

CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements
 , , and
, , and 
Abstract
:1. Introduction
2. Results
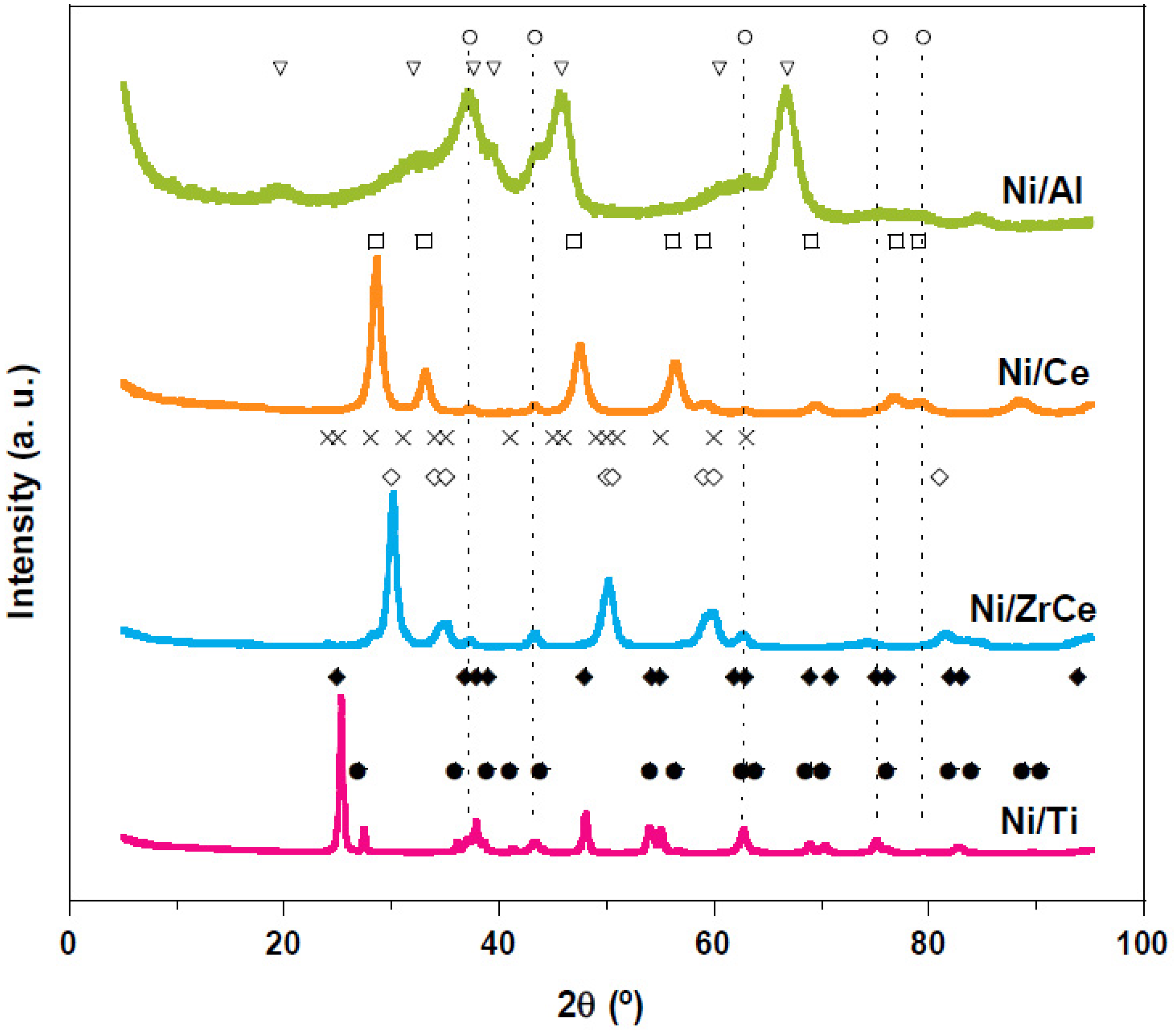
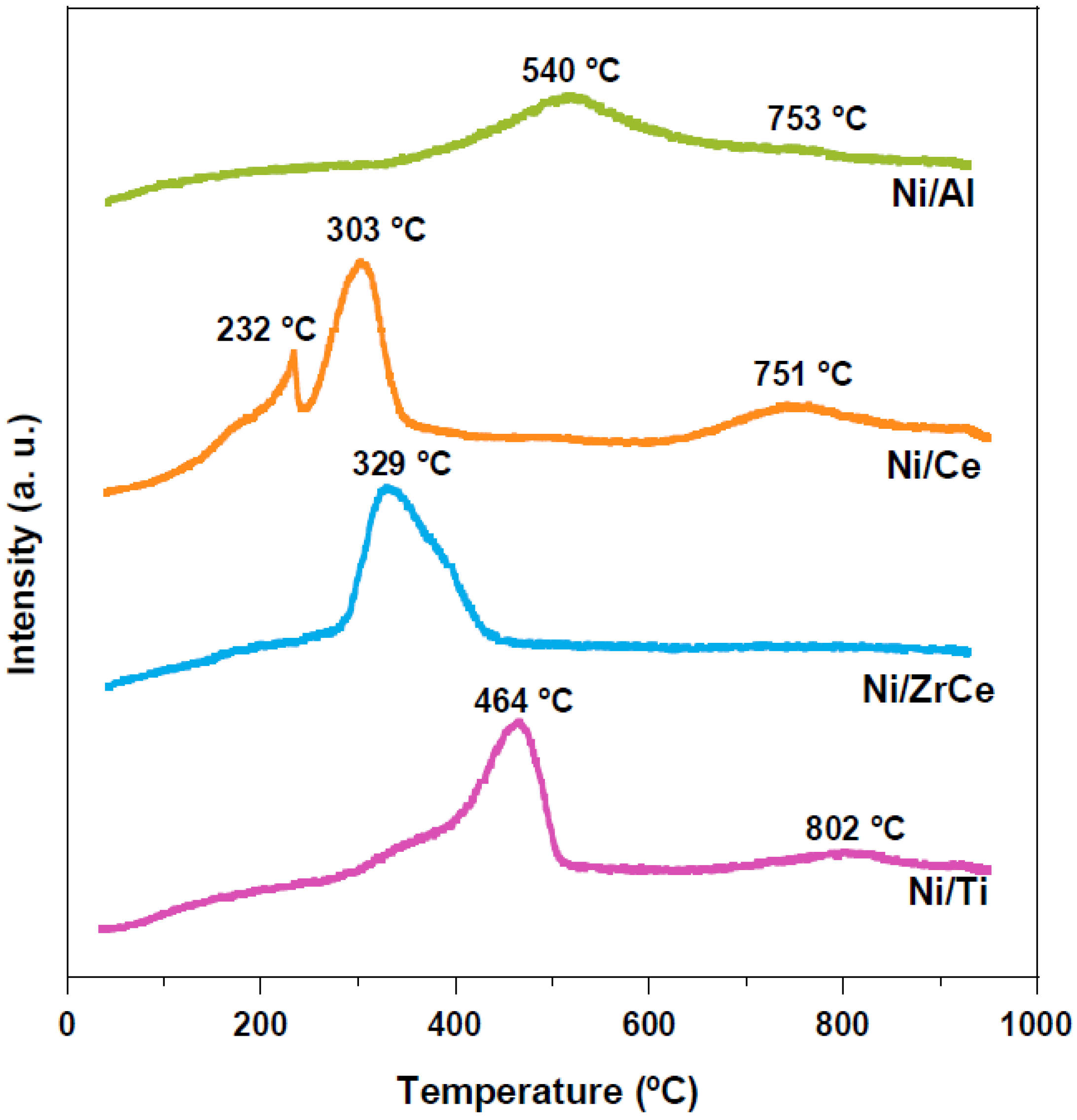
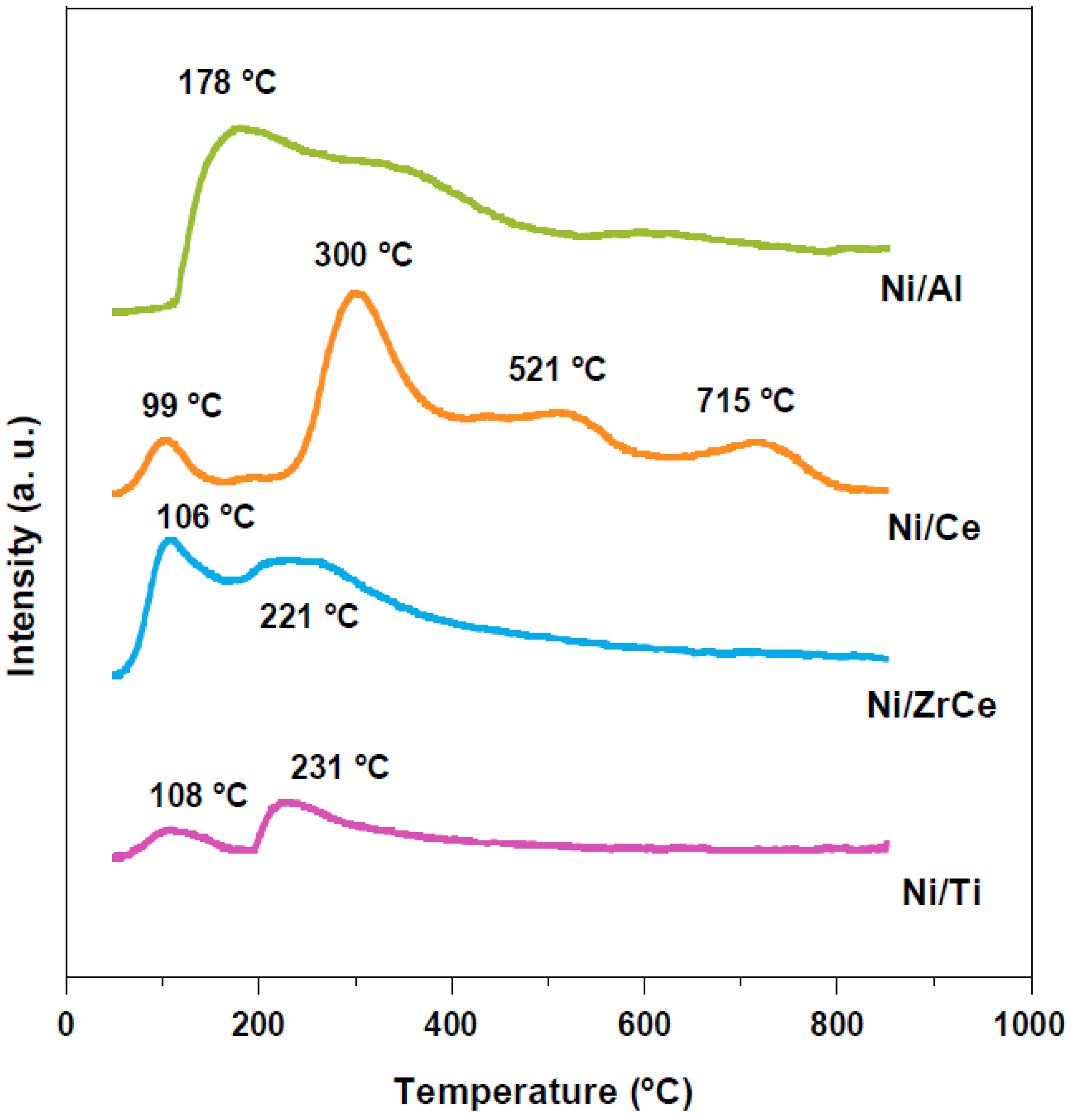
2.1. Physicochemical Characterization of the Catalysts
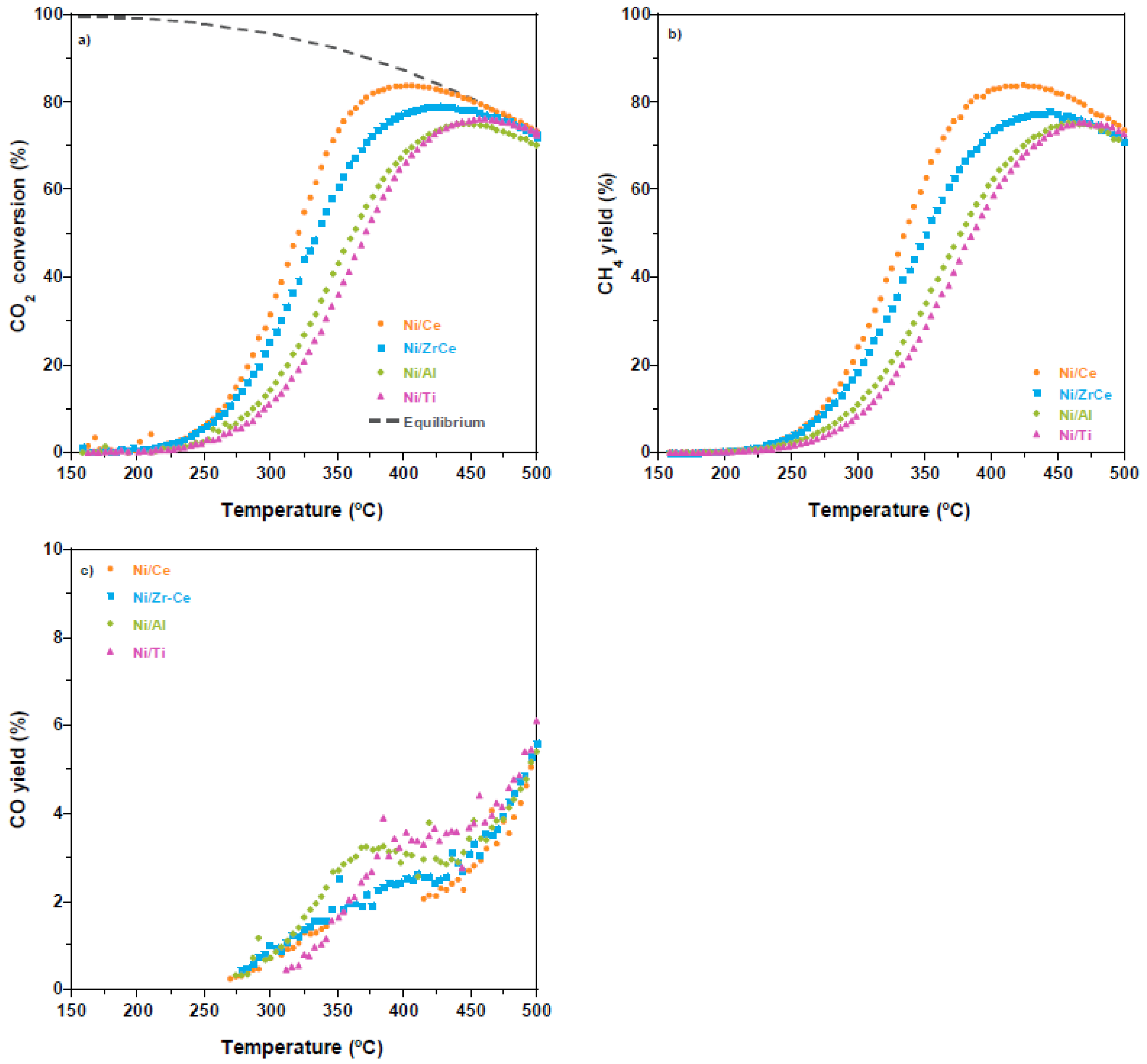
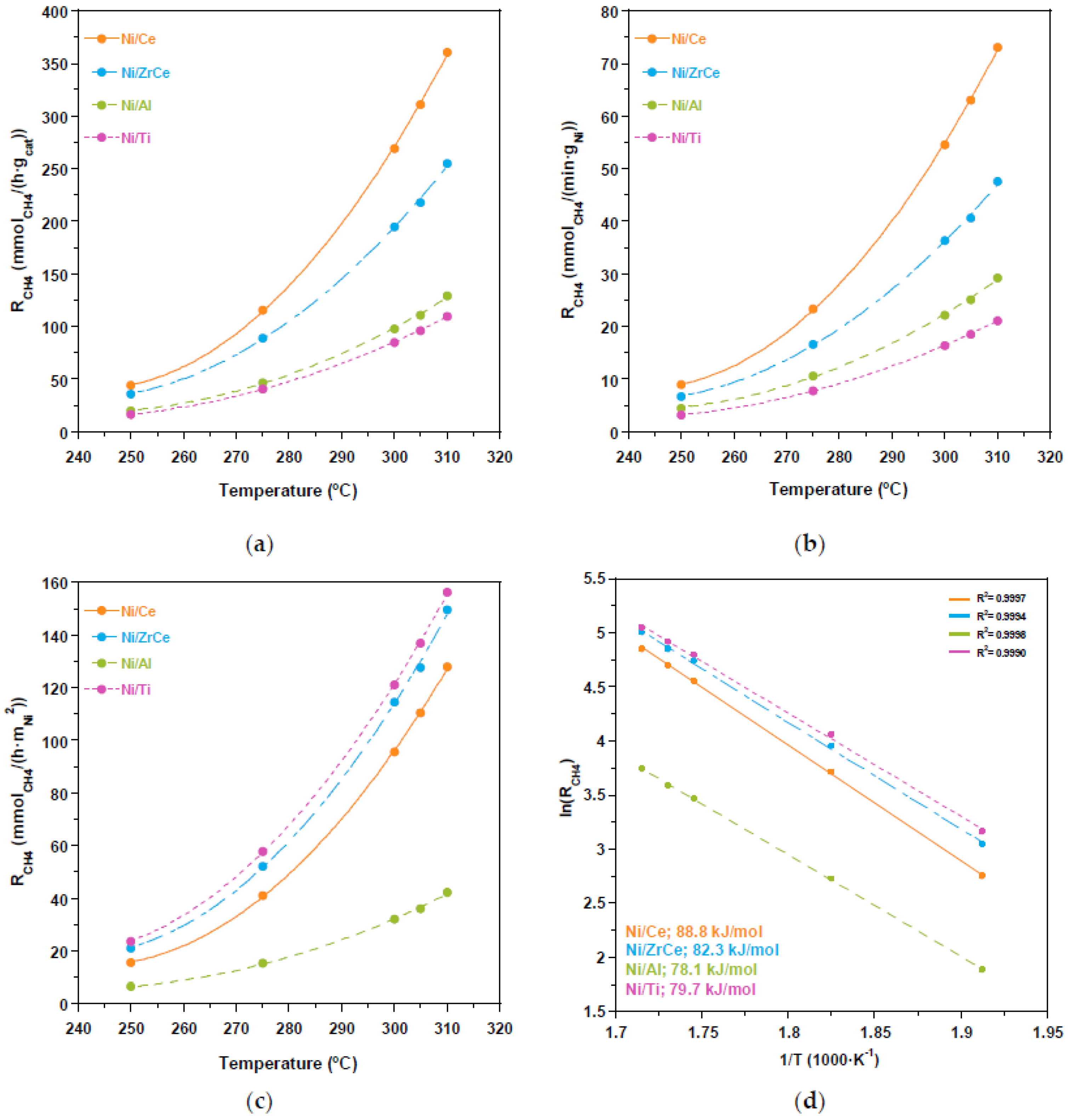
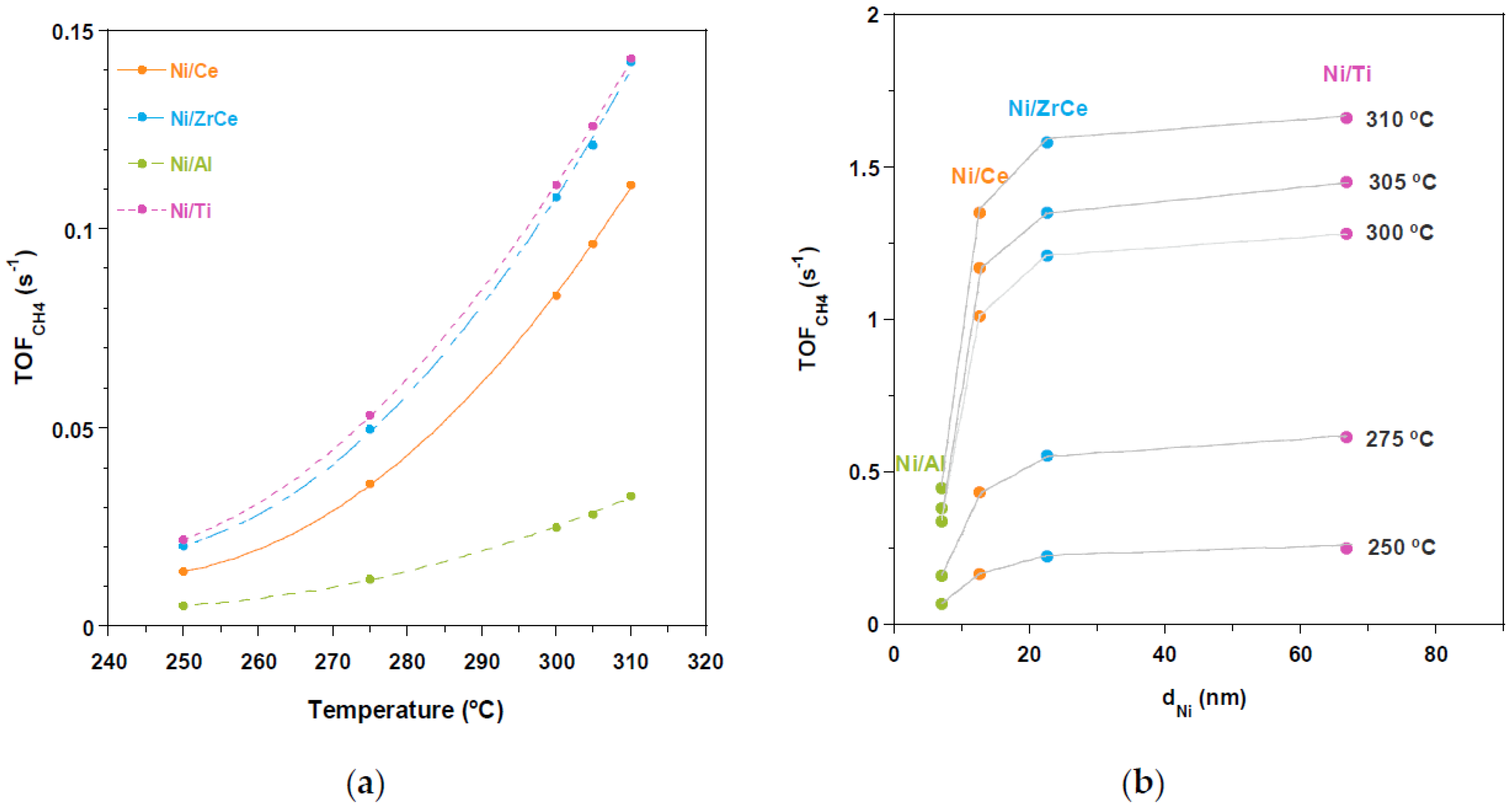
2.2. Catalytic Performance
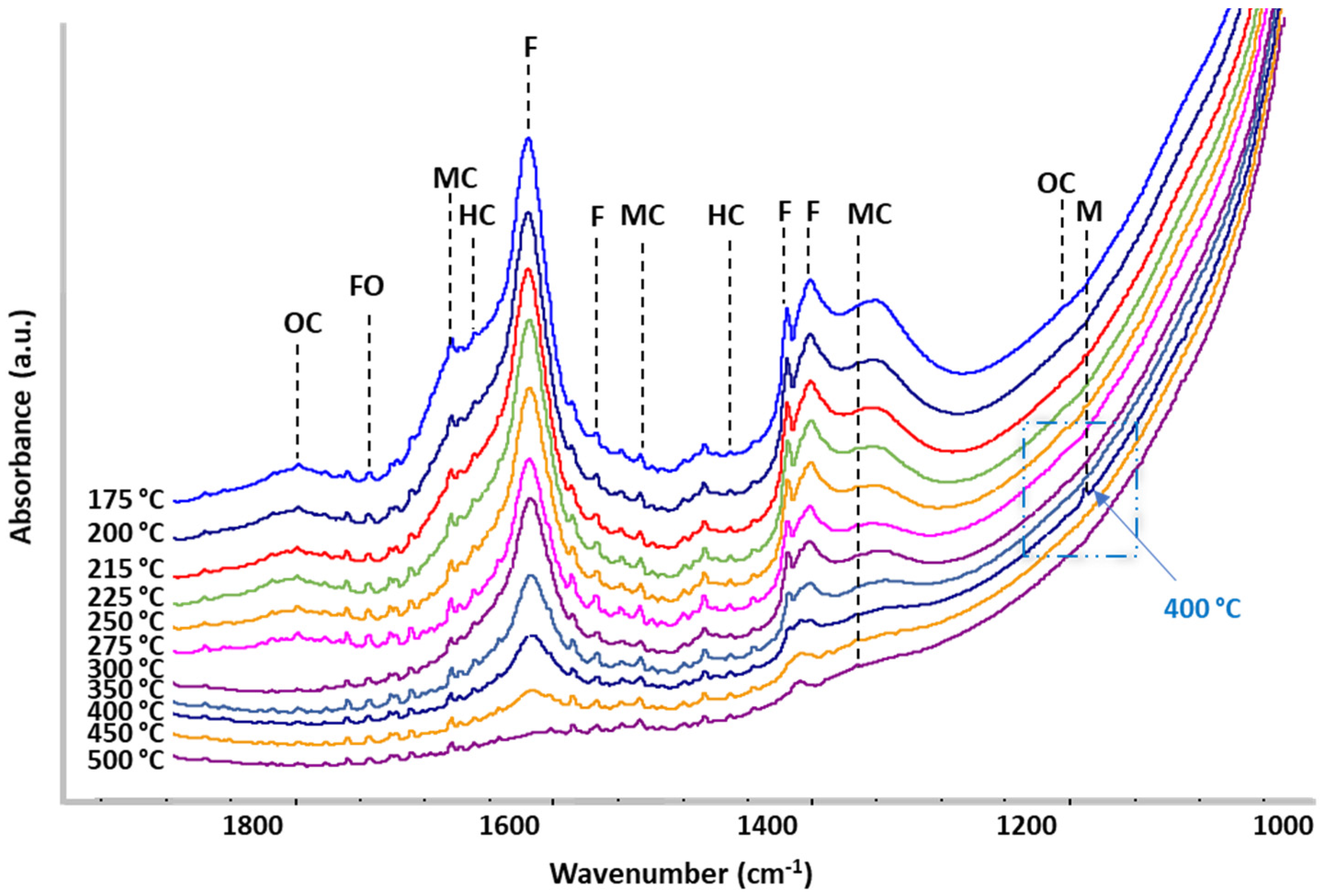
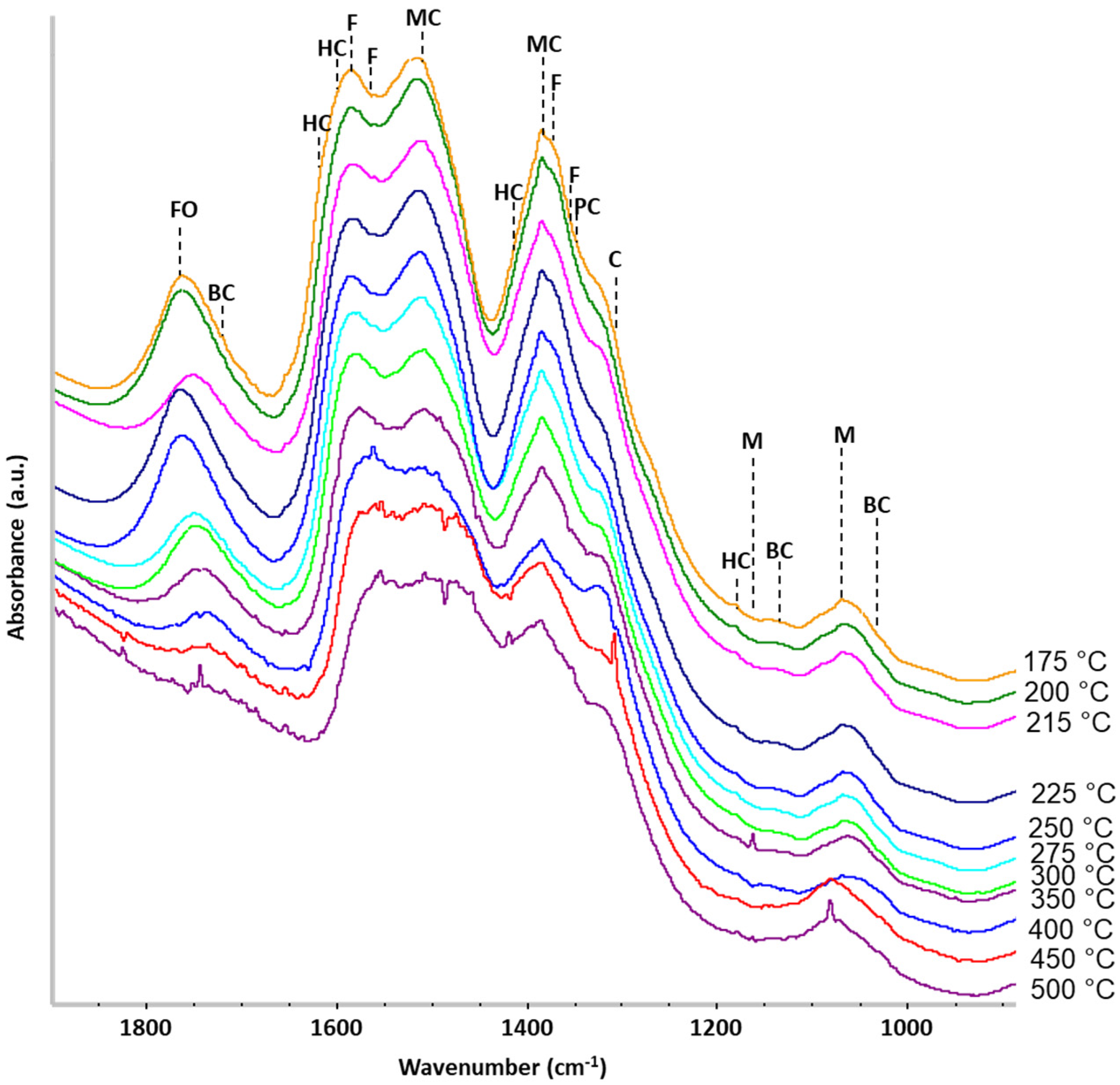
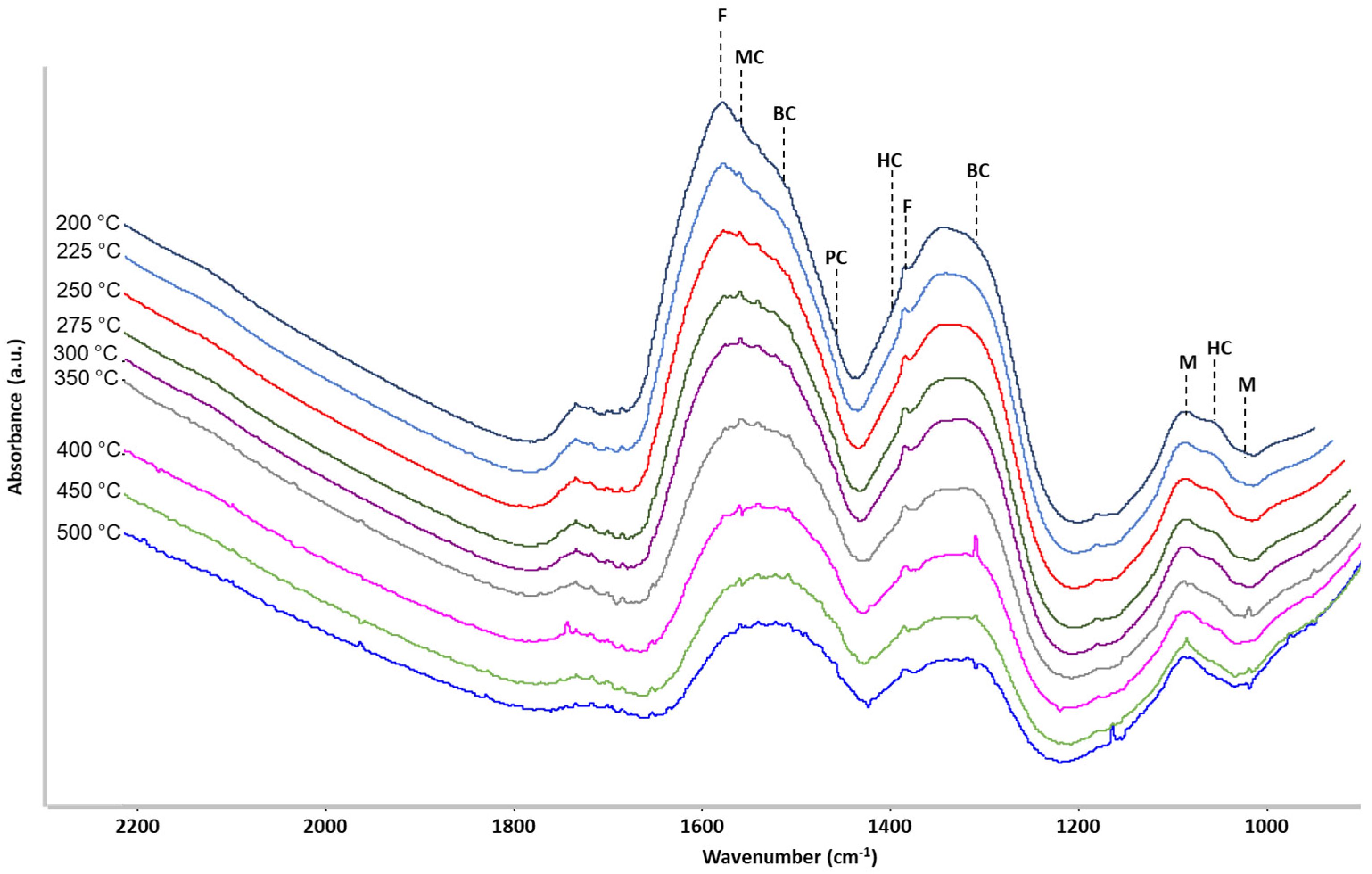
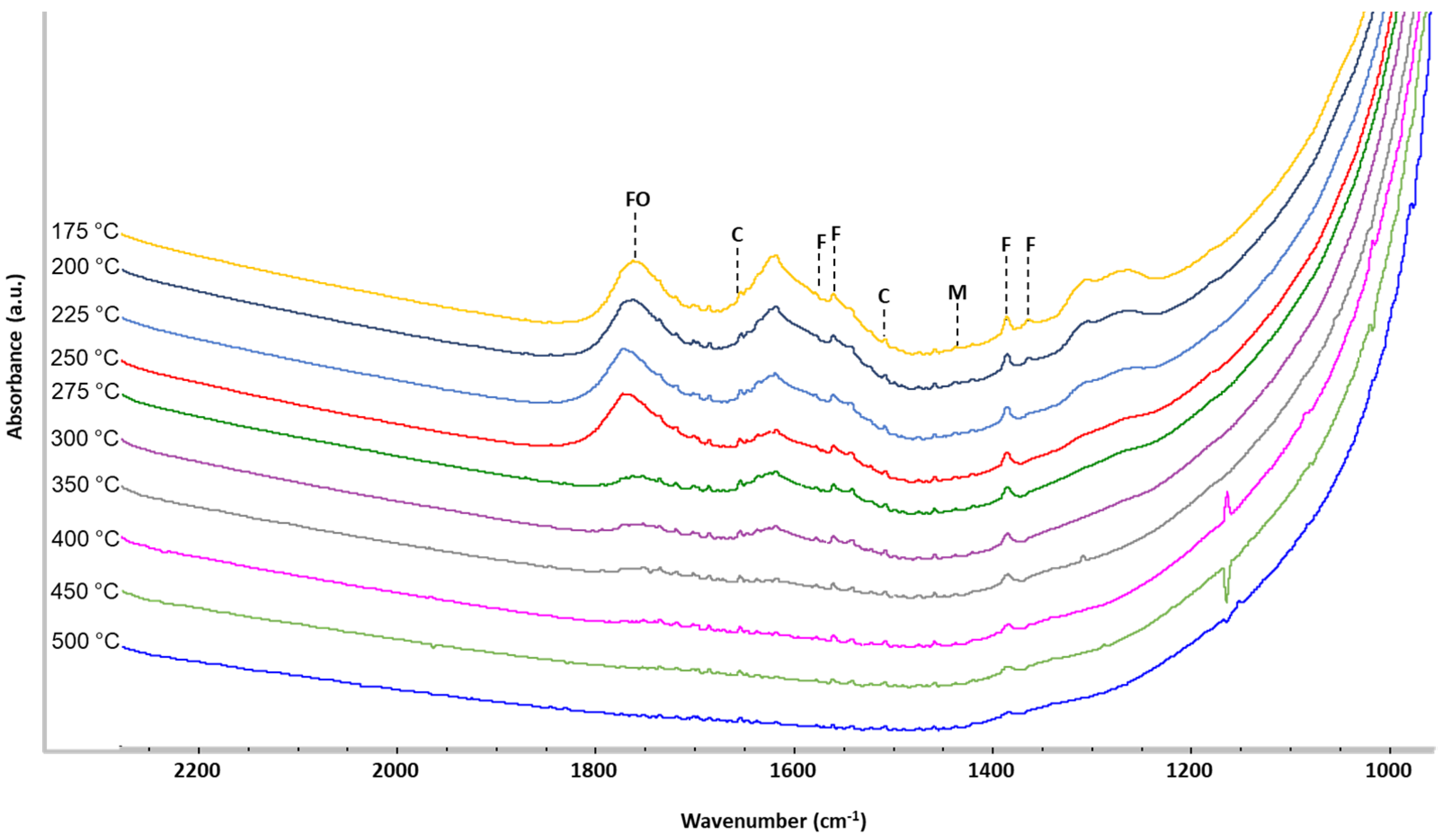
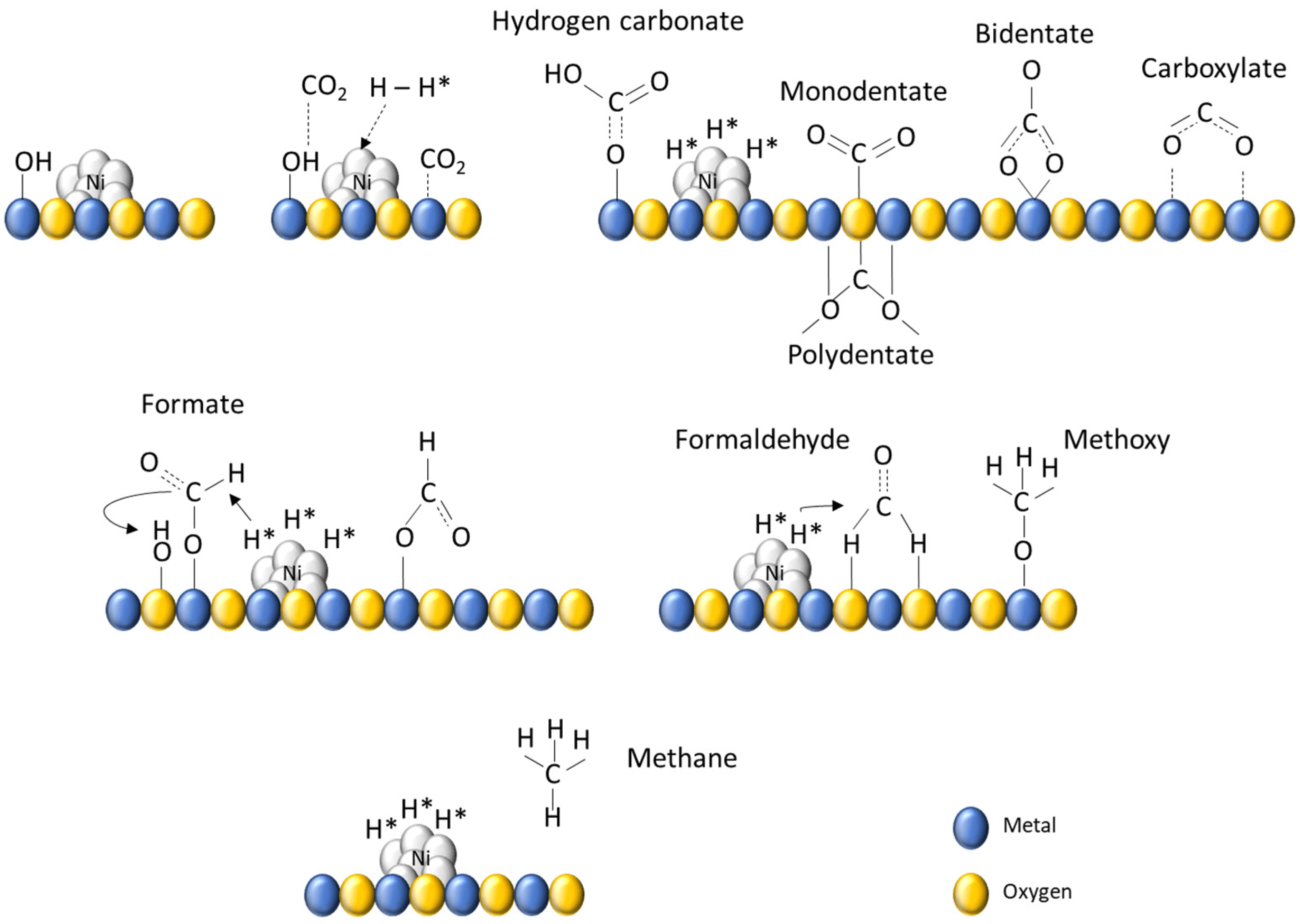
2.3. Operando IR Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Catalysts Preparation
4.2. Physicochemical Characterization
4.3. Catalytic Tests
4.4. Operando IR Spectroscopy
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, B.B.; Twigg, M.V.; Woodward, C. Methanation. In Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Manson Publishing: Frome, UK, 1996; pp. 340–383. [Google Scholar]
- Seglin, L.; Geosits, R.; Franko, B.R.; Gruber, G. Survey of Methanation Chemistry and Processes. In Methanation of Synthesis Gas. Advances in Chemistry Vol. 146; Seglin, L., Ed.; American Chemical Society: Washington, DC, USA, 1975; pp. 1–30. [Google Scholar]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass—A technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Duret, A.; Friedli, C.; Maréchal, F. Process design of Synthetic Natural Gas (SNG) production using wood gasification. J. Clean Prod. 2005, 13, 1434–1446. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Gong, J.; English, N.J.; Pont, D.; Patzke, G.R.; Protti, S.; Zhang, T. Power-to-X: Lighting the Path to a Net-Zero-Emission Future. ACS Sustain. Chem. Eng. 2021, 9, 7179–7181. [Google Scholar] [CrossRef]
- Incer-Valverde, J.; Patiño-Arévalo, L.J.; Tsatsaronis, G.; Morosuk, T. Hydrogen-driven Power-to-X: State of the art and multicriteria evaluation of a study case. Energy Convers. Manag. 2022, 266, 115814. [Google Scholar] [CrossRef]
