
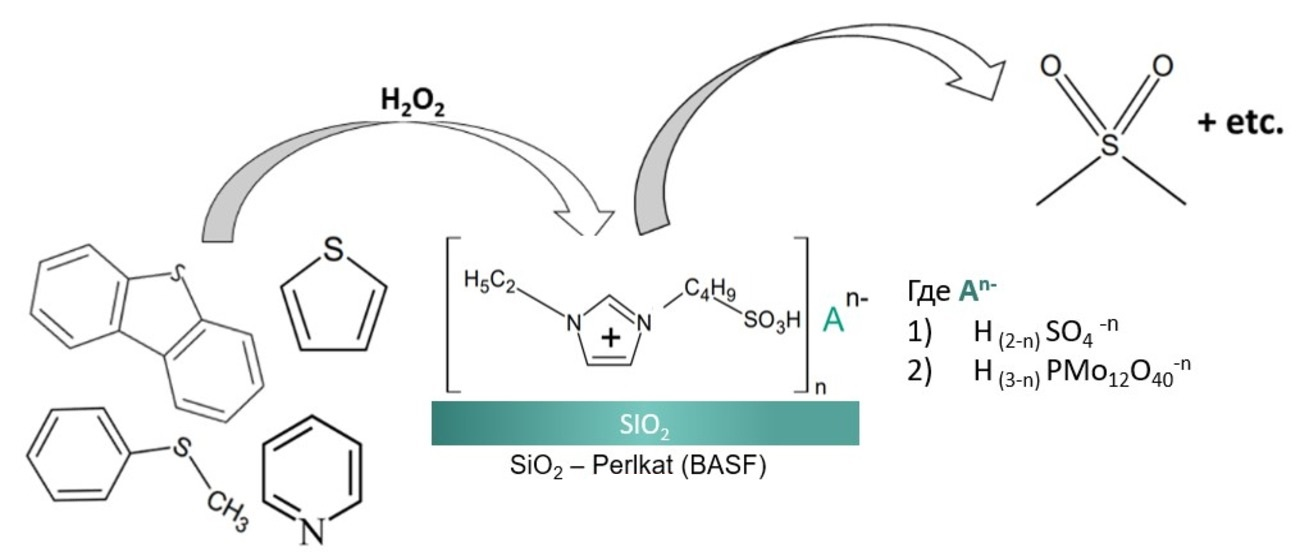
Supported Ionic Liquid Catalysts for the Oxidation of S- and N-Containing Compounds—The Effect of Bronsted Sites and Heteropolyacid Concentration
 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
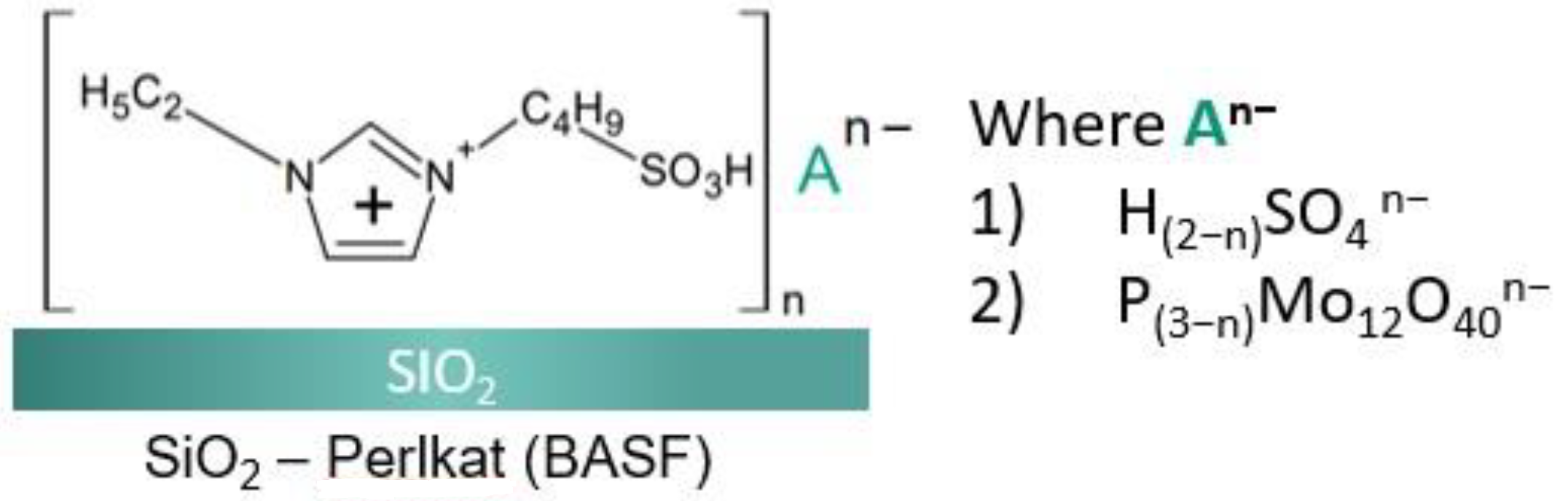
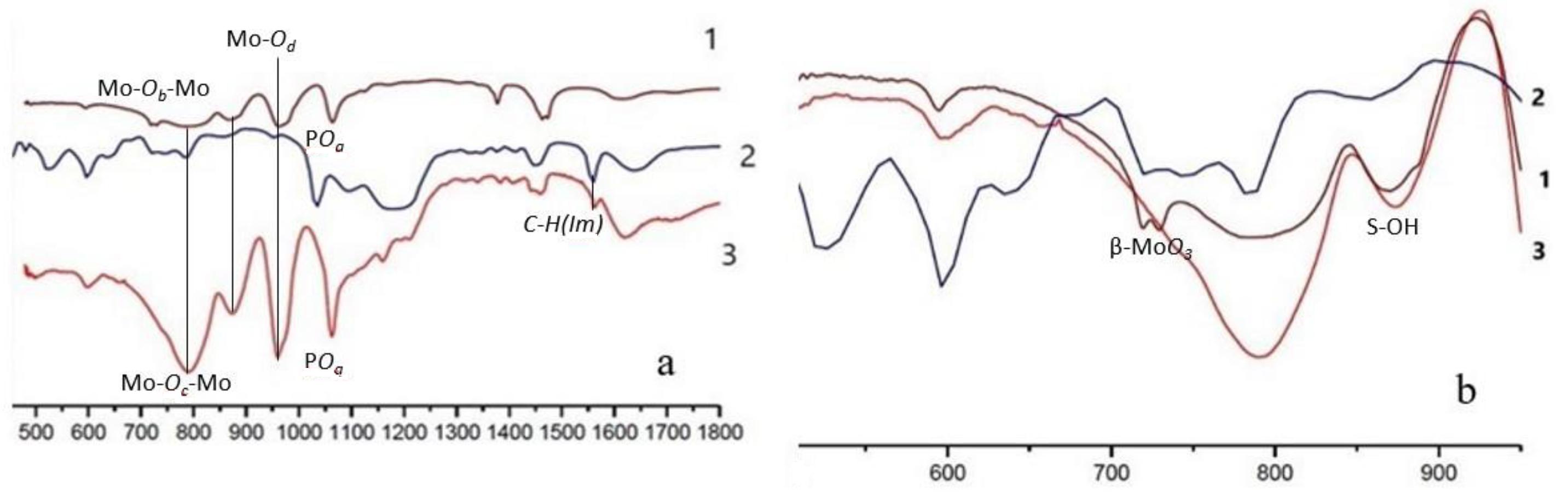
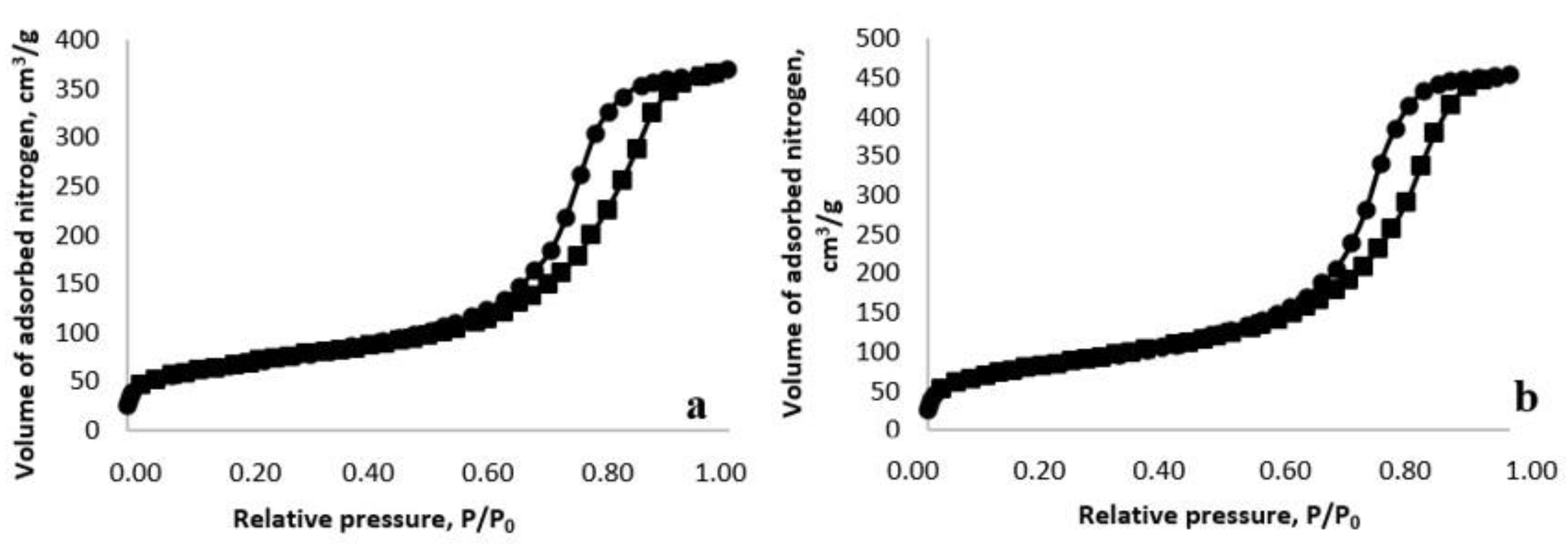
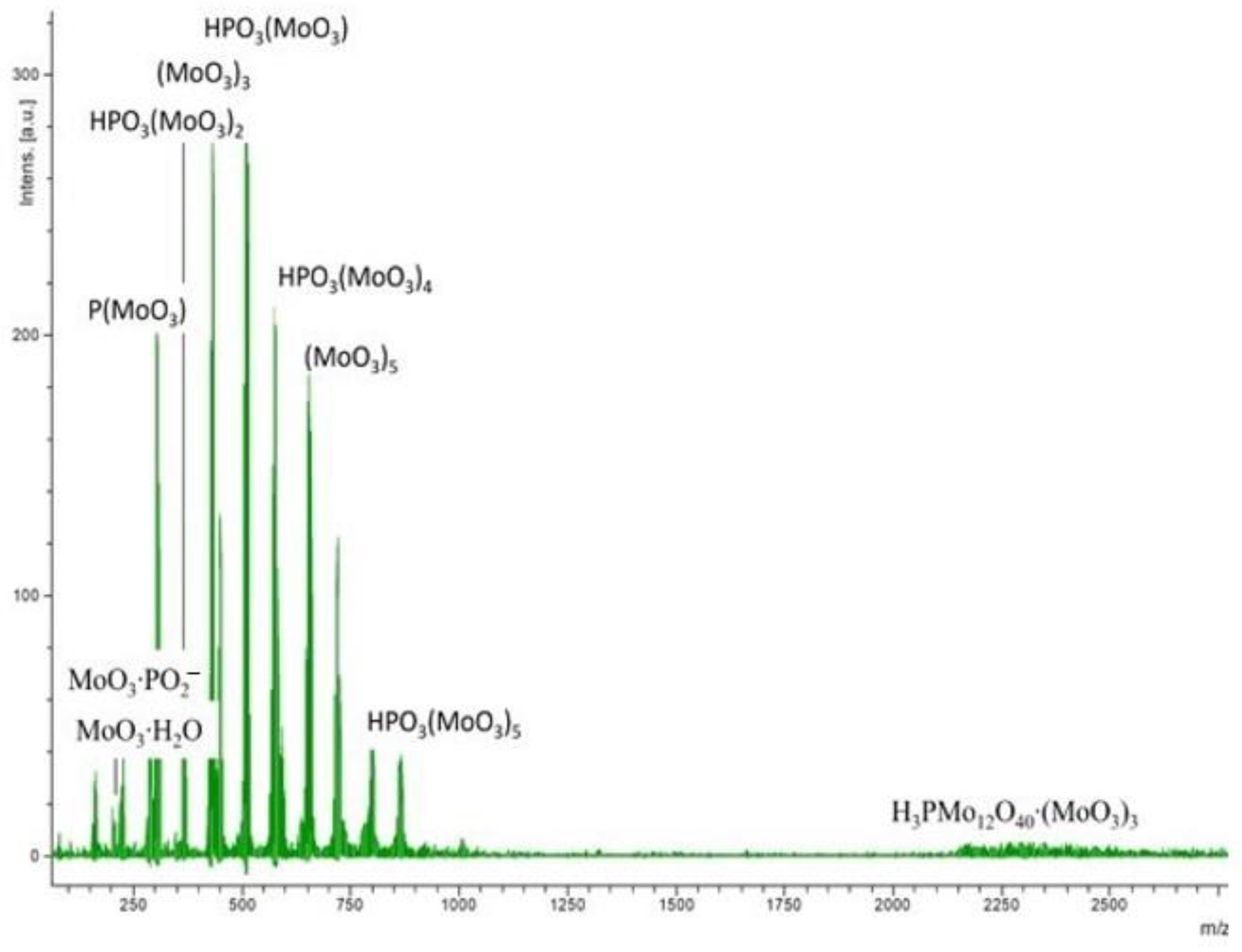
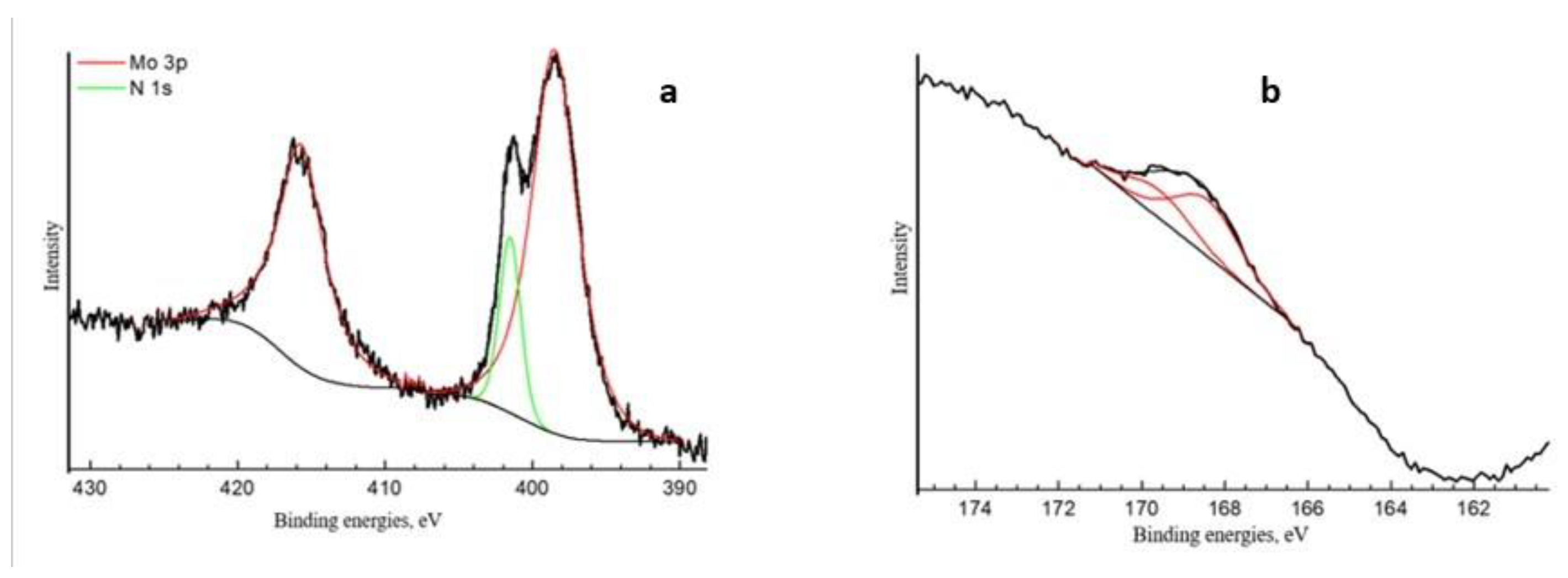
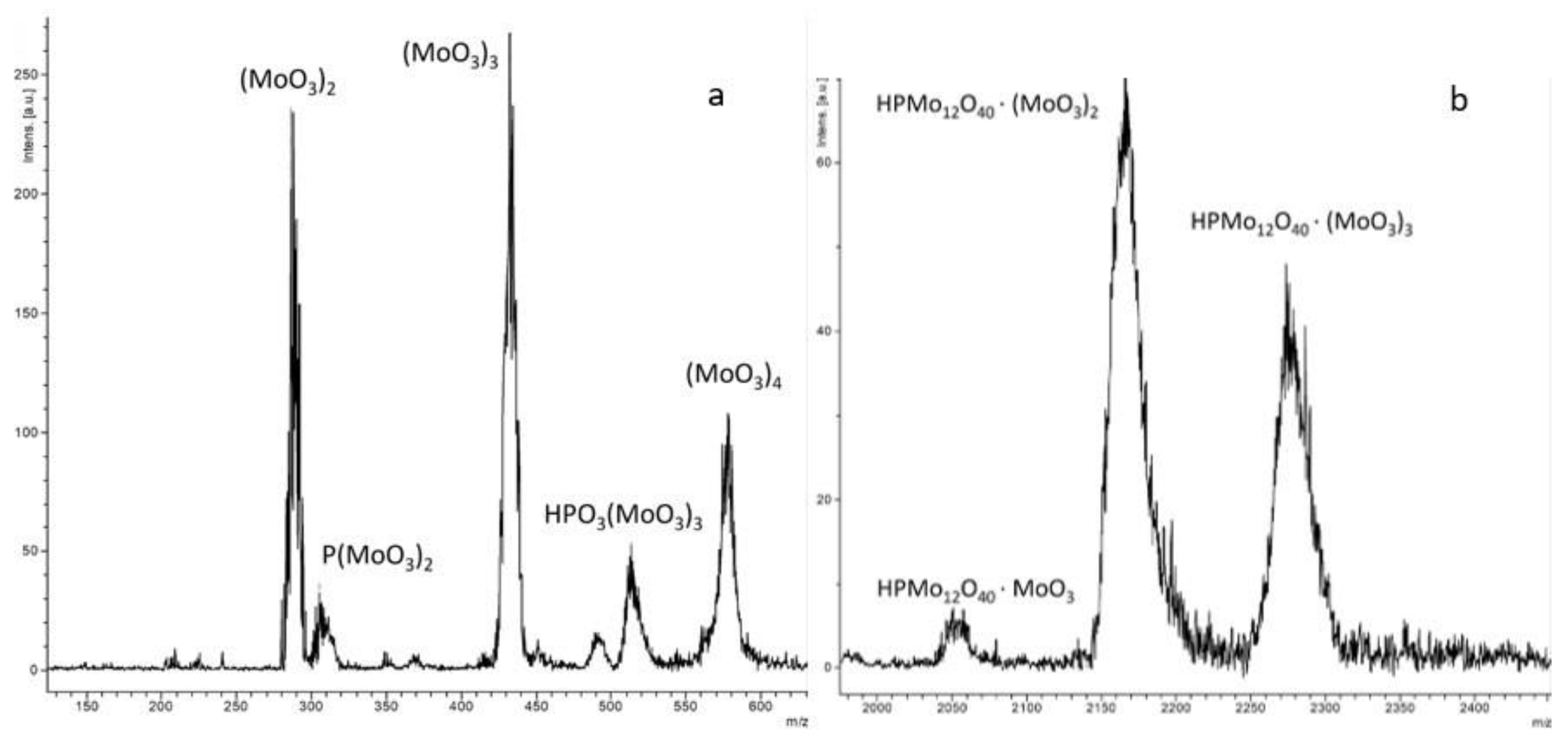
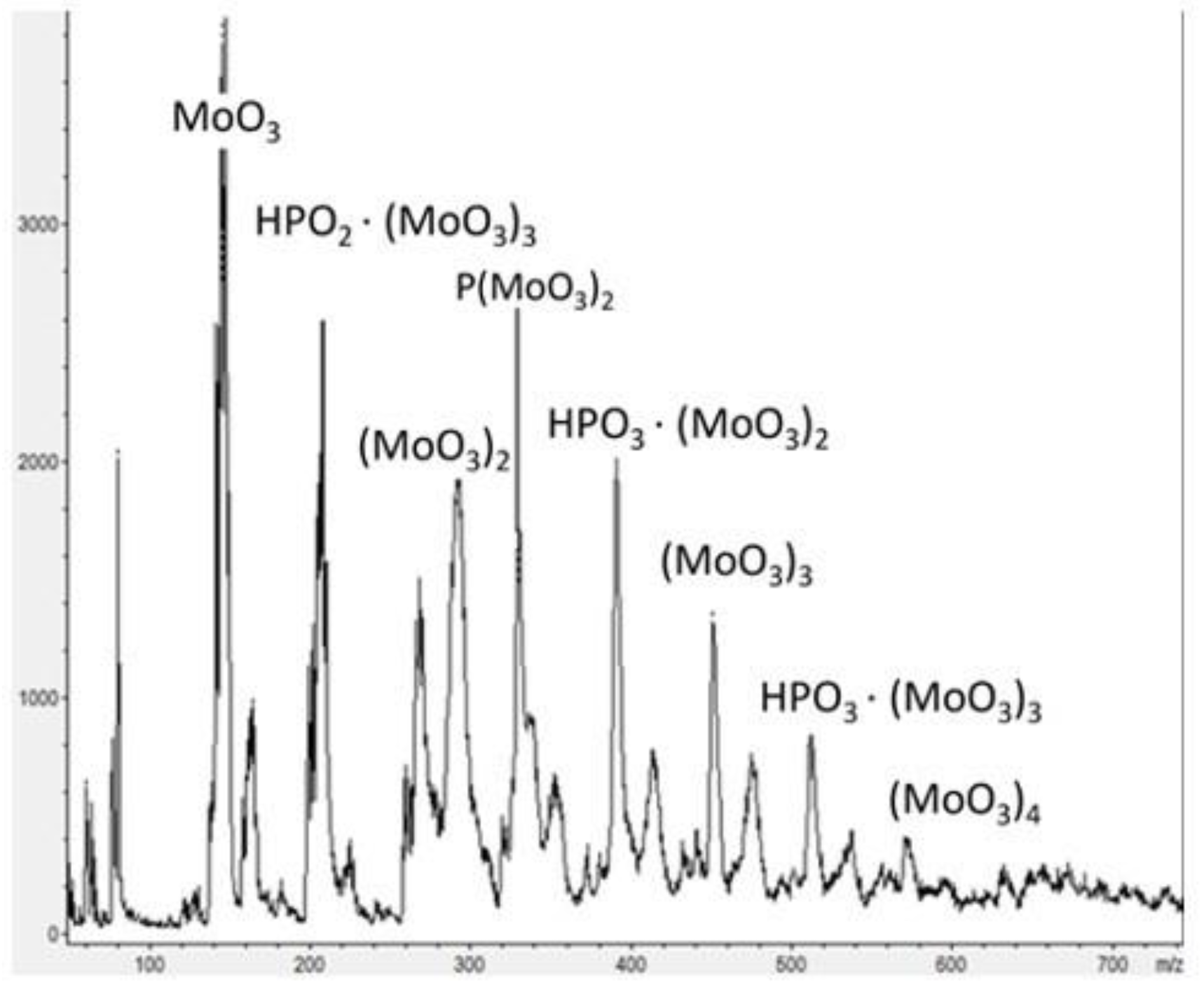
2.1. Physico-Chemical Characteristics of the Synthesized Ionic Liquids and Heterogeneous Catalysts
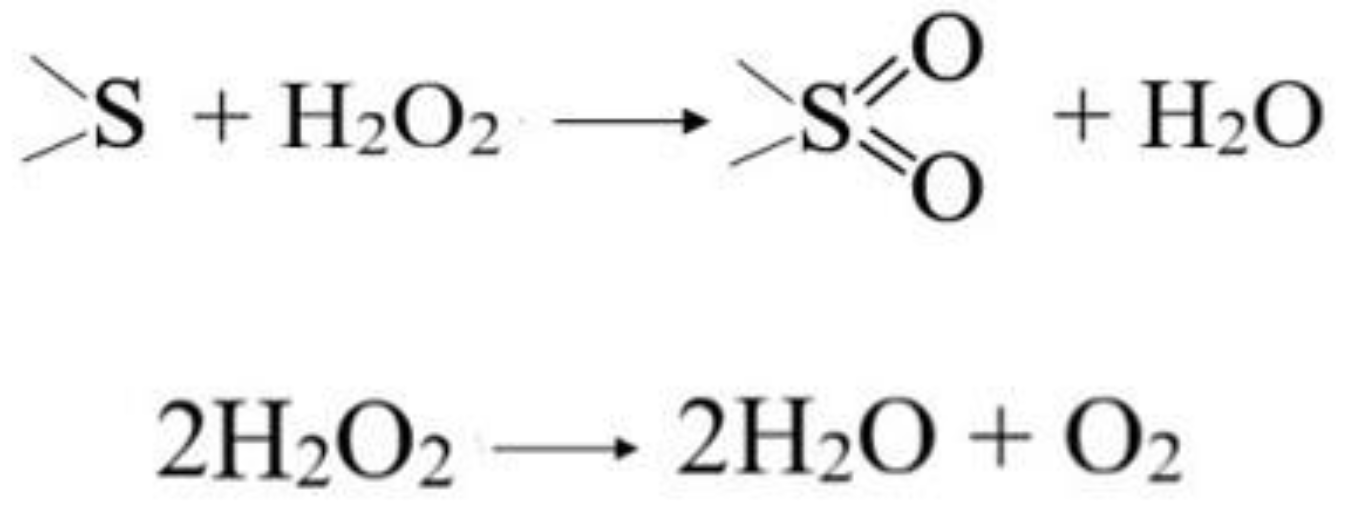
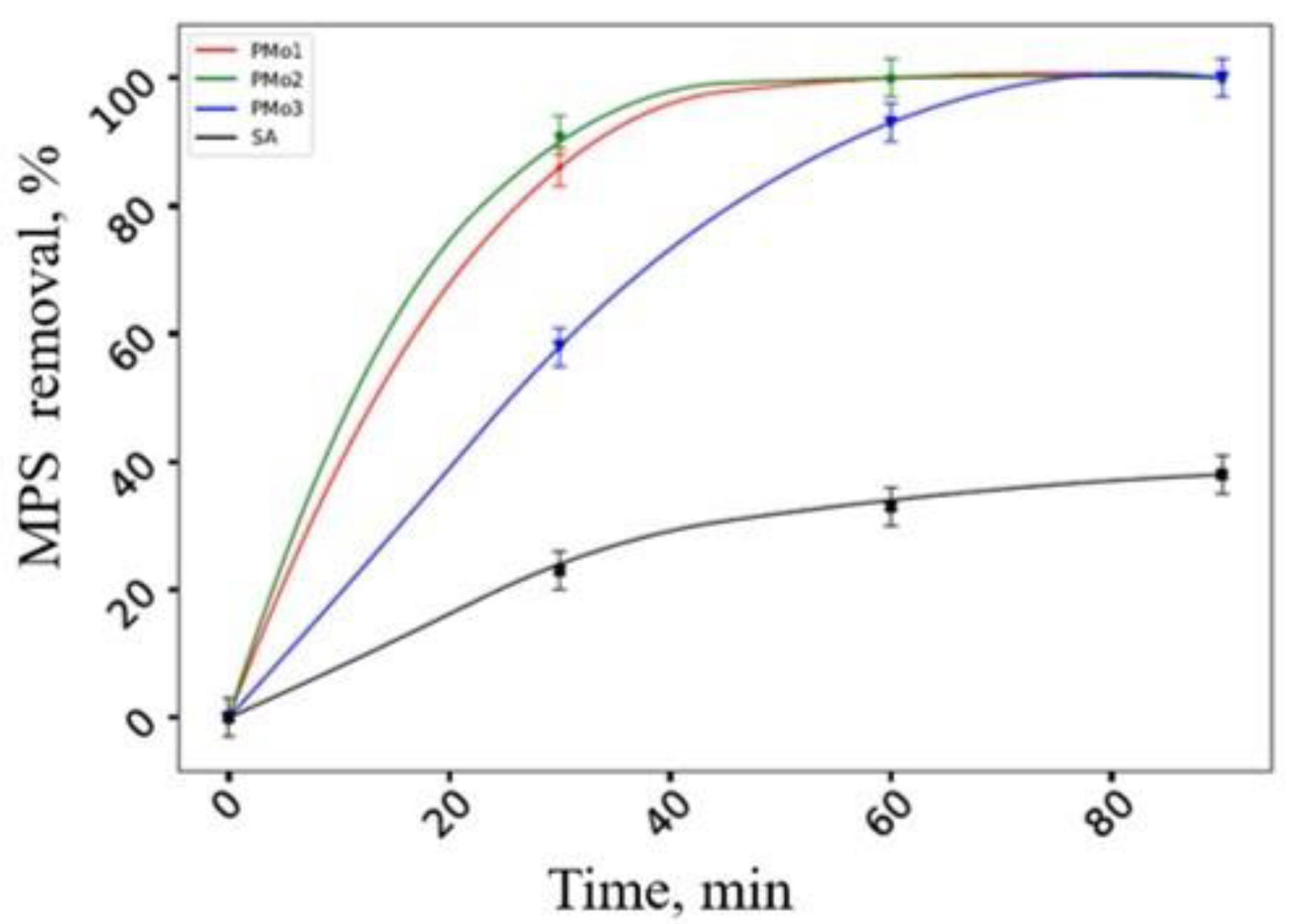
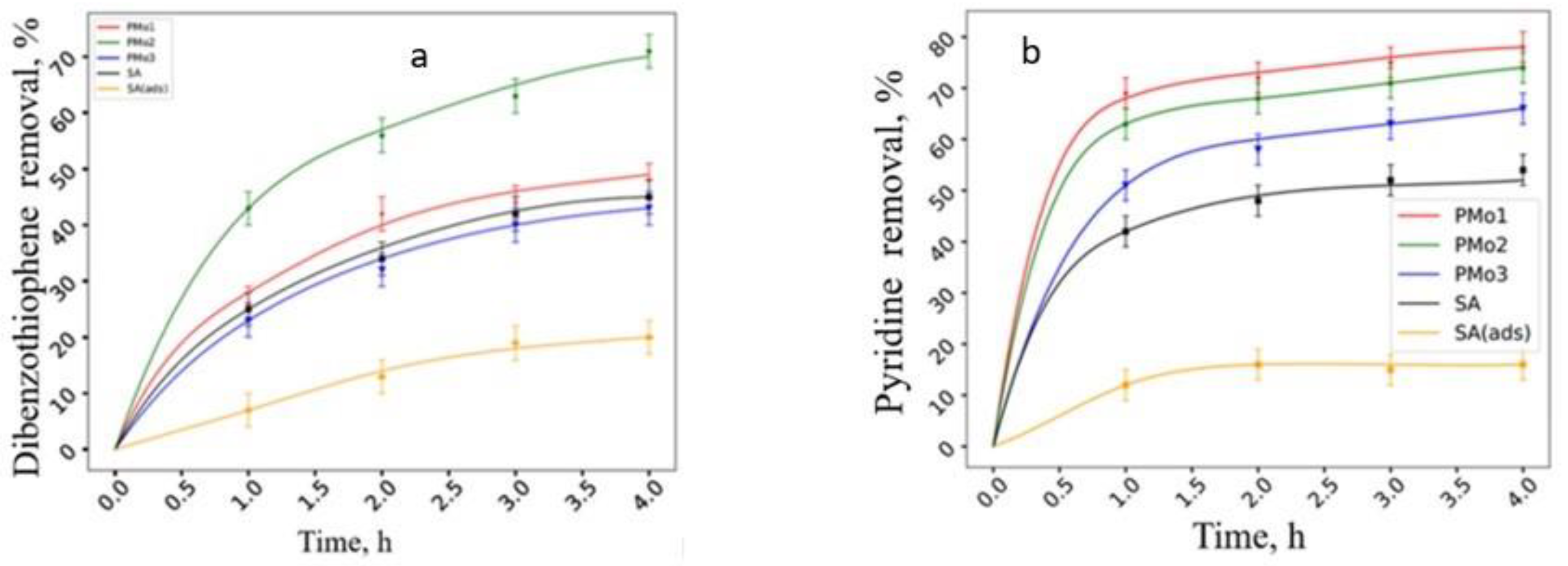
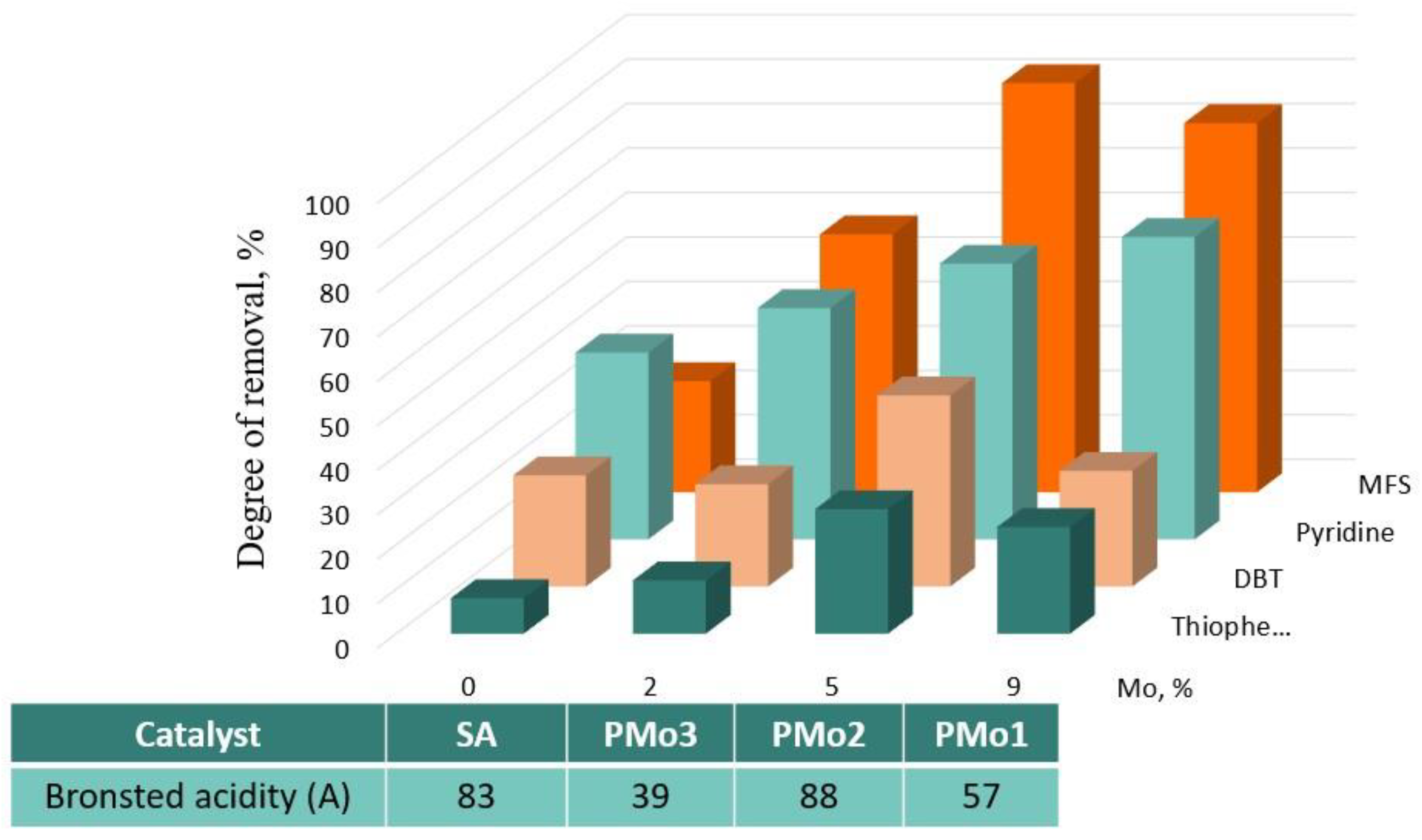
2.2. Catalytic Properties of Heterogeneous Compositions
3. Materials and Methods
3.1. Materials
3.2. Preparation of Ionic Liquids
3.3. Preparation of Catalysts
3.4. Methods for the Analysis of Ionic Liquids and Catalysts
3.5. Catalytic Test
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
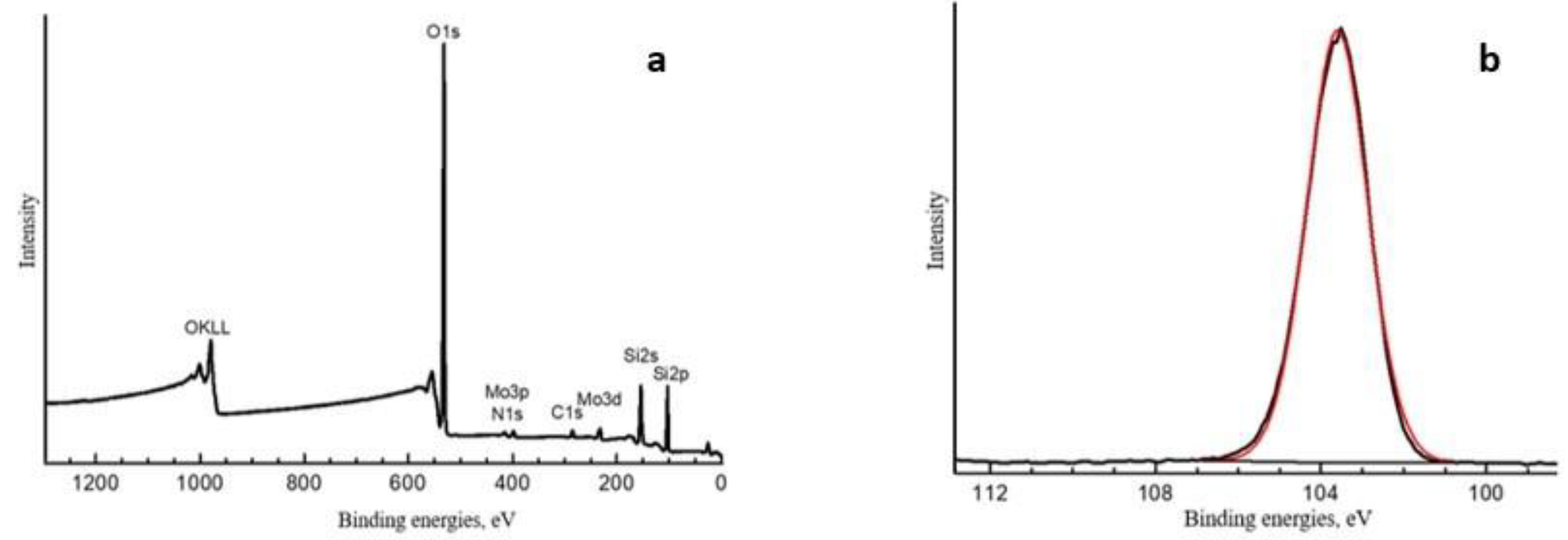
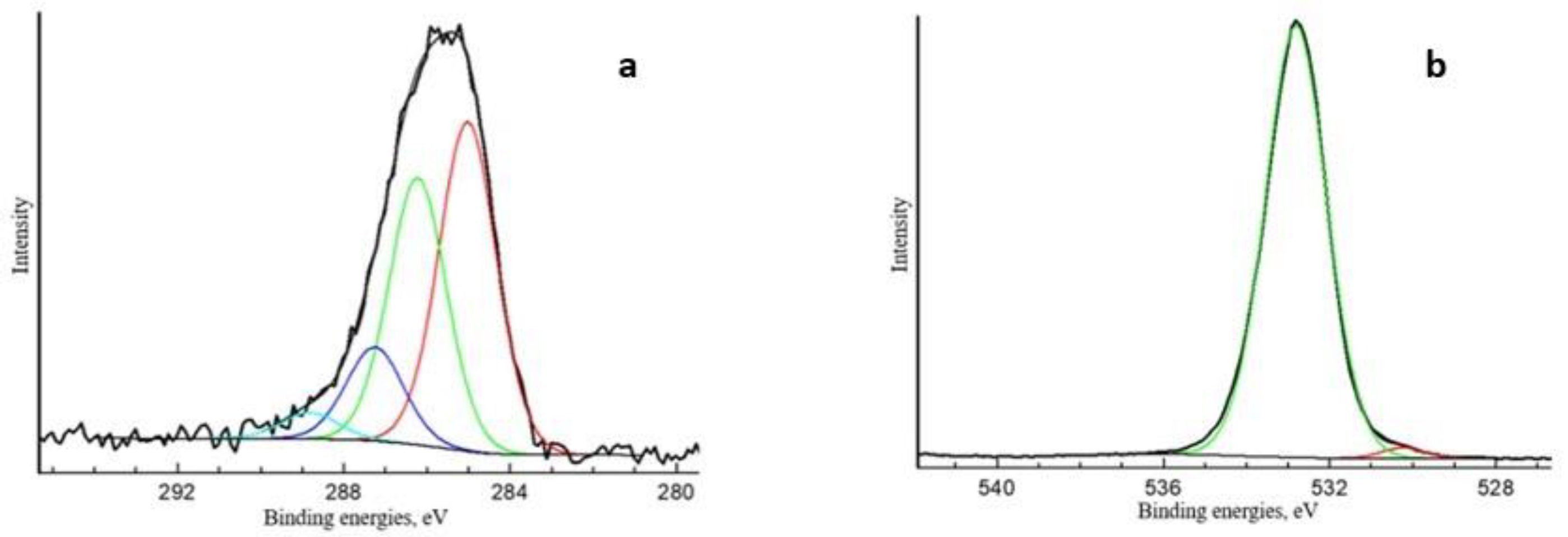
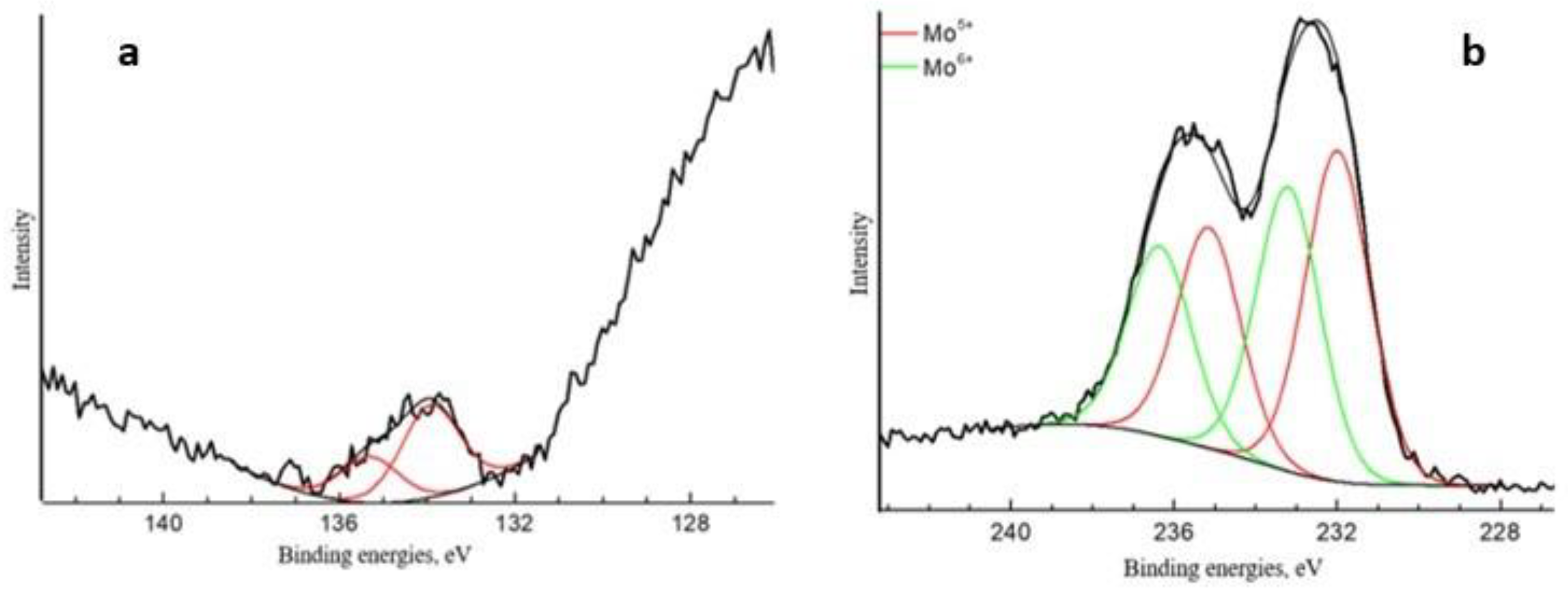
Appendix A. XPS and SALDI Spectrums






References
- Koolen, C.D.; Rothenberg, G. Air Pollution in Europe. ChemSusChem 2019, 12, 164–172. [Google Scholar] [CrossRef]
- Cui, T.; Fan, H.; Yang, Z.; Feng, J.; Li, W. A comprehensive review on oxidative desulfurization catalysts targeting clean energy and environment. J. Mater. Chem. A 2020, 8, 2246–2285. [Google Scholar]
