

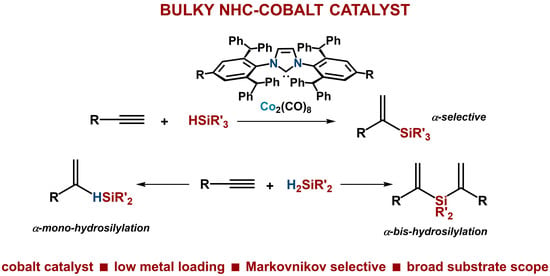
Bulky NHC–Cobalt Complex-Catalyzed Highly Markovnikov-Selective Hydrosilylation of Alkynes

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Reaction Conditions
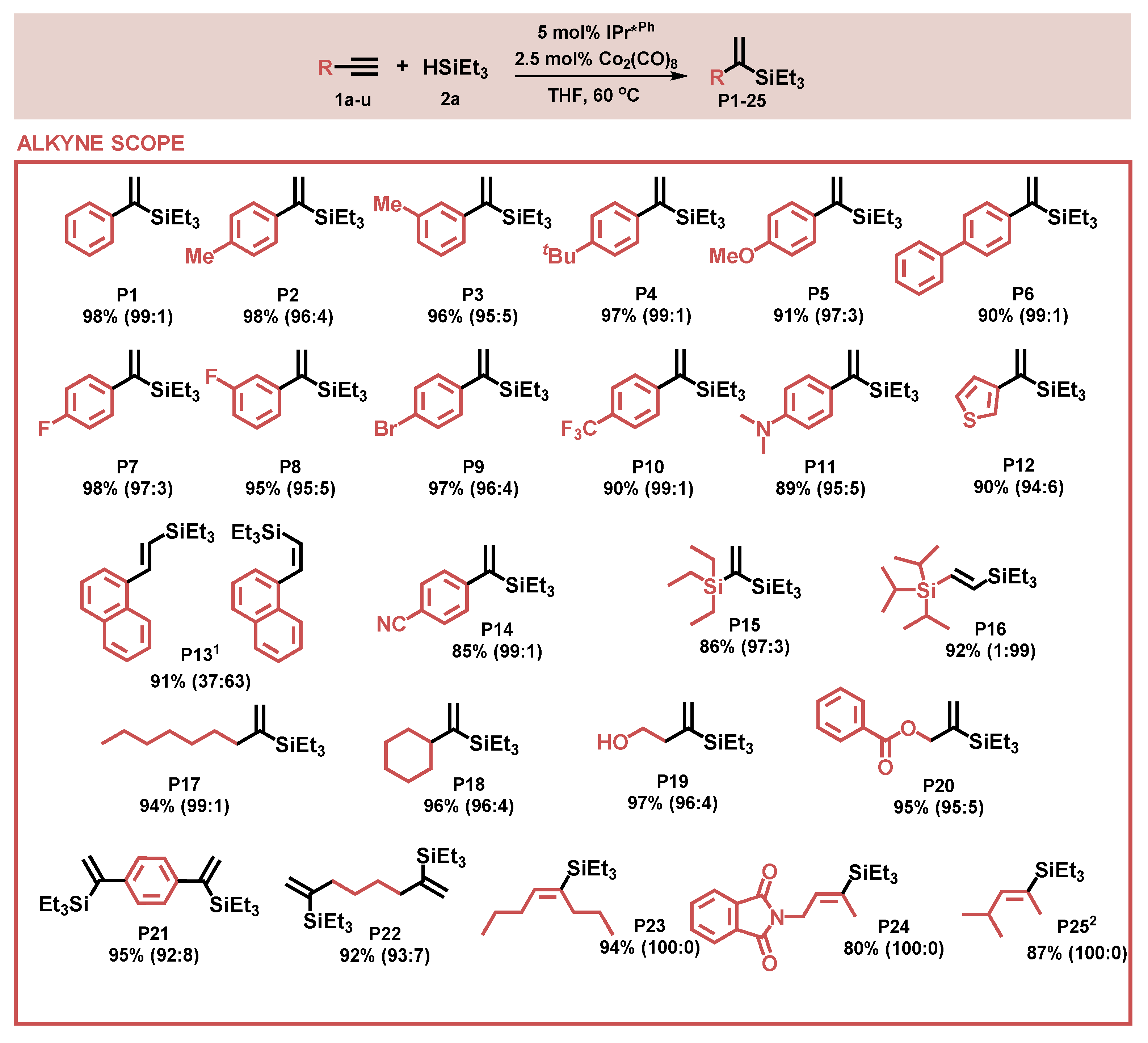
2.2. Substrate Scope—Alkynes
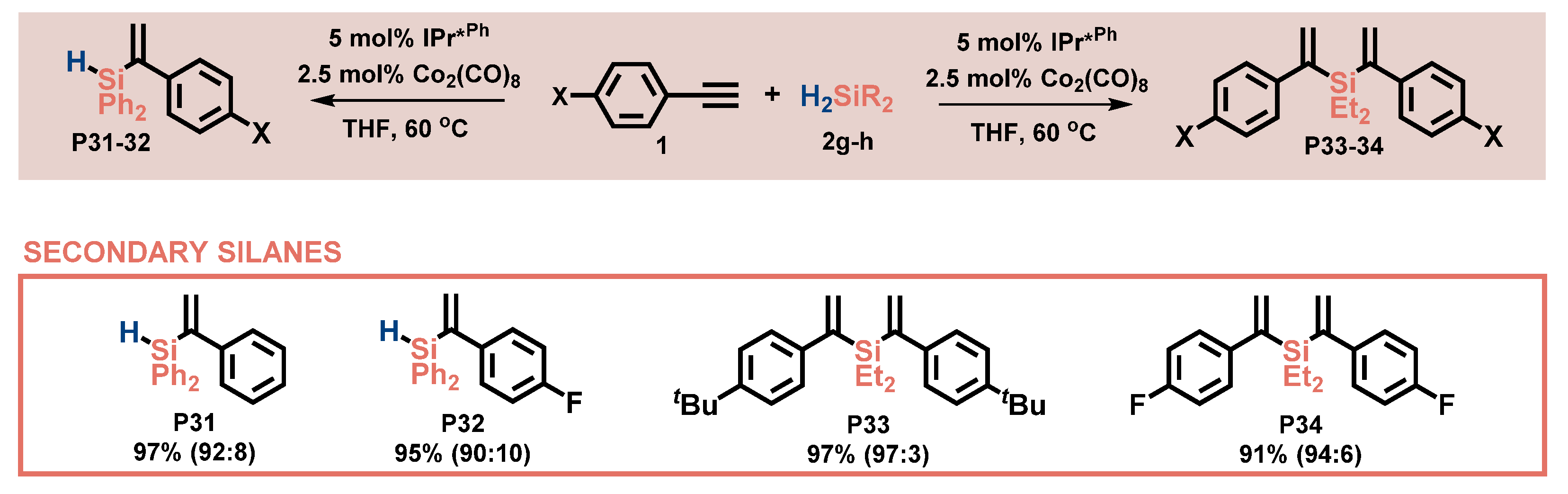
2.3. Substrate Scope—Silanes
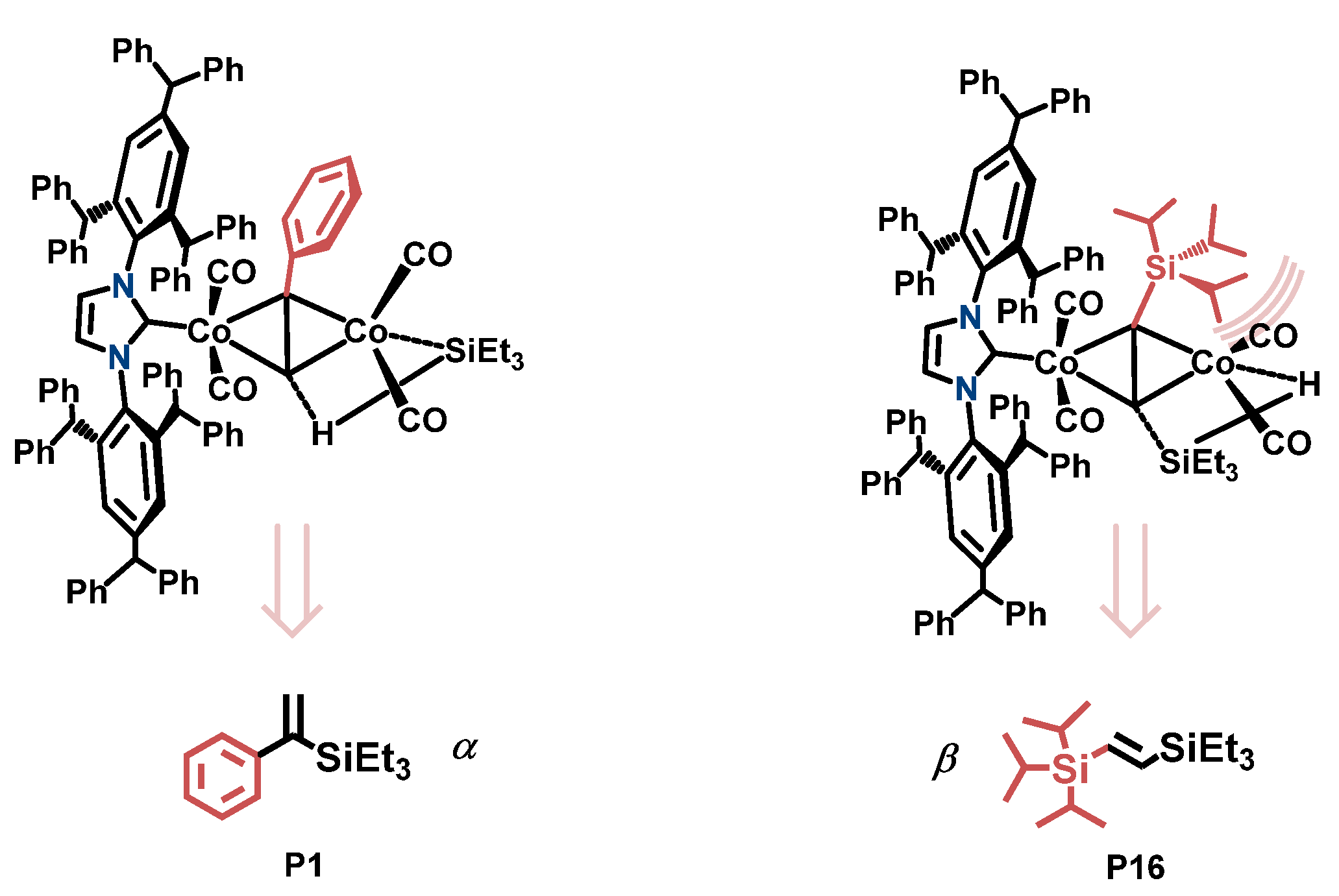
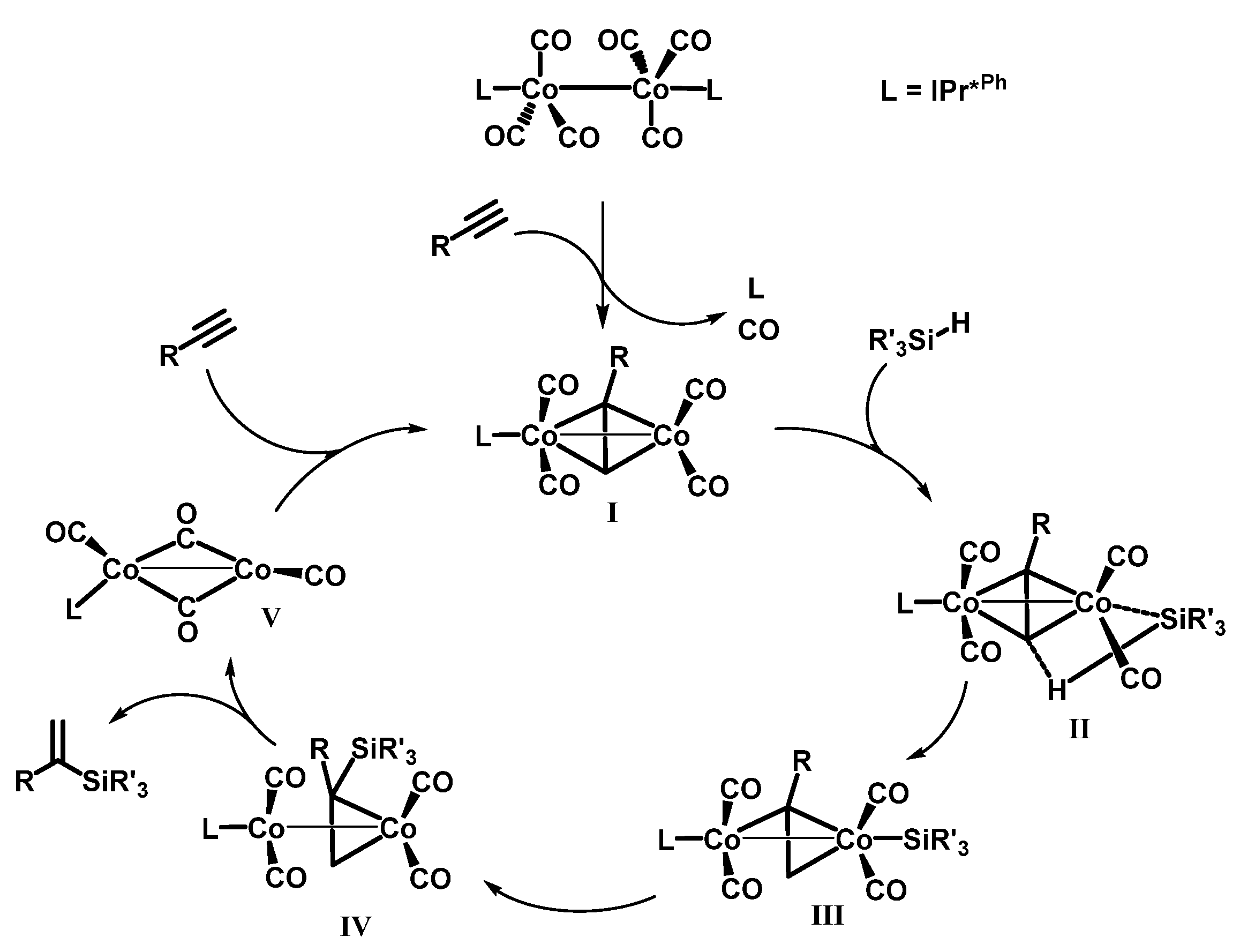
2.4. Mechanistic Investigation
3. Materials and Methods
3.1. Instruments and Reagents
3.2. Experimental Procedures
3.2.1. Catalytic Tests
3.2.2. Synthesis of Products P1–P30
3.2.3. Synthesis of Products P31–P34
3.2.4. Mercury Poisoning Experiment
3.2.5. Hot Filtration Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marciniec, B. (Ed.) Hydrosilylation: A Comprehensive Review on Recent Advances; Advances in silicon science; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 978-1-4020-8171-2. [Google Scholar]
- Nakajima, Y.; Shimada, S. Hydrosilylation Reaction of Olefins: Recent Advances and Perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Fleming, I.; Barbero, A.; Walter, D. Stereochemical Control in Organic Synthesis Using Silicon-Containing Compounds. Chem. Rev. 1997, 97, 2063–2192. [Google Scholar] [CrossRef]
- Bracegirdle, S.; Anderson, E.A. Recent Advances in the Use of Temporary Silicon Tethers in Metal-Mediated Reactions. Chem. Soc. Rev. 2010, 39, 4114. [Google Scholar] [CrossRef]
- Nakao, Y.; Hiyama, T. Silicon-Based Cross-Coupling Reaction: An Environmentally Benign Version. Chem. Soc. Rev. 2011, 40, 4893. [Google Scholar] [CrossRef]