- Bargiacchi, E.; Candelaresi, D.; Spazzafumo, G. Power to methane. In Power to Fuel. How to Speed Up a Hydrogen Economy; Spazzafumo, G., Ed.; Academic Press—Elsevier: London, UK, 2021; pp. 75–101. [Google Scholar]
- Bargiacchi, E. Power-to-Fuel existing plants and pilot projects. In Power to Fuel. How to Speed Up a Hydrogen Economy; Spazzafumo, G., Ed.; Academic Press—Elsevier: London, UK, 2021; pp. 211–237. [Google Scholar]
- Ghaib, K.; Ben-Fares, F.-Z. Power-to-Methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Navajas, A.; Mendiara, T.; Gandía, L.M.; Abad, A.; García-Labiano, F.; de Diego, L.F. Life cycle assessment of power-to-methane systems with CO2 supplied by the chemical looping combustion of biomass. Energy Convers. Manag. 2022, 267, 115866. [Google Scholar] [CrossRef]
- Ursúa, A.; Gandía, L.M.; Sanchis, P. Hydrogen Production From Water Electrolysis: Current Status and Future Trends. Proc. IEEE 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Blanco, H.; Nijs, W.; Ruf, J.; Faaij, A. Potential of Power-to-Methane in the EU energy transition to a low carbon system using cost optimization. Appl. Energy 2018, 232, 323–340. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef] [Green Version]
- Gassner, M.; Maréchal, F. Thermo-economic optimisation of the integration of electrolysis in synthetic natural gas production from wood. Energy 2008, 33, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Hausberger, A.L.; Knight, C.B.; Atwood, K. Development of Methanation Catalysts for the Synthetic Natural Gas Processes. In Methanation of Synthesis Gas. Advances in Chemistry Vol. 146; Seglin, L., Ed.; American Chemical Society: Washington, DC, USA, 1975; pp. 47–70. [Google Scholar]
- Mills, G.A.; Steffgen, F.W. Catalytic Methanation. Catal. Rev. 1973, 8, 159–210. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on Group VIII Metals, I. Specific Activity of Ni/SiO2. J. Catal. 1981, 68, 67–76. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Younas, M.; Kong, L.L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P.L. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Ducamp, J.; Bengaouer, A.; Baurens, P.; Fechete, I.; Turek, P.; Garin, F. Statu quo sur la methanation du dioxide de carbone: Une revue de la littérature. Comptes Rendus Chim. 2018, 21, 427–469. [Google Scholar] [CrossRef]
