

Polydopamine-Coated Polyurethane Foam as a Structured Support for the Development of an Easily Reusable Heterogeneous Photocatalyst Based on Eosin Y

Abstract
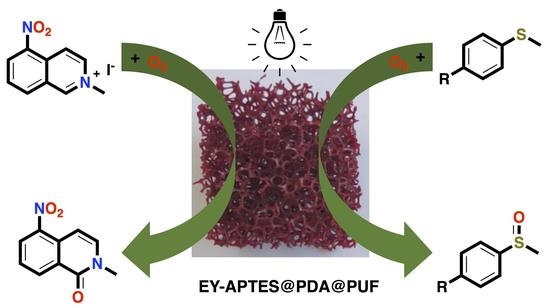
:
1. Introduction
2. Results and Discussion
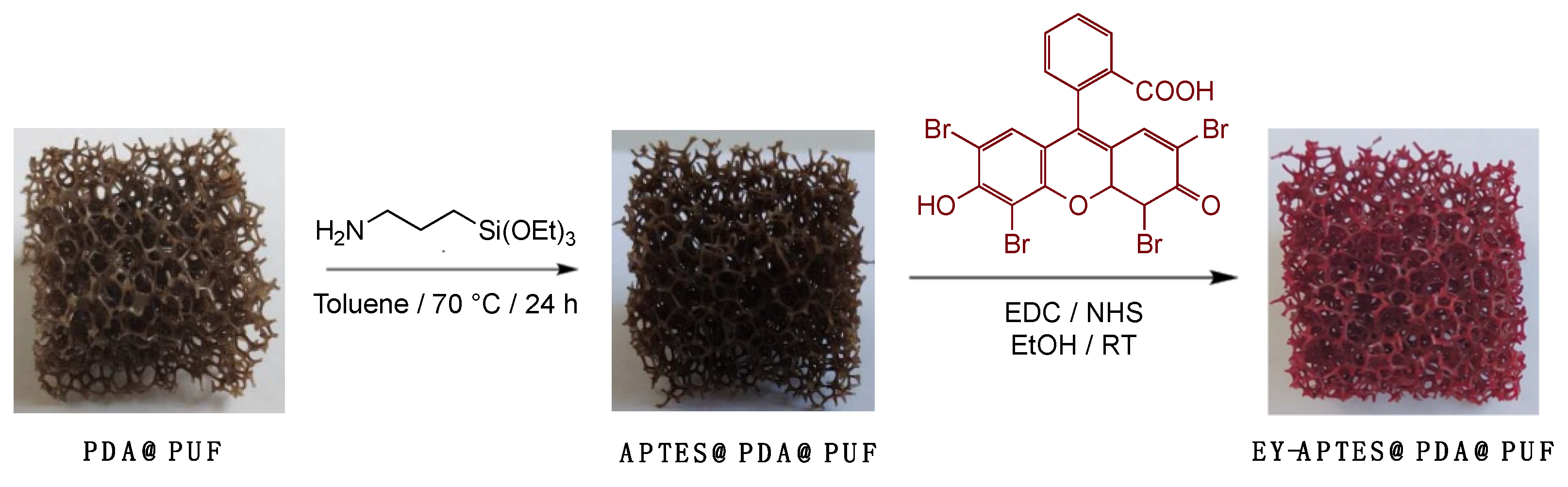
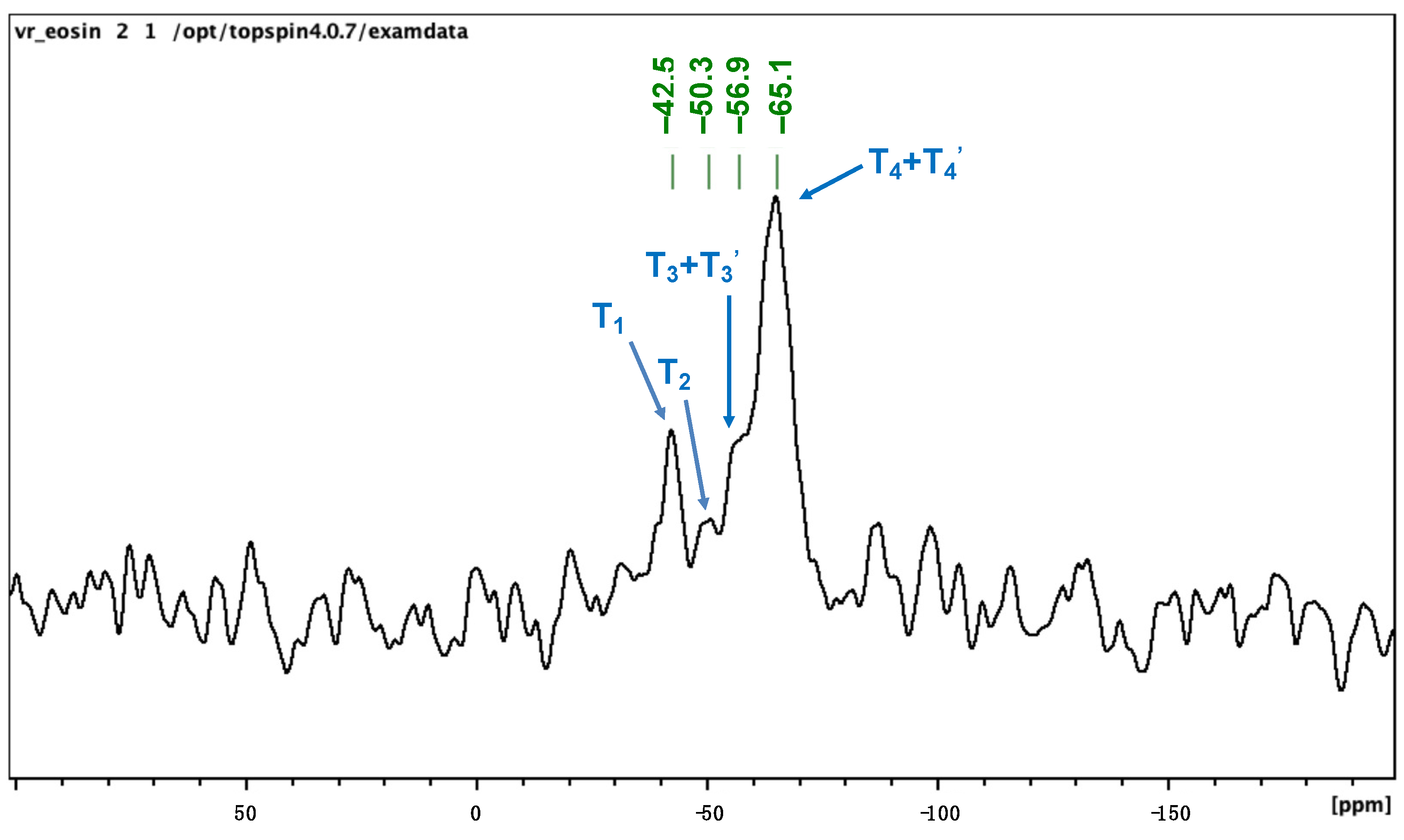
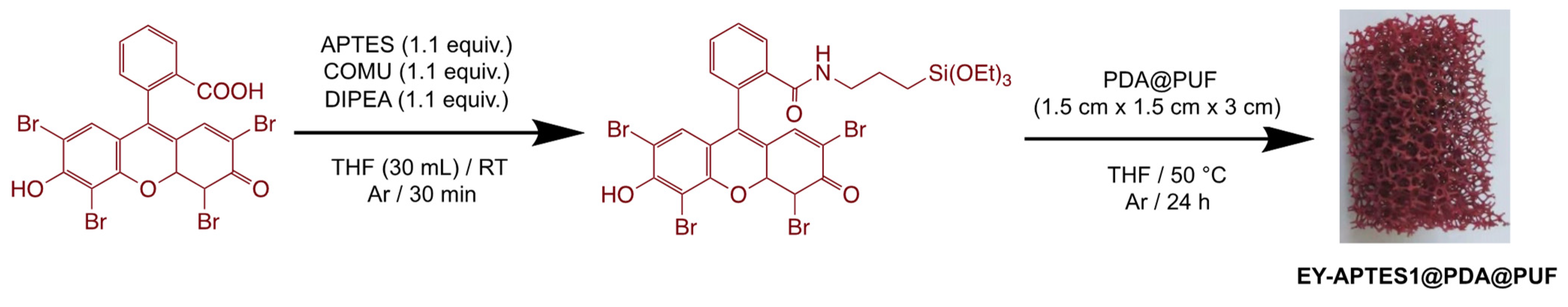
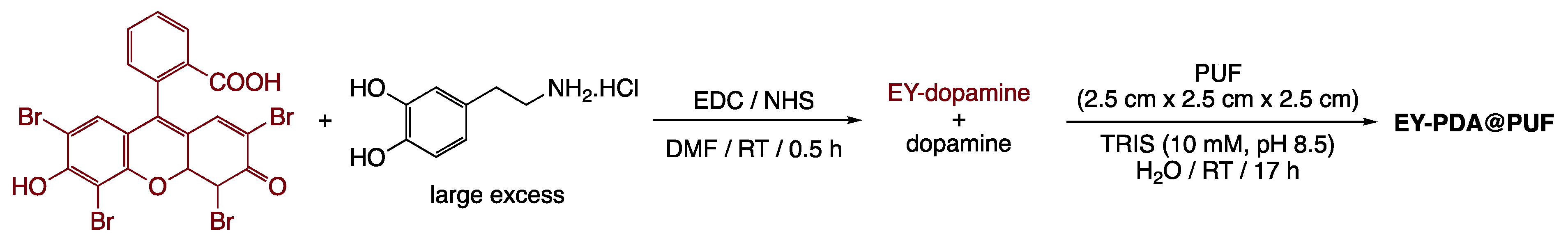
2.1. Preparation and Characterization of EY-APTES@PDA@PUF Based on Post-Functionalization of PDA@PUF
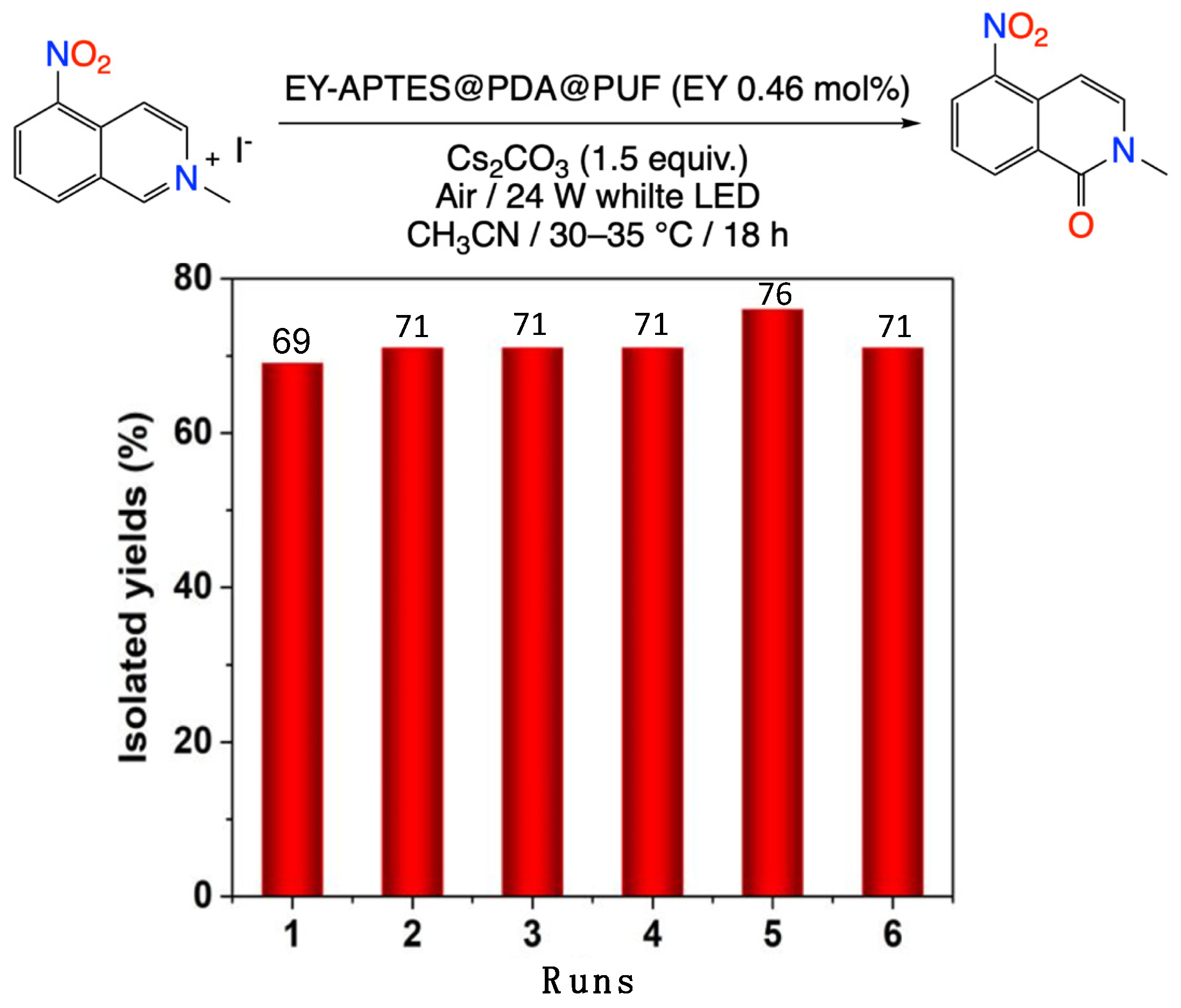
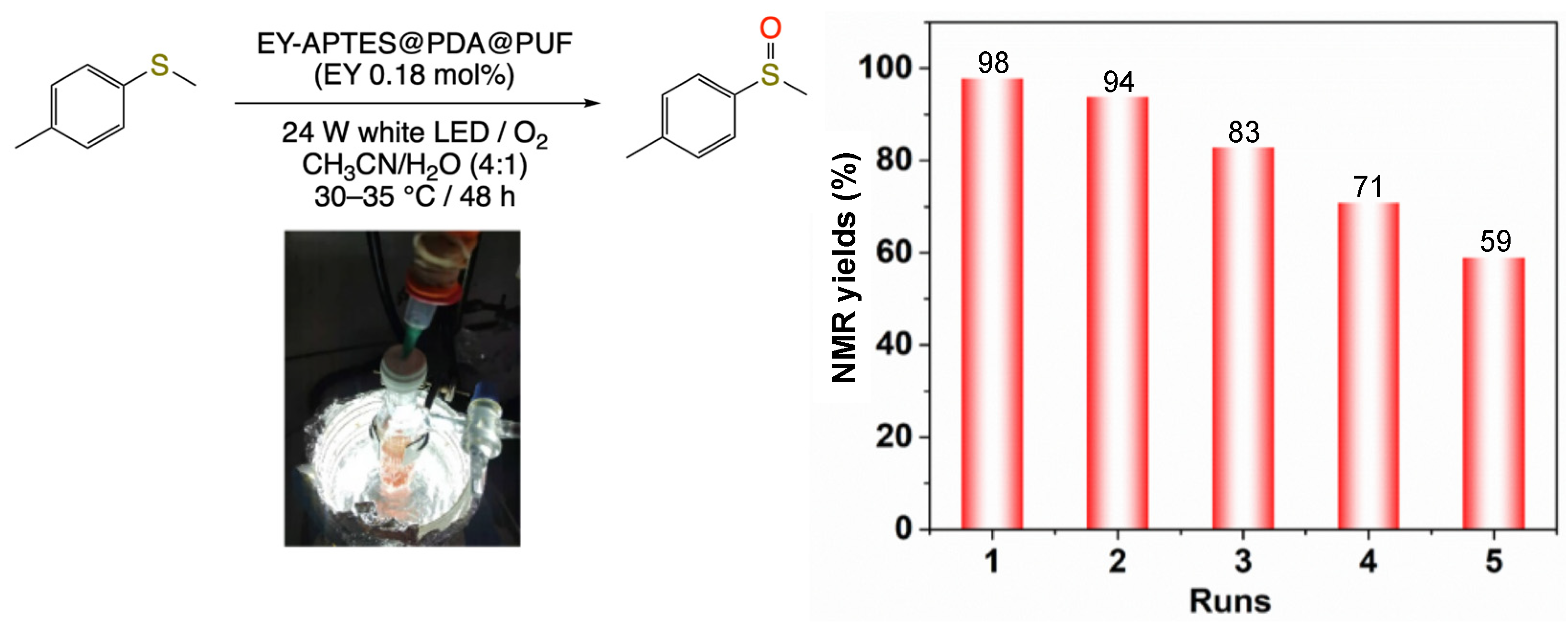
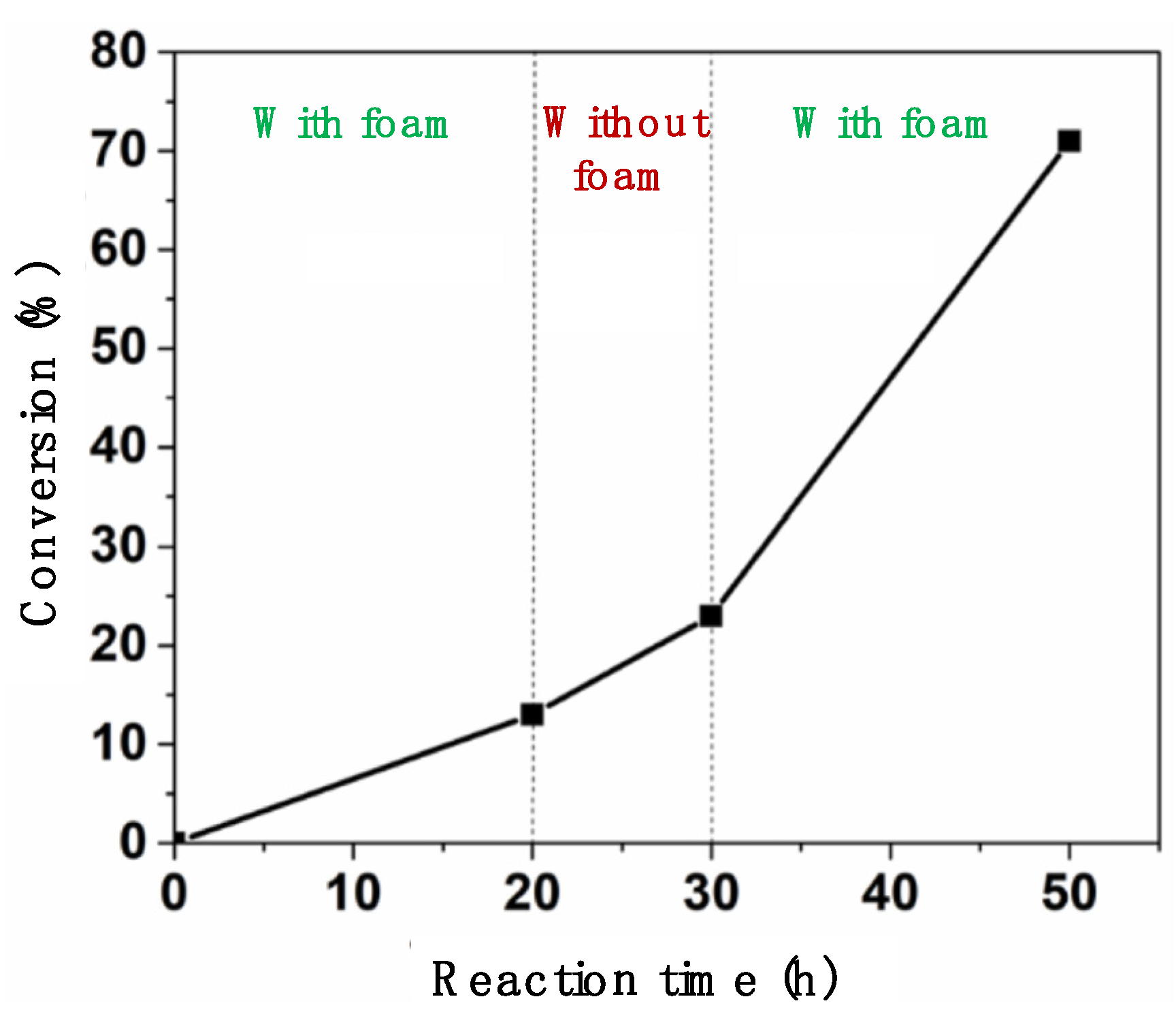
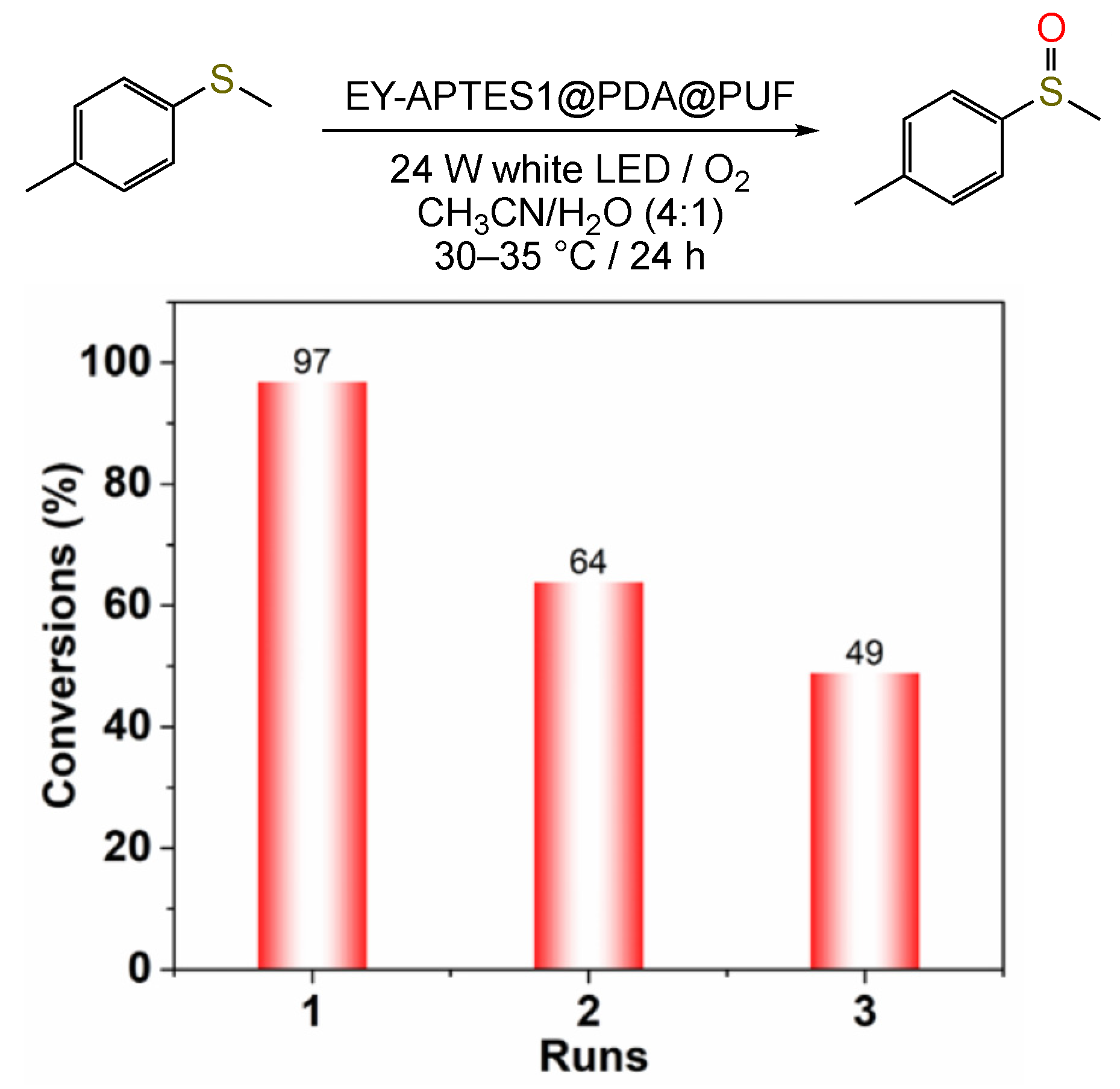
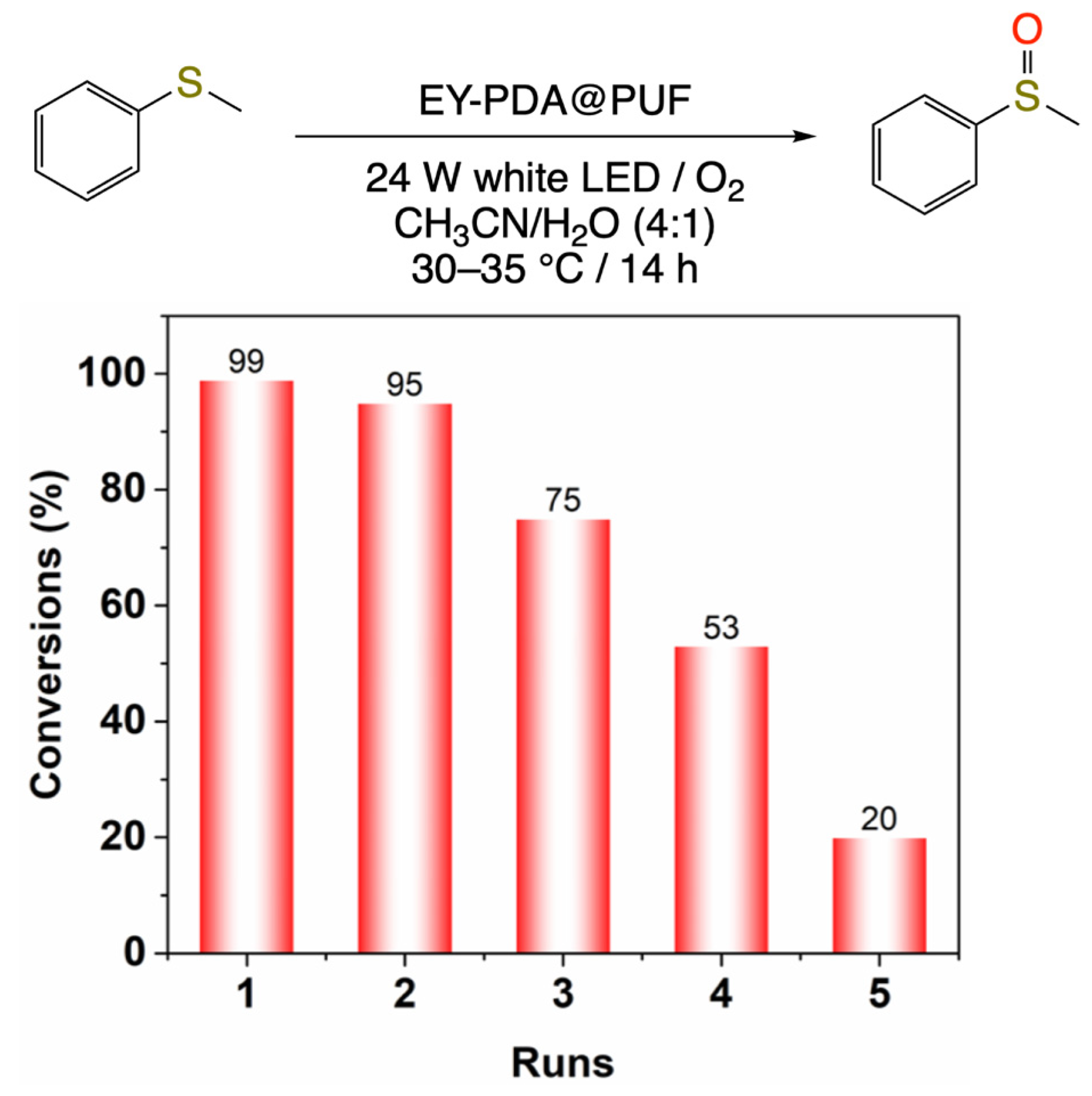
2.2. Photocatalytic Studies
3. Experimental Section
3.1. Materials and Methods
3.2. Experimental Procedures
3.2.1. Open Cell Polyurethane Foam (PUF) Coating with Polydopamine (PDA)
3.2.2. Functionalization of PDA@PUF with APTES
3.2.3. Coupling of Eosin Y with APTES@PDA@PUF
3.2.4. Preparation of EY-APTES1@PDA@PUF in a One-Pot Procedure from PDA@PUF
3.2.5. Preparation of EY-PDA@PUF
3.2.6. General Procedure for the Oxidation of 2-methyl-5-nitro-isoquinolin-2-ium Iodide Photocatalyzed by EY-APTES@PDA@PUF
3.2.7. General Procedures for the Oxidation of Sulfides to Sulfoxides Photocatalyzed by EY-APTES@PDA@PUF, EY-APTES1@PDA@PUF or EY-PDA@PUF
3.2.8. Reusability Procedure
3.2.9. Filtration Test for the Oxidation of p-Tolyl Methyl Sulfide
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hari, D.P.; König, B. Synthetic applications of eosin Y in photoredox catalysis. Chem. Commun. 2014, 50, 6688–6699. [Google Scholar] [CrossRef]
- Pitre, S.P.; McTiernan, C.D.; Ismaili, H.; Scaiano, J.C. Mechanistic Insights and Kinetic Analysis for the Oxidative Hydroxylation of Arylboronic Acids by Visible Light Photoredox Catalysis: A Metal-Free Alternative. J. Am. Chem. Soc. 2013, 135, 13286–13289. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhao, J.; Guo, S.; Zhang, C.; Ma, J. Bodipy Derivatives as Organic Triplet Photosensitizers for Aerobic Photoorganocatalytic Oxidative Coupling of Amines and Photooxidation of Dihydroxylnaphthalenes. J. Org. Chem. 2013, 78, 5627–5637. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Yan, J.; Tang, H.; Kuek, E.J.R.; Shi, X.; Liu, C.; Zhang, M.; Piper, J.L.; Duan, S.; Wu, J. Divergent functionalization of aldehydes photocatalyzed by neutral eosin Y with sulfone reagents. Nat. Commun. 2021, 12, 7214. [Google Scholar] [CrossRef]
