


Dynamic EPR Studies of the Formation of Catalytically Active Centres in Multicomponent Hydrogenation Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
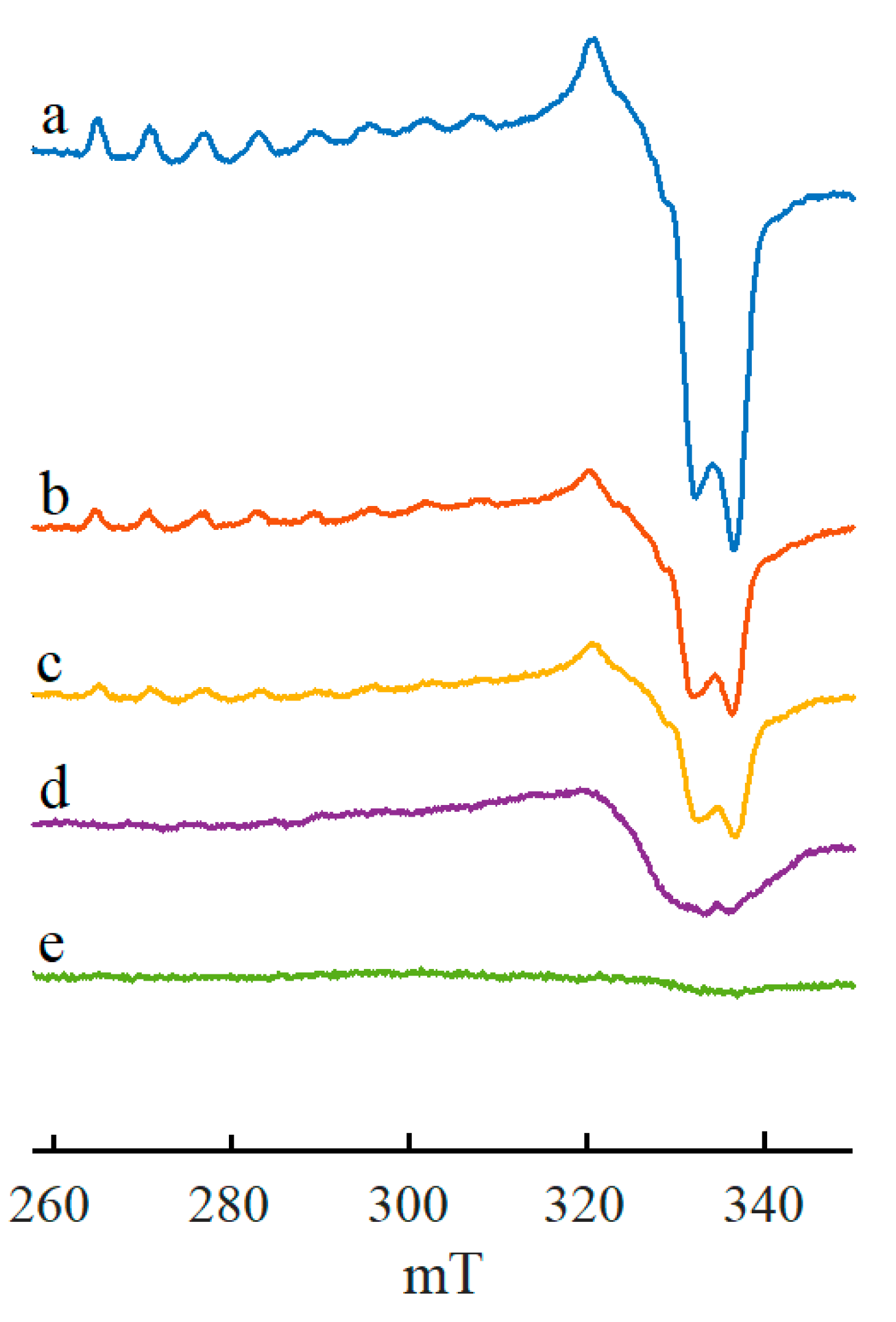{kind=link}
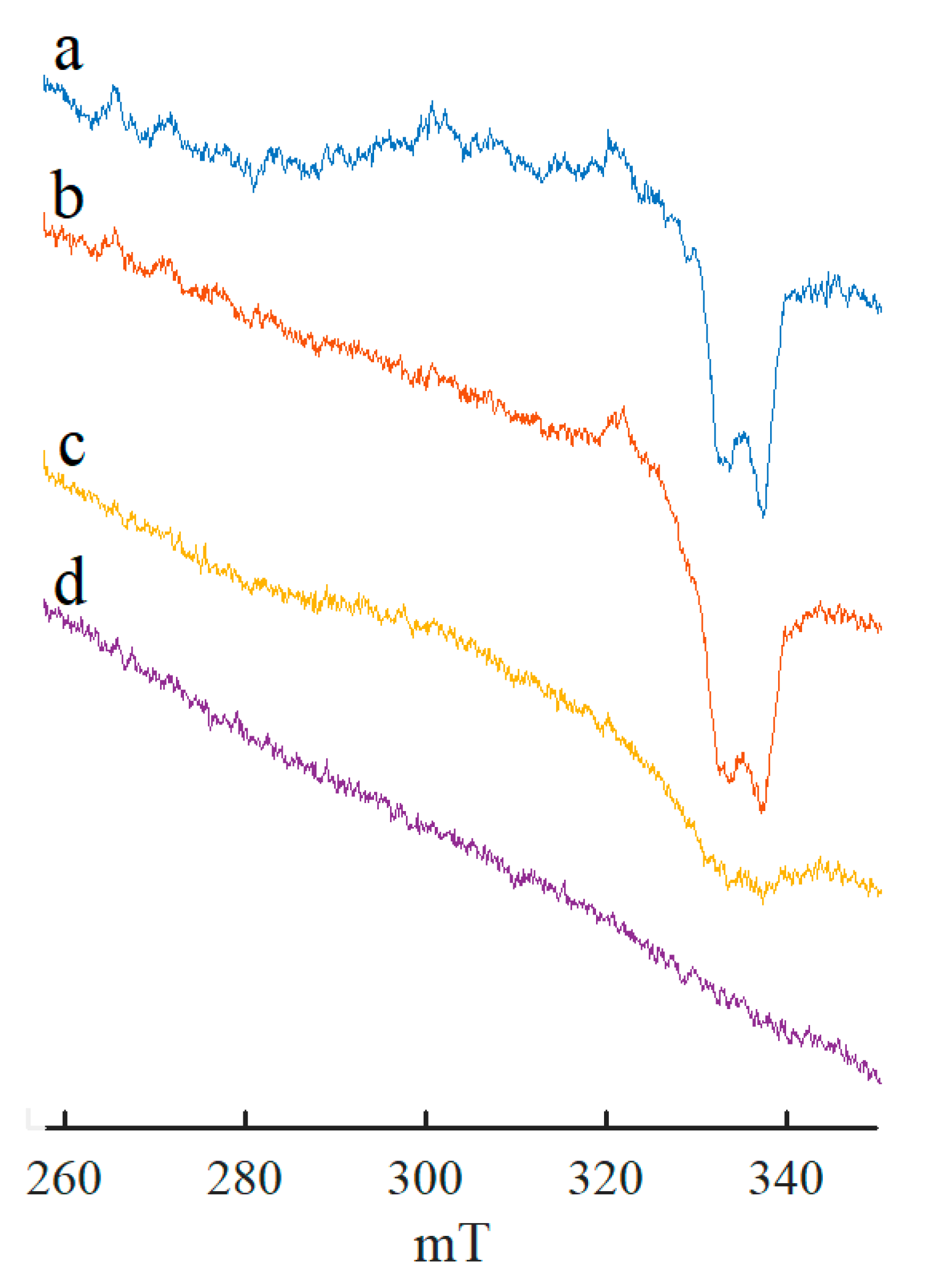{kind=link}
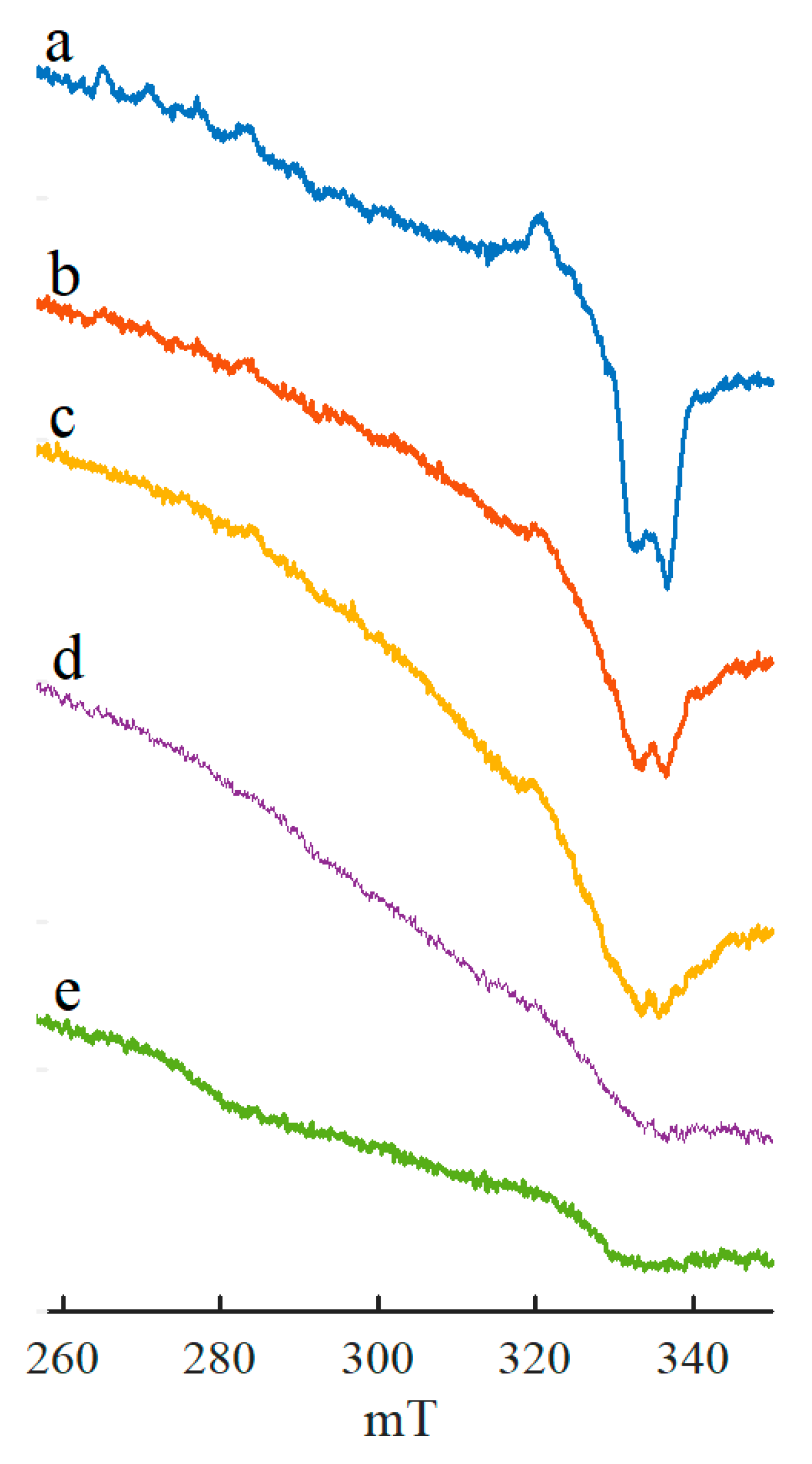{kind=link}
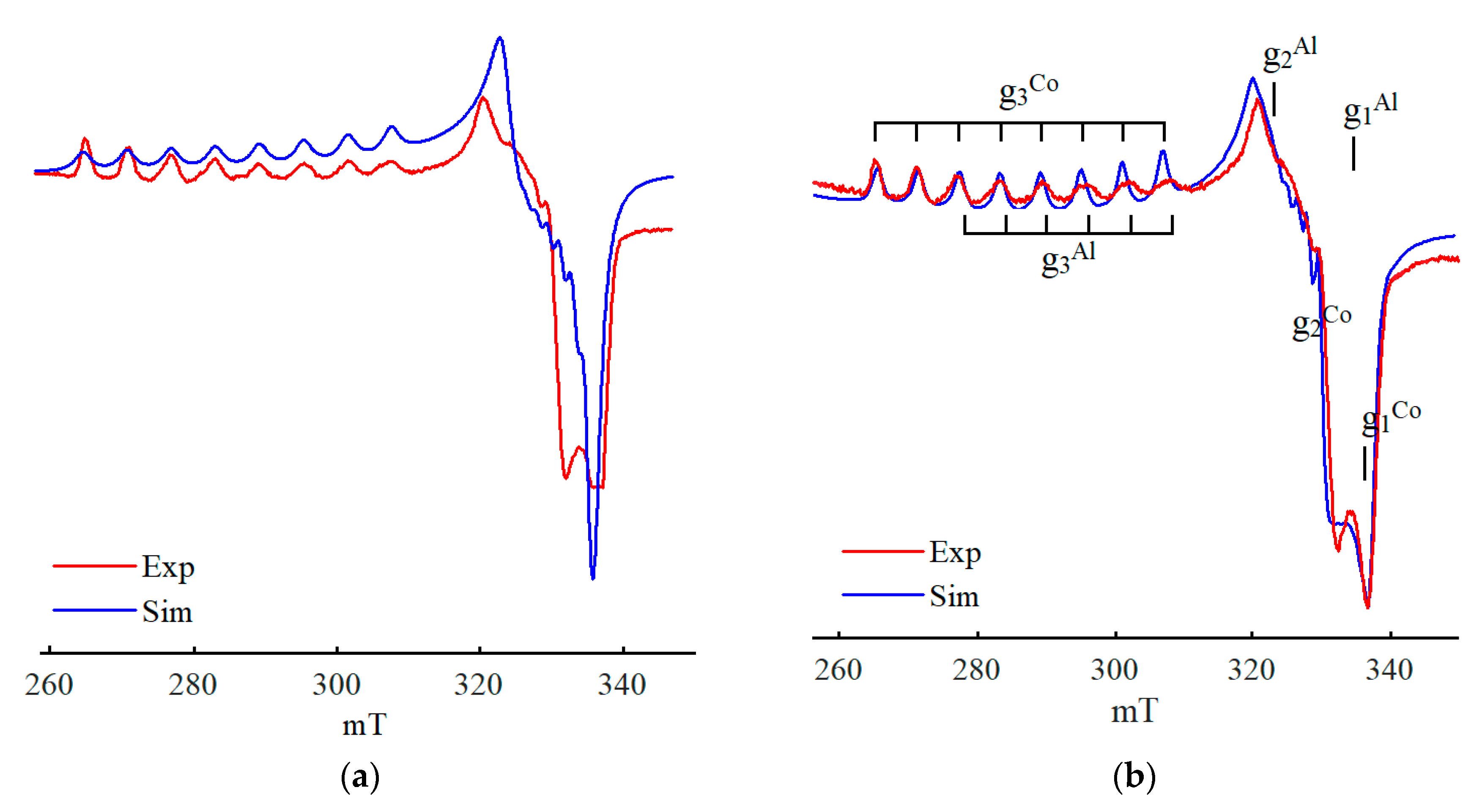{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion

4. Models of Paramagnetic Site
5. Materials and Methods
6. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertrand, P. Electron Paramagnetic Resonance Spectroscopy; Springer Nature: Basel, Switzerland, 2020; pp. 1–420. [Google Scholar]
- Perera, K.P.U.; Smith, D.W. In Situ EPR Spectroscopy of Aromatic Diyne Cyclopolymerization. J. Org. Chem. 2004, 49, 6124–6127. [Google Scholar] [CrossRef]
- Liu, F.; Karoui, H.; Rockenbauer, A.; Liu, S.; Ouari, O.; Bardelang, D. EPR Spectroscopy: A Powerful Tool to Analyze Supramolecular Host—Guest Complexes of Stable Radicals with Cucurbiturils. Molecules 2020, 25, 776. [Google Scholar] [CrossRef] [Green Version]
- Peyrot, F.; Lajnef, S.; Versace, D. Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes. Catalysts 2022, 12, 772. [Google Scholar] [CrossRef]
- Pietrzyk, P.; Mazur, T.; Sojka, Z. Electron Paramagnetic Resonance Spectroscopy of Inorganic Materials. In Local Structural Characterisation: Inorganic Materials Series; Bruce, D.W., O’Hare, D., Walton, R., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 225–301. [Google Scholar]
- Boere, T. Inorganic and Organometallic Radicals of Main Group Elements. In Electron Paramagnetic Resonance; RSC Publishing: London, UK, 2013; Volume 23, pp. 22–57. [Google Scholar] [CrossRef]
- Bracci, M.; Van Doorslaer, S.; García, I. EPR of Compound I: An Illustrated Revision of the Theoretical Model; Springer: Vienna, Austria, 2020; Volume 51, pp. 1559–1589. ISBN 0123456789. [Google Scholar]
- Prosser, K.E.; Walsby, C.J. Electron Paramagnetic Resonance as a Tool for Studying the Mechanisms of Paramagnetic Anticancer Metallodrugs. Eur. J. Inorg. Chem. 2017, 2017, 1573–1585. [Google Scholar] [CrossRef]
- Cutsail, G.E. Applications of Electron Paramagnetic Resonance Spectroscopy to Heavy Main-Group Radicals. Dalt. Trans. 2020, 49, 12128–12135. [Google Scholar] [CrossRef]
- Dasgupta, A.; Richards, E.; Melen, R.L. Frustrated Radical Pairs: Insights from EPR Spectroscopy. Angew. Chem. Int. Ed. 2021, 133, 53–65. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Fedoseev, A.; Summerer, D.; Drescher, M. Genetically Encoded Spin Labels for In Vitro and In-Cell EPR Studies of Native Proteins. In Electron Paramagnetic Resonance Investigations of Biological Systems by Using Spin Labels, Spin Probes, and Intrinsic Metal Ions, Part A, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 563, pp. 483–502. [Google Scholar]