- Denmark, S.E.; Sweis, R.F. Organosilicon Compounds in Cross-Coupling Reactions. In Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, Germany, 2014; pp. 475–532. ISBN 978-3-527-65558-8. [Google Scholar]
- Markó, I.E.; Stérin, S.; Buisine, O.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J.-P. Selective and Efficient Platinum(0)-Carbene Complexes as Hydrosilylation Catalysts. Science 2002, 298, 204–206. [Google Scholar] [CrossRef]
- Berthon-Gelloz, G.; Schumers, J.-M.; De Bo, G.; Markó, I.E. Highly β-(E)-Selective Hydrosilylation of Terminal and Internal Alkynes Catalyzed by a (IPr)Pt(Diene) Complex. J. Org. Chem. 2008, 73, 4190–4197. [Google Scholar] [CrossRef]
- Żak, P.; Bołt, M.; Kubicki, M.; Pietraszuk, C. Highly Selective Hydrosilylation of Olefins and Acetylenes by Platinum(0) Complexes Bearing Bulky N-Heterocyclic Carbene Ligands. Dalton Trans. 2018, 47, 1903–1910. [Google Scholar] [CrossRef]
- Puerta-Oteo, R.; Munarriz, J.; Polo, V.; Jiménez, M.V.; Pérez-Torrente, J.J. Carboxylate-Assisted β-(Z) Stereoselective Hydrosilylation of Terminal Alkynes Catalyzed by a Zwitterionic Bis-NHC Rhodium(III) Complex. ACS Catal. 2020, 10, 7367–7380. [Google Scholar] [CrossRef]
- Gao, W.; Ding, S. Progress on Iridium-Catalyzed Hydrosilylation of Alkenes and Alkynes. Synthesis 2020, 52, 3549–3563. [Google Scholar] [CrossRef]
- Karstedt, B.D. Platinum Complexes of Unsaturated Siloxanes and Platinum Containing Organopolysiloxanes. U.S. Patent No 3,775,452, 27 November 1973. [Google Scholar]
- Hitchcock, P.B.; Lappert, M.F.; Warhurst, N.J.W. Synthesis and Structure of Arac-Tris(Divinyldisiloxane) Diplatinum(0) Complex and Its Reaction with Maleic Anhydride. Angew. Chem. Int. Ed. Engl. 1991, 30, 438–440. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Chirik, P.J. Earth-Abundant Transition Metal Catalysts for Alkene Hydrosilylation and Hydroboration. Nat. Rev. Chem. 2018, 2, 15–34. [Google Scholar] [CrossRef]