- Sreedhar, I.; Varun, Y.; Singh, S.A.; Venugopal, A.; Reddy, B.M. Developmental trends in CO2 methanation using various catalysts. Catal. Sci. Technol. 2019, 9, 4478. [Google Scholar] [CrossRef]
- Mebrahtu, C.; Krebs, F.; Abate, S.; Perathoner, S.; Centi, G.; Palkovits, R. CO2 Methanation: Principles and Challenges. Stud. Surf. Sci. Catal. 2019, 178, 85–103. [Google Scholar]
- Ashok, J.; Pati, S.; Hongmaronon, P.; Tianxi, Z.; Junmei, Z.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Le, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charision, N.D.; Yentekakis, I.V.; Goula, M.A. Bimetallic Ni-Based Catalysts for CO2 Methanation: A Review. Nanomaterials 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, L.; Cui, Y.; Xing, Y.; Su, W. Research on nickel-based catalysts for carbon dioxide methanation combined with literature measurements. J. CO2 Util. 2022, 63, 102117. [Google Scholar] [CrossRef]
- Vance, C.K.; Bartholomew, C.H. Hydrogenation of carbon dioxide on group VIII metals. III, Effects of support on Activity/selectivity and adsorption properties of nickel. Appl. Catal. 1983, 7, 169–177. [Google Scholar] [CrossRef]
- Abate, S.; Mebrahtu, C.; Giglio, E.; Deorsola, F.; Bensaid, S.; Perathoner, S.; Pirone, R.; Centi, G. Catalytic Performance of γ-Al2O3−ZrO2−TiO2−CeO2 Composite Oxide Supported Ni-Based Catalysts for CO2 Methanation. Ind. Eng. Chem. Res. 2016, 55, 4451–4460. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293–294, 89–96. [Google Scholar] [CrossRef]
- Martínez, J.; Hernández, E.; Alfaro, S.; López Medina, R.; Valverde Aguilar, G.; Albiter, E.; Valenzuela, M.A. High Selectivity and Stability of Nickel Catalysts for CO2 Methanation: Support Effects. Catalysts 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Gac, W.; Zawadzki, W.; Rotko, M.; Greluk, M.; Słowik, G.; Kolb, G. Effects of support composition on the performance of nickel catalysts in CO2 methanation reaction. Catal. Today 2020, 357, 468–482. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Aldana, P.A.U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Muroyama, H.; Tsuda, Y.; Asakoshi, T.; Masitah, H.; Okanishi, T.; Matsui, T.; Eguchi, K. Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J. Catal. 2016, 343, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Traitangwong, A.; Hu, M.; Zuo, C.; Meeyoo, V.; Peng, Z.; Li, C. Carbon Dioxide Methanation over Nickel-Based Catalysts Supported on Various Mesoporous Material. Energy Fuels 2018, 32, 3681–3689. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, L.; Zheng, Y.; Zhang, S.; Liu, Q.; Gao, G.; Dong, D.; Wang, Y.; Xu, L.; Hu, X. Methanation of CO2 over nickel catalysts: Impacts of acidic/basic sites on formation of the reaction intermediates. Fuel 2020, 262, 116521. [Google Scholar] [CrossRef]
- Ilsemann, J.; Murshed, M.M.; Gesing, T.M.; Kopyscinski, J.; Bäumer, M. On the support dependency of the CO2 methanation—Decoupling size and support effects. Catal. Sci. Technol. 2021, 11, 4098. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Z.; Huang, Q.; Jiang, M.; Askari, S.; Dewangan, N.; Kawi, S. State-of-art modifications of heterogeneous catalysts for CO2 methanation—Active sites, surface basicity and oxygen defects. Catal. Today 2022, 402, 88–103. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts. Appl. Catal. B Environ. 2020, 265, 118538. [Google Scholar] [CrossRef]
- Pu, T.; Shen, L.; Liu, X.; Cao, X.; Xu, J.; Wachs, I.E.; Zhu, M. Formation and influence of surface hydroxyls on product selectivity during CO2 hydrogenation by Ni/SiO2 catalysts. J. Catal. 2021, 400, 154–196. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S.; Lee, S.M. Role of oxide support in Ni based catalysts for CO2 methanation. RSC Adv. 2021, 11, 17648. [Google Scholar] [CrossRef]
- Solis-Garcia, A.; Zepeda, T.A.; Fierro-Gonzalez, J.C. Spectroscopic evidence of surface species during CO2 methanation catalyzed by supported metals: A review. Catal. Today 2022, 394–396, 2–12. [Google Scholar] [CrossRef]