- Klare, J.P. Electron Paramagnetic Resonance of Membrane Proteins, 3rd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; pp. 442–446. ISBN 9780124095472. [Google Scholar]
- Universit, A.M. Probing Conformational Changes and Interfacial Recognition Site of Lipases with Surfactants and Inhibitors. Methods Enzymol. 2017, 583, 279–307. [Google Scholar] [CrossRef]
- Yordanov, N.D. Quantitative EPR Spectrometry—“State of the Art”. Appl. Magn. Reson. 1994, 6, 241–257. [Google Scholar] [CrossRef]
- Nagy, V. Quantitative EPR: Some of the Most Difficult Problems. Appl. Magn. Reson. 1994, 6, 259–285. [Google Scholar] [CrossRef]
- Mazúr, M.; Valko, M.; Pelikán, P. Quantitative EPR Spectroscopy in Solid State Chemistry. Chem. Pap. 1997, 51, 134–136. [Google Scholar]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR; Springer: Vienna, Austria, 2010; p. 1985. [Google Scholar]
- Fendorf, S.E.; Sparks, D.L.; Franz, J.A.; Camaioni, D.M. Electron Paramagnetic Resonance Stopped-Flow Kinetic Study of Manganese (II) Sorption—Desorption on Birnessite. Soil Sci. Soc. Am. J. 1985, 57, 57–62. [Google Scholar] [CrossRef]
- Hu, Y.; Norton, J.R. Kinetics and Thermodynamics of H–/H•/H+ Transfer from a Rhodium(III) Hydride. J. Am. Chem. Soc. 2014, 136, 5938–5948. [Google Scholar] [CrossRef] [PubMed]
- Schubert, E.; Hett, T.; Schiemann, O.; Nejatyjahromy, Y. EPR Studies on the Kinetics of the α-Hydroxyethyl Radical Generated by Fenton-like Chemistry. J. Magn. Reson. 2016, 265, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. EPR Techniques for Studying Radical Enzymes. Biochim. Biophys. Acta 2005, 1707, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Chirila, A.; Rebreyend, C. EPR Spectroscopy as a Tool in Homogeneous Catalysis Research. Top. Catal. 2015, 58, 719–750. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, C.J. The Role of ESR Spectroscopy in Advancing Catalytic Science: Some Recent Developments. Prog. React. Kinet. Mech. 2015, 40, 201–248. [Google Scholar] [CrossRef]
- Behrens, M.; Zander, S.; Kurr, P.; Jacobsen, N.; Senker, J.J.; Koch, G.; Ressler, T.; Fischer, R.W.; Schlögl, R.; Schlo, R.; et al. Performance Improvement of Nanocatalysts by Promoter-Induced Defects in the Support Material: Methanol Synthesis over Cu/ZnO:Al. J. Am. Chem. Soc. 2013, 135, 6061–6068. [Google Scholar] [CrossRef]
- Selim, M.M.; Abd El-Maksoud, I.H.; El-maksoud, I.H.A. Spectroscopic and Catalytic Characterization of Ni Nano-Size Catalyst for Edible Oil Hydrogenation. Microporous Mesoporous Mater. 2005, 85, 273–278. [Google Scholar] [CrossRef]
- Shim, H.; Dutta, P.; Seehra, M.S.; Bonevich, J. Size dependence of the blocking temperatures and electron magnetic resonance spectra in NiO nanoparticles. Solid State Commun. 2008, 145, 192–196. [Google Scholar] [CrossRef]
