
Tuning the Structure and Acidity of Pt/Hierarchical SSZ-32 Catalysts to Boost the Selective Hydroisomerization of n-Hexadecane
Abstract
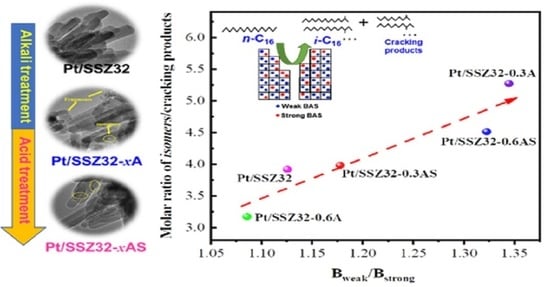
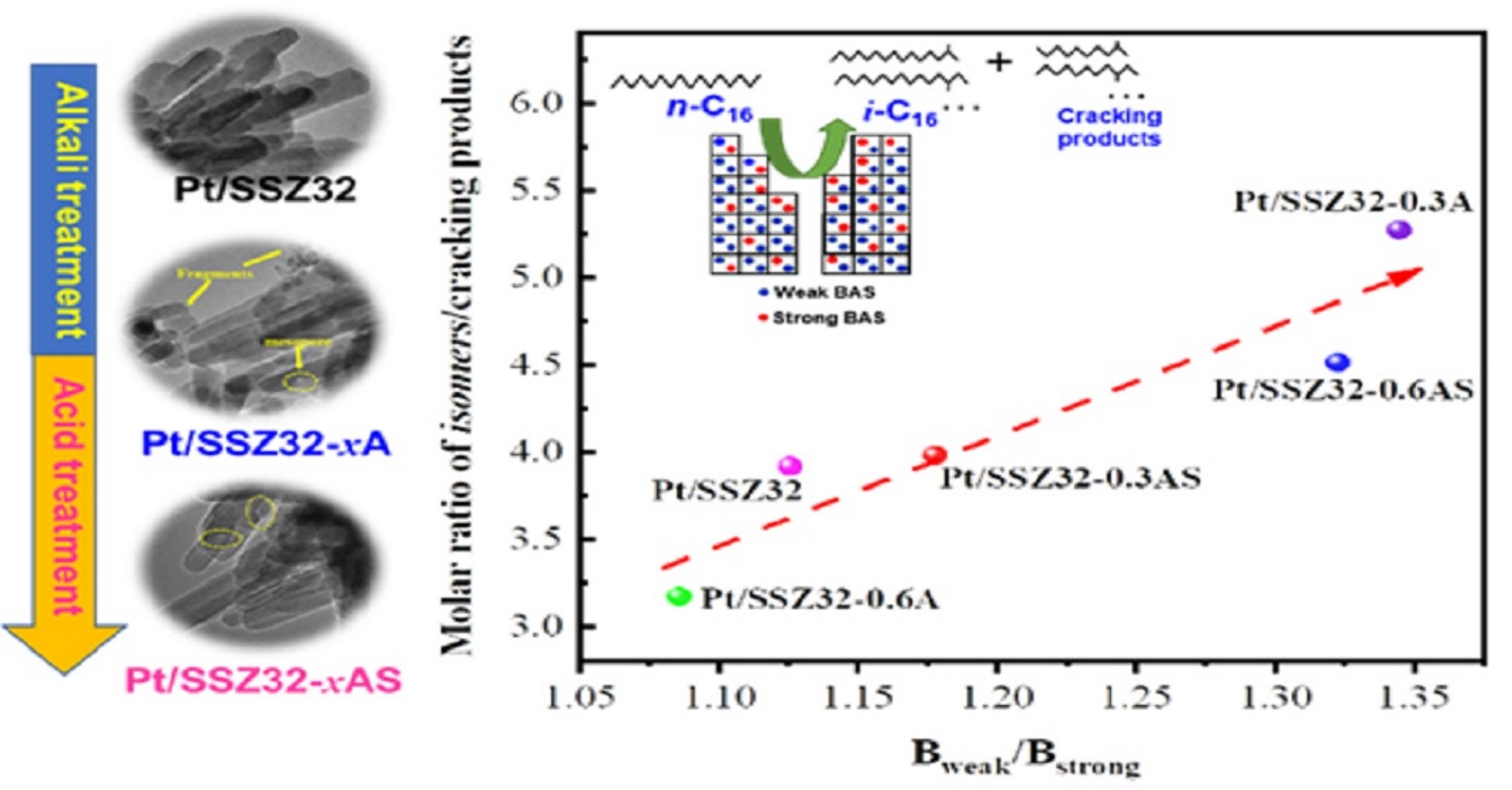
:
1. Introduction
2. Results and Discussion
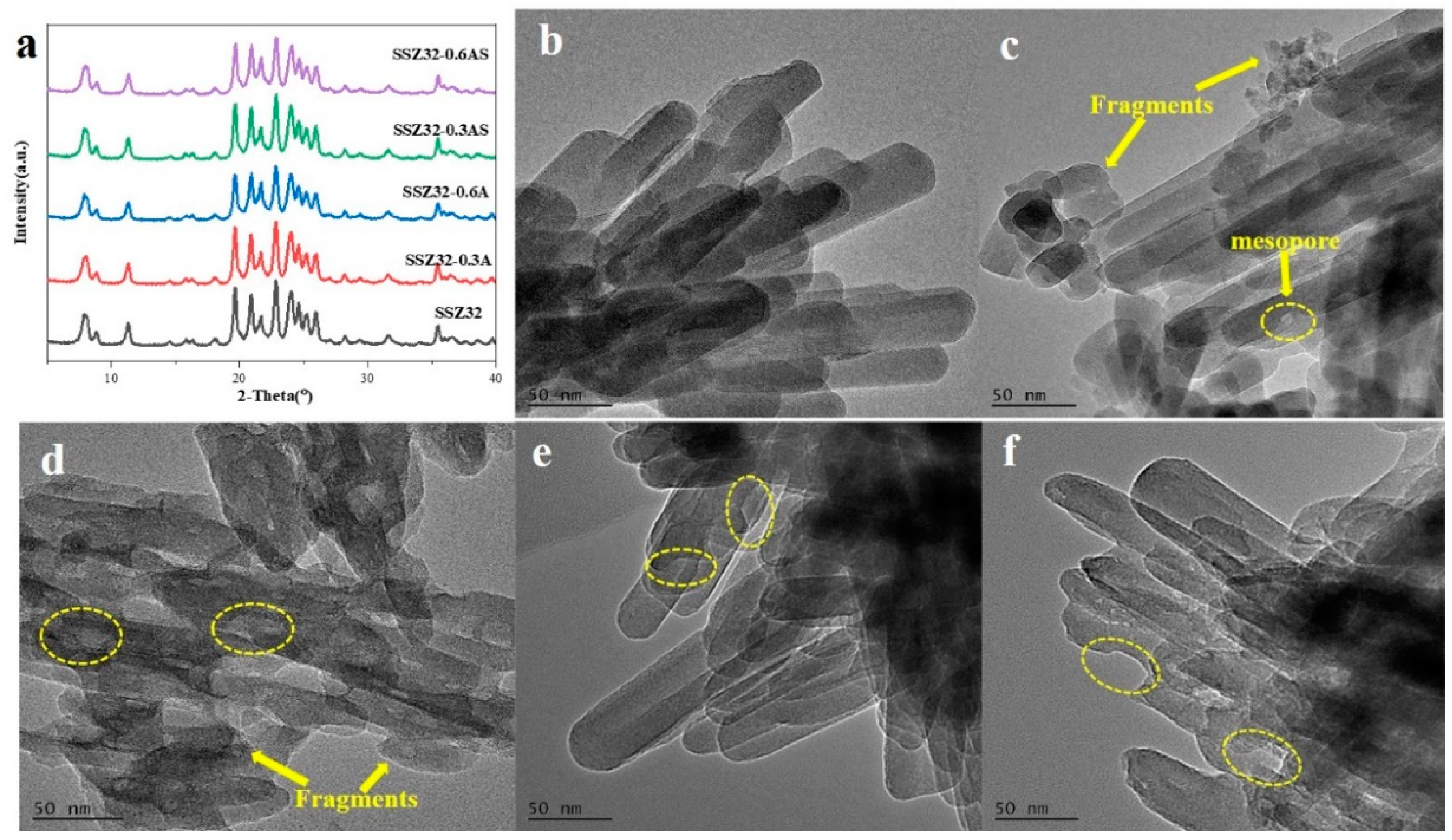
2.1. Structure and Morphology of Pt/Hierarchical SSZ-32 Catalysts
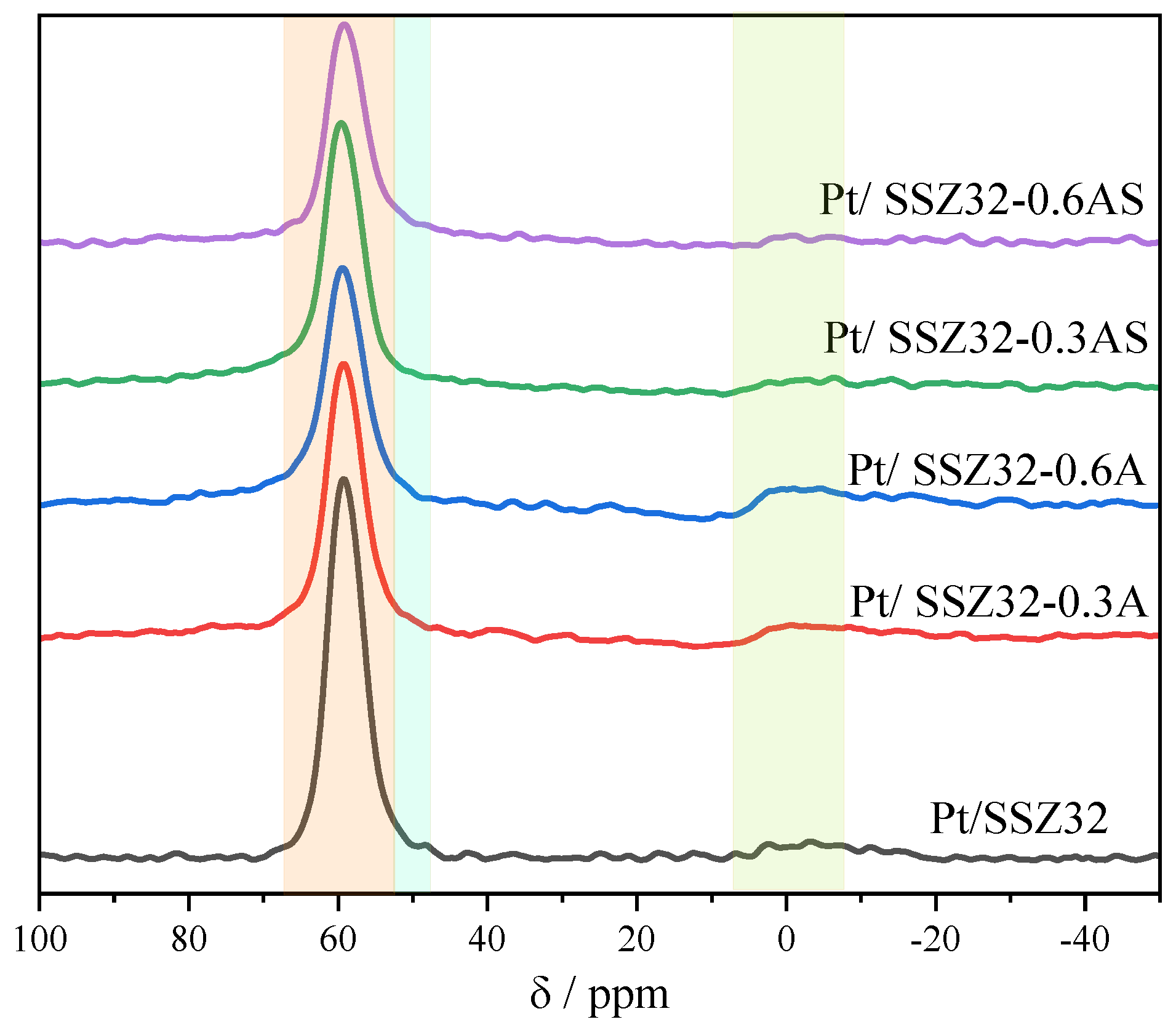
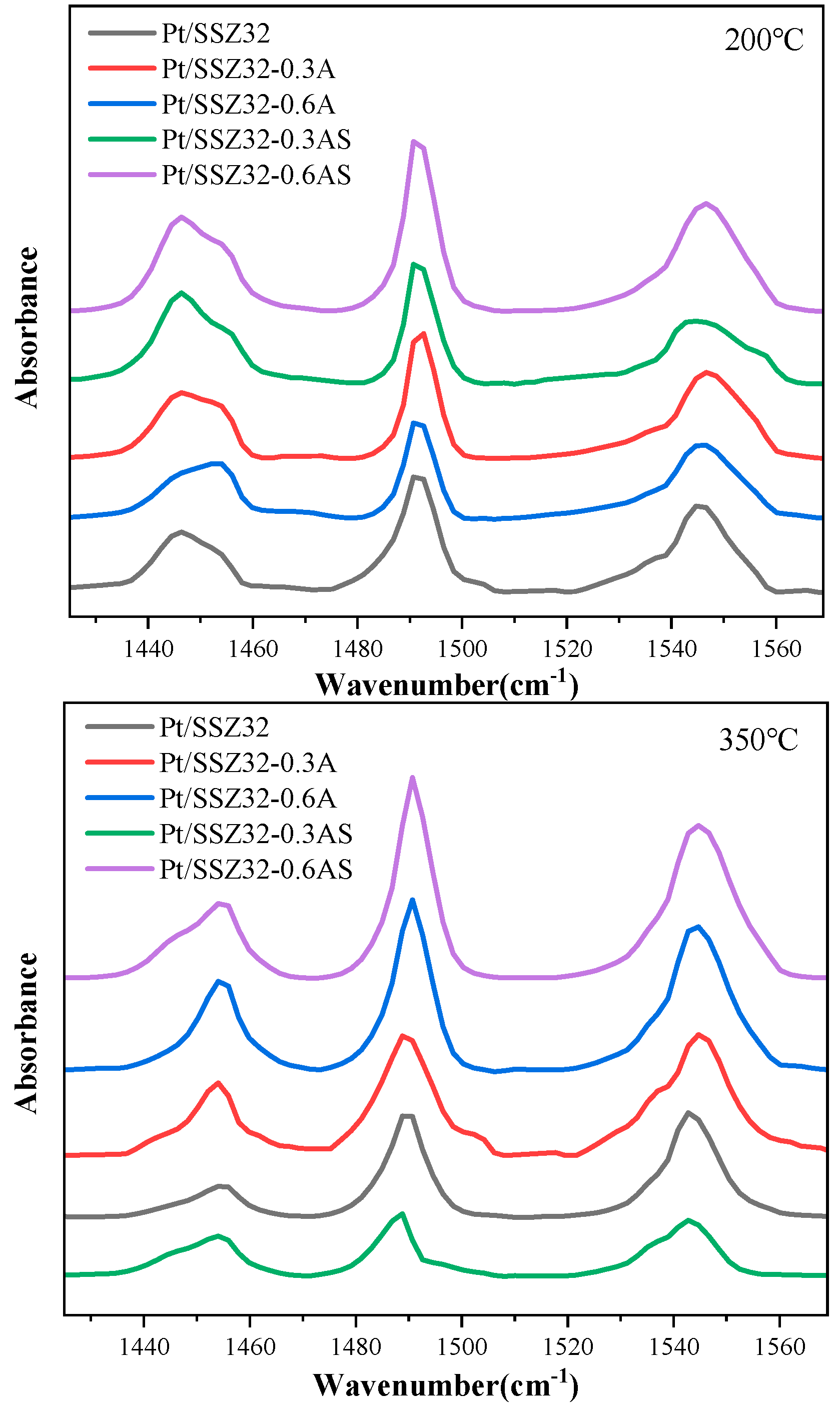
Acidity Properties
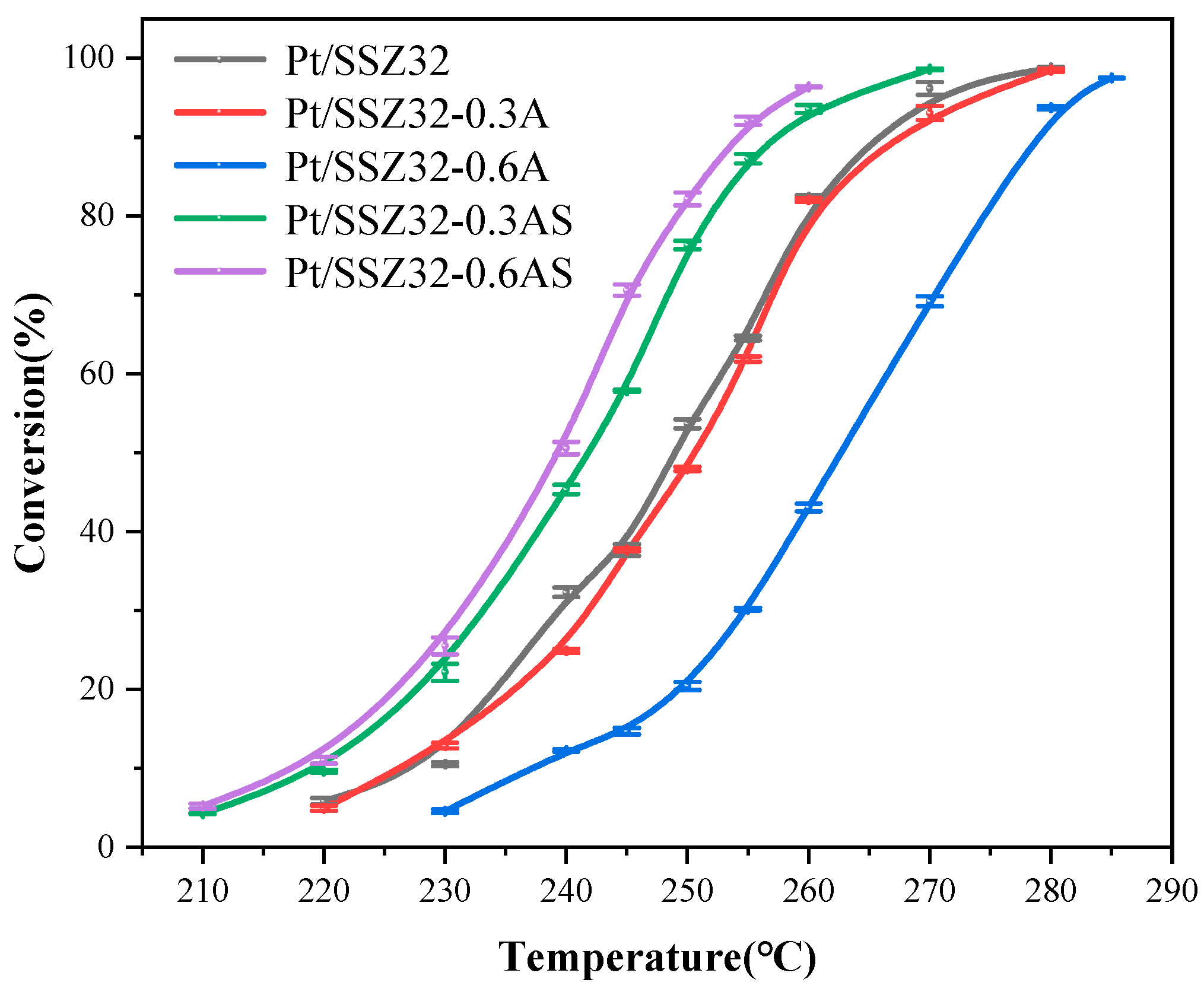
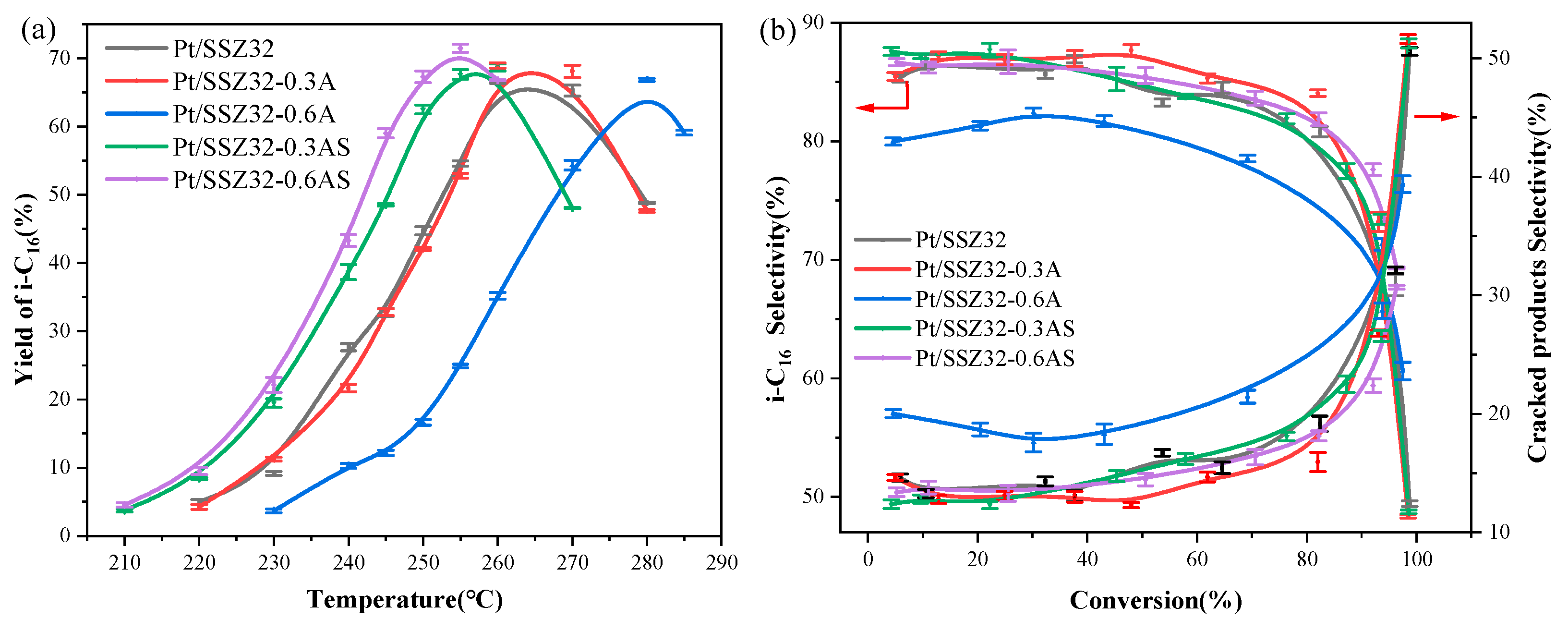
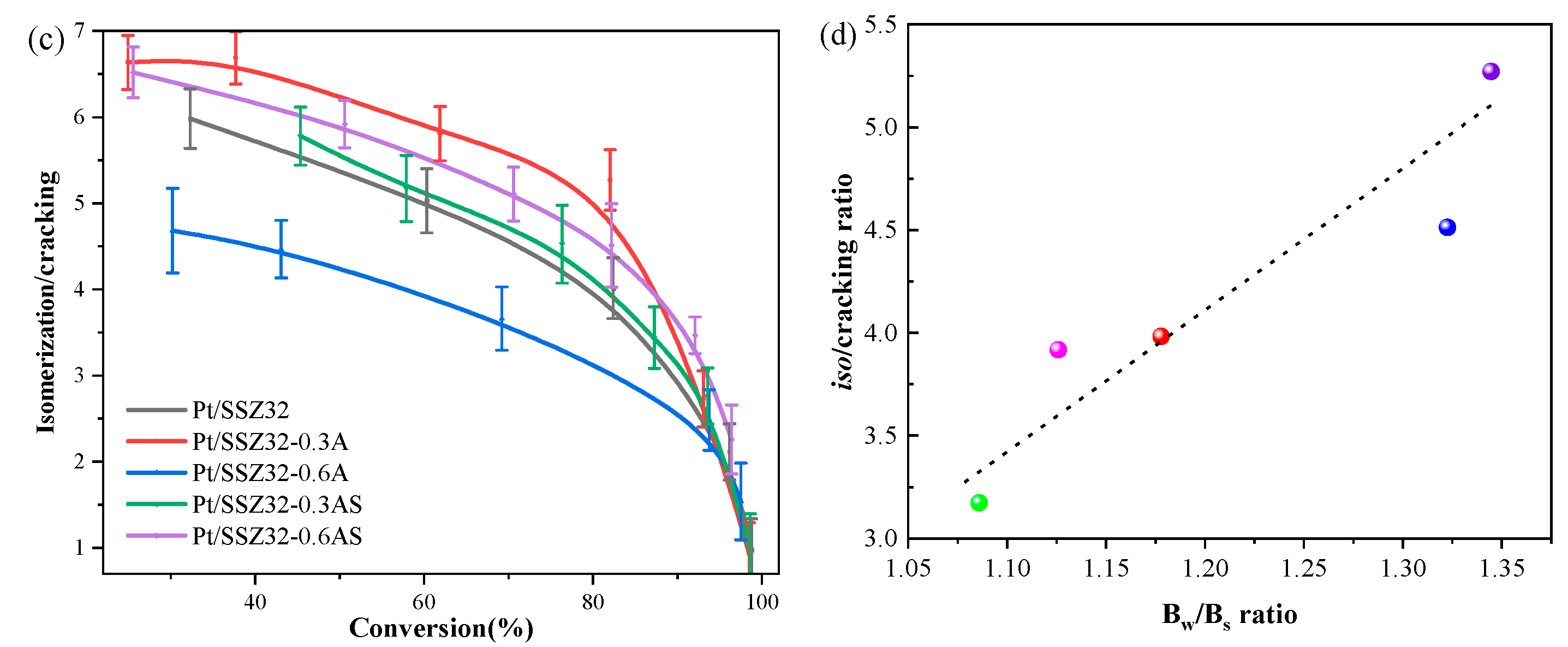
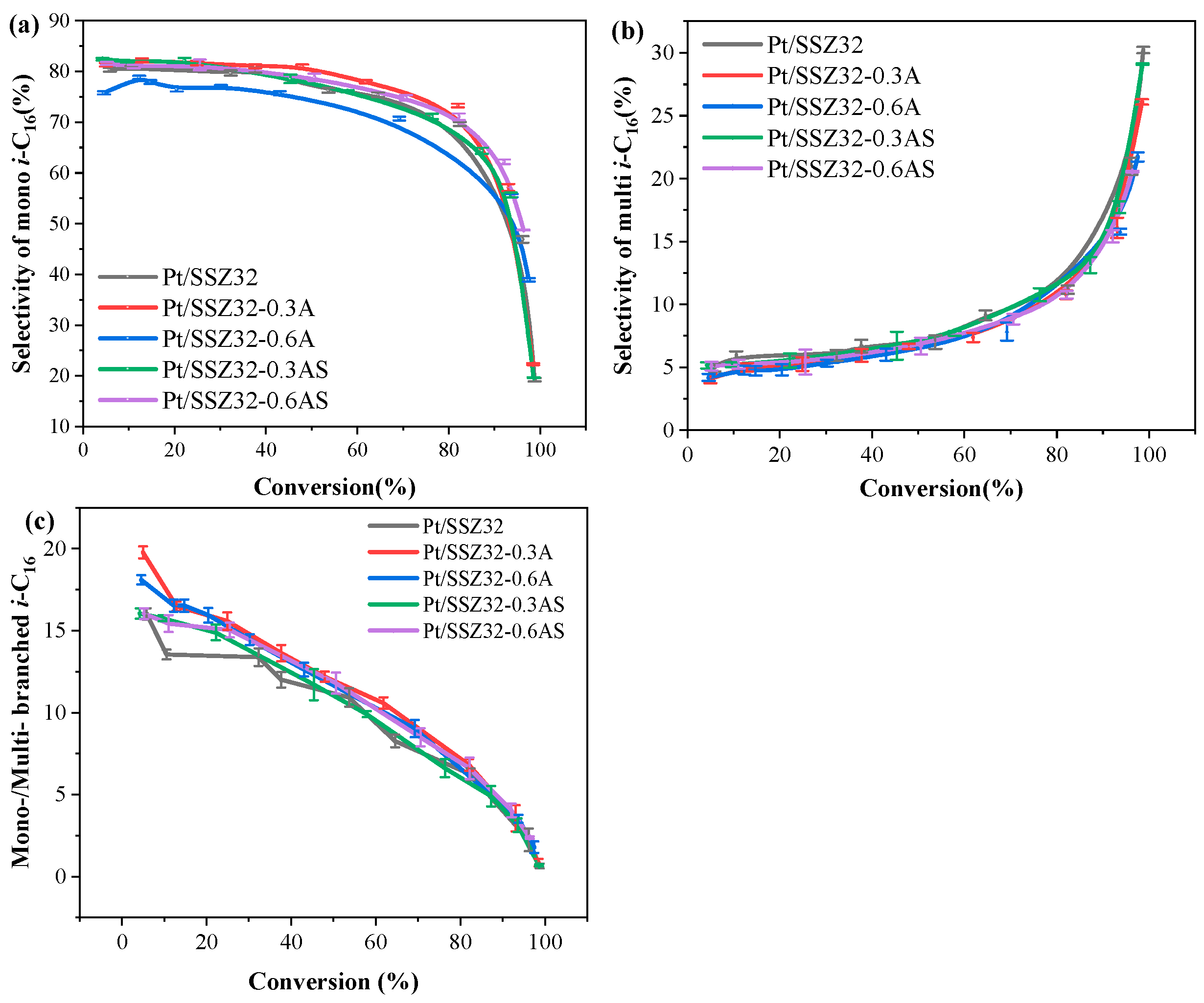
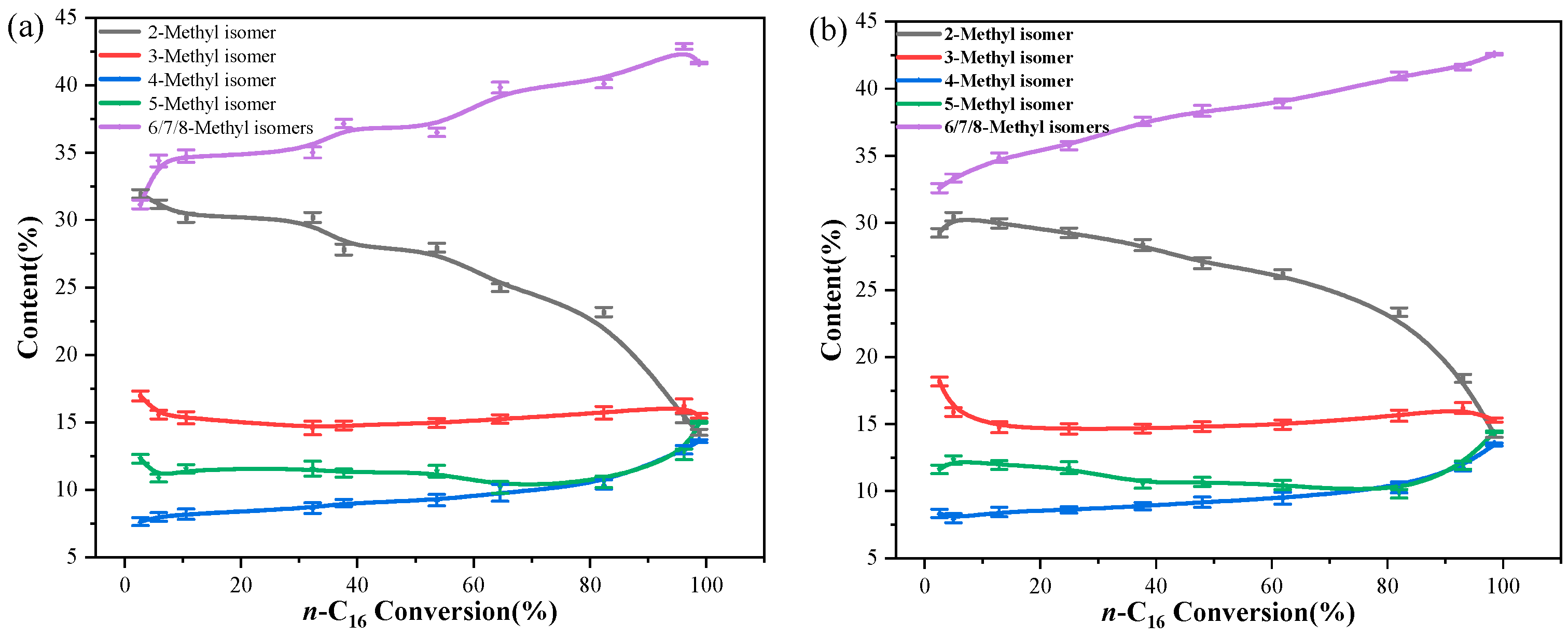
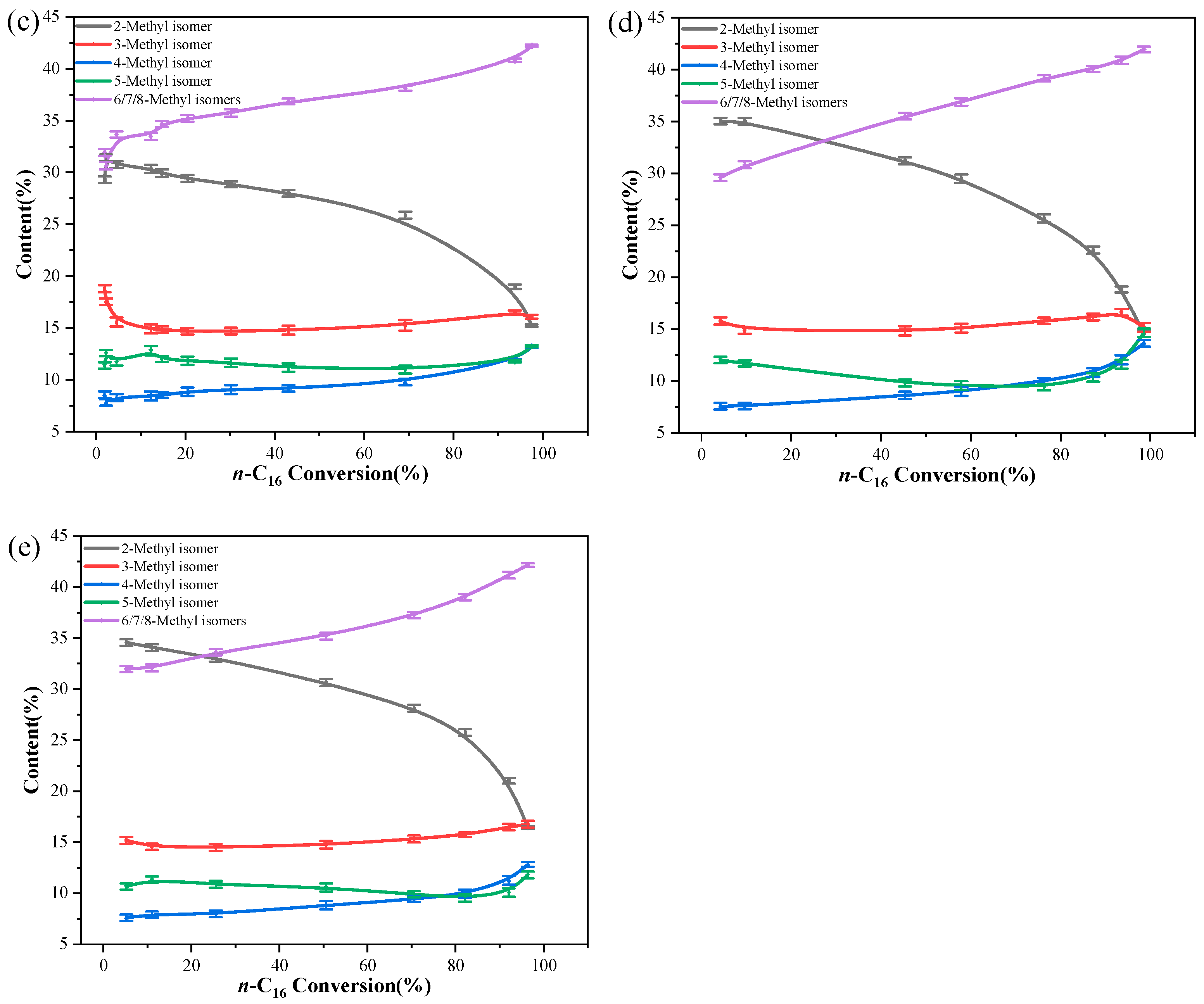
2.2. Catalytic Hydroisomerization of n-Hexadecane over Different Catalysts
3. Experimental Section
3.1. Catalysts Preparation
3.1.1. Synthesis of Parent SSZ-32
3.1.2. Synthesis of Hierarchical SSZ-32
3.2. Catalyst Characterization
3.3. Catalytic Hydroisomerization of n-Hexadecane
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benitez, V.M.; Yori, J.C.; Grau, J.M.; Pieck, C.L.; Vera, C.R. Hydroisomerization and Cracking of n-Octane and n-Hexadecane over Zirconia Catalysts. Energy Fuels 2006, 20, 422–426. [Google Scholar] [CrossRef]
- Corma, A. Transformation of hydrocarbons on zeolite catalysts. Catal. Lett. 1993, 22, 33–52. [Google Scholar] [CrossRef]
- Ward, J.A. Hydrocracking processes and catalysts. Fuel Process. Technol. 1993, 35, 55–85. [Google Scholar] [CrossRef]
- Gao, L.; Shi, Z.Y.; Etim, U.J.; Wu, P.P.; Xing, W.; Zhang, Y.; Bai, P.; Yan, Z.F. Superior catalytic performance of micro-mesoporous Beta-SBA-15 composite with a high indexed isomerization factor in hydroisomerization of n-heptane. Fuel 2019, 252, 653–665. [Google Scholar] [CrossRef]
- Yu, R.; Tan, Y.; Yao, H.; Xu, Y.; Huang, J.; Zhao, B.; Du, Y.; Hua, Z.; Li, J.; Shi, J. Toward n-Alkane Hydroisomerization Reactions: High-Performance Pt–Al2O3/SAPO-11 Single-Atom Catalysts with Nanoscale Separated Metal-Acid Centers and Ultralow Platinum Content. ACS Appl. Mater. Inter. 2022, 14, 44377–44388. [Google Scholar] [CrossRef]