- Herbrik, F.; Camarero González, P.; Krstic, M.; Puglisi, A.; Benaglia, M.; Sanz, M.A.; Rossi, S. Eosin Y: Homogeneous Photocatalytic In-Flow Reactions and Solid-Supported Catalysts for In-Batch Synthetic Transformations. Appl. Sci. 2020, 10, 5596. [Google Scholar] [CrossRef]
- Yang, X.-J.; Chen, B.; Zheng, L.-Q.; Wu, L.-Z.; Tung, C.-H. Highly efficient and selective photocatalytic hydrogenation of functionalized nitrobenzenes. Green Chem. 2014, 16, 1082–1086. [Google Scholar] [CrossRef]
- Sridhar, A.; Rangasamy, R.; Selvaraj, M. Polymer-supported eosin Y as a reusable photocatalyst for visible light mediated organic transformations. New J. Chem. 2019, 43, 17974–17979. [Google Scholar] [CrossRef]
- Rueping, M.; Zhu, S.; Koenigs, R.M. Photoredox catalyzed C–P bond forming reactions—Visible light mediated oxidative phosphonylations of amines. Chem. Commun. 2011, 47, 8679–8681. [Google Scholar] [CrossRef] [PubMed]
- Mangi, F.; Abdul Sattar, F.; Lu, C.Z. Metal Free Synthesis of Organosulfur Compounds Employing Eosin Y as Photoredox Catalyst. Chem. Methodol. 2019, 3, 535–542. [Google Scholar] [CrossRef]
- Hari, D.P.; Schroll, P.; König, B. Metal-Free, Visible-Light-Mediated Direct C–H Arylation of Heteroarenes with Aryl Diazonium Salts. J. Am. Chem. Soc. 2012, 134, 2958–2961. [Google Scholar] [CrossRef]
- Cantillo, D.; de Frutos, O.; Rincón, J.A.; Mateos, C.; Kappe, C.O. Continuous Flow α-Trifluoromethylation of Ketones by Metal-Free Visible Light Photoredox Catalysis. Org. Lett. 2014, 16, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.V.; Nagib, D.A.; MacMillan, D.W.C. Photoredox Catalysis: A Mild, Operationally Simple Approach to the Synthesis of α-Trifluoromethyl Carbonyl Compounds. Angew. Chem. Int. Ed. 2011, 50, 6119–6122. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shanmugam, S.; Duong, H.T.; Boyer, C. Organo-photocatalysts for photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization. Polym. Chem. 2015, 6, 5615–5624. [Google Scholar] [CrossRef]