- Lucarini, M.; Pasquato, L. ESR Spectroscopy as a Tool to Investigate the Properties of Self-Assembled Monolayers Protecting Gold Nanoparticles. Nanoscale 2010, 2, 668–676. [Google Scholar] [CrossRef]
- Alonso, F.; Riente, P.; Sirvent, J.A.; Yus, M. Nickel Nanoparticles in Hydrogen-Transfer Reductions: Characterisation and Nature of the Catalyst. Appl. Catal. A Gen. 2010, 378, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Alley, W.M.; Hamdemir, I.K.; Johnson, K.A.; Finke, R.G. Ziegler-Type Hydrogenation Catalysts Made from Group 8–10 Transition Metal Precatalysts and AlR3 Cocatalysts: A Critical Review of the Literature. J. Mol. Catal. A Chem. 2010, 315, 1–27. [Google Scholar] [CrossRef]
- Claverie, J.P.; Schaper, F. Ziegler-Natta Catalysis: 50 Years after the Nobel Prize. MRS Bull. 2013, 38, 213–218. [Google Scholar] [CrossRef]
- Nifant, I.; Ivchenko, P.; Tavtorkin, A.; Vinogradov, A. Non-Traditional Ziegler-Natta Catalysis in α -Olefin Transformations: Reaction Mechanisms and Product Design. Pure Appl. Chem. 2017, 89, 1017–1032. [Google Scholar] [CrossRef]
- Klaue, A.; Kruck, M.; Friederichs, N.; Bertola, F.; Wu, H.; Morbidelli, M. Insight into the Synthesis Process of an Industrial Ziegler-Natta Catalyst. Ind. Eng. Chem. Res. 2019, 58, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Woo, T.K.; Cavallo, L.; Margl, P.M.; Ziegler, T.; January, R.V.; Re, V.; Recei, M.; April, V. The Role of Bulky Substituents in Brookhart-Type Ni(II) Diimine Catalyzed Olefin Polymerization: A Combined Density Functional Theory and Molecular Mechanics Study. J. Am. Chem. Soc. 1997, 119, 6177–6186. [Google Scholar] [CrossRef]
- Jeon, M.; Kim, S.Y. Ethylene Polymerizations with Unsymmetrical (α-Diimine)Nickel(II) Catalysts. Polym. J. 2008, 40, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chen, C. A Continuing Legend: The Brookhart-Type α-Diimine Nickel and Palladium Catalysts. Polym. Chem. 2019, 10, 2354–2369. [Google Scholar] [CrossRef] [Green Version]
- Bruckner, A. In Situ Electron Paramagnetic Resonance: A Unique Tool for Analyzing Structure–Reactivity Relationships in Heterogeneous Catalysis. Chem. Soc. Rev. 2010, 39, 4673–4684. [Google Scholar] [CrossRef] [Green Version]
- Rabeah, J.; Radnik, J.; Briois, V.; Maschmeyer, D.; Stochniol, G.; Peitz, S.; Reeker, H.; La Fontaine, C.; Brückner, A.; Rabeah, J.; et al. Tracing Active Sites in Supported Ni Catalysts during Butene Oli-Gomerization by Operando Spectroscopy under Pressure Tracing Active Sites in Supported Ni Catalysts during Butene Oligomerization by Operando Spectroscopy under Pressure. ACS Catal. 2016, 6, 8224–8228. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, R. In Situ Electron Paramagnetic Resonance Spectroscopy in Catalysis. In Heterogeneous Catalysts: Advanced Design, Characterization and Applications; Teoh, W.Y., Urakawa, A., Ng, Y.H., Sit, P., Eds.; Wiley-VCH: Weinheim, Germany, 2021; pp. 295–310. [Google Scholar]