- Yang, L.; Franco, V.; Mock, P.; Kolke, R.; Zhang, S.; Wu, Y.; German, J. Experimental Assessment of NO x Emissions from 73 Euro 6 Diesel Passenger Cars. Environ. Sci. Technol. 2015, 49, 14409–14415. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.; Tang, X.; Zhang, Q.; Dai, S.; Peng, H.; Lin, Y.; Tian, Z.; Lu, Z.; Chen, L. Ligand Defect Density Regulation in Metal–Organic Frameworks by Functional Group Engineering on Linkers. Nano Lett. 2022, 22, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Abro, R.; Abro, M.; Gao, S.; Bhutto, A.W.; Ali, Z.M.; Shah, A.; Chen, X.; Yu, G. Extractive Denitrogenation of Fuel Oils Using Ionic Liquids: A Review. RSC Adv. 2016, 6, 93932–93946. [Google Scholar] [CrossRef]
- Glotov, A.P.; Vutolkina, A.V.; Vinogradov, N.A.; Pimerzin, A.A.; Vinokurov, V.A.; Pimerzin, A.A. Enhanced HDS and HYD Activity of Sulfide Co-PMo Catalyst Supported on Alumina and Structured Mesoporous Silica Composite. Catal. Today 2021, 377, 82–91. [Google Scholar] [CrossRef]
- Tanimu, A.; Alhooshani, K. Advanced Hydrodesulfurization Catalysts: A Review of Design and Synthesis. Energy Fuels 2019, 33, 2810–2838. [Google Scholar] [CrossRef]
- Liu, F.; Yu, J.; Qazi, A.B.; Zhang, L.; Liu, X. Metal-Based Ionic Liquids in Oxidative Desulfurization: A Critical Review. Environ. Sci. Technol. 2021, 55, 1419–1435. [Google Scholar] [CrossRef]
- Houda, S.; Lancelot, C.; Blanchard, P.; Poinel, L.; Lamonier, C. Oxidative Desulfurization of Heavy Oils with High Sulfur Content: A Review. Catalysts 2018, 8, 344. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Anbia, M. Highly efficient oxidative desulfurization catalyzed by copper-based materials using hydrogen peroxide as oxidant. Fuel 2022, 324, 124471. [Google Scholar] [CrossRef]