- Yeow, J.; Chapman, R.; Xu, J.; Boyer, C. Oxygen tolerant photopolymerization for ultralow volumes. Polym. Chem. 2017, 8, 5012–5022. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, S.; Adnan, N.N.M.; Boyer, C. Heterogeneous Photocatalysis as a Means for Improving Recyclability of Organocatalyst in “Living” Radical Polymerization. Macromolecules 2018, 51, 779–790. [Google Scholar] [CrossRef]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Zeitler, K. A Cooperative Hydrogen-Bond-Promoted Organophotoredox Catalysis Strategy for Highly Diastereoselective, Reductive Enone Cyclization. Chem. Eur. J. 2013, 19, 6950–6955. [Google Scholar] [CrossRef]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef]
- Herbrik, F.; Rossi, S.; Sanz, M.; Puglisi, A.; Benaglia, M. Immobilised eosin Y for the photocatlytic oxidation of tetrahydroisoquinolines in flow. ChemCatChem 2022, 14, e202200461. [Google Scholar] [CrossRef]
- Douglas, J.J.; Sevrin, M.J.; Stephenson, C.R.J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Proc. Res. Dev. 2016, 20, 1134–1147. [Google Scholar] [CrossRef]
- Mou, Z.; Dong, Y.; Li, S.; Du, Y.; Wang, X.; Yang, P.; Wang, S. Eosin Y functionalized graphene for photocatalytic hydrogen production from water. Int. J. Hydrogen Energy 2011, 36, 8885–8893. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Li, Y.; Xia, L.; Jiang, M.; Wu, P. Highly Efficient Visible-Light-Driven H2 Production via an Eosin Y-Based Metal–Organic Framework. Inorg. Chem. 2018, 57, 7495–7498. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Solanki, P.; Nazish, M.; Neogi, S.; Kureshy, R.I.; Khan, N.-u.H. Covalently hooked EOSIN-Y in a Zr(IV) framework as visible-light mediated, heterogeneous photocatalyst for efficient CH functionalization of tertiary amines. J. Catal. 2019, 371, 298–304. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Z.; Qiu, B.; Guo, S.; Yang, P.; Yu, B.; Zhang, H.; Zhao, Y.; Yang, X.; Han, B.; et al. Eosin Y-Functionalized Conjugated Organic Polymers for Visible-Light-Driven CO2 Reduction with H2O to CO with High Efficiency. Angew. Chem. Int. Ed. 2019, 58, 632–636. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, S.; Xia, J.; Huang, Y.; Zhang, Y.C.; Li, X.; Cai, T. Hydrophilic ultrafiltration membranes with surface-bound eosin Y for an integrated synthesis-separation system of aqueous RAFT photopolymerization. J. Mater. Chem. A 2020, 8, 9825–9831. [Google Scholar] [CrossRef]