- Kuciński, K.; Hreczycho, G. Hydrosilylation and Hydroboration in a Sustainable Manner: From Earth-Abundant Catalysts to Catalyst-Free Solutions. Green Chem. 2020, 22, 5210–5224. [Google Scholar] [CrossRef]
- Guo, J.; Shen, X.; Lu, Z. Regio- and Enantioselective Cobalt-Catalyzed Sequential Hydrosilylation/Hydrogenation of Terminal Alkynes. Angew. Chem. Int. Ed. 2017, 56, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Chakraborty, U.; Jacobi von Wangelin, A. Regiocontrol in the Cobalt-Catalyzed Hydrosilylation of Alkynes. Chem. Commun. 2018, 54, 12322–12325. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shen, X.; Lu, Z. Cobalt-Catalyzed Markovnikov Selective Sequential Hydrogenation/Hydrohydrazidation of Aliphatic Terminal Alkynes. J. Am. Chem. Soc. 2020, 142, 14455–14460. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.; Shen, X.; Lu, Z. Ligand Relay Catalysis for Cobalt-Catalyzed Sequential Hydrosilylation and Hydrohydrazidation of Terminal Alkynes. Nat. Commun. 2022, 13, 650. [Google Scholar] [CrossRef]
- Bera, S.S.; Szostak, M. Cobalt–N-Heterocyclic Carbene Complexes in Catalysis. ACS Catal. 2022, 12, 3111–3137. [Google Scholar] [CrossRef]
- Park, J.-W. Cobalt-Catalyzed Alkyne Hydrosilylation as a New Frontier to Selectively Access Silyl-Hydrocarbons. Chem. Commun. 2022, 58, 491–504. [Google Scholar] [CrossRef]
- Guo, J.; Lu, Z. Highly Chemo-, Regio-, and Stereoselective Cobalt-Catalyzed Markovnikov Hydrosilylation of Alkynes. Angew. Chem. Int. Ed. 2016, 55, 10835–10838. [Google Scholar] [CrossRef]