- Zichittella, G.; Polyhach, Y.; Tschaggelar, R.; Jeschke, G.; Pérez-Ramírez, J. Quantification of Redox Sites during Catalytic Propane Oxychlorination by Operando EPR Spectroscopy. Angew. Chem. Int. Ed. 2021, 60, 3596–3602. [Google Scholar] [CrossRef] [PubMed]
- Bryliakov, K.P.; Talsi, E.P. Frontiers of Mechanistic Studies of Coordination Polymerization and Oligomerization of α-Olefins. Coord. Chem. Rev. 2012, 256, 2994–3007. [Google Scholar] [CrossRef]
- Dong, Q.; Yang, X.-J.; Gong, S.; Luo, Q.; Li, Q.-S.; Su, J.-H.; Zhao, Y.; Wu, B. Distinct Stepwise Reduction of a Nickel-Nickel-Bonded Compound Containing an α-Diimine Ligand: From Perpendicular to Coaxial Structures. Chem. A Eur. J. 2013, 19, 15240–15247. [Google Scholar] [CrossRef]
- Takeuchi, D.; Osakada, K. Oligomerization of Olefins. In Organometallic Reactions and Polymerization; Osakada, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 169–215. [Google Scholar]
- Shmidt, F.K.; Titova, Y.Y.; Belykh, L.B. The Role of Phosphine and 1,2-Diimine Complexes of Nickel in the Oxidation States 0, +1, and +2 in the Catalyzed Di-, Oligo-, and Polymerization of Ethylene. Kinet. Catal. 2016, 57, 61–71. [Google Scholar] [CrossRef]
- Olivier-Bourbigou, H.; Breuil, P.A.R.; Magna, L.; Michel, T.; Espada Pastor, M.F.; Delcroix, D. Nickel Catalyzed Olefin Oligomerization and Dimerization. Chem. Rev. 2020, 120, 7919–7983. [Google Scholar] [CrossRef]
- Lee, H.; Bçrgel, J.; Ritter, T. Carbon—Fluorine Reductive Elimination from Nickel (III) Complexes. Angew. Chem. Int. Ed. 2017, 56, 6966–6969. [Google Scholar] [CrossRef]
- Matsubara, K.; Fukahori, Y.; Inatomi, T.; Tazaki, S.; Yamada, Y.; Koga, Y.; Kanegawa, S.; Nakamura, T. Monomeric Three-Coordinate N-Heterocyclic Carbene Nickel(I) Complexes: Synthesis, Structures, and Catalytic Applications in Cross-Coupling Reactions. Organometallics 2016, 35, 3281–3287. [Google Scholar] [CrossRef]
- Mondal, P.; Pirovano, P.; Das, A.; Farquhar, E.R.; McDonald, A.R. Hydrogen Atom Transfer by a High-Valent Nickel-Chloride Complex. J. Am. Chem. Soc. 2018, 140, 1834–1841. [Google Scholar] [CrossRef]
- Gao, W.; Xin, L.; Hao, Z.; Li, G.; Su, J.-H.; Zhou, L.; Mua, Y.; Mu, Y. The Ligand Redox Behavior and Role in 1,2-Bis[(2,6-Diisopropylphenyl)Imino]-Acenaphthene Nickel–TMA(MAO) Systems for Ethylene Polymerization. Chem. Commun. 2015, 51, 7004–7007. [Google Scholar] [CrossRef]