- Wang, C.-A.; Li, Y.-W.; Cheng, X.-L.; Zhang, J.-P.; Han, Y.-F. Eosin Y dye-based porous organic polymers for highly efficient heterogeneous photocatalytic dehydrogenative coupling reaction. RSC Adv. 2017, 7, 408–414. [Google Scholar] [CrossRef]
- Teixeira, R.I.; de Lucas, N.C.; Garden, S.J.; Lanterna, A.E.; Scaiano, J.C. Glass wool supported ruthenium complexes: Versatile, recyclable heterogeneous photoredox catalysts. Catal. Sci. Technol. 2020, 10, 1273–1280. [Google Scholar] [CrossRef]
- Chu, Y.; Huang, Z.; Liu, R.; Boyer, C.; Xu, J. Scalable and Recyclable Heterogeneous Organo-photocatalysts on Cotton Threads for Organic and Polymer Synthesis. ChemPhotoChem 2020, 4, 5201–5208. [Google Scholar] [CrossRef]
- Singh, P.; Yadav, R.K.; Kumar, K.; Lee, Y.; Gupta, A.K.; Kumar, K.; Yadav, B.C.; Singh, S.N.; Dwivedi, D.K.; Nam, S.-H.; et al. Eosin-Y and sulfur-codoped g-C3N4 composite for photocatalytic applications: The regeneration of NADH/NADPH and the oxidation of sulfide to sulfoxide. Catal. Sci. Technol. 2021, 11, 6401–6410. [Google Scholar] [CrossRef]
- Li, P.; Wang, G.-W.; Zhu, X.; Wang, L. Magnetic nanoparticle-supported eosin Y ammonium salt: An efficient heterogeneous catalyst for visible light oxidative C–C and C–P bond formation. Tetrahedron 2019, 75, 3448–3455. [Google Scholar] [CrossRef]
- Jarrahi, M.; Maleki, B.; Tayebee, R. Magnetic nanoparticle-supported eosin Y salt [SB- DABCO@eosin] as an efficient heterogeneous photocatalyst for the multi-component synthesis of chromeno [4,3-b]chromene in the presence of visible light. RSC Adv. 2022, 12, 28886–28901. [Google Scholar] [CrossRef]
- Bakker, J.J.; Groendijk, W.J.; de Lathouder, K.M.; Kapteijn, F.; Moulijn, J.A.; Kreutzer, M.T.; Wallin, S.A. Enhancement of catalyst performance using pressure pulses on macroporous structured catalysts. Ind. Eng. Chem. Res. 2007, 46, 8574–8583. [Google Scholar] [CrossRef]
- Giani, L.; Groppi, G.; Tronconi, E. Mass-transfer characterization of metallic foams as supports for structured catalysts. Ind. Eng. Chem. Res. 2005, 44, 4993–5002. [Google Scholar] [CrossRef]