- Zuo, Z.; Yang, J.; Huang, Z. Cobalt-Catalyzed Alkyne Hydrosilylation and Sequential Vinylsilane Hydroboration with Markovnikov Selectivity. Angew. Chem. Int. Ed. 2016, 55, 10839–10843. [Google Scholar] [CrossRef]
- Kong, D.; Hu, B.; Chen, D. Highly Regio- and Stereoselective Hydrosilylation of Alkynes Catalyzed by Tridentate Cobalt Complexes. Chem. Asian J. 2019, 14, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Skrodzki, M.; Patroniak, V.; Pawluć, P. Schiff Base Cobalt(II) Complex-Catalyzed Highly Markovnikov-Selective Hydrosilylation of Alkynes. Org. Lett. 2021, 23, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Skrodzki, M.; Ortega Garrido, V.; Csáky, A.G.; Pawluć, P. Searching for Highly Active Cobalt Catalysts Bearing Schiff Base Ligands for Markovnikov-Selective Hydrosilylation of Alkynes with Tertiary Silanes. J. Catal. 2022, 411, 116–121. [Google Scholar] [CrossRef]
- Sahoo, M.K.; Kim, D.; Chang, S.; Park, J.-W. Regioselective Access to α-Vinylsilanes and α-Vinylgermanes by Cobalt-Catalyzed Migratory Hydrofunctionalization of 2-Alkynes. ACS Catal. 2021, 11, 12777–12784. [Google Scholar] [CrossRef]
- Wang, D.; Lai, Y.; Wang, P.; Leng, X.; Xiao, J.; Deng, L. Markovnikov Hydrosilylation of Alkynes with Tertiary Silanes Catalyzed by Dinuclear Cobalt Carbonyl Complexes with NHC Ligation. J. Am. Chem. Soc. 2021, 143, 12847–12856. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-H.; Isobe, M. Highly Regioselective Hydrosilylation of Unsymmetric Alkynes Using a Phenylthio Directing Group: Highly Regioselective Hydrosilylation of Unsymmetric Alkynes. Eur. J. Org. Chem. 2014, 2014, 4733–4740. [Google Scholar] [CrossRef]