- Schwab, M.M.; Himmel, D.; Kacprzak, S.; Radtke, V.; Kratzert, D.; Weis, P.; Wernet, M.; Peter, A.; Yassine, Z.; Schmitz, D.; et al. Synthesis, Characterisation and Reactions of Truly Cationic Ni(I) -Phosphine Complexes. Chem. A Eur. J. 2018, 98, 1823–1833. [Google Scholar] [CrossRef]
- Do, L.H.; Labinger, J.A.; Bercaw, J.E. Spectral Studies of a Cr(PNP)–MAO System for Selective Ethylene Trimerization Catalysis: Searching for the Active Species. ACS Catal. 2013, 3, 2582–2585. [Google Scholar] [CrossRef] [Green Version]
- Friedfeld, M.R.; Margulieux, G.W.; Schaefer, B.A.; Chirik, P.J. Bis(Phosphine)Cobalt Dialkyl Complexes for Directed Catalytic Alkene Hydrogenation. J. Am. Chem. Soc. 2014, 136, 13178–13181. [Google Scholar] [CrossRef]
- Lin, T.-P.; Peters, J.C. Boryl–Metal Bonds Facilitate Cobalt/Nickel-Catalyzed Olefin Hydrogenation. J. Am. Chem. Soc. 2014, 136, 13672–13683. [Google Scholar] [CrossRef] [Green Version]
- Noveron, C.; Herradora, R.; Olmstead, M.M.; Mascharak, P.K. Low-Spin Iron(III) Complexes with N,S Coordination: Syntheses, Structures, and Properties of Bis(N-2-Mercaptophenyl-2′-Pyridylmethyleniminato)Iron(III) Tetraphenylborate and Bis(N-2-Mercapto-2-Methylpropyl-2′-Pyridylmethyleniminato)Iron(III) Tetraphenylbo. Inorg. Chim. Acta 1999, 285, 269–276. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Greer, S.M.; Freedman, D.E. Chemical Science Splitting Is Resistant to Structural Variation. Chem. Sci. 2016, 7, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaki, S.; Yanase, Y.; Hagiwara, N.; Takeshita, T.; Naganuma, H.; Ohyoshi, A.; Ohkubo, K. ESR and MO Studies of Some C4v Symmetrical Ruthenium(III) Complexes. J. Phys. Chem. 1982, 86, 1038–1043. [Google Scholar] [CrossRef]
- Frantz, S.; Weber, M.; Scheiring, T.; Fiedler, J.; Duboc, C.; Kaim, W. Mechanism and Product Characterization from the Electroreduction of Heterodinuclear Complexes [(C5Me5)ClM(m-L)Re(CO)3X](PF6), M=Rh or Ir, L=2,2-Azobispyridine or 2,2-Azobis(5-Chloropyrimidine), X=halide. Inorg. Chim. Acta 2004, 357, 2905–2914. [Google Scholar] [CrossRef]
- Knijnenburg, Q.; Horton, A.D.; van der Heijden, H.; Kooistra, T.M.; Hetterscheid, D.G.H.; Smits, J.M.M.; De Bruin, B.; Budzelaar, P.H.M.; Gal, A.W. Olefin Hydrogenation Using Diimine Pyridine Complexes of Co and Rh. J. Mol. Catal. A Chem. 2005, 232, 151–159. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Skatova, A.A.; Ketkov, S.Y.; Eremenko, O.V.; Piskunov, A.V.; Fukin, G.K. [(Dpp-Bian)Zn-Zn(Dpp-Bian)]: A Zinc–Zinc-Bonded Compound Supported by Radical-Anionic Ligands. Angew. Chemie Int. Ed. 2007, 46, 4302–4305. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.P.; Darmon, J.M.; Milsmann, C.; Margulieux, G.W.; Stieber, S.C.E.; DeBeer, S.; Chirik, P.J. Catalytic Hydrogenation Activity and Electronic Structure Determination of Bis(Arylimidazol-2-Ylidene)Pyridine Cobalt Alkyl and Hydride Complexes. J. Am. Chem. Soc. 2013, 135, 13168–13184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokorin, A.I.; Landgraf, S.; Shapiro, A.B.; Grampp, G. EPR Spectroscopy of Mercury-Organic Compounds with Nitroxide Radicals. Appl. Magn. Reason. 2014, 45, 125–133. [Google Scholar] [CrossRef]