- Richardson, J.; Peng, Y.; Remue, D. Properties of ceramic foam catalyst supports: Pressure drop. Appl. Catal. A 2000, 204, 19–32. [Google Scholar] [CrossRef]
- Lacroix, M.; Nguyen, P.; Schweich, D.; Pham-Huu, C.; Savin-Poncet, S.; Edouard, D. Pressure drop measurements and modeling on SiC foams. Chem. Eng. Sci. 2007, 62, 3259–3267. [Google Scholar] [CrossRef]
- Lacroix, M.; Dreibine, L.; de Tymowski, B.; Vigneron, F.; Edouard, D.; Bégin, D.; Nguyen, P.; Pham, C.; Savin-Poncet, S.; Luck, F.; et al. Silicon carbide foam composite containing cobalt as a highly selective and re-usable Fischer–Tropsch synthesis catalyst. Appl. Catal. A 2011, 397, 62–72. [Google Scholar] [CrossRef]
- Engels, H.W.; Pirkl, H.G.; Albers, R.; Albach, R.W.; Krause, J.; Hoffmann, A.; Casselmann, H.; Dormish, J. Polyurethanes: Versatile materials and sustainable problem solvers for today’s challenges. Angew. Chem. Int. Ed. 2013, 52, 9422–9441. [Google Scholar] [CrossRef]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef]
- Edouard, D.; Ritleng, V.; Jierry, L.; Chau, N.T.T. Method for Modifying the Surface Properties of Elastomer Cellular Foams. WO 2016012689A2, 2016. Available online: https://patents.google.com/patent/WO2016012689A3/en (accessed on 23 February 2023).
- Pardieu, E.; Chau, N.T.T.; Dintzer, T.; Romero, T.; Favier, D.; Roland, T.; Edouard, D.; Jierry, L.; Ritleng, V. Polydopamine-coated open cell polyurethane foams as an inexpensive, flexible yet robust catalyst support: A proof of concept. Chem. Commun. 2016, 52, 4691–4693. [Google Scholar] [CrossRef]
- Lefebvre, L.; Kelber, J.; Jierry, L.; Ritleng, V.; Edouard, D. Polydopamine-coated open cell polyurethane foam as an efficient and easy-to-regenerate soft structured catalytic support (S2CS) for the reduction of dye. J. Environ. Chem. Eng. 2017, 5, 79–85. [Google Scholar] [CrossRef]
- Lefebvre, L.; Kelber, J.; Mao, X.; Ponzio, F.; Agusti, G.; Vigier-Carrière, C.; Ball, V.; Jierry, L.; Ritleng, V.; Edouard, D. Borohydride-functionalized polydopamine-coated open cell polyurethane foam as a reusable soft structured material for reduction reactions: Application to the removal of a dye. Environ. Prog. Sustain. Energy 2019, 38, 329–335. [Google Scholar] [CrossRef]