- Clavier, H.; Nolan, S.P. Percent Buried Volume for Phosphine and N-Heterocyclic Carbene Ligands: Steric Properties in Organometallic Chemistry. Chem. Commun. 2010, 46, 841. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and Understanding the Steric Properties of N-Heterocyclic Carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [Green Version]
- Vanden Broeck, S.M.P.; Nahra, F.; Cazin, C.S.J. Bulky-Yet-Flexible Carbene Ligands and Their Use in Palladium Cross-Coupling. Inorganics 2019, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Meng, G.; Li, G.; Flach, C.; Mendelsohn, R.; Lalancette, R.; Szostak, R.; Szostak, M. IPr#—Highly Hindered, Broadly Applicable N-Heterocyclic Carbenes. Chem. Sci. 2021, 12, 10583–10589. [Google Scholar] [CrossRef]
- Żak, P.; Bołt, M.; Lorkowski, J.; Kubicki, M.; Pietraszuk, C. Platinum Complexes Bearing Bulky N-Heterocyclic Carbene Ligands as Efficient Catalysts for the Fully Selective Dimerization of Terminal Alkynes. ChemCatChem 2017, 9, 3627–3631. [Google Scholar] [CrossRef]
- Żak, P.; Bołt, M.; Pietraszuk, C. Selective Synthesis of E -Vinylsilanes and E, E-Divinylsilanes via Platinum-Catalyzed Hydrosilylation of Alkynes with Secondary Silanes. RSC Adv. 2018, 8, 40016–40021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bołt, M.; Żak, P. Application of Bulky NHC–Rhodium Complexes in Efficient S–Si and S–S Bond Forming Reactions. Inorg. Chem. 2021, 60, 17579–17585. [Google Scholar] [CrossRef]
- Bołt, M.; Delaude, L.; Żak, P. Rhodium Catalysts with Superbulky NHC Ligands for the Selective α-Hydrothiolation of Alkynes. Dalton Trans. 2022, 51, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Bołt, M.; Mermela, A.; Żak, P. Influence of Bis-NHC Ligand on Platinum-Catalyzed Hydrosilylation of Internal Alkynes. Eur. J. Inorg. Chem. 2022, 26, e202200622. [Google Scholar] [CrossRef]