- García-Pérez, D.; Blanco-Brieva, G.; Alvarez-Galvan, M.C.; Campos-Martin, J.M. Influence of W loading, support type, and preparation method on the performance of zirconia or alumina-supported Pt catalysts for n-dodecane hydroisomerization. Fuel 2022, 319, 123704. [Google Scholar] [CrossRef]
- Jokar, F.; Alavi, S.M.; Rezaei, M. Investigating the hydroisomerization of n- pentane using Pt supported on ZSM-5, desilicated ZSM-5, and modified ZSM-5/MCM-41. Fuel 2022, 324, 124511. [Google Scholar] [CrossRef]
- Tan, Y.; Hu, W.; Du, Y.; Li, J. Species and impacts of metal sites over bifunctional catalyst on long chain n-alkane hydroisomerization: A review. Appl. Catal. A Gen. 2021, 611, 117916. [Google Scholar] [CrossRef]
- Deldari, H. Suitable catalysts for hydroisomerization of long-chain normal paraffins. Appl. Catal. A Gen. 2005, 293, 1–10. [Google Scholar] [CrossRef]
- Jin, D.; Li, L.; Ye, G.; Ding, H.; Zhao, X.; Zhu, K.; Coppens, M.-O.; Zhou, X. Manipulating the mesostructure of silicoaluminophosphate SAPO-11 via tumbling-assisted, oriented assembly crystallization: A pathway to enhance selectivity in hydroisomerization. Catal. Sci. Technol. 2018, 8, 5044–5061. [Google Scholar] [CrossRef] [Green Version]
- Niu, P.; Xi, H.; Ren, J.; Lin, M.; Wang, Q.; Chen, X.; Wang, P.; Jia, L.; Hou, B.; Li, D. Micropore blocked core–shell ZSM-22 designed via epitaxial growth with enhanced shape selectivity and high n-dodecane hydroisomerization performance. Catal. Sci. Technol. 2018, 8, 6407–6419. [Google Scholar] [CrossRef]
- Liu, S.; He, Y.; Zhang, H.; Chen, Z.; Lv, E.; Ren, J.; Yun, Y.; Wen, X.; Li, Y.-W. Design and synthesis of Ga-doped ZSM-22 zeolites as highly selective and stable catalysts for n-dodecane isomerization. Catal. Sci. Technol. 2019, 9, 2812–2827. [Google Scholar] [CrossRef]