- Meunier, F.C. Hydrogenation of CO and CO2: Contributions of IR operando studies. Catal. Today 2022, in press. [CrossRef]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048. [Google Scholar] [CrossRef]
- Lin, W.; Stocker, K.M.; Schatz, G.C. Mechanisms of Hydrogen-Assisted CO2 Reduction on Nickel. J. Am. Chem. Soc. 2017, 139, 4663–4666. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Deo, G. A potential descriptor for the CO2 hydrogenation to CH4 over Al2O3 supported Ni and Ni-based alloy catalysts. Appl. Catal. B Environ. 2019, 18, 525–537. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- Lozano-Reis, P.; Prats, H.; Gamallo, P.; Illas, F.; Sayós, R. Multiscale Study of the Mechanism of Catalytic CO2 Hydrogenation: Role of the Ni(111) Facets. ACS. Catal. 2020, 10, 8077–8089. [Google Scholar] [CrossRef]
- Villagra-Soza, F.; Godoy, S.; Karelovic, A.; Jiménez, R. Scrutinizing the mechanism of CO2 hydrogenation over Ni, Co and bimetallic NiCo surfaces: Isotopic measurements, operando-FTIR experiments and kinetics modelling. J. Catal. 2022, 414, 1–15. [Google Scholar] [CrossRef]
- Bentaleb, F.; Marceau, E. Influence of the textural properties of porous aluminas on the reducibility of Ni/Al2O3 catalysts. Microporous Mesoporous Mater. 2012, 146, 40–44. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Gandía, L.M.; Montes, M. Effect of the reduction temperature on the selectivity of the high temperature reaction of acetone and hydrogen over alumina and titania supported nickel and cobalt catalysts. J. Mol. Catal. 1994, 94, 347–367. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Tang, X.; Xu, Y.; Shen, W. Hydrogen production from methane decomposition over Ni/CeO2 catalysts. Catal. Commun. 2006, 7, 380–386. [Google Scholar] [CrossRef]
- Zheng, H.; Liao, W.; Ding, J.; Xu, F.; Jia, A.; Huang, W.; Zhang, Z. Unveiling the Key Factors in Determining Activity and Selectivity of CO2 Hydrogenation over Ni/CeO2 Catalysts. ACS Catal. 2022, 12, 15451–15462. [Google Scholar] [CrossRef]
