

Straightforward and Efficient Deuteration of Terminal Alkynes with Copper Catalysis

Abstract
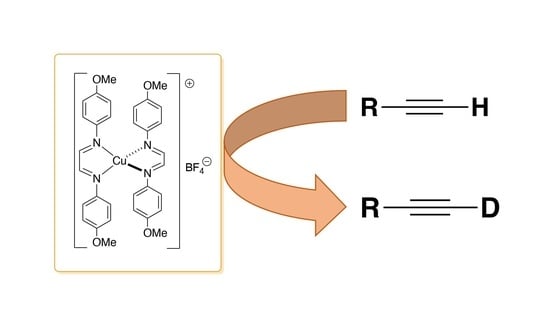
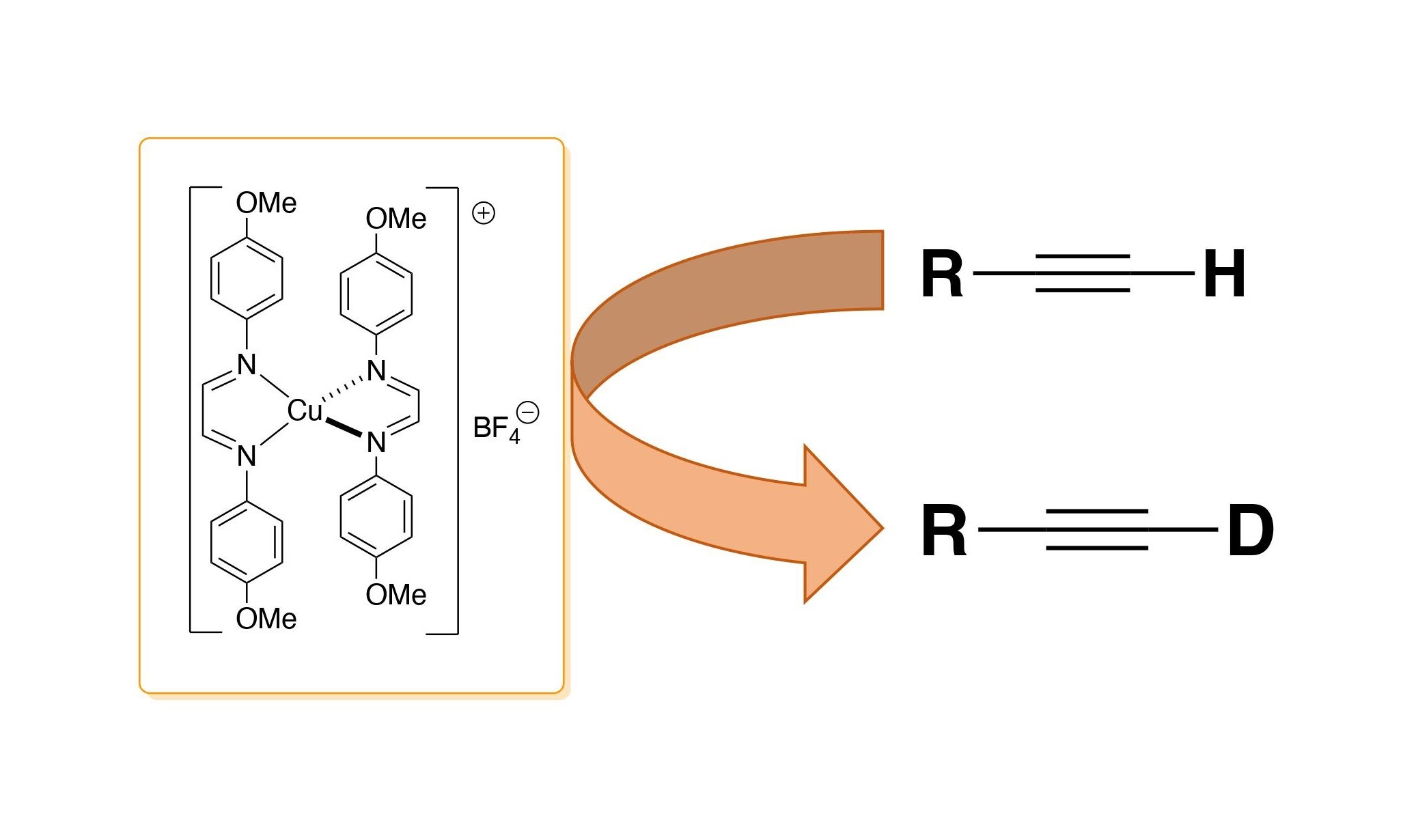
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Copper-Mediated Deuteration of Alkynes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Atzrodt, J.; Derdau, V.; Fey, T.; Zimmermann, J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 2007, 46, 7744–7765. [Google Scholar] [CrossRef] [PubMed]
- Lappin, G.; Temple, S. (Eds.) Radiotracers in Drug Development, 1st ed.; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar] [CrossRef]
- Voges, R.; Heys, J.R.; Moenius, T. Preparation of Compounds Labelled with Tritium and Carbon-14; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar] [CrossRef]
- Isin, E.M.; Elmore, C.S.; Nilsson, G.N.; Thompson, R.A.; Weidolf, L. Use of Radiolabeled Compounds in Drug Metabolism and Pharmacokinetic Studies. Chem. Res. Toxicol. 2012, 25, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Yabe, Y.; Sawama, Y.; Monguchi, Y.; Sajiki, H. Site-selective deuterated-alkene synthesis with palladium on boron nitride. Chem.-Eur. J. 2013, 19, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.K.; Mallik, R. Transition metal-catalyzed hydroalkoxylation of alkynes: An overview. Chem.-Eur. J. 2021, 27, 15571–15604. [Google Scholar] [CrossRef]
- Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Malacria, M. Cotrimerization of acetylenic compounds. In Organic Reactions; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar] [CrossRef]