- Li, T.; Wang, W.; Feng, Z.; Bai, X.; Su, X.; Yang, L.; Jia, G.; Guo, C.; Wu, W. The hydroisomerization of n-hexane over highly selective Pd/ZSM-22 bifunctional catalysts: The improvements of metal-acid balance by room temperature electron reduction method. Fuel 2020, 272, 117717. [Google Scholar] [CrossRef]
- Niu, P.; Xi, H.; Ren, J.; Lin, M.; Wang, Q.; Jia, L.; Hou, B.; Li, D. High selectivity for n-dodecane hydroisomerization over highly siliceous ZSM-22 with low Pt loading. Catal. Sci. Technol. 2017, 7, 5055–5068. [Google Scholar] [CrossRef]
- Woo Lee, S.; Ki Ihm, S. Hydroisomerization and hydrocracking over platinum loaded ZSM-23 catalysts in the presence of sulfur and nitrogen compounds for the dewaxing of diesel fuel. Fuel 2014, 134, 237–243. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, Y.; Wang, L.; Zhang, Q.; Tsang, C.-W.; Liang, C. Shape Selectivity in Hydroisomerization of Hexadecane over Pt Supported on 10-Ring Zeolites: ZSM-22, ZSM-23, ZSM-35, and ZSM-48. Ind. Eng. Chem. Res. 2016, 55, 6069–6078. [Google Scholar] [CrossRef]
- Zhang, M.; Li, C.; Chen, Y.; Tsang, C.-W.; Zhang, Q.; Liang, C. Hydroisomerization of hexadecane over platinum supported on EU-1/ZSM-48 intergrowth zeolite catalysts. Catal. Sci. Technol. 2016, 6, 8016–8023. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, M.; Wang, L.; Zhang, X.; Li, G. Construction of ordered mesopores outside MTT zeolite for efficient hydroisomerization. Appl. Catal. A Gen. 2020, 602, 117664. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, M.; Wang, L.; Zhang, X.; Li, G. Modulating acid site distribution in MTT channels for controllable hy-droi-som-erization of long-chain n-alkanes. Fuel Process. Technol. 2023, 241, 107605. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Wang, Q. N-dodecane hydroisomerization over Pt/ZSM-22: Controllable microporous Brönsted acidity distribution and shape-selectivity. Appl. Catal. A Gen. 2020, 590, 117335. [Google Scholar] [CrossRef]
