
Bimetal–Organic Framework-Derived CoMn@C Catalysts for Fischer–Tropsch Synthesis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. FTS Catalytic Performance
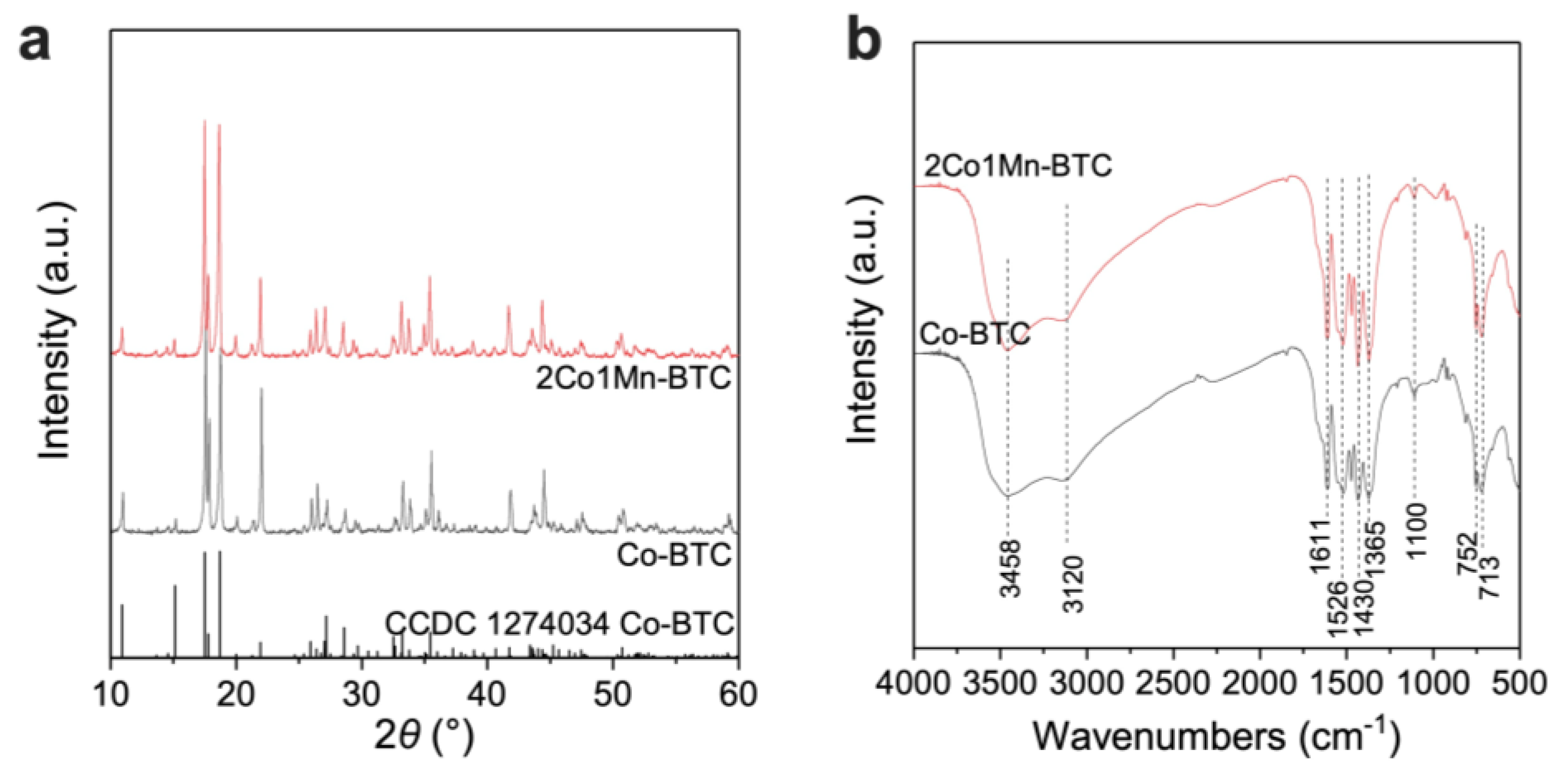
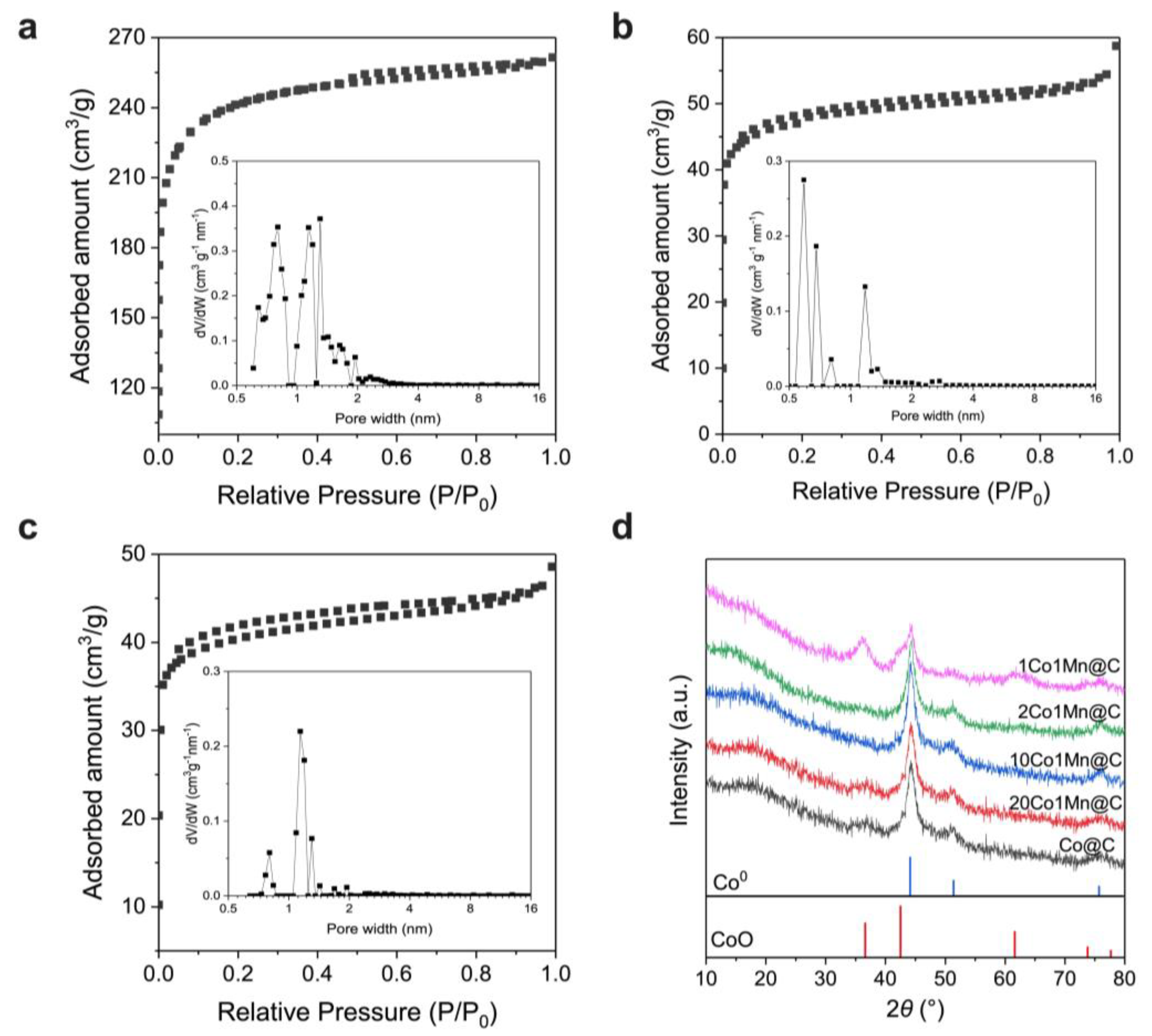
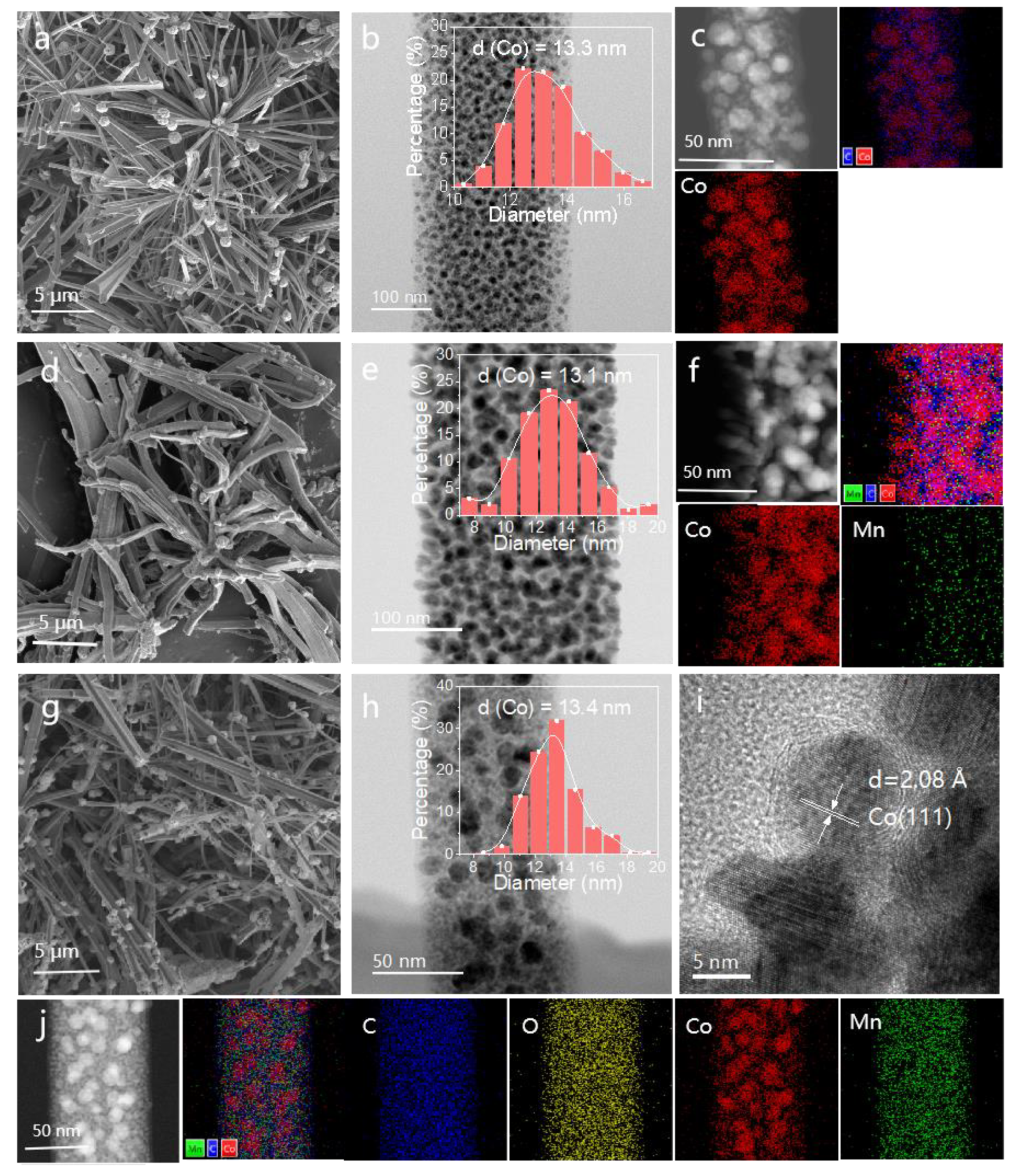
2.2. Morphology and Crystal Structure of Co-BTC and 2Co1Mn-BTC Precursors
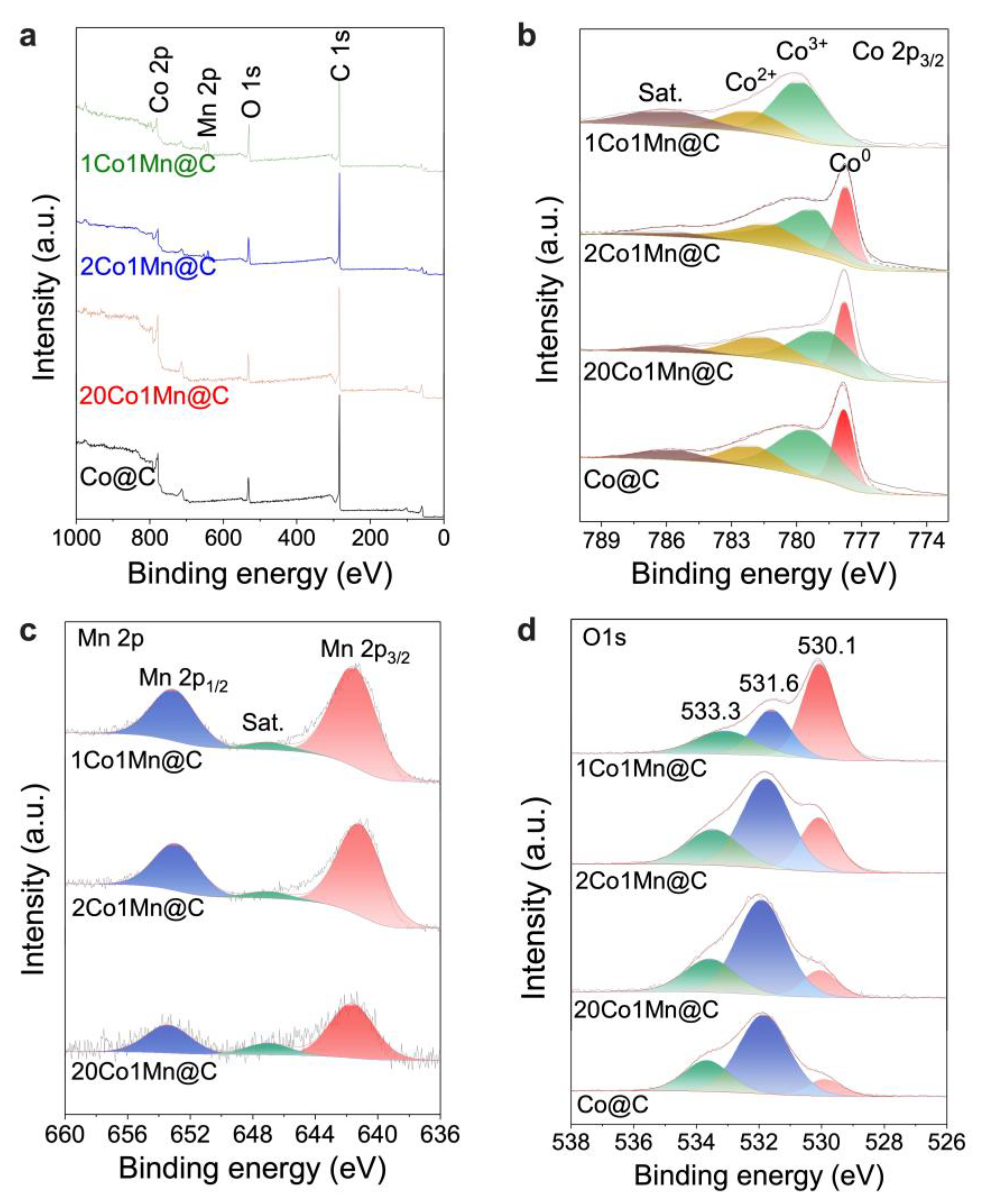
2.3. Mn Promotion Effect on the Catalysts
2.4. Theoretical Investigations
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Evaluation
3.3. Catalyst Characterization
3.4. Computational and Modeling Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bao, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Significant Advances in C1 Catalysis: Highly Efficient Catalysts and Catalytic Reactions. ACS Catal. 2019, 9, 3026–3053. [Google Scholar] [CrossRef]
- Mesters, C. A Selection of Recent Advances in C1 Chemistry. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 223–238. [Google Scholar] [CrossRef]
- Yang, J.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch Synthesis: A Review of the Effect of CO Conversion on Methane Selectivity. Appl. Catal. A Gen. 2014, 470, 250–260. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer–Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Zhang, Q.; Cheng, K.; Kang, J.; Deng, W.; Wang, Y. Fischer–Tropsch Catalysts for the Production of Hydrocarbon Fuels with High Selectivity. ChemSusChem 2014, 7, 1251–1264. [Google Scholar] [CrossRef]