- Cintrat, J.-C.; Pillon, F.; Rousseau, B. 9-Phenylfluorene: A powerful labeling agent. Tetrahedron Lett. 2001, 42, 5001–5003. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Rudolph, M.; Siehl, H.-U.; Tanaka, M.; Bats, J.W.; Frey, W. Gold catalysis: Deuterated substrates as the key for an experimental insight into the mechanism and selectivity of the phenol synthesis. Chem.-Eur. J. 2008, 14, 3703–3708. [Google Scholar] [CrossRef]
- Sabot, C.; Kumar, K.A.; Antheaume, C.; Mioskowski, C. Triazabicyclodecene: An effective isotope exchange catalyst in CDCl3. J. Org. Chem. 2007, 72, 5001–5004. [Google Scholar] [CrossRef]
- Bew, S.P.; Hiatt-Gipson, G.D.; Lovell, J.A.; Poullain, C. Mild reaction conditions for the terminal deuteration of alkynes. Org. Lett. 2012, 14, 456–459. [Google Scholar] [CrossRef]
- Yamada, T.; Park, K.; Monguchi, Y.; Sawama, Y.; Sajiki, H. Mild deuteration method of terminal alkynes in heavy water using reusable basic resin. RSC Adv. 2015, 5, 92954–92957. [Google Scholar] [CrossRef]
- Kumar, S.; Patel, M.; Verma, A.K. Base-catalyzed selective deuteration of alkynes. Asian J. Org. Chem. 2021, 10, 2365–2369. [Google Scholar] [CrossRef]
- Chatterjee, B.; Gunanathan, C. The ruthenium-catalysed selective synthesis of mono-deuterated terminal alkynes. Chem. Commun. 2016, 52, 4509–4512. [Google Scholar] [CrossRef] [PubMed]
- £65.90 for 250 mg (£161/mmol) or £131 for 1 g (£79/mmol). Available online: https://www.sigmaaldrich.com/GB/en/product/aldrich/739103 (accessed on 27 February 2023).