- Bołt, M.; Żak, P. Solvent-Free Hydroboration of Alkynes Catalyzed by an NHC–Cobalt Complex. RSC Adv. 2022, 12, 18572–18577. [Google Scholar] [CrossRef]
- Inoue, A.; Kondo, J.; Shinokubo, H.; Oshima, K. Facile Synthesis of Acylsilanes via Aerobic Oxidation of g Em- Disilylalkylcopper Compounds. J. Am. Chem. Soc. 2001, 123, 11109–11110. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, X.; Xie, X.; Ding, S. Construction of Highly Sterically Hindered 1,1-Disilylated Terminal Alkenes. Chem. Commun. 2018, 54, 12958–12961. [Google Scholar] [CrossRef]
- Inoue, A.; Kondo, J.; Shinokubo, H.; Oshima, K. Facile Synthesis of Ketones from 1,1-Disilylethenes via Oxidation of Gem-Disilylalkanes. Chem. Commun. 2002, 2, 114–115. [Google Scholar] [CrossRef]
- Li, H.; Yang, C.; Wang, D.; Deng, L. Cobalt-Catalyzed Regio- and Stereoselective Hydrosilylation of Alk-2-Ynes with Tertiary Silanes. Organometallics 2023. [Google Scholar] [CrossRef]
- Meiries, S.; Speck, K.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P. [Pd(IPr* OMe)(Acac)Cl]: Tuning the N-Heterocyclic Carbene in Catalytic C–N Bond Formation. Organometallics 2013, 32, 330–339. [Google Scholar] [CrossRef]
- Saberov, V.S.H.; Evans, D.A.; Korotkikh, N.I.; Cowley, A.H.; Pekhtereva, T.M.; Popov, A.F.; Shvaika, O.P. Exceptionally Efficient Catalytic Hydrodechlorination of Persistent Organic Pollutants: Application of New Sterically Shielded Palladium Carbene Complexes. Dalton Trans. 2014, 43, 18117–18122. [Google Scholar] [CrossRef] [Green Version]
