
One-Pot Synthesis of Benzoxazole/Benzothiazole-Substituted Esters by Michael Addition: A Selective Construction of C-N/C-S Bonds
Abstract
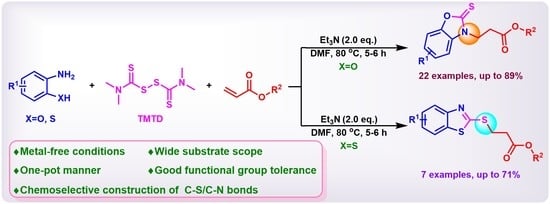
:
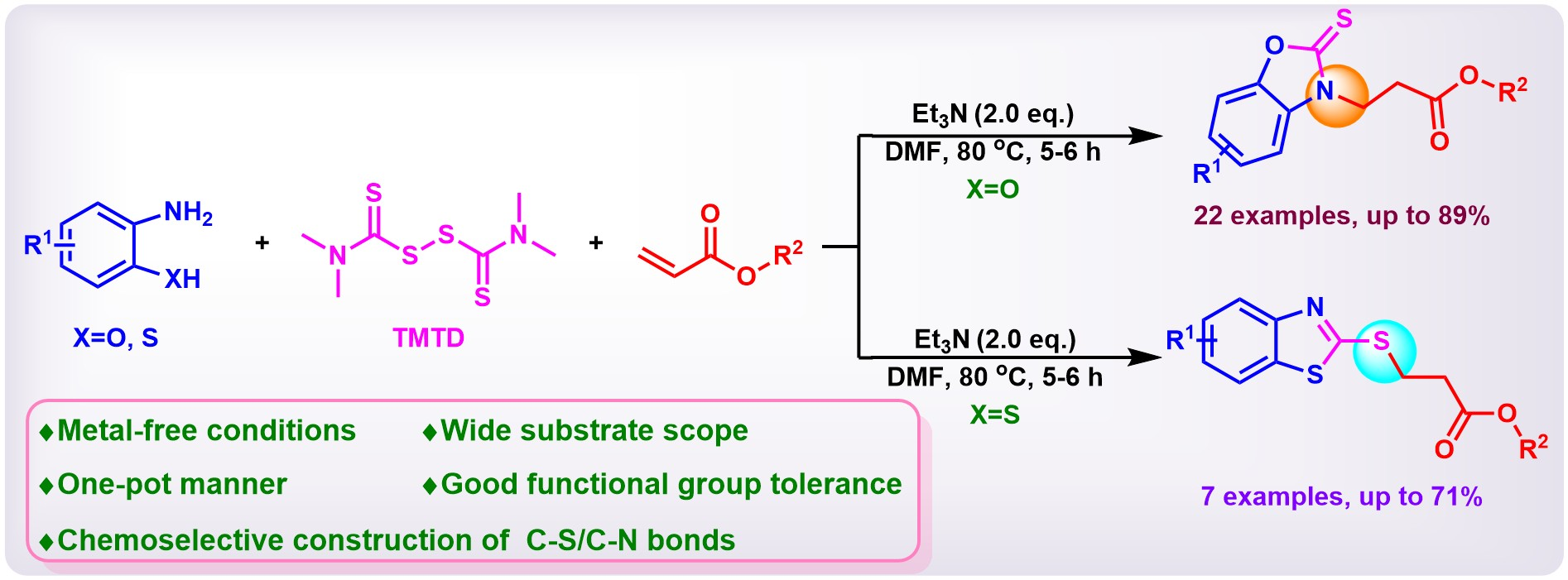
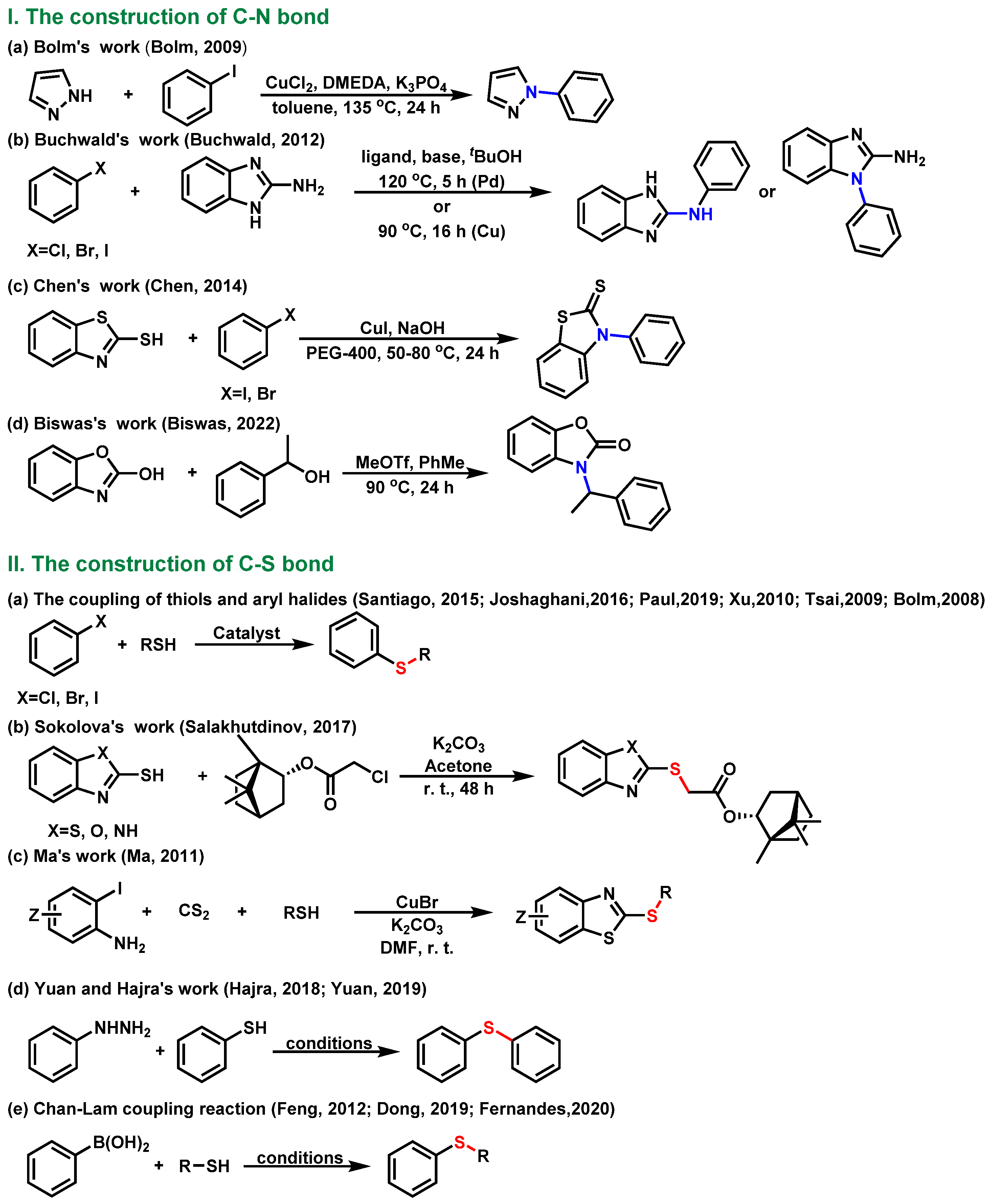
1. Introduction
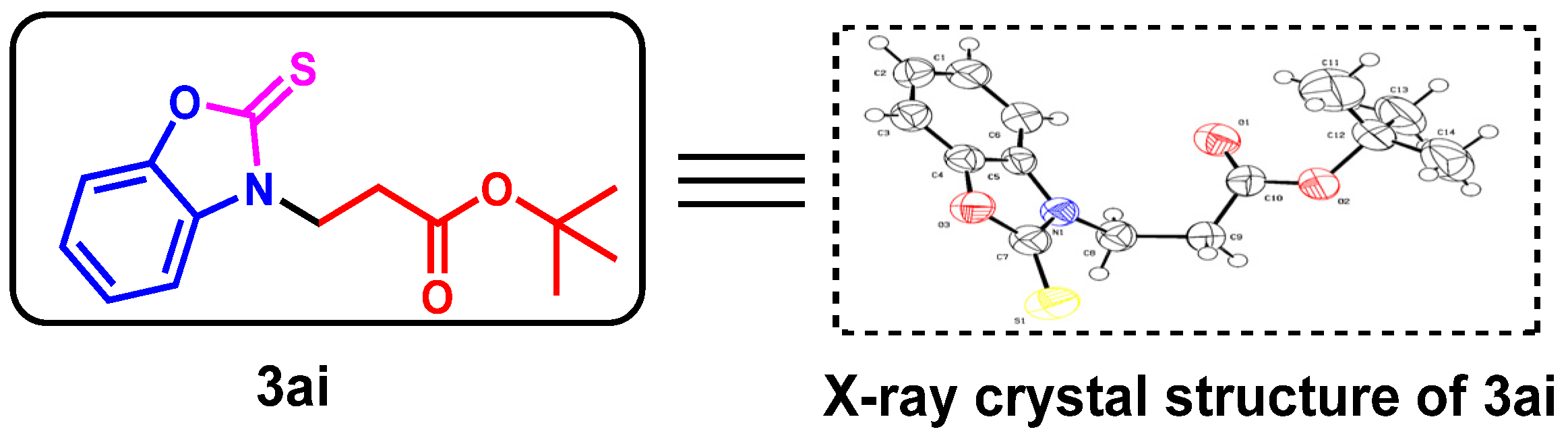
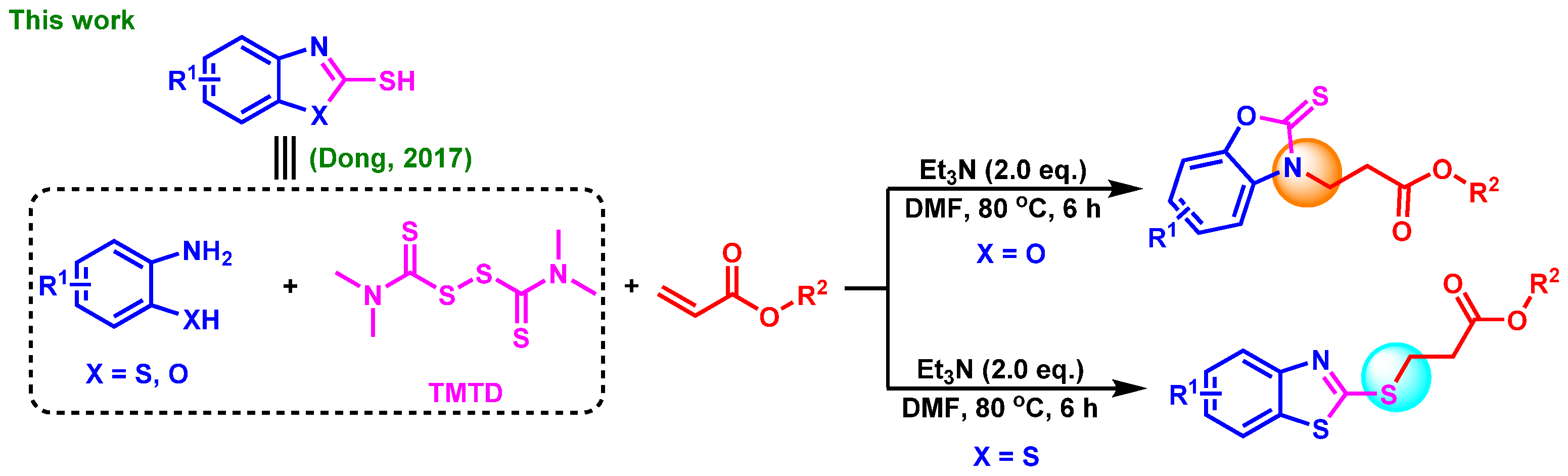
2. Results and Discussion
3. Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, W.; Yang, G.F. Microwave-Assisted, One-Pot Syntheses and Fungicidal Activity of Polyfluorinated 2-Benzylthiobenzothiazoles. Bioorg. Med. Chem. 2006, 14, 8280–8285. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Segal, S. Study of Fungicidal Activities of Some Benzothiazoles. Indian J. Chem. 1988, 27B, 941–943. [Google Scholar]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.A.; Loddo, R.; Colla, P. In Vitro Antitumor and Antiviral Activities of New Benzothiazole and 1, 3, 4-Oxadiazole-2-Thione Derivatives. Acta Pharm. 2008, 58, 135–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.T.; Hsei, I.J.; Chen, C. Synthesis and Anticancer Evaluation of Bis (Benzimidazoles), Bis (Benzoxazoles), and Benzothiazoles. Bioorg. Med. Chem. 2006, 14, 6106–6119. [Google Scholar]
- Palmer, P.J.; Trigg, R.B.; Warrington, J.V. Benzothiazolines as Antituberculous Agents. J. Med. Chem. 1971, 14, 248–251. [Google Scholar] [CrossRef]
- Rao, A.J.; Rao, P.V.; Rao, V.K.; Mohan, C.; Raju, C.N.; Reddy, C.S. Microwave Assisted One-Pot Synthesis of Novel a-Aminophosphonates and Their Biological Activity. Bull. Korean Chem. Soc. 2010, 31, 1863–1868. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Singh, S.K.; Gangwar, M.; Nath, G.; Singh, S.K. Design, Synthesis and Mode of Action of Some Benzothiazole Derivatives Bearing an Amide Moiety as Antibacterial Agents. RSC Adv. 2014, 4, 19013–19023. [Google Scholar] [CrossRef]