- Carter, E.; Sharples, K.M.; Platts, J.A.; Murphy, D.M. Structure Determination of Bound Nitrogen-Based Adducts with Copper(II) Acetylacetonato; an EPR, ENDOR and DFT Study. Phys. Chem. Chem. Phys. 2015, 17, 11445–11454. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Buisson, P.; Joseph, N. Application Des Hydroborures Alcalins a La Preparation de Catalyseurs D’hydrogenation. Comptes Rendus Hebd. Seances Acad. Sci. 1951, 232, 627–629. [Google Scholar]
- Polkovnikov, B.D.; Freidlin, L.K.; Balandin, A.A. Selective hydrogenation of adiponitrile over a cobalt boride catalyst. Bull. Acad. Sci. USSR Div. Chem. Sci. 1959, 8, 1436–1488. [Google Scholar] [CrossRef]
- Brown, H.C.; Brown, C.A. A Simple Preparation of Highly Active Platinum Metal Catalysts for Catalytic Hydrogenation. J. Am. Chem. Soc. 1962, 84, 1494–1495. [Google Scholar] [CrossRef]
- Alley, W.M.; Hamdemir, I.K.; Wang, Q.; Frenkel, A.I.; Li, L.; Yang, J.C.; Menard, L.D.; Nuzzo, R.G.; Ozkar, S.; Yih, K.-H.; et al. Industrial Ziegler-Type Hydrogenation Catalysts Made from Co(Neodecanoate)2 or Ni(2-Ethylhexanoate)2 and AlEt3: Evidence for Nanoclusters and Sub-Nanocluster or Larger Ziegler-Nanocluster Based Catalysis. Langmuir 2011, 27, 6279–6294. [Google Scholar] [CrossRef]
- Shmidt, F.K.; Nindakova, L.O.; Shainyanb, B.A.; Saraev, V.V.; Chipaninab, N.N.; Umanetz, V.A. Hydrogenation Catalysts Formation in the System AlEt3-Co(Acac)2,3. J. Mol. Catal. A Chem. 2005, 235, 161–172. [Google Scholar] [CrossRef]
- Crooks, A.B.; Yih, K.-H.; Li, L.; Yang, J.C.; Özkar, S.; Finke, R.G. Unintuitive Inverse Dependence of the Apparent Turnover Frequency on Precatalyst Concentration: A Quantitative Explanation in the Case of Ziegler-Type Nanoparticle Catalysts Made from [(1,5-COD)Ir(μ-O2C8H15)]2. ACS Catal. 2015, 5, 3342–3353. [Google Scholar] [CrossRef]
- Titova, Y.Y.; Belykh, L.B.; Shmidt, F.K. Ziegler-Type Nickel-Based Hydrogenation Catalysts: The Effect of the Water Content of the Nickel Precursor on the Size and Nature of the Resulting Particles. Kinet. Catal. 2016, 57, 388–393. [Google Scholar] [CrossRef]
- Titova, Y.Y. Physico-Chemical Aspects of the Formation and Nature of the Activity of Systems Based on Cobalt, Nickel or Palladium Complexes in Hydrogenation and Oligomerization Reactions. Ph.D. Thesis, Irkutsk State University, Irkutsk, Russia, 2018. Available online: http://old.isu.ru/filearchive/dissert/ar_Titova.pdf (accessed on 1 February 2018).