- Hans, M.; Lorkowski, J.; Demonceau, A.; Delaude, L. Efficient Synthetic Protocols for the Preparation of Common N-Heterocyclic Carbene Precursors. Beilstein J. Org. Chem. 2015, 11, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Żak, P.; Bołt, M.; Dudziec, B.; Kubicki, M. Synthesis of (E)-1,4-Disilsesquioxylsubstituted but-1-En-3-Ynes via Platinum-Catalyzed Dimerization of Ethynylsiloxysilsesquioxanes. Dalton Trans. 2019, 48, 2657–2663. [Google Scholar] [CrossRef]
- Rivero-Crespo, M.A.; Leyva-Pérez, A.; Corma, A.A.; Rivero-Crespo, M.A.; Leyva-Pérez, A.; Corma, A.A. A Ligand-Free Pt3 Cluster Catalyzes the Markovnikov Hydrosilylation of Alkynes with up to 106 Turnover Frequencies. Chem. Eur. J. 2017, 23, 1702–1708. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, B.P.S.; Sarka, A. Functionalized vinylsilanes via highly efficient and recyclable Pt-nanoparticle catalysed hydrosilylation of alkynes. Dalton Trans. 2017, 46, 8709–8715. [Google Scholar] [CrossRef]
- Gee, J.C.; Fuller, B.A.; Lockett, H.-M.; Sedghi, G.; Robertson, C.M.; Luzyanin, K.V. Visible light accelerated hydrosilylation of alkynes using platinum–[acyclic diaminocarbene] photocatalysts. Chem. Commun. 2018, 54, 9450–9453. [Google Scholar] [CrossRef]
- Battace, A.; Zair, T.; Doucet, H.; Santelli, M.J. Selective synthesis of (E)-triethyl (2-arylethenyl) silane derivatives by reaction of aryl bromides with triethyl vinylsilane catalysed by a palladium–tetraphosphine complex. Organomet. Chem. 2005, 690, 3790–3802. [Google Scholar] [CrossRef]
- Miura, H.; Sasaki, S.; Ogawa, R.; Shishido, T. Hydrosilylation of Allenes Over Palladium–Gold Alloy Catalysts: Enhancing Activity and Switching Selectivity by the Incorporation of Palladium into Gold Nanoparticles. Eur. J. Org. Chem. 2018, 2018, 1858–1862. [Google Scholar] [CrossRef]