- Suresh, C.H.; Rao, J.V.; Jayaveera, K.N.; Subudhi, S.K. Synthesis and Anthelminitc Activity of 3 (2-Hydrazino Benzothiazoles)-Substituted Indole-2-One. Int. Res. J. Pharm. 2011, 2, 257–261. [Google Scholar]
- Reddy, P.; Lin, Y.; Chang, H. Synthesis of Novel Benzothiazole Compounds with an Extended Conjugated System. Arkivoc 2007, 16, 113–122. [Google Scholar] [CrossRef]
- Heo, Y.; Song, Y.S.; Kim, B.T.; Heo, J.N. A Highly Regioselective Synthesis of 2-Aryl-6-Chlorobenzothiazoles Employing Microwave-Promoted Suzuki-Miyaura Coupling Reaction. Tetrahedron Lett. 2006, 47, 3091–3094. [Google Scholar] [CrossRef]
- Azam, M.A.; Suresh, B. Biological Activities of 2-Mercaptobenzothiazole Derivatives: A Review. Sci. Pharm. 2012, 80, 789–824. [Google Scholar] [CrossRef] [Green Version]
- Dumas, J.; Brittelli, D.; Chen, J.; Dixon, B.; Hatoum-Mokdad, H.; Konig, G.; Sibley, R.; Witowsky, J.; Wong, S. Synthesis and Structure Activity Relationships of Novel Small Molecule Cathepsin D Inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2531–2536. [Google Scholar] [CrossRef]
- Pejin, B.; Iodice, C.; Tommonaro, G.; Rosa, S.D. Synthesis and Biological Activities of Thio-Avarol Derivatives. J. Nat. Prod. 2008, 71, 1850–1853. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.Q.; Li, X.F.; Huang, L.J. The Importance of Indole and Azaindole Scaffold in the Development of Antitumor Agents. Eur. J. Med. Chem. 2020, 203, 112506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fan, J.; Vu, K.; Hong, K.; Le Brazidec, J.Y.; Shi, J.; Biamonte, M.; Busch, D.J.; Lough, R.E.; Grecko, R.; et al. 7′-Substituted Benzothiazolothio-and Pyridinothiazolothio-Purines as Potent Heat Shock Protein 90 Inhibitors. J. Med. Chem. 2006, 49, 5352–5362. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.Y.; Jiang, L.Y.; Li, T.; Song, L.L.; Wu, L.J.; Cao, H.; Yang, C. Natural Product-Based Semisynthesis and Biological Evaluation of Thiol/Amino-Michael Adduct of Xanthatin Derived from Xanthium Strumarium as Potential Pesticidal Agents. Bioorg. Chem. 2020, 97, 103696. [Google Scholar] [CrossRef]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Chin, J.E.; Liu, X.; Striepen, B.; Makowska-Grzyska, M.; Kim, Y.; Joachimiak, A.; Hedstrom, L.; et al. Optimization of Benzoxazole-Based Inhibitors of Cryptosporidium Parvum Inosine 5′-Monophosphate Dehydrogenase. J. Med. Chem. 2013, 56, 4028–4043. [Google Scholar] [CrossRef] [Green Version]
- Naya, A.; Kobayashi, K.; Ishikawa, M.; Ohwaki, K.; Saeki, T.; Noguchi, K.; Ohtake, N. Discovery of a Novel CCR3 Selective Antagonist. Bioorg. Med. Chem. Lett. 2001, 11, 1219–1223. [Google Scholar] [CrossRef]
- Choudhary, A.N.; Kumar, A.; Juyal, V. Quantitative Structure Activity Relationship (QSAR) Analysis of Substituted 4-Oxothiazolidines and 5-Arylidines as Lipoxygenase Inhibitors. Mini-Rev. Med. Chem. 2010, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Chekal, B.P.; Guinness, S.M.; Lillie, B.M.; McLaughlin, R.W.; Palmer, C.W.; Post, R.J.; Sieser, J.E.; Singer, R.A.; Sluggett, G.W.; Vaidyanathan, R.; et al. Development of an Efficient Pd-Catalyzed Coupling Process for Axitinib. Org. Process Res. Dev. 2013, 18, 266–274. [Google Scholar] [CrossRef]
- Klimešová, V.; Kočí, J.; Pour, M.; Stachel, J.; Waisser, K.; Kaustová, J. Synthesis and Preliminary Evaluation of Benzimidazole Derivatives as Antimicrobial Agents. Eur. J. Med. Chem. 2002, 37, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.F.; Correa, A.; Carril, M.; Norrby, P.O.; Bolm, C. Copper-Catalyzed Cross-Couplings with Part-per-Million Catalyst Loadings. Angew. Chem. Int. Ed. 2009, 48, 5691. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Buchwald, S.L. Catalyst-Controlled Chemoselective Arylation of 2-Aminobenzimidazoles. Angew. Chem. Int. Ed. 2012, 51, 10364–10367. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yuan, T.; Yang, Y.; Chen, J. Novel Copper/PEG-400 Catalyst Systems for Chemoselective S- and N-arylation of 2-Mercaptobenzothiazole. Tetrahedron 2014, 70, 9652–9660. [Google Scholar] [CrossRef]
- Duari, S.; Biswas, S.; Roy, A.; Maity, S.; Mishra, A.K.; Souza, A.R.; Elsharif, A.M.; Morgon, N.H.; Biswas, S. Regioselective N-Functionalization of Tautomerizable Heterocycles through Methyl Trifluoromethanesulfonate-Catalyzed Substitution of Alcohols and Alkyl Group Migrations. Adv. Synth. Catal. 2022, 364, 865–872. [Google Scholar] [CrossRef]
- Herrera Cano, N.; Ballari, M.S.; Lopez, A.G.; Santiago, A.N. New Synthesis and Biological Evaluation of Benzothiazole Derivates as Antifungal Agents. J. Agric. Food. Chem. 2015, 63, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi-Shekhi Abadi, P.; Rafiee, E.; Joshaghani, M. Pd-PVP-Fe (Palladium-Poly(N-vinylpyrrolidone)-iron) Catalyzed S-arylation of Thiols with Aryl Halides in Aqueous Media. Inorg. Chim. Acta. 2016, 451, 162–170. [Google Scholar] [CrossRef]
- Sikari, R.; Sinha, S.; Das, S.; Saha, A.; Chakraborty, G.; Mondal, R.; Paul, N.D. Achieving Nickel Catalyzed C-S Cross-Coupling under Mild Conditions Using Metal-Ligand Cooperativity. J. Org. Chem. 2019, 84, 4072–4085. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Li, Y.-Y.; Tang, L.; Wu, W.; Xu, H.-J. Efficient Ligand-Free Copper-Catalyzed C-S Cross-Coupling of Thiols with Aryl Iodides Using KF/Al2O3 as Base. Tetrahedron Lett. 2010, 51, 2489–2492. [Google Scholar] [CrossRef]
- Wu, W.-Y.; Wang, J.-C.; Tsai, F.-Y. A Reusable FeCl3-6H2O/Cationic 2,2′-Bipyridyl Catalytic System for the Coupling of Aryl Iodides with Thiols in Water under Aerobic Conditionst. Green Chem. 2009, 11, 326–329. [Google Scholar] [CrossRef]