- Titova, Y.Y.; Schmidt, F.K. Directed Design of Hydrogenation Ziegler Systems. New J. Chem. 2021, 45, 4525–4533. [Google Scholar] [CrossRef]
- Schmidt, F.K. Hydrogenation and Dimerization Catalyzed by Complexes of First Row Transition Metals; Gos. University: Irkutsk, Russia, 1986; p. 231. [Google Scholar]
- Muetterties, E.L.L.; Rakowski, M.C.C.; Hirsekorn, F.J.J.; Larson, W.D.D.; Basus, V.J.J.; Anet, F.A.L.A.L. Hydrogenation of Arenes with Discrete Coordination Catalysts. III. I Synthesis and Nuclear Magnetic Resonance Spectrum of All-Cis-Cyclohexane-D6. J. Am. Chem. Soc. 1975, 97, 1266–1267. [Google Scholar] [CrossRef]
- Stuhl, L.S.; DuBois; Rakowski, M.; Hirsekorn, F.J.; Bleeke, J.R.; Stevens, A.E.; Muetterties, E.L.; DuBois, M.R.; Hirsekorn, F.J.; Bleeke, J.R.; et al. Catalytic Homogeneous Hydrogenation of Arenes. 6. Reaction Scope for the (Tetta.3-2-Propenyl)Tris(Trimethyl Phosphite-P)Cobalt Catalyst. J. Am. Chem. Soc. 1978, 100, 2405–2410. [Google Scholar] [CrossRef]
- Bleeke, J.R.; Muetterties, E.L. Catalytic Hydrogenation of Aromatic Hydrocarbons. Stereochemical Definition of the Catalytic Cycle for H3-C3H5Co(P(OCH3)3)3. J. Am. Chem. Soc. 1981, 103, 556–564. [Google Scholar] [CrossRef]
- Jupp, A.R.; Stephan, D.W. New Directions for Frustrated Lewis Pair Chemistry. Trends Cogn. Sci. 2019, 1, 35–48. [Google Scholar] [CrossRef]
- Paradies, J. From Structure to Novel Reactivity in Frustrated Lewis Pairs. Coord. Chem. Rev. 2019, 380, 170–183. [Google Scholar] [CrossRef]
- Titova, Y.Y.; Belykh, L.B.; Shmidt, F.K. Preparation Method Effect on the Properties of Ziegler-Type Hydrogenation Catalysts Based on Bis(Acetylacetonato)Cobalt. Kinet. Catal. 2016, 57, 344–353. [Google Scholar] [CrossRef]
- Titova, Y.Y.; Schmidt, F.K. Multicomponent Catalytic Systems for Di- and Oligomerization of Ethylene: New Mechanistic Aspects. Catalysts 2021, 11, 1489. [Google Scholar] [CrossRef]
- Bauschlicher, C.W.; Partridge, H.; Langhoff, S.R. Theoretical Study of Transition-Metal Ions Bound to Benzene. J. Phys. Chem. 1992, 96, 3273–3278. [Google Scholar] [CrossRef]
- Pandey, R.; Rao, B.K.; Jena, P.; Blanco, M.A. Electronic Structure and Properties of Transition Metal—Benzene Complexes. J. Am. Chem. Soc. 2001, 123, 3799–3808. [Google Scholar] [CrossRef]
- Chaquin, P.; Costa, D.; Lepetit, C.; Che, M. Structure and Bonding in a Series of Neutral and Cationic Transition Metal−Benzene η6 Complexes [M(η6-C6H6)]N+ (M = Ti, V, Cr, Fe, Co, Ni, and Cu). Correlation of Charge Transfer with the Bathochromic Shift of the E1 Ring Vibration. J. Phys. Chem. A 2001, 105, 4541–4545. [Google Scholar] [CrossRef]
- Taylor, P.; Dargel, T.K.; Hertwig, R.H.; Koch, W. How Do Coinage Metal Ions Bind to Benzene? Mol. Phys. 1999, 96, 583–591. [Google Scholar] [CrossRef]
- Pampaloni, G. Aromatic Hydrocarbons as Ligands. Recent Advances in the Synthesis, the Reactivity and the Applications of Bis(η6-Arene) Complexes. Coord. Chem. Rev. 2010, 254, 402–419. [Google Scholar] [CrossRef]
- Shvydkiy, N.V.; Perekalin, D.S. Reactions of Arene Replacement in Transition Metal Complexes. Coord. Chem. Rev. 2020, 411, 213238. [Google Scholar] [CrossRef]
- Pörschke, K.-R.; Kleimann, W.; Tsay, Y.-H.; Krüger, C.; Wilke, G. Zur Lewis-Acidität von Nickel(0). XII. Dimethylaluminiumhydrid-Komplexe von Nickel(0). Chem. Ber. 1990, 123, 1267–1273. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Steinbock, O.; Neumann, B.; Cage, B.; Saltiel, J.; Mu, S.C.; Dalal, N.S. A Demonstration of Principal Component Analysis for EPR Spectroscopy: Identifying Pure Component Spectra from Complex Spectra. Anal. Chem. 1997, 69, 3708–3713. [Google Scholar] [CrossRef]
- Rieger, P. Electron Spin Resonance—Analysis and Interpretation, 1st ed.; Royal Society of Chemistry: London, UK, 2007; p. 173. [Google Scholar]
- Li, D.; Ma, F.; Guo, L.; Huang, J.; Zhang, Y.; Li, F.; Li, C. Polynuclear (α-Diimine) Nickel(II) Complex as Catalyst for Ethylene Oligomerization. Appl. Organomet. Chem. 2022, 36, e6509. [Google Scholar] [CrossRef]
- Frenking, G.; Fro, N. The Nature of the Bonding in Transition-Metal Compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models of Main-Group Compounds. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef]
- Schwab, M.M.; Himmel, D.; Kacprzak, S.; Radtke, V.; Weis, P.; Ray, K.; Scheidt, E.W.; Scherer, W.; de Bruin, B.; Krossing, W.S. [Ni(cod)2][Al(OR(F))4], a Source for Naked Nickel(I) Chemistry. Chem. A Eur. J. 2015, 54, 14706–14709. [Google Scholar] [CrossRef] [Green Version]