- Ait Khouya, A.; Mendez Martinez, M.L.; Bertani, P.; Romero, T.; Favier, D.; Roland, T.; Guidal, V.; Bellière-Baca, V.; Edouard, D.; Jierry, L.; et al. Coating of polydopamine on polyurethane open cell foams to design soft structured supports for molecular catalysts. Chem. Commun. 2019, 55, 11960–11963. [Google Scholar] [CrossRef]
- Birba, L.; Ritleng, V.; Jierry, L.; Agusti, G.; Fongarland, P.; Edouard, D. An efficient bio-inspired catalytic tool for hydrogen release at room temperature from a stable borohydride solution. Int. J. Energy Res. 2020, 44, 10612–10627. [Google Scholar] [CrossRef]
- Ponzio, F.; Kelber, J.; Birba, L.; Rekab, K.; Ritleng, V.; Jierry, L.; Edouard, D. Polydopamine film coating on polyurethane foams as efficient “sunscreen”. Application to photocatalysis under UV irradiation. Environ. Technol. Innov. 2021, 23, 101618. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, X.; Papaefthimiou, V.; Pham-Huu, C.; Ritleng, V. Pd-functionalized polydopamine-coated polyurethane foam: A readily prepared and highly reusable structured catalyst for selective alkyne semi-hydrogenation and Suzuki coupling under air. Green Chem. 2023, 25, 264–279. [Google Scholar] [CrossRef]
- Saiz-Poseu, J.; Mancebo-Aracil, J.; Nador, F.; Busqué, F.; Ruiz-Molina, D. The chemistry behind catechol-based adhesion. Angew. Chem. Int. Ed. 2019, 58, 696–714. [Google Scholar] [CrossRef]
- Jin, Y.; Ou, L.; Yang, H.; Fu, H. Visible-Light-Mediated Aerobic Oxidation of N-Alkylpyridinium Salts under Organic Photocatalysis. J. Am. Chem. Soc. 2017, 139, 14237–14243. [Google Scholar] [CrossRef]
- Sandaroos, R.; Maleki, B.; Naderi, S.; Peiman, S. Efficient synthesis of sulfones and sulfoxides from sulfides by cobalt-based. Schiff complex supported on nanocellulose as catalyst and Oxone as the terminal oxidant. Inorg. Chem. Commun. 2023, 148, 110294. [Google Scholar] [CrossRef]
- Peng, H. Supports Catalytiques Structurés à Base de Mousses de Polyuréthane Fonctionnalisées: Applications en Photocatalyse et en Semi-hydrogénation D’alcynes. Ph.D. Thesis, Université de Strasbourg, Strasbourg, France, 2023. [Google Scholar]
- Vejayakumaran, P.; Rahman, I.A.; Sipaut, C.S.; Ismail, J.; Chee, C.K. Structural and thermal characterizations of silica nanoparticles grafted with pendant maleimide and epoxide groups. J. Colloid Interface Sci. 2008, 328, 81–91. [Google Scholar] [CrossRef]