- Maesen, T.; Schenk, M.; Vlugt, T.; Jonge, J.; Smit, B. The Shape Selectivity of Paraffin Hydroconversion on TON-, MTT-, and AEL-Type Sieves. J. Catal. 1999, 188, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Lv, G.; Wang, C.; Wang, P.; Sun, L.; Liu, H.; Qu, W.; Wang, D.; Ma, H.; Tian, Z. Pt/ZSM-22 with Partially Filled Micropore Channels as Excellent Shape-Selective Hydroisomerization Catalyst. ChemCatChem 2019, 11, 1431–1436. [Google Scholar] [CrossRef]
- Ahmed, M.H.M.; Muraza, O.; Al-Amer, A.M.; Yamani, Z.H. Investigation of crucial synthesis parameters of rich Al-MTT framework zeolite: Toward more determination for synthesis zone of SSZ-32. Microporous Mesoporous Mater. 2016, 227, 48–56. [Google Scholar] [CrossRef]
- Ojo, A.F.; Lei, G.; Zhang, Y.; Krishna, K.R. Processes for Producing Diesel from Unconventional Feedstoks. U.S. Patent 2,022,204,867-A1, 30 June 2022. [Google Scholar]
- Dai, X.; Cheng, Y.; Si, M.; Wei, Q.; Chen, D.; Huang, W.; Zhou, Y. SAPO-11 molecular sieves synthesized in alcohol-water concentrated gel system with improved acidity, mesoporous volume and hydroisomerization performance. Fuel 2022, 314, 123131. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Chen, X.; Liu, Y.; Liang, C. Synthesis of ZSM-23 zeolite with dual structure directing agents for hydroisomerization of n-hexadecane. Microporous Mesoporous Mater. 2018, 268, 216–224. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Chen, X.; Liu, Y.; Tsang, C.-W.; Liang, C. Synthesis and Characterization of Iron-Substituted ZSM-23 Zeolite Catalysts with Highly Selective Hydroisomerization of n-Hexadecane. Ind. Eng. Chem. Res. 2018, 57, 13721–13730. [Google Scholar] [CrossRef]
- Chen, L.-H.; Sun, M.-H.; Wang, Z.; Yang, W.; Xie, Z.; Su, B.-L. Hierarchically Structured Zeolites: From Design to Application. Chem. Rev. 2020, 120, 11194–11294. [Google Scholar] [CrossRef]
- Serrano, D.P.; Escola, J.M.; Pizarro, P. Synthesis strategies in the search for hierarchical zeolites. Chem. Soc. Rev. 2013, 42, 4004–4035. [Google Scholar] [CrossRef]