- Hao, Z.; Shen, J.; Lin, S.; Han, X.; Chang, X.; Liu, J.; Li, M.; Ma, X. Decoupling the effect of Ni particle size and surface oxygen deficiencies in CO2 methanation over ceria supported Ni. Appl. Catal. B Environ. 2021, 286, 119922. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, J.; Wu, X.; Wen, J.; Zhao, R.; Li, Z.; Tian, G.; Zhang, Q.; Ning, P.; Hao, J. Frustrated Lewis Pairs Boosting Low-Temperature CO2 Methanation Performance over Ni/CeO2 Nanocatalysts. ACS Catal. 2022, 12, 10587–10602. [Google Scholar] [CrossRef]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tange, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B Environ. 2020, 268, 118474. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Ferraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B Environ. 2020, 264, 118494. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev.-Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.-C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol–gel method. Appl. Catal. A Gen. 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; van Helden, J.W.A.; Parastaev, A.; Groeneveld, E.; Pidko, E.A.; Hensen, E.J.M. Ceria–zirconia encapsulated Ni nanoparticles for CO2 methanation. Catal. Sci. Technol. 2019, 9, 5001. [Google Scholar] [CrossRef] [Green Version]
- Unwiset, P.; Chanapattharapol, K.C.; Kidkhunthod, P.; Poo-arporn, Y.; Ohtani, B. Catalytic activities of titania-supported nickel for carbon-dioxide methanation. Chem. Eng. Sci. 2020, 228, 115955. [Google Scholar] [CrossRef]
- van de Loosdrecht, J.; van der Kraan, A.M.; van Dillen, A.J.; Geus, J.W. Metal–Support Interaction: Titania-Supported and Silica-Supported Nickel Catalysts. J. Catal. 1997, 170, 217–226. [Google Scholar] [CrossRef]
- Ho, S.-W.; Chu, C.-Y.; Chen, S.-G. Effect of Thermal Treatment on the Nickel State and CO Hydrogenation Activity of Titania-Supported Nickel Catalysts. J. Catal. 1998, 178, 34–48. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Crystal structures, acid–base properties, and reactivities of CexZr1−xO2 catalysts. Catal. Today 2009, 148, 323–328. [Google Scholar] [CrossRef]