- Wu, D.-C.; Bai, J.-W.; Guo, L.; Hu, G.-Q.; Liu, K.H.; Sheng, F.-F.; Zhang, H.-H.; Sun, Z.-Y.; Shen, K.; Liu, X. A practical and efficient method for late-stage deuteration of terminal alkynes with silver salt as catalyst. Tetrahedron Lett. 2021, 66, 152807. [Google Scholar] [CrossRef]
- Lewandos, G.S.; Maki, J.W.; Ginnebaugh, J.P. Kinetics of terminal alkyne sp carbon–hydrogen bond activation catalyzed by silver(I). Organometallics 1982, 1, 1700–1705. [Google Scholar] [CrossRef]
- Chang, X.; Cheng, X.; Wang, C.-J. Catalytic asymmetric synthesis of enantioriched α-deuterated pyrrolidine derivatives. Chem. Sci. 2022, 13, 4041–4049. [Google Scholar] [CrossRef]
- Reyes, A.; Rivera Torres, E.; Vang, Z.P.; Clark, J.R. Highly regioselective copper-catalyzed transfer hydrodeuteration of unactivated terminal alkenes. Chem.-Eur. J. 2022, 28, e202104340. [Google Scholar] [CrossRef]
- Li, W.; Qu, R.; Liu, W.; Bourriquen, F.; Bartling, S.; Rockstroh, N.; Junge, K.; Beller, M. Copper-catalysed low-temperature water-gas shift reaction for selective deuteration of aryl halides. Chem. Sci. 2021, 12, 14033–14038. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Yang, K.; Song, Q. Highly selective copper-catalyzed trifunctionalization of alkynyl carboxylic acids: An efficient route to bis-deuterated β-borylated α,β-styrene. Chem. Commun. 2015, 51, 15394–15397. [Google Scholar] [CrossRef]
- Sloane, S.E.; Reyes, A.; Vang, Z.P.; Li, L.; Behlow, K.T.; Clark, J.R. Copper-catalyzed formal transfer hydrogenation/deuteration of aryl alkynes. Org. Lett. 2020, 22, 9139–9144. [Google Scholar] [CrossRef]
- Hefner, J.G.; Zizelman, P.M.; Durfee, L.D.; Lewandos, G.S. Syntheses and metal-catalyzed C–H bond activation of alkyne π complexes of copper(I) trifluoromethanesulfonate. J. Organomet. Chem. 1984, 260, 369–380. [Google Scholar] [CrossRef]