- Verboekend, D.; Pérez-Ramírez, J. Design of hierarchical zeolite catalysts by desilication. Catal. Sci. Technol. 2011, 1, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Silaghi, M.-C.; Chizallet, C.; Raybaud, P. Challenges on molecular aspects of dealumination and desilication of zeolites. Microporous Mesoporous Mater. 2014, 191, 82–96. [Google Scholar] [CrossRef]
- Bai, D.; Meng, J.; Li, C.; Zhang, M.; Liang, C. Mesoporosity and Acidity Manipulation in ZSM-23 and their n-Hexadecane Hydroisomerization Performance. Chemistryselect 2022, 7, e202200839. [Google Scholar] [CrossRef]
- Guo, K.; Ma, A.; Wang, Z.; Li, J.; Wu, B.; Liu, T.; Li, D. Investigation of n-heptane hydroisomerization over alkali-acid-treated hierarchical Pt/ZSM-22 zeolites. New J. Chem. 2022, 46, 16752–16763. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Wang, S.; Wu, Z.W.; Qin, Z.F.; Dong, M.; Wang, J.G.; Fan, W.B. Aromatization of n-C7–n-C9 alkanes on a Pt/KZSM-5(deAl) catalyst. Catal. Sci. Technol. 2023, 13, 1009. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zhang, X.W.; Li, G.Z. Constructing optimally hierarchical HY zeolite for the synthesis of high-energy-density tricyclic hydrocarbon fuel. Mol. Catal. 2023, 535, 112871. [Google Scholar] [CrossRef]
- Mihályi, R.M.; Kollár, M.; Király, P.; Karolyc, Z.; Mavrodinova, V. Effect of extra-framework Al formed by successive steaming and acid leaching of zeolite MCM-22 on its structure and catalytic performance. Appl. Catal. A Gen. 2012, 417, 76–86. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Groen, J.C.; Moulijn, J.A.; Perez-Ramirez, J. Desilication: On the controlled generation of mesoporosity in MFI zeolites. J. Mater. Chem. 2006, 16, 2121–2131. [Google Scholar] [CrossRef]
- Chen, K.; Horstmeier, S.; Nguyen, V.T.; Wang, B.; Crossley, S.P.; Pham, T.; Gan, Z.; Hung, I.; White, J.L. Structure and Catalytic Characterization of a Second Framework Al(IV) Site in Zeolite Catalysts Revealed by NMR at 35.2 T. J. Am. Chem. Soc. 2020, 142, 7514–7523. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Liu, K.; Chen, W.; Li, J.; Xu, S.; Li, C.; Xiao, Y.; Liu, H.; Guo, X.; Liu, S.