- Mao, Z.; Campbell, C.T. Apparent Activation Energies in Complex Reaction Mechanisms: A Simple Relationship via Degrees of Rate Control. ACS Catal. 2019, 9, 9465–9473. [Google Scholar] [CrossRef]
- Slot, T.K.; Riley, N.; Shiju, N.R.; Medlin, J.W.; Rothenberg, G. An experimental approach for controlling confinement effects at catalyst interfaces. Chem. Sci. 2020, 11, 11024. [Google Scholar] [CrossRef]
- Vannice, M.A. Kinetics of Catalytic Reactions; Springer: New York, NY, USA, 2010; p. 145. [Google Scholar]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Finocchio, E.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3: Catalytic activity and infrared study. Catal. Today 2016, 277, 21–28. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E.; MacDonald, S.M.; Affrossman, S. Comparative Study of Carbon Dioxide Reforming of Methane to Synthesis Gas over Ni/La2O3 and Conventional Nickel-Based Catalysts. J. Phys. Chem. 1996, 100, 744–754. [Google Scholar] [CrossRef]
- Montanari, T.; Castoldi, L.; Lietti, L.; Busca, G. Basic catalysis and catalysis assisted by basicity: FT-IR and TPD characterization of potassium-doped alumina. Appl. Catal. A Gen. 2011, 400, 61–69. [Google Scholar] [CrossRef]
- Davydov, A. Molecular Spectroscopy of Oxide Catalyst Surfaces; Sheppard, N.T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2003. [Google Scholar]
- Bechoux, K.; Marie, O.; Daturi, M.; Delahay, G.; Petitto, C.; Rousseau, S.; Blanchard, G. Infrared evidence of room temperature dissociative adsorption of carbon monoxide over Ag/Al2O3. Catal. Today 2012, 197, 155–161. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces. Mater. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- Busca, G.; Lamotte, J.; Lavalley, J.; Lorenzelli, V. FT-IR Study of the Adsorption and Transformation of Formaldehyde on Oxide Surfaces. J. Am. Chem. Soc. 1987, 109, 5197–5202. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology dependence of catalytic properties of Ni/CeO2 for CO2 methanation: A kinetic and mechanism study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Finocchio, E.; Daturi, M.; Binet, C.; Lavalley, J.C.; Blanchard, G. Thermal evolution of the adsorbed methoxy species on CexZr1−xO2 solid solution samples: A FT-IR study. Catal. Today 1999, 52, 53–63. [Google Scholar] [CrossRef]