- Correa, A.; Carril, M.; Bolm, C. Iron-Catalyzed S-Arylation of Thiols with Aryl Iodides. Angew. Chem. Int. Ed. 2008, 47, 2880–2883. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Shtro, A.A.; Borisova, M.S.; Morozova, E.A.; Tolstikova, T.G.; Zarubaev, V.V.; Salakhutdinov, N.F. Synthesis and Biological Activity of Heterocyclic Borneol Derivatives. Chem. Heterocycl. Compd. 2017, 53, 371–377. [Google Scholar] [CrossRef]
- Shi, L.; Liu, X.; Zhang, H.; Jiang, Y.; Ma, D. Synthesis of 2-Thio-Substituted Benzothiazoles via a Domino Condensation/S-Arylation/Heterocyclization Process. J. Org. Chem. 2011, 76, 4200–4204. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, G.; Mondal, S.; Hajra, A. Visible-Light-Mediated Synthesis of Unsymmetrical Diaryl Sulfides via Oxidative Coupling of Arylhydrazine with Thiol. Org. Lett. 2018, 20, 7740–7743. [Google Scholar] [CrossRef]
- Ren, X.; Tang, S.; Li, L.; Li, J.; Liang, H.; Li, G.; Yang, G.; Li, H.; Yuan, B. Surfactant-Type Catalyst for Aerobic Oxidative Coupling of Hydrazine with Thiol in Water. J. Org. Chem. 2019, 84, 8683–8690. [Google Scholar] [CrossRef]
- Xu, H.J.; Zhao, Y.Q.; Feng, T.; Feng, Y.S. Chan-Lam-Type S-Arylation of Thiols with Boronic Acids at Room Temperature. J. Org. Chem. 2012, 77, 2878–2884. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Z.B. Chemoselective Chan-Lam Coupling Reactions between Benzimidazoline-2-thiones and Arylboronic Acids. J. Org. Chem. 2019, 84, 11524–11532. [Google Scholar] [CrossRef]
- Bhowmik, A.; Yadav, M.; Fernandes, R.A. Room Temperature Nickel-Catalyzed Cross-Coupling of Aryl-Boronic Acids with Thiophenols: Synthesis of Diarylsulfides. Org. Biomol. Chem. 2020, 18, 2447–2458. [Google Scholar] [CrossRef]