- Chen, W.; Pestman, R.; Chiang, F.K.; Hensen, E.J. Silver Addition to a Cobalt Fischer–Tropsch Catalyst. J. Catal. 2018, 366, 107–114. [Google Scholar] [CrossRef]
- Zhang, X.; Su, H.; Zhang, Y.; Gu, X. Effect of CeO2 Promotion on the Catalytic Performance of Co/ZrO2 Catalysts for Fischer–Tropsch Synthesis. Fuel 2016, 184, 162–168. [Google Scholar] [CrossRef]
- Piao, Y.; Jiang, Q.; Li, H.; Matsumoto, H.; Liang, J.; Liu, W.; Pham-Huu, C.; Liu, Y.; Wang, F. Identify Zr Promotion Effects in Atomic Scale for Co-based Catalysts in Fischer–Tropsch Synthesis. ACS Catal. 2020, 10, 7894–7906. [Google Scholar] [CrossRef]
- Xiang, Y.; Kruse, N. Tuning the Catalytic CO Hydrogenation to Straight- and Long-Chain Aldehydes/Alcohols and Olefins/Paraffins. Nat. Commun. 2016, 7, 13058. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, X.; Gao, J.; Wang, J.; Ma, G.; Wen, X.; Yang, Y.; Li, Y.; Ding, M. A Hydrophobic FeMn@Si Catalyst Increases Olefins from Syngas by Suppressing C1 By-Products. Science 2021, 371, 610–613. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Radstake, P.B.; Falke, U.; Oosterbeek, H.P.; Kuipers, H.P.; Van Dillen, A.J.; De Jong, K.P. Investigation of Promoter Effects of Manganese Oxide on Carbon Nanofiber-Supported Cobalt Catalysts for Fischer–Tropsch Synthesis. J. Catal. 2006, 237, 152–161. [Google Scholar] [CrossRef]