- Zelenay, B.; Munton, P.; Tian, X.; Díez-González, S. A commercially available and user-friendly catalyst for hydroamination reactions under technical conditions. Eur. J. Org. Chem. 2019, 2019, 4725–4730. [Google Scholar] [CrossRef]
- Zelenay, B. Structural and Catalytic Studies on Novel Copper Complexes. Ph.D. Thesis, Imperial College London, London, UK, 2017. [Google Scholar]
- Müller, T.E.; Grosche, M.; Herdtweck, E.; Pleier, A.-K.; Walter, E.; Yan, Y.-K. Developing transition-metal catalysts for the intramolecular hydroamination of alkynes. Organometallics 2000, 19, 170–183. [Google Scholar] [CrossRef]
- Díez-González, S. Copper(I)-acetylides: Access, structure and relevance in catalysis. Adv. Organomet. Chem. 2016, 66, 93–141. [Google Scholar] [CrossRef]
- Zelenay, B.; Frutos-Pedreño, R.; Markalain-Barta, J.; Vega-Isa, E.; White, A.J.P.; Díez-González, S. Homo- and heteroleptic copper(I) complexes with diazabutadiene ligands: Synthesis, solution- and solid-state structural studies. Eur. J. Inorg. Chem. 2016, 2016, 4649–4658. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, B.; Besora, M.; Monasterio, Z.; Ventura-Espinosa, D.; White, A.J.P.; Maseras, F.; Díez-González, S. Copper-mediated reduction of azides under seemingly oxidising conditions: Catalytic and computational studies. Catal. Sci. Technol. 2018, 8, 5763–5773. [Google Scholar] [CrossRef] [Green Version]
- Gujadhur, R.; Venkataraman, D.; Kintigh, J.T. Formation of aryl nitrogen bonds using a soluble copper(I) catalyst. Tetrahedron Lett. 2001, 42, 4791–4793. [Google Scholar] [CrossRef]
- Jurkauskas, V.; Sadighi, J.P.; Buchwald, S.L. Conjugated reduction of α,β-unsaturated carbonyl compounds catalyzed by a copper carbene complex. Org. Lett. 2003, 5, 2417–2420. [Google Scholar] [CrossRef]