- Ma, S.; Bao, W.; Liu, B.; Zhang, C.; Wang, C.; Liu, Y.; Guo, P. Mo11V polyoxometalate encapsulated into hollow mesoporous carbon spheres: A highly efficient and ultrastable catalyst for oxidative desulfurization. Appl. Surf. Sci. 2022, 606, 154781. [Google Scholar] [CrossRef]
- Bryzhin, A.A.; Gantman, M.G.; Buryak, A.K.; Tarkhanova, I.G. Brønsted Acidic SILP-Based Catalysts with H3PMo12O40 or 334 H3PW12O40 in the Oxidative Desulfurization of Fuels. Appl. Catal. B Environ. 2019, 257, 117938. [Google Scholar] [CrossRef]
- Kumar, S.; Chandra Srivastava, V.; Kumar, A.; Nanoti, S.M. Effect of Gas Oil Composition on Performance Parameters of the Extractive Desulfurization Process. RSC Adv. 2016, 6, 25293–25301. [Google Scholar] [CrossRef]
- Valenzuela, C.; Donoso, C.; Guzmán-Beckmann, L. Extraction of Sulfur from Commercial Gasoline Using 1-Butyl-3-Methylimidazolium Tetrafluoroborate [BMIM] [BF4] as the Extraction Solvent. Key Eng. Mater. 2020, 834, 42–48. [Google Scholar] [CrossRef]
- Ahmed, O.U.; Mjalli, F.S.; Talal, A.-W.; Al-Wahaibi, Y.; Al Nashef, I.M. Extractive Desulfurization of Liquid Fuel Using Modified Pyrollidinium and Phosphonium Based Ionic Liquid Solvents. J. Solut. Chem. 2018, 47, 468–483. [Google Scholar] [CrossRef]
- Aslam, S.; Subhan, F.; Yan, Z.; Etim, U.J.; Zeng, J. Dispersion of Nickel Nanoparticles in the Cages of Metal-Organic Framework: An Efficient Sorbent for Adsorptive Removal of Thiophene. Chem. Eng. J. 2017, 315, 469–480. [Google Scholar] [CrossRef]
- Zhao, Z.; Zuhra, Z.; Qin, L.; Zhou, Y.; Zhang, L.; Tang, F.; Mu, C. Confinement of Microporous MOF-74(Ni) within Mesoporous y-Al2O3 Beads for Excellent Ultra-Deep and Selective Adsorptive Desulfurization Performance. Fuel Process. Technol. 2018, 276–282. [Google Scholar] [CrossRef]