- Feltes, T.E.; Espinosa-Alonso, L.; Smit, E.D.; D’Souza, L.; Meyer, R.J.; Weckhuysen, B.M.; Regalbuto, J.R. Selective Adsorption of Manganese onto Cobalt for Optimized Mn/Co/TiO2 Fischer–Tropsch Catalysts. J. Catal. 2010, 270, 95–102. [Google Scholar] [CrossRef]
- Johnson, G.R.; Werner, S.; Bell, A.T. An Investigation into the Effects of Mn Promotion on the Activity and Selectivity of Co/SiO2 for Fischer–Tropsch Synthesis: Evidence for Enhanced CO Adsorption and Dissociation. ACS Catal. 2015, 5, 5888–5903. [Google Scholar] [CrossRef] [Green Version]
- Lyu, S.; Wu, Q.; Li, Z.; Zhang, Y.; Li, J.; Wang, L. Cobalt Clusters Decorated CoxMn1−xO Nanocomposites for Improving the Efficiency of Syngas to Lower Olefins with Lower CO2 Emission. Appl. Catal. B 2022, 325, 122347. [Google Scholar] [CrossRef]
- Koshy, D.M.; Johnson, G.R.; Bustillo, K.C.; Bell, A.T. Scanning Nanobeam Diffraction and Energy Dispersive Spectroscopy Characterization of a Model Mn-Promoted Co/Al2O3 Nanosphere Catalyst for Fischer–Tropsch Synthesis. ACS Catal. 2020, 10, 12071–12079. [Google Scholar] [CrossRef]
- Morales, F.; de Groot, F.M.; Gijzeman, O.L.; Mens, A.; Stephan, O.; Weckhuysen, B.M. Mn Promotion Effects in Co/TiO2 Fischer–Tropsch Catalysts as Investigated by XPS and STEM-EELS. J. Catal. 2005, 230, 301–308. [Google Scholar] [CrossRef]
