. 2010, 46, 338–346. [Google Scholar] [CrossRef]
- Kamal, A.; Ramana, K.; Ramana, A.; Babu, A. Chemoenzymatic enantioselective synthesis of 3-hydroxy-2-pyrrolidinones and 3-hydroxy-2-piperidinones. Tetrahedron Asymm. 2003, 14, 2587–2594. [Google Scholar] [CrossRef]
- Huang., P.-Q.; Zheng, X.; Wei, H. Synthesis of (S)-Vasicol and (S)-3-hydroxy-2-pyrrolidinone. Heterocycles 2003, 60, 1833–1841. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate (mmol) | Conc. (mol/L) | Enzyme (mg/mL) | H2O (eq.) | Time (h) | Temperature (°C) | Conversion a (%) | [α]D20 | ee (%) | E |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.17 | 0.2 | 20 | 2 | 24 | 45 | 3 b | Nd | ≥99 c | 205 d |
| 2 | 0.15 | 0.2 | 20 | 4 | 72 | 45 | 13 e | Nd | ≥99 c | 230 d |
| 3 | 0.25 f | 0.4 | 100 | 3 | 72 | 60 | 29 b | +45.2 g | ≥99 c | 383 d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Ibañez, J.; Ortega-Rojas, M.A.; Valdéz-Camacho, J.R.; Hernández-Vázquez, L.G.; Sartillo-Piscil, F.; Escalante, J. Asymmetric Synthesis of trans-3-Alkoxyamino-4-Oxygenated-2-Piperidones Mediated by Transition-Metal-Free Dual C-H Oxidation and Its CAL-B-Assisted Enzymatic Resolution. Catalysts 2023, 13, 703. https://doi.org/10.3390/catal13040703
Romero-Ibañez J, Ortega-Rojas MA, Valdéz-Camacho JR, Hernández-Vázquez LG, Sartillo-Piscil F, Escalante J. Asymmetric Synthesis of trans-3-Alkoxyamino-4-Oxygenated-2-Piperidones Mediated by Transition-Metal-Free Dual C-H Oxidation and Its CAL-B-Assisted Enzymatic Resolution. Catalysts. 2023; 13(4):703. https://doi.org/10.3390/catal13040703
Chicago/Turabian StyleRomero-Ibañez, Julio, Marina A. Ortega-Rojas, Jonathan R. Valdéz-Camacho, Luis G. Hernández-Vázquez, Fernando Sartillo-Piscil, and Jaime Escalante. 2023. "Asymmetric Synthesis of trans-3-Alkoxyamino-4-Oxygenated-2-Piperidones Mediated by Transition-Metal-Free Dual C-H Oxidation and Its CAL-B-Assisted Enzymatic Resolution" Catalysts 13, no. 4: 703. https://doi.org/10.3390/catal13040703