- Wang, S.; Tong, H.; Li, H.; Shi, X.; Liu, D.; Li, J.; Guo, K.; Zhao, L.; Song, S.; Chen, L.; et al. Synthesis of a Phosphomolybdic Acid/Nanocrystalline Titanium Silicalite-1 Catalyst in the Presence of Hydrogen Peroxide for Effective Adsorption-Oxidative Desulfurization. New J. Chem. 2022, 46, 2559–2568. [Google Scholar] [CrossRef]
- Vafaee, F.; Mandizadeh, S.; Amiri, O.; Jahangiri, M.; Salavati-Niasari, M. Correction: Synthesis and Characterization of AFe2O4 (A: Ni, Co, Mg)–Silica Nanocomposites and Their Application for the Removal of Dibenzothiophene (DBT) by an Adsorption Process: Kinetics, Isotherms and Experimental Design. RSC Adv. 2022, 12, 20973–20974. [Google Scholar] [CrossRef] [PubMed]
- Crandall, B.S.; Zhang, J.; Stavila, V.; Allendorf, M.D.; Li, Z. Desulfurization of Liquid Hydrocarbon Fuels with Microporous and Mesoporous Materials: Metal-Organic Frameworks, Zeolites, and Mesoporous Silicas. Ind. Eng. Chem. Res. 2019, 58, 19322–19352. [Google Scholar] [CrossRef]
- Khudoyarova, O.; Gordienko, O.; Sydoruk, T.; Titov, T.; Petruk, R.; Prokopchuk, S. Adsorptive desulfurization of industrial wastewater. Environ. Probl. 2020, 5, 102–105. [Google Scholar] [CrossRef]
- Singh, S.K.; Savoy, A.W. Ionic Liquids Synthesis and Applications: An Overview. J. Mol. Liq. 2020, 297, 112038. [Google Scholar] [CrossRef]
- Moreno, D.; Gonzalez-Miquel, M.; Ferro, V.R.; Palomar, J. Molecular and Thermodynamic Properties of Zwitterions versus Ionic Liquids: A Comprehensive Computational Analysis to Develop Advanced Separation Processes. ChemPhysChem 2018, 19, 794. [Google Scholar] [CrossRef]