-B.; et al. Origin and Structural Characteristics of Tri-coordinated Extra-framework Aluminum Species in Dealuminated Zeolites. J. Am. Chem. Soc. 2018, 140, 10764–10774. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fan, B.; Zhi, Y.; Liu, C.; Xu, S.; Yu, Z.; Liu, Z. Dynamic Evolution of Aluminum Coordination Environments in Mordenite Zeolite and Their Role in the Dimethyl Ether (DME) Carbonylation Reaction. Angew. Chem. Int. Edit. 2022, 61, e202210658. [Google Scholar]
- Ravenelle, R.M.; Schüβler, F.; D’ Amico, A.; Danilina, N.; van Bokhoven, J.A.; Lercher, J.A.; Jones, C.W.; Sievers, C. Stability of Zeolites in Hot Liquid Water. J. Phys. Chem. C 2010, 114, 19582–19595. [Google Scholar] [CrossRef]
- van Donk, S.; Bitter, J.H.; Verberckmoes, A.; VersluijsHelder, M.; Broersma, A.; de Jong, K.P. Physicochemical characterization of porous materials: Spatially resolved accessibility of zeolite crystals. Angew. Chem. Int. Edit. 2005, 44, 1360–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, M.; Sushkevich, V.L.; van Bokhoven, J.A. On the location of Lewis acidic aluminum in zeolite mordenite and the role of framework-associated aluminum in mediating the switch between Brønsted and Lewis acidity. Chem. Sci. 2021, 12, 4094–4103. [Google Scholar] [CrossRef]
- Ravi, M.; Sushkevich, V.L.; van Bokhoven, J.A. Towards a better understanding of Lewis acidic aluminium in zeolites. Nat. Mater. 2020, 19, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Palčić, A.; Valtchev, V. Analysis and control of acid sites in zeolites. Appl. Catal. A Gen. 2020, 606, 117795. [Google Scholar] [CrossRef]
- Ono, Y. A survey of the mechanism in catalytic isomerization of alkanes. Catal. Today 2003, 81, 3–16. [Google Scholar] [CrossRef]
- Campelo, J.M.; Lafont, F.; Marinas, J.M. Comparison of the activity and selectivity of Pt/SAPO-5 and Pt/SAPO-11 in n-hexane and n-heptane hydroconversion. Appl. Catal. A Gen. 1997, 152, 53–62. [Google Scholar] [CrossRef]
- Blomsma, E.; Martens, J.A.; Jacobs, P.A. Reaction Mechanisms of Isomerization and Cracking of Heptane on Pd/H-Beta Zeolite. J. Catal. 1995, 155, 141–147. [Google Scholar] [CrossRef]