- Quan, Z.J.; Ren, R.G.; Da, Y.X.; Zhang, Z.; Wang, X.C. Alkylation of SH-Heterocycles with Diethyl Phosphite Using Tetrachloroethylene as an Efficient Solvent. Heteroat. Chem. 2011, 22, 653–658. [Google Scholar] [CrossRef]
- Anan’Eva, K.V.; Rozhkova, N.K. Benzazolin-2-Thiones in the Michael Reaction. 2. Reaction of Benzothiazolin-and Benzoxazolin-2-Thiones with Acrylonitrile, Acrylamide, and Methyl Acrylate in the Presence of Basic Catalysts. Chem. Heterocycl. Compd. 1986, 22, 564–567. [Google Scholar] [CrossRef]
- Dong, Z.B.; Liu, X.; Bolm, C. Copper-Catalyzed C(sp2)-S Coupling Reactions for the Synthesis of Aryl Dithiocarbamates with Thiuram Disulfide Reagents. Org. Lett. 2017, 19, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.B.; Balkenhohl, M.; Tan, E.; Knochel, P. Synthesis of Functionalized Diaryl Sulfides by Cobalt-Catalyzed Coupling between Arylzinc Pivalates and Diaryl Disulfides. Org. Lett. 2018, 20, 7581–7584. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Y.; Li, J.H.; Zhang, S.B.; Chen, L.J.; Li, Y.S.; Dong, Z.B. A Mild Synthesis of 2-Substituted Benzothiazoles via Nickel-Catalyzed Intramolecular Oxidative C-H Functionalization. J. Org. Chem. 2020, 85, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Peng, H.Y.; Yang, M.M.; Hao, E.J.; Li, Y.S.; Dong, Z.B. Cs2CO3-Promoted Hydrothiolation of Alkynes with Aryl Thioureas: Stereoselective Synthesis of (Z)-Vinyl Sulfides. J. Org. Chem. 2021, 86, 8457–8464. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, M.; Xu, W.; Zeng, M.T.; Zhu, H.; Chang, C.Z.; Dong, Z.B. An Environmentally Benign and Efficient Synthesis of Substituted Benzothiazole-2-thiols, Benzoxazole-2-thiols, and Benzimidazoline-2-thiones in Water. Green Chem. 2017, 19, 5591–5598. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cat. | Base | Temp. (°C) | Solvent | Yield b (%) |
|---|---|---|---|---|---|
| 1 | CuI | Et3N (2.0 eq.) | 60 | EtOH | 19 |