- Fu, R.; Baumann, T.F.; Cronin, S.; Dresselhaus, G.; Dresselhaus, M.S.; Satcher, J.H. Formation of Graphitic Structures in Cobalt- and Nickel-Doped Carbon Aerogels. Langmuir 2005, 21, 2647–2651. [Google Scholar] [CrossRef]
- Shen, K.; Chen, X.; Chen, J.; Li, Y. Development of MOF-Derived Carbon-Based Nanomaterials for Efficient Catalysis. ACS Catal. 2016, 6, 5887–5903. [Google Scholar] [CrossRef]
- Yang, L.; Zeng, X.; Wang, W.; Cao, D. Recent Progress in MOF-Derived, Heteroatom-Doped Porous Carbons as Highly Efficient Electrocatalysts for Oxygen Reduction Reaction in Fuel Cells. Adv. Funct. Mater. 2018, 28, 1704537. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, J.; Fu, H.; Li, W.; Fan, X.; Xin, G.; Zheng, J.; Li, X. MOF Derived Catalysts for Electrochemical Oxygen Reduction. J. Mater. Chem. A 2014, 2, 14064–14070. [Google Scholar] [CrossRef]
- Qi, X.; Tian, H.; Dang, X.; Fan, Y.; Zhang, Y.; Zhao, H. A Bimetallic Co/Mn Metal-Organic-Framework with a Synergistic Catalytic Effect as Peroxidase for the Colorimetric Detection of H2O2. Anal. Methods 2019, 11, 1111–1124. [Google Scholar] [CrossRef]
- Luo, Q.X.; Guo, L.P.; Yao, S.Y.; Bao, J.; Liu, Z.T.; Liu, Z.W. Cobalt Nanoparticles Confined in Carbon Matrix for Probing the Size Dependence in Fischer–Tropsch Synthesis. J. Catal. 2019, 369, 143–156. [Google Scholar] [CrossRef]
- Pei, Y.; Li, Z.; Li, Y. Highly Active and Selective Co-Based Fischer–Tropsch Catalysts Derived from Metal-Organic Frameworks. AIChE J. 2017, 63, 2935–2944. [Google Scholar] [CrossRef]