- Paul, Z.; Zhan, Z.; Manninger, I.; Sachtler, W.M.H. Skeletal Reactions of n-Hexane over Pt-NaY, Pt/SiO2, Hy, and Mixed Pt/Sio2 + HY Catalysts. J. Catal. 1995, 155, 43–51. [Google Scholar] [CrossRef]
- Martens, J.A.; Jacobs, P.A.; Weitkamp, J. Attempts to rationalize the distribution of hydrocracked products. II. Relative rates of primary hydrocracking modes of long chain paraffins in open zeolites. Appl. Catal. 1986, 20, 283–303. [Google Scholar] [CrossRef]
- Parton, R.; Uytterhoeven, L.; Martens, J.A.; Jacobs, P.A.; Froment, G.F. Synergism of ZSM-22 and Y zeolites in the bifunctional conversion of n-alkanes. Appl. Catal. 1991, 76, 131–142. [Google Scholar] [CrossRef]
- Alvarez, F.; Ribeiro, F.R.; Perot, G.; Thomazeau, C.; Guisnet, M. Hydroisomerization and Hydrocracking of Alkanes: 7. Influence of the Balance between Acid and Hydrogenating Functions on the Transformation ofn-Decane on PtHY Catalysts. J. Catal. 1996, 162, 179–189. [Google Scholar] [CrossRef]
- Claude, M.C.; Martens, J.A. Monomethyl-Branching of Long n-Alkanes in the Range from Decane to Tetracosane on Pt/H-ZSM-22 Bifunctional Catalyst. J. Catal. 2000, 190, 39–48. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | RC a (%) | Specific Surface Area (m2/g) | Volume (cm3/g) | ||||
|---|---|---|---|---|---|---|---|
| SBET | Smicro | Smeso | Vtotal | Vmicro | Vmeso | ||
| Pt/SSZ32 | 100 | 103.73 | 71.65 | 32.08 | 0.158 | 0.0400 | 0.118 |
| Pt/SSZ32-0.3A | 95.5 | 92.92 | 42.05 | 50.87 | 0.147 | 0.0203 | 0.127 |
| Pt/SSZ32-0.6A | 84.7 | 98.50 | 33.48 | 65.01 | 0.162 | 0.0184 | 0.144 |
| Pt/SSZ32-0.3AS | 99.7 | 134.85 | 62.30 | 72.55 | 0.178 | 0.0271 | 0.151 |
| Pt/SSZ32-0.6AS | 88.6 | 160.75 | 68.63 | 92.12 | 0.218 | 0.0342 | 0.184 |
| Catalysts | The Proportion of Different Al Species | Si/Al | ||
|---|---|---|---|---|
| FAl % (57 ppm) | Distorted FAl(~52 ppm) | EFAl % (0 ppm) | ||
| Pt/SSZ32 | 83.6 | 6.1 | 10.3 | 20.6 |
| Pt/SSZ32-0.3A | 80.6 | 5.4 | 14.0 | 17.4 |
| Pt/SSZ32-0.6A | 71.5 | 3.7 | 24.8 | 14.3 |