- Krylov, A.I. The Quantum Chemistry of Open-Shell Species. In Reviews in Computational Chemistry; Parrill, A.L., Lipkowitz, K.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; Volume 30, pp. 151–224. [Google Scholar]
- Yamasaki, A. Cobalt-59 Nuclear Magnetic Resonance Spectroscopy in Coordination Chemistry. J. Coord. Chem. 1991, 24, 211–260. [Google Scholar] [CrossRef]
- Chan, J.C.C.; Au-Yeung, S.C.F. Cobalt-59 NMR Spectroscopy. Annu. Rep. NMR Spectrosc. 2000, 41, 1–54. [Google Scholar] [CrossRef]
- Martineau, C.; Taulelle, F.; Group, T.; Lavoisier, I.; Umr, D.V.; De, U. The Use of 27Al NMR to Study Aluminum Compounds: A Survey of the Last 25 Years. In PATAI’S Chemistry of Functional Groups; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 1–51. [Google Scholar] [CrossRef]
- Haouas, M.; Taulelle, F.; Martineau, C. Recent Advances in Application of Science Al NMR Spectroscopy to Materials. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 94–95, 11–36. [Google Scholar] [CrossRef]
- Bond, S.P.; Gelder, A.; Homer, J.; Mcwhinnie, W.R.; Perry, M.C. 6Li Magic Angle Spinning Nuclear Magnetic Resonance Spectroscopy: A Powerful Probe for the Study of Lithium-Containing Materials. J. Mater. Chem. 1991, 1, 327–330. [Google Scholar] [CrossRef]
- Davies, R.P. The Structures of Lithium and Magnesium Organocuprates and Related Species. Coord. Chem. Rev. 2011, 255, 1226–1251. [Google Scholar] [CrossRef]
- Berm, B.R.; Lehmkuhl, H.; Mehler, K.; Rujinska, A. 25Mg-NMR: A Method for the Characterization of Organomagnesium Compounds, Their Complexes, and Schlenk Equilibria. Angen. Chrm. lnt. Ed. 1984, 23, 534–535. [Google Scholar] [CrossRef]
- Freitas, J.C.C.; Smith, M.E. Recent Advances in Solid-State 25Mg NMR Spectroscopy. Annu. Rep. NMR Spectrosc. 2012, 75, 25–114. [Google Scholar] [CrossRef]
- Gordon, A.J.; Ford, R.A. Handbook of Practical Data, Techniques, and References; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Mitchell, J.; Smith, D. Aquametry: A Treatise on Methods for the Determination of Water, 2nd ed.; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon: Oxford, UK, 1988. [Google Scholar]
- Preparativnaya Organicheskaya Khimiya (Preparative Organic Chemistry); Vul’fson, N.S. (Ed.) Khimiya: Moscow, Russia, 1964. [Google Scholar]
- Ioffe, S.T.; Nesmeyanov, A.N. Metody Elementoorganicheskoi Khimii: Magnii, Berillii, Kal’tsii, Strontsii, Barii. In Methods of Organoelement Chemistry: Magnesium, Beryllium, Calcium, Strontium, and Barium; Akad. Nauk SSSR: Moscow, Russia, 1963. [Google Scholar]
- Gilman, H.; Haubein, A.H. The Quantitative Analysis of Alkyllithium Compounds1. J. Am. Chem. Soc. 1944, 66, 1515–1516. [Google Scholar] [CrossRef]
- Schuldt, B.; Heller, B.; Storz, F.; Madeja, K. Ziegler systems as hydrogenation catalysts. Part 1. CoBr2/Li[AlH(O-/-butyl)3]3 system as a catalyst for the hydrogenation of anthracene. J. Mol. Cat. 1993, 81, 195–206. [Google Scholar] [CrossRef]
- Ellern, J.B.; Ragsdale, R. Hexacoordinate Complexes of Bis (2, 4-Pentanedionato) Cobalt (II):[Bis (Acetylacetonato) Cobalt (II)]. Inorg. Synth. 1968, 11, 82–89. [Google Scholar]
- Davis, C.M.; Curran, K.A. Manipulation of a Schlenk line: Preparation of tetrahydrofuran complexes of transition-metal chlorides. J. Chem. Educ. 2007, 84, 1822. [Google Scholar] [CrossRef]
- McGarvey, B.R. Spin hamiltonian for Cr III complexes. Calculation from crystal field and molecular orbital models and ESR determination for some ethylenediammine complexes. J. Chem. Phys. 1964, 41, 3743–3758. [Google Scholar] [CrossRef]
- Titova, Y.Y.; Shmidt, F.K. Role of Water in the Catalysis of Ethylene Di-and Oligomerization and Toluene Alkylation Reactions Based on Nickel Bis (Acetylacetonate) Systems. Kinet. Catal. 2017, 58, 749–757. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titova, Y.Y. Dynamic EPR Studies of the Formation of Catalytically Active Centres in Multicomponent Hydrogenation Systems. Catalysts 2023, 13, 653. https://doi.org/10.3390/catal13040653
Titova YY. Dynamic EPR Studies of the Formation of Catalytically Active Centres in Multicomponent Hydrogenation Systems. Catalysts. 2023; 13(4):653. https://doi.org/10.3390/catal13040653
Chicago/Turabian StyleTitova, Yuliya Yu. 2023. "Dynamic EPR Studies of the Formation of Catalytically Active Centres in Multicomponent Hydrogenation Systems" Catalysts 13, no. 4: 653. https://doi.org/10.3390/catal13040653