- Hatab, F.A.; Darwish, A.S.; Lemaoui, T.; Warrag, S.E.E.; Benguerba, Y.; Kroon, M.C.; AlNashef, I.M. Extraction of Thiophene, Pyridine, and Toluene from n-Decane as a Diesel Model Using Betaine-Based Natural Deep Eutectic Solvents. J. Chem. Eng. Data 2020, 65, 5443–5457. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, Y.; Ren, J.; Wu, B.; Wang, X.; Zhao, D.; Li, F. Extractive/Catalytic Oxidative Mechanisms over [Hnmp]ClxFeCl3 Ionic Liquids towards the Desulfurization of Model Oils. New J. Chem. 2019, 43, 7725–7732. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.A.; de la Fuente, N.; Zhou, S.; Jiang, P.; Song, Y.; Zhou, X. Roles of the Structural Defects and the Combined Acidity of H 3 PW 12 O 40 /Zr-MCM-41 Catalysts in Ultralow Sulfur Diesel Production. New J. Chem. 2022, 46, 2081–2093. [Google Scholar] [CrossRef]
- Vafaeezadeh, M.; Alinezhad, H. Brønsted Acidic Ionic Liquids: Green Catalysts for Essential Organic Reactions. J. Mol. Liq. 2016, 218, 95–105. [Google Scholar] [CrossRef]
- Ibrahim, M.H.; Hayyan, M.; Hashim, M.A.; Hayyan, A. The Role of Ionic Liquids in Desulfurization of Fuels: A Review. Renew. Sustain. Energy Rev. 2017, 76, 1534–1549. [Google Scholar] [CrossRef]
- Mai, N.L.; Ahn, K.; Koo, Y.-M. Methods for Recovery of Ionic Liquids—A Review. Process Biochem. 2014, 49, 872–881. [Google Scholar] [CrossRef]
- Ali-Zade, A.G.; Buryak, A.K.; Zelikman, V.M.; Oskolok, K.V.; Tarkhanova, I.G. SILCs in Oxidative Desulfurization: Effect of Support and Heteropolyanion. New J. Chem. 2020, 44, 6402–6410. [Google Scholar] [CrossRef]
- Gorbunov, V.S.; Bryzhin, A.A.; Popov, A.G.; Tarkhanova, I.G. Immobilized Acid Catalysts in the Oxidation of Sulfur-Containing Compounds with Hydrogen Peroxide. Pet. Chem. 2021, 61, 1260–1269. [Google Scholar] [CrossRef]