- Qiu, B.; Yang, C.; Guo, W.; Xu, Y.; Liang, Z.; Ma, D.; Zou, R. Highly Dispersed Co-Based Fischer–Tropsch Synthesis Catalysts from Metal-Organic Frameworks. J. Mater. Chem. A 2017, 5, 8081–8086. [Google Scholar] [CrossRef]
- You, B.; Jiang, N.; Sheng, M.; Drisdell, W.S.; Yano, J.; Sun, Y. Bimetal-Organic Framework Self-Adjusted Synthesis of Support-Free Nonprecious Electrocatalysts for Efficient Oxygen Reduction. ACS Catal. 2015, 5, 7068–7076. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.C.; Ortuño, M.A.; Bernales, V.; Gaggioli, C.A.; Cramer, C.J.; Bhan, A.; Gagliardi, L. C–H Bond Activation on Bimetallic Two-Atom Co-M Oxide Clusters Deposited on Zr-Based MOF Nodes: Effects of Doping at the Molecular Level. ACS Catal. 2018, 8, 2864–2869. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, Y.J.; Kim, D.H.; Lee, Y.J. Bimetallic Metal-Organic Frameworks as Efficient Cathode Catalysts for Li-O2 Batteries. ACS Appl. Mater. Interfaces 2018, 10, 660–667. [Google Scholar] [CrossRef]
- Ji, Z.; Li, T.; Yaghi, O.M. Sequencing of Metals in Multivariate Metal-Organic Frameworks. Science 2020, 369, 674–680. [Google Scholar] [CrossRef]
- Wu, L.L.; Wang, Z.; Long, Y.; Li, J.; Liu, Y.; Wang, Q.S.; Wang, X.; Song, S.Y.; Liu, X.; Zhang, H.J. Multishelled NixCo3−xO4 Hollow Microspheres Derived from Bimetal-Organic Frameworks as Anode Materials for High-Performance Lithium-Ion Batteries. Small 2017, 13, 1604270. [Google Scholar] [CrossRef]