- Albert, K.; Pfleiderer, B.; Bayer, E.; Schnabel, R. Characterization of chemically modified glass surfaces by 13C and 29Si CP/MAS NMR spectroscopy. J. Colloid Interface Sci. 1991, 142, 35–40. [Google Scholar] [CrossRef]
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef]
- Claassen, G.; Brin, E.; Crogan-Grundy, C.; Vaillancourt, M.T.; Zhang, H.Z.; Cai, S.X.; Drewe, J.; Tseng, B.; Kasibhatla, S. Selective activation of apoptosis by a novel set of 4-aryl-3-(3-aryl-1-oxo-2-propenyl)-2(1H)-quinolinones through a Myc-dependent pathway. Cancer Lett. 2009, 274, 243–249. [Google Scholar] [CrossRef]
- Forbis, R.M.; Rinehart, K.L. Nybomycin. VII. Preparative routes to nybomycin and deoxynybomycin. J. Am. Chem. Soc. 1973, 95, 5003–5013. [Google Scholar] [CrossRef]
- Afonso, A.; Weinstein, J.; Gentles, M.J. Antiviral Compounds and Antihypertensive Compounds. Patent WO92004327, 19 March 1992. [Google Scholar]
- Bai, L.-G.; Zhou, Y.; Zhuang, X.; Zhang, L.; Xue, J.; Lin, X.-L.; Cai, T.; Luo, Q.-L. Base-promoted aerobic oxidation of N-alkyl iminium salts derived from isoquinolines and related heterocycles. Green Chem. 2020, 22, 197–203. [Google Scholar] [CrossRef]
- Goncharov, N.; Orekhov, A.N.; Voitenko, N.; Ukolov, A.; Jenkins, R.; Avdonin, P. Organosulfur Compounds as Nutraceuticals. In Nutraceuticals; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 555–568. [Google Scholar] [CrossRef]
- Huang, X.; Maulide, N. Sulfoxide-Mediated α-Arylation of Carbonyl Compounds. J. Am. Chem. Soc. 2011, 133, 8510–8513. [Google Scholar] [CrossRef]
- Carreno, M.C. Applications of Sulfoxides to Asymmetric Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Shi, J.-L.; Lang, X. Assembling polydopamine on TiO2 for visible light photocatalytic selective oxidation of sulfides with aerial O2. Chem. Eng. J. 2020, 392, 123632. [Google Scholar] [CrossRef]
- Wang, G.; Hu, W.; Hu, Z.; Zhang, Y.; Yao, W.; Li, L.; Fu, Z.; Huang, W. Carbene-catalyzed aerobic oxidation of isoquinolinium salts: Efficient synthesis of isoquinolinones. Green Chem. 2018, 20, 3302–3307. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Light | Time (h) | Isolated Yield (%) |