- Akopyan, A.; Eseva, E.; Polikarpova, P.; Kedalo, A.; Vutolkina, A.; Glotov, A. Deep Oxidative Desulfurization of Fuels in the Presence of Brönsted Acidic Polyoxometalate-Based Ionic Liquids. Molecules 2020, 25, 536. [Google Scholar] [CrossRef] [Green Version]
- Prado, G.H.C.; Rao, Y.; de Klerk, A. Nitrogen Removal from Oil: A Review. Energy Fuels 2017, 31, 14–36. [Google Scholar] [CrossRef]
- Julião, D.; Gomes, A.C.; Pillinger, M.; Gonçalves, I.S.; Balula, S.S. Desulfurization and Denitrogenation Processes to Treat Diesel 388 Using Mo(VI)-Bipyridine Catalysts. Chem. Eng. Technol. 2020, 43, 1774–1783. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, M.; Liu, Z.; Li, Y.; Tian, S. Direct and Rapid Quantitative Analysis of Alkyldibenzothiophenes in Deeply Hy- 390 drodesulfurized Diesel Fuel by Gas Chromatography Quadrupole Time-of-Flight Mass Spectrometry. Energy Fuels 2017, 31, 9125–9131. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Aouissi, A.; Bettahar, M.; Launay, S.; Fournier, M. Catalysis by 12-Molybdophosphates. J. Catal. 1996, 164, 16–27. [Google Scholar] [CrossRef]
- Langer, R.; Phys, J. FT-IR investigation of polarizable, strong hydrogen bonds in sulfonic acid-sulfoxide, phosphine oxide, and arsine oxide complexes in the middle and far-infrared region. J. Phys. Chem. 1995, 99, 12214–12219. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Mendoza-Sanchez, B.; Fernandez, V.; Veenstra, R.; Dukstiene, N.; Roberts, A.; Fairley, N. Generalized Molybdenum Oxide Surface Chemical State XPS Determination via Informed Amorphous Sample Model. Appl. Surf. Sci. 2015, 326, 151–161. [Google Scholar] [CrossRef]
- Kozhevnikov, I. Catalysts for Fine Chemical Synthesis. In Catalysis by Polyoxometalates 2; 2002; Volume 2, p. 220. Wiley: Hoboken, NJ, USA. [Google Scholar]