- Fang, R.; Luque, R.; Li, Y. Selective Aerobic Oxidation of Biomass-Derived HMF to 2,5-Diformylfuran Using a MOF-Derived Magnetic Hollow Fe-Co Nanocatalyst. Green Chem. 2016, 18, 3152–3157. [Google Scholar] [CrossRef]
- Liu, H.; Xia, G.; Zhang, R.; Jiang, P.; Chen, J.; Chen, Q. MOF-Derived RuO2/Co3O4 Heterojunctions as Highly Efficient Bifunctional Electrocatalysts for HER and OER in Alkaline Solutions. RSC Adv. 2017, 7, 3686–3694. [Google Scholar] [CrossRef] [Green Version]
- Yaghi, O.M.; Li, H.; Groy, T.L. Construction of Porous Solids from Hydrogen-Bonded Metal Complexes of 1,3,5-Benzenetricarboxylic Acid. J. Am. Chem. Soc. 1996, 118, 9096–9101. [Google Scholar] [CrossRef]
- Abbasi, A.; Soleimani, M.; Najafi, M.; Geranmayeh, S. New Interpenetrated Mixed (Co/Ni) Metal-Organic Framework for Dye Removal Under Mild Conditions. Inorg. Chim. Acta 2016, 439, 18–23. [Google Scholar] [CrossRef]
- Kirkland, E.J.; Loane, R.F.; Silcox, J. Simulation of Annular Dark Field STEM Images Using a Modified Multislice Method. Ultramicroscopy 1987, 23, 77–96. [Google Scholar] [CrossRef]
- Dong, Z.; Liu, G.; Zhou, S.; Zhang, Y.; Zhang, W.; Fan, A.; Zhang, X.; Dai, X. Restructured Fe−Mn Alloys Encapsulated by N-doped Carbon Nanotube Catalysts Derived from Bimetallic MOF for Enhanced Oxygen Reduction Reaction. ChemCatChem 2018, 10, 5475–5486. [Google Scholar] [CrossRef]