| 1 a | Eosin Y | THF | 40 W CFL | 6 | 94 |
| 2 b | Eosin Y | THF | 24 W white LED | 18 | 71 |
| 3 b | Eosin Y | EtOH | 24 W white LED | 18 | 76 |
| 4 b | Eosin Y | CH3CN | 24 W white LED | 18 | 96 |
| 5 c | EY-APTES@PDA@PUF | CH3CN | 24 W white LED | 18 | 70 |
| 6 d | - | CH3CN | 24 W white LED | 18 | 44 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yield (%) | TON d | TOF (h−1) e |
| 1 a | Eosin Y | CH3CN/H2O (3:1) | 6 | 84 | 168 | 28 |
| 2 b | EY-APTES@PDA@PUF | CH3CN/H2O (3:1) | 18 | 36 | 200 | 11.1 |
| 3 b | EY-APTES@PDA@PUF | CH3CN | 18 | 0 | 0 | 0 |
| 4 b | EY-APTES@PDA@PUF | CH3CN/H2O (1:1) | 18 | 9 | 50 | 2.8 |
| 5 c | EY-APTES@PDA@PUF | CH3CN/H2O (4:1) | 48 | 98 | 544 | 11.3 |
| 6 c | PDA@PUF | CH3CN/H2O (4:1) | 48 | 0 | 0 | 0 |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (EY mol%) | Solvent | Light | Temp. | Atmosph. | R1 | R2 | Time (h) | Yield (%) | TON a | TOF (h−1) b |
| 1 c | EY-APTES@ PDA@PUF (0.18) | CH3CN/H2O (4:1) | White LED 24 W | 30–35 °C | O2 balloon | Me | Me | 48 | 98 | 544 | 11.3 |
| 2 d | PSEY (3) | CH3CN/H2O (4:1) | Green LED 3 W | RT | Open air | H | Me | 6 | 91 | 30.3 | 5.1 |
| 3 e | EY@Cotton thread (3.6) | CH3CN/H2O (5:2) | Green light 0.45 mW/cm2 | RT | Air bubbling | H | Me | 28 | 45 | 12.5 | 0.4 |
| 4 f | EY-s-g-C3N4 | EtOH HCl (0.05 M) | Blue LED | RT | Open air | Cl | C6H4Cl | - | 99.6 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, H.; Romero, T.; Bertani, P.; Ritleng, V. Polydopamine-Coated Polyurethane Foam as a Structured Support for the Development of an Easily Reusable Heterogeneous Photocatalyst Based on Eosin Y. Catalysts 2023, 13, 589. https://doi.org/10.3390/catal13030589
Peng H, Romero T, Bertani P, Ritleng V. Polydopamine-Coated Polyurethane Foam as a Structured Support for the Development of an Easily Reusable Heterogeneous Photocatalyst Based on Eosin Y. Catalysts. 2023; 13(3):589. https://doi.org/10.3390/catal13030589
Chicago/Turabian StylePeng, Han, Thierry Romero, Philippe Bertani, and Vincent Ritleng. 2023. "Polydopamine-Coated Polyurethane Foam as a Structured Support for the Development of an Easily Reusable Heterogeneous Photocatalyst Based on Eosin Y" Catalysts 13, no. 3: 589. https://doi.org/10.3390/catal13030589