- Díez-González, S.; Scott, N.M.; Nolan, S.P. Cationic copper(I) complexes as efficient pre-catalysts for the hydrosilylation of carbonyl compounds. Organometallics 2006, 25, 2355–2358. [Google Scholar] [CrossRef]
- Kubas, G.J.; Monzyk, B.; Crumbliss, A.L. Tetrakis(acetonitrile)copper(I)hexafluorophosphate. Inorg. Synth. 1979, 19, 90–92. [Google Scholar] [CrossRef]
- Brown, J.M.; Lloyd-Jones, G.C. Vinylborane formation in rhodium-catalyzed hydroboration of vinylarenes. Mechanism versus borane structure and relationship to silation. J. Am. Chem. Soc. 1994, 116, 866–878. [Google Scholar] [CrossRef]
- Gelinas, B.S.; Jaye, J.; Mattos, G.; Fort, E.H. Rapid and efficient desilylation and deuteration of alkynylpyridines. Tetrahedron Lett. 2015, 56, 4232–4233. [Google Scholar] [CrossRef]




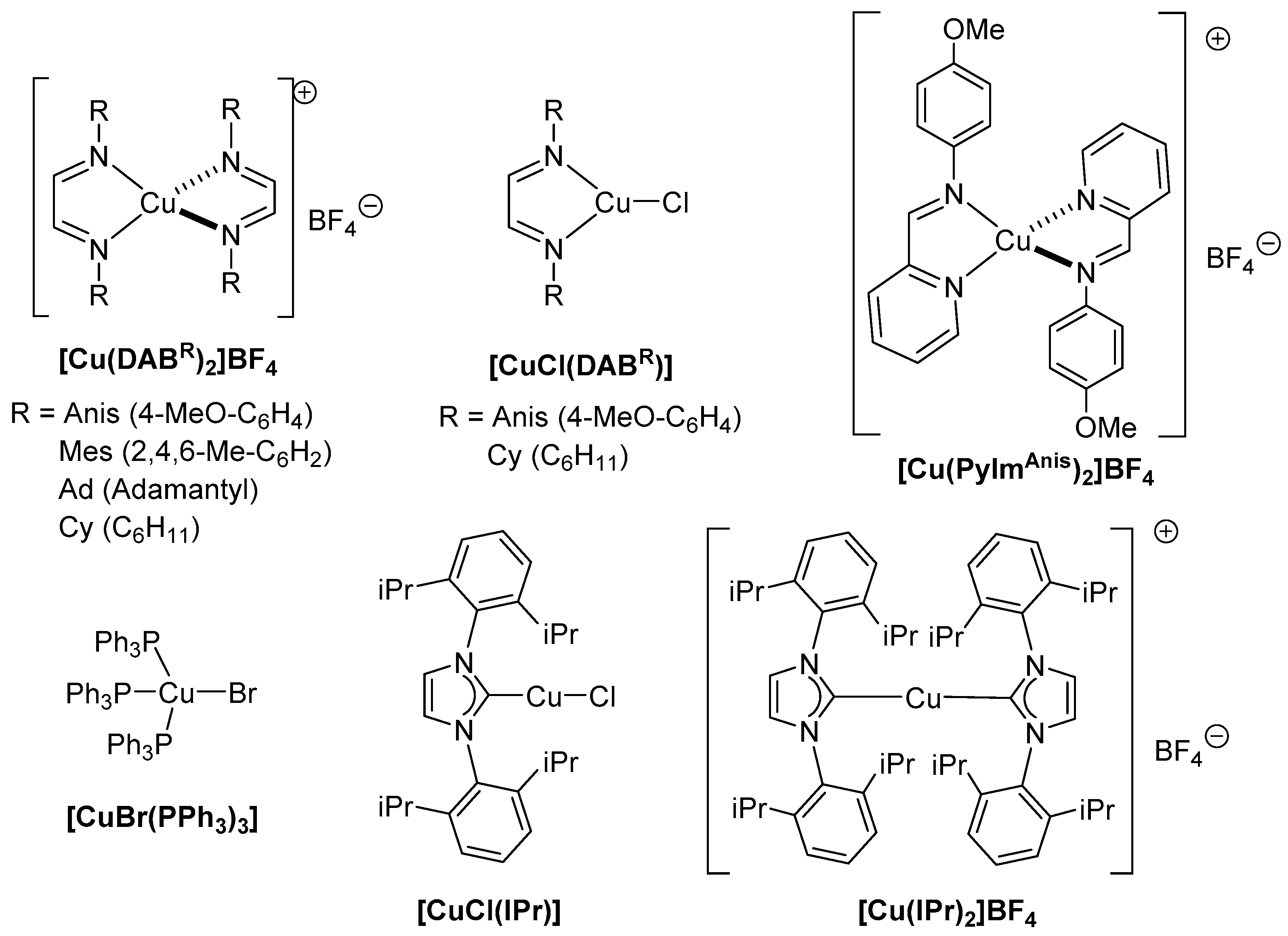
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | [Cu] | 1a-D (%) b | Entry | [Cu] | 1a-D (%) b |
|---|---|---|---|---|---|
| 1 | [Cu(DABAnis)2]BF4 | 92 | 8 | [CuCl(DABAnis)] | 7 |
| 2 | [Cu(DABMes)2]BF4 | 29 | 9 | [CuCl(DABCy)] | 54 |
| 3 | Cu(DABAd)2]BF4 | 10 | 10 | [CuBr(PPh3)3] | 11 |
| 4 | [Cu(DABCy)2]BF4 | 69 | 11 | [CuCl(IPr)] | 22 |
| 5 | [Cu(PyImAnis)2]BF4 | 23 | 12 | CuCl | 19 |
| 6 | [Cu(IPr)2]BF4 | 54 | 13 | -- | 15 |
| 7 | [Cu(NCMe)4]BF4 | 18 | 14 c | [Cu(DABAnis)2]BF4 | 67 |
 | |||
| Entry | Solvent | T (°C) | 1a-D (%) b |
|---|---|---|---|
| 1 | Acetone-d6 | 50 | 92 |
| 2 | Acetone-d6 | RT | 16 |
| 3 | Methanol-d4 | 50 | N.D. |
| 4 | CDCl3 | 50 | 63 |
| 5 | Toluene-d8 | 50 | 12 |
| 6 | Acetonitrile-d3 | 50 | N.D. |
 | |||||
| Entry | Deuterated Alkyne | [Cu] (mol%) | T (°C) | 1X-D(%) b | |
|---|---|---|---|---|---|
| 1 |  | 1a-D | 2 | 50 | 92 |
| 2 |  | 1b-D | 2 | 50 | 85 |