- Liu, D.; Li, M.; Li, X.; Ren, F.; Sun, P.; Zhou, L. Core-Shell Zn/Co MOFs Derived Co3O4/CNTs as an Efficient Magnetic Heterogeneous Catalyst for Persulfate Activation and Oxytetracycline Degradation. Chem. Eng. J. 2020, 387, 124008. [Google Scholar] [CrossRef]
- Pedersen E, Ø.; Svenum, I.H.; Blekkan, E.A. Mn Promoted Co Catalysts for Fischer–Tropsch Production of Light Olefins-An Experimental and Theoretical Study. J. Catal. 2018, 361, 23–32. [Google Scholar] [CrossRef]
- Ma, X.; Su, H.Y.; Deng, H.; Li, W.X. Carbon Monoxide Adsorption and Dissociation on Mn-Decorated Rh (111) and Rh (553) Surfaces: A First-Principles Study. Catal. Today 2011, 160, 228–233. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Gao, Y.; Guo, Y.; Li, Z.; Cen, J.; Yao, N.; Li, X. Bimetal–Organic Framework-Derived CoMn@C Catalysts for Fischer–Tropsch Synthesis. Catalysts 2023, 13, 633. https://doi.org/10.3390/catal13030633
Yang L, Gao Y, Guo Y, Li Z, Cen J, Yao N, Li X. Bimetal–Organic Framework-Derived CoMn@C Catalysts for Fischer–Tropsch Synthesis. Catalysts. 2023; 13(3):633. https://doi.org/10.3390/catal13030633
Chicago/Turabian StyleYang, Linyan, Yu Gao, Yupeng Guo, Zhengjia Li, Jie Cen, Nan Yao, and Xiaonian Li. 2023. "Bimetal–Organic Framework-Derived CoMn@C Catalysts for Fischer–Tropsch Synthesis" Catalysts 13, no. 3: 633. https://doi.org/10.3390/catal13030633