- Smolin, R.A. Catalytic Peroxide Decomposition of a Publicly Available Oxo-Peroxo Molybdenum Compound. Bull. Kazan Technol. Univ. 2011, 15, 57–61. [Google Scholar]
- Tarkhanova, I.G.; Bryzhin, A.A.; Gantman, M.G.; Yarovaya, T.P.; Lukiyanchuk, I.V.; Nedozorov, P.M.; Rudnev, V.S. Ce-, Zr-Containing Oxide Layers Formed by Plasma Electrolytic Oxidation on Titanium as Catalysts for Oxidative Desulfurization. Surf. Coat. Technol. 2019, 362, 132–140. [Google Scholar] [CrossRef]
- Bryzhin, A.A.; Tarkhanova, I.G.; Gantman, M.G.; Rudnev, V.S.; Vasilyeva, M.S.; Lukiyanchuk, I.V. Titanium-Supported W-Containing PEO Layers Enriched with Mn or Zn in Oxidative Desulfurization and the Zwitterionic Liquid Effect. Surf. Coat. Technol. 2020, 393, 125746. [Google Scholar] [CrossRef]
- Tamura, M.; Shimizu, K.; Satsuma, A. Comprehensive IR Study on Acid/Base Properties of Metal Oxides. Appl. Catal. A Gen. 2012, 433–434, 135–145. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | (keV) | Weight, % | Sigma | Atom, % | K |
|---|---|---|---|---|---|
| C K | 0.277 | 16.13 | 0.18 | 23.23 | 3.06 |
| N K | 0.392 | 5.94 | 0.24 | 7.36 | 4.67 |
| O K | 0.525 | 52.00 | 0.37 | 56.22 | 43.59 |
| Si K | 1.739 | 19.49 | 0.11 | 12.00 | 38.16 |
| P K * | 2.010 | 0.04 | 0.02 | 0.02 | 0.07 |
| S K | 2.307 | 0.03 | 0.02 | 0.02 | 0.83 |
| Mo L | 2.293 | 6.37 | 0.10 | 1.15 | 10.44 |
| Element | (keV) | Weight, % | Sigma | Atom, % | K |
|---|---|---|---|---|---|
| C K | 0.277 | 11.09 | 0.18 | 23.23 | 3.06 |
| N K | 0.392 | 3.91 | 0.24 | 7.37 | 4.67 |
| O K | 0.525 | 57.56 | 0.37 | 56.22 | 43.59 |
| Si K | 1.739 | 26.09 | 0.11 | 12.00 | 38.16 |
| S K | 2.307 | 0.54 | 0.02 | 0.29 | 0.83 |