| 3 |  | 1c-D | 4 | 50 | 94 |
| 4 |  | 1d-D | 2 | 50 | 87 |
| 5 |  | 1e-D | 2 | 50 | 48 |
| 6 |  | 1f-D | 2 | 50 | 15 |
| 7 |  | 1g-D | 2 | 50 | 93 |
| 8 |  | 1h-D | 2 | 50 | 56 |
| 9 |  | 1i-D | 5 | 50 | 91 |
| 10 |  | 1j-D | 2 | 50 | 64 |
| 4 | 50 | 81 | |||
| 11 |  | 1k-D | 4 | 50 | 90 |
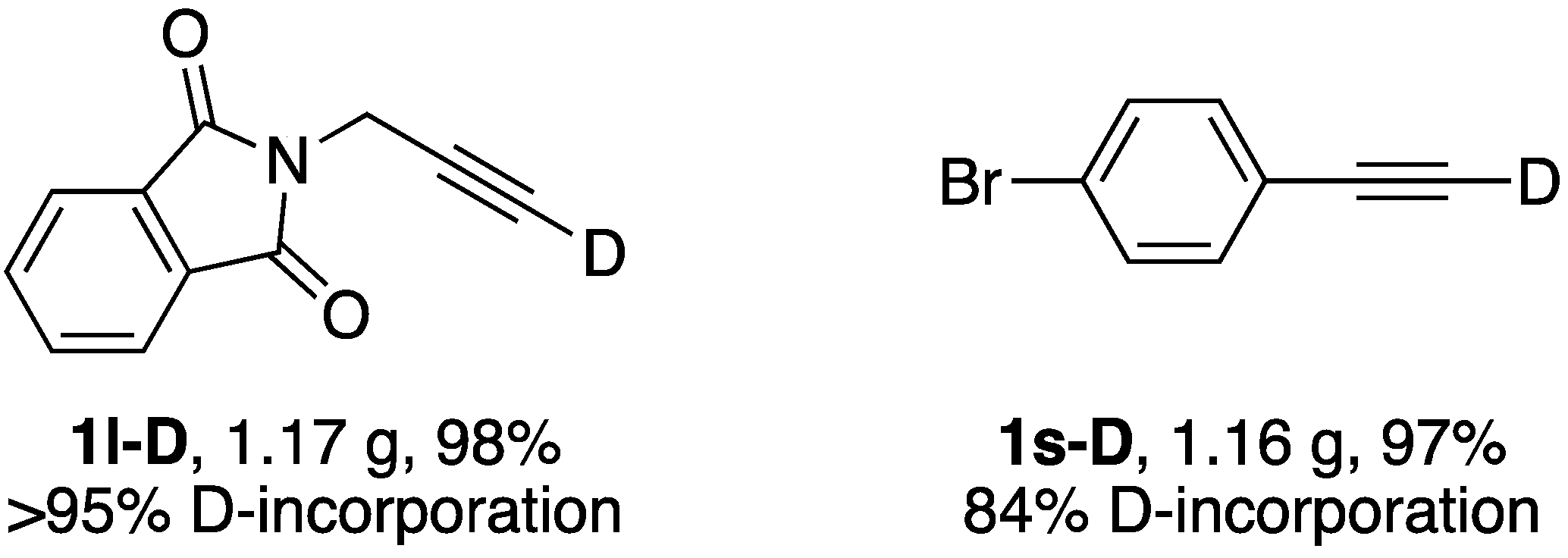
| 12 |  | 1l-D | 2 | 50 | 21 |
| 4 | 50 | 41 | |||
| 2 | rfx | >95 | |||
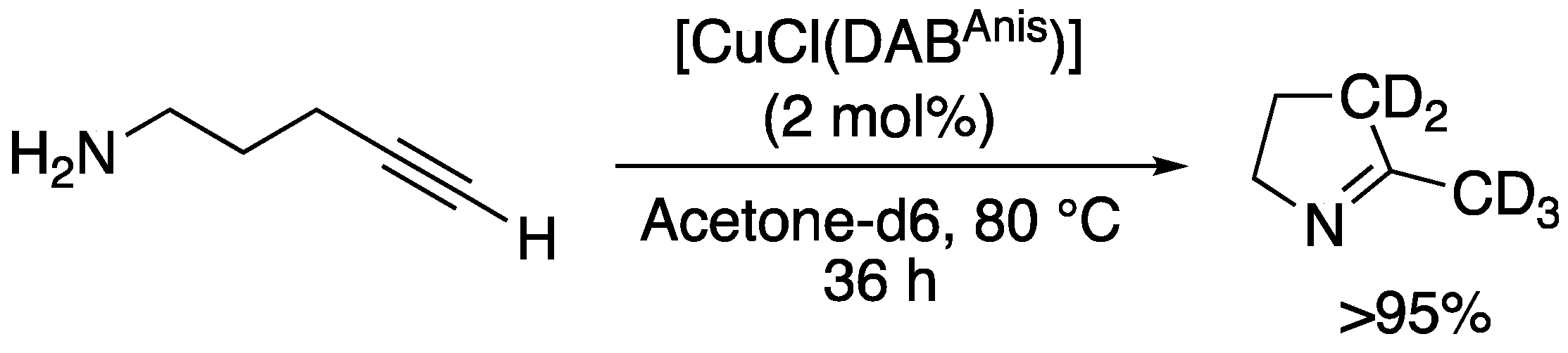
| 13 c |  | 1m-D | 2 | 50 | >95 |
| 14 c |  | 1n-D | 2 | rfx | 77 |
| 15 d |  | 1o-D | 2 | 60 | 93 (66) |
| 2 | 50 | 84 (<5) c | |||
| 2 | rt | 16 (<5) | |||
| 16 |  | 1p-D | 2 | rfx | 33 |
| 2 | 50 | 20 | |||
| 17 |  | 1q-D | 2 | 50 | 85 |
| 18 |  | 1r-D | 2 | rfx | 94 |
| 2 | 50 | 51 | |||
| 4 | 50 | 53 | |||
| 19 |  | 1s-D | 2 | rfx | 94 |
| 2 | 50 | 46 | |||
| 20 c |  | 1t-D | 2 | 50 | 93 |
| 21 |  | 1u-D | 2 | 50 | 95 |
| 22 |  | 1v-D | 2 | 50 | 91 |
 | |||
| Entry | Acetone-d6/Toluene (v:v) | Acetone-d6 (equiv) | 1s-D (%) b |
|---|---|---|---|
| 1 | 1:0 | 17 | 92 |
| 2 | 1:1 | 8 | 88 |
| 3 | 3:7 | 6 | 88 |
| 4 | 1:16 | 1 | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarrach, X.; Yang, J.; Soleiman-Beigi, M.; Díez-González, S. Straightforward and Efficient Deuteration of Terminal Alkynes with Copper Catalysis. Catalysts 2023, 13, 648. https://doi.org/10.3390/catal13040648
Tarrach X, Yang J, Soleiman-Beigi M, Díez-González S. Straightforward and Efficient Deuteration of Terminal Alkynes with Copper Catalysis. Catalysts. 2023; 13(4):648. https://doi.org/10.3390/catal13040648
Chicago/Turabian StyleTarrach, Xènia, Jingzhou Yang, Mohammad Soleiman-Beigi, and Silvia Díez-González. 2023. "Straightforward and Efficient Deuteration of Terminal Alkynes with Copper Catalysis" Catalysts 13, no. 4: 648. https://doi.org/10.3390/catal13040648