| 2 | CuO | Et3N (2.0 eq.) | 60 | EtOH | 17 |
| 3 | CuBr2 | Et3N (2.0 eq.) | 60 | EtOH | 15 |
| 4 | FeCl3 | Et3N (2.0 eq.) | 60 | EtOH | <10 |
| 5 | CuI | Et3N (2.0 eq.) | 60 | DMF | 48 |
| 6 | - | Et3N (2.0 eq.) | 60 | DMF | 68 |
| 7 | - | Na2CO3 (2.0 eq.) | 60 | DMF | 53 |
| 8 | - | NaHCO3 (2.0 eq.) | 60 | DMF | 52 |
| 9 | - | Et2NH (2.0 eq.) | 60 | DMF | 60 |
| 10 | - | Et3N (2.0 eq.) | 80 | DMF | 85 |
| 11 | - | Et3N (2.0 eq.) | 100 | DMF | 84 |
| 12 | - | Et3N (2.0 eq.) | 110 | DMF | 80 |
| 13 | - | Et3N (1.0 eq.) | 80 | DMF | 70 |
| 14 | - | Et3N (3.0 eq.) | 80 | DMF | 72 |
| 15 | - | Et3N (2.0 eq.) | 80 | H2O | 36 |
| 16 | - | Et3N (2.0 eq.) | 80 | DMSO | 79 |
 | |||
 3aa, 85% |  3ab, 84% |  3ac, 82% |  3ad, 78% |
 3ae, 88% |  3af, 53% |  3ag, 70% |  3ah, 28% |
 3ai, 86% |  3aj, 29% |  3ak, 89% |  3al, 73% |
 3am, 84% |  3an, 70% |  3ao, 89% |  3ap, 69% |
 3aq, 70% |  3ar, 36% |  3as, 80% |  3at, 88% |
 3au, 56% |  3av, 87% | ||
 | ||
 5aa, 66% |  5ab, 71% |  5ac, 62% |
 5ad, 44%  5ag, 70% |  5ae, 61% |  5af, 62% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Z.-Y.; Yang, C.-L.; Wang, D.; Huang, L.; Dong, Z.-B. One-Pot Synthesis of Benzoxazole/Benzothiazole-Substituted Esters by Michael Addition: A Selective Construction of C-N/C-S Bonds. Catalysts 2023, 13, 658. https://doi.org/10.3390/catal13040658
Gong Z-Y, Yang C-L, Wang D, Huang L, Dong Z-B. One-Pot Synthesis of Benzoxazole/Benzothiazole-Substituted Esters by Michael Addition: A Selective Construction of C-N/C-S Bonds. Catalysts. 2023; 13(4):658. https://doi.org/10.3390/catal13040658
Chicago/Turabian StyleGong, Zhi-Ying, Cheng-Li Yang, Dan Wang, Lang Huang, and Zhi-Bing Dong. 2023. "One-Pot Synthesis of Benzoxazole/Benzothiazole-Substituted Esters by Michael Addition: A Selective Construction of C-N/C-S Bonds" Catalysts 13, no. 4: 658. https://doi.org/10.3390/catal13040658