| Catalyst | SBET (m2/g) | Dp (nm) | Vp (m3/g) | Mo, wt.% | BAS * | LAS * |
|---|---|---|---|---|---|---|
| SiO2 | 300 | 10.0 | 0.75 | |||
| PMo1 | 246 | 8.6 | 0.60 | 9.0 | 57 | 17 |
| PMo2 | 276 | 9.1 | 0.62 | 5.0 | 88 | 13 |
| PMo3 | 291 | 9.8 | 0.66 | 2.4 | 39 | 3 |
| SA | 297 | 9.1 | 0.72 | 83 | 0 |
| Spectrum | Content Element, at. % | Binding Energies, eV | Fraction, at.% | Type of Chemical Bond |
|---|---|---|---|---|
| O1s | 69.01 | 530.2 532.8 | 1.20 67.81 | O-Met O-C, SiO2 |
| N1s | 0.52 | 401.5 | 0.52 | NR4+ |
| P2p3/2 | 0.12 | 133.9 | 0.12 | P5+ |
| Si2p | 25.24 | 103.6 | 25.24 | SiO2 |
| Mo3d5/2 | 0.77 | 232.0 233.2 | 0.42 0.35 | Mo5+ Mo6+ |
| C1s | 4.11 | 285.0 286.2 287.3 | 1.89 1.53 0.54 | C-C C-O, C-N N-C=N, C=O |
| 288.8 | 0.15 | O-C=O | ||
| S2p3/2 | 0.23 | 168.4 | 0.23 | S6+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbunov, V.; Buryak, A.; Oskolok, K.; Popov, A.G.; Tarkhanova, I. Supported Ionic Liquid Catalysts for the Oxidation of S- and N-Containing Compounds—The Effect of Bronsted Sites and Heteropolyacid Concentration. Catalysts 2023, 13, 664. https://doi.org/10.3390/catal13040664
Gorbunov V, Buryak A, Oskolok K, Popov AG, Tarkhanova I. Supported Ionic Liquid Catalysts for the Oxidation of S- and N-Containing Compounds—The Effect of Bronsted Sites and Heteropolyacid Concentration. Catalysts. 2023; 13(4):664. https://doi.org/10.3390/catal13040664
Chicago/Turabian StyleGorbunov, Vladislav, Aleksey Buryak, Kirill Oskolok, Andrey G. Popov, and Irina Tarkhanova. 2023. "Supported Ionic Liquid Catalysts for the Oxidation of S- and N-Containing Compounds—The Effect of Bronsted Sites and Heteropolyacid Concentration" Catalysts 13, no. 4: 664. https://doi.org/10.3390/catal13040664