- Kita, Y.; Tobisu, M.; Chatani, N. Rhodium-Catalyzed Alkenylation of Nitriles via Silicon-Assisted C-CN Bond Cleavage. Org. Lett. 2010, 12, 1864–1867. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | NHC | [Co] (%) | Time (h) | Conv. 1 (%) | Selectivity 2 α:β-(E):β-(Z) |
| 1 | - | 5 | 24 | 76 | 32:68:0 |
| 2 | IPr | 5 | 5 | 94 | 95:5:0 |
| 3 | IMes | 5 | 5 | 90 | 90:10:0 |
| 4 | IPr*OMe | 5 | 3 | 99 | 99:1:0 |
| 5 | IPr*Et | 5 | 3 | 96 | 98:2:0 |
| 6 | IPr*Ph | 5 | 3 | 97 | 99:1:0 |
| 7 | IPr*Ph | 2.5 | 5 | 95 | 99:1:0 |
| 8 | IPr*Ph | 1 | 24 | 70 | 97:3:0 |
| 9 | IPr*Ph | 0.5 | 24 | 19 | 93:7:0 |
 | ||||
|---|---|---|---|---|
| Entry | Temp. (°C) | Solvent | Conv. 1 (%) | Selectivity 2 α:β-(E):β-(Z) |
| 1 | 60 | THF | 99 | 99:1:0 |
| 2 | 40 | THF | 36 | 95:5:0 |
| 3 | RT | THF | 10 | 92:8:0 |
| 4 | 60 | Toluene | 99 | 99:1:0 |
| 5 | 60 | DCM | 90 | 97:3:0 |
| 6 | 60 | MTBE | 75 | 84:16:0 |
| 7 | 60 | Acetonitrile | 16 | 80:20:0 |
| 8 | 60 | iPrOH | <5 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bołt, M.; Żak, P. Bulky NHC–Cobalt Complex-Catalyzed Highly Markovnikov-Selective Hydrosilylation of Alkynes. Catalysts 2023, 13, 510. https://doi.org/10.3390/catal13030510
Bołt M, Żak P. Bulky NHC–Cobalt Complex-Catalyzed Highly Markovnikov-Selective Hydrosilylation of Alkynes. Catalysts. 2023; 13(3):510. https://doi.org/10.3390/catal13030510
Chicago/Turabian StyleBołt, Małgorzata, and Patrycja Żak. 2023. "Bulky NHC–Cobalt Complex-Catalyzed Highly Markovnikov-Selective Hydrosilylation of Alkynes" Catalysts 13, no. 3: 510. https://doi.org/10.3390/catal13030510