| Pt/SSZ32-0.3AS | 85.4 | 3.8 | 10.8 | 22.5 |
| Pt/SSZ32-0.6AS | 85.9 | 8.9 | 5.2 | 19.6 |
| Samples | Brønsted Acidity (µmol g−1) | Lewis Acidity (µmol g−1) | Total Acidity (µmol g−1) | B/L Ratio | ||||
|---|---|---|---|---|---|---|---|---|
| 200 °C | 350 °C | 200 °C | 350 °C | 200 °C | 350 °C | 200 °C | 350 °C | |
| Pt/SSZ32 | 86.8 | 40.8 | 47.0 | 13.1 | 133.8 | 53.9 | 1.85 | 3.12 |
| Pt/SSZ32-0.3A | 83.0 | 35.4 | 47.8 | 29.6 | 130.8 | 65.0 | 1.74 | 1.20 |
| Pt/SSZ32-0.6A | 72.5 | 34.8 | 53.7 | 35.4 | 126.3 | 70.2 | 1.35 | 0.98 |
| Pt/SSZ32-0.3AS | 95.5 | 43.8 | 39.1 | 27.9 | 134.6 | 71.7 | 2.44 | 1.57 |
| Pt/SSZ32-0.6AS | 116.1 | 50.0 | 32.8 | 20.2 | 149.0 | 70.2 | 3.54 | 2.47 |
| i-C16 Products | Yield (%) | ||||
|---|---|---|---|---|---|
| Pt/SSZ32 | Pt/SSZ32-0.3A | Pt/SSZ32-0.6A | Pt/SSZ32-0.3AS | Pt/SSZ32-0.6AS | |
| 2-Methyl isomer (pore mouth) | 13.30 | 14.03 | 11.19 | 13.27 | 15.04 |
| 3-Methyl isomer (pore mouth) | 9.02 | 9.40 | 8.07 | 8.85 | 9.19 |
| 4-Methyl isomer | 5.97 | 6.18 | 5.52 | 5.77 | 5.82 |
| 5-Methyl isomer | 6.08 | 5.90 | 5.78 | 5.48 | 5.57 |
| 6/7/8-Methyl isomers (key–lock) | 23.03 | 24.62 | 20.02 | 21.92 | 22.80 |
| Pore mouth/key–lock | 0.969 | 0.952 | 0.962 | 1.009 | 1.063 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhao, W.; Liu, L.; Niu, X.; Wang, Q. Tuning the Structure and Acidity of Pt/Hierarchical SSZ-32 Catalysts to Boost the Selective Hydroisomerization of n-Hexadecane. Catalysts 2023, 13, 702. https://doi.org/10.3390/catal13040702
Yang X, Zhao W, Liu L, Niu X, Wang Q. Tuning the Structure and Acidity of Pt/Hierarchical SSZ-32 Catalysts to Boost the Selective Hydroisomerization of n-Hexadecane. Catalysts. 2023; 13(4):702. https://doi.org/10.3390/catal13040702
Chicago/Turabian StyleYang, Xinyue, Wenli Zhao, Linlin Liu, Xiaopo Niu, and Qingfa Wang. 2023. "Tuning the Structure and Acidity of Pt/Hierarchical SSZ-32 Catalysts to Boost the Selective Hydroisomerization of n-Hexadecane" Catalysts 13, no. 4: 702. https://doi.org/10.3390/catal13040702