- Konishcheva, M.V.; Potemkin, D.I.; Badmaev, S.D.; Snytnikov, P.V.; Paukshtis, E.; Sobyanin, V.; Parmon, V.N. On the Mechanism of CO and CO2 Methanation Over Ni/CeO2 Catalysts. Top. Catal. 2016, 59, 1424–1430. [Google Scholar] [CrossRef]
- Lustemberg, P.G.; Bosco, M.V.; Bonivardi, A.; Busnengo, H.F.; Ganduglia-pirovano, M.V. Insights into the Nature of Formate Species in the Decomposition and Reaction of Methanol over Cerium Oxide Surfaces: A Combined Infrared Spectroscopy and Density Functional Theory Study. J. Phys. Chem. C 2015, 119, 21452–21464. [Google Scholar] [CrossRef]
- Bazin, P.; Thomas, S.; Marie, O.; Daturi, M. New insights into the methanol oxidation mechanism over Au/CeO2 catalyst through complementary kinetic and FTIR operando SSITKA approaches. Catal. Today 2012, 182, 3–11. [Google Scholar] [CrossRef]
- Daturi, M.; Binet, C.; Lavalley, J.C.; Galtayries, A.; Sporken, R. Surface investigation on Ce(x)Zr(1−x)O2 compounds. Phys. Chem. Chem. Phys. 1999, 1, 5717–5724. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Rousseau, S.; Marie, O.; Bazin, P.; Daturi, M.; Verdier, S.; Harlé, V. Investigation of Methanol Oxidation over Au/Catalysts Using Operando IR Spectroscopy: Determination of the Active Sites, Intermediate/Spectator Species, and Reaction Mechanism. J. Am. Chem. Soc. 2010, 132, 10832–10841. [Google Scholar] [CrossRef]
- Riguetto, B.A.; Damyanova, S.; Gouliev, G.; Marques, C.M.P.; Petrov, L.; Bueno, J.M.C. Surface Behavior of Alumina-Supported Pt Catalysts Modified with Cerium as Revealed by X-ray Diffraction, X-ray Photoelectron Spectroscopy, and Fourier Transform Infrared Spectroscopy of CO Adsorption. J. Phys. Chem. B 2004, 108, 5349–5358. [Google Scholar] [CrossRef]
- Daturi, M.; Binet, C.; Lavalley, J.C.; Blanchard, G. Surface FTIR investigations on CexZr1−xO2 system. Surf. Interface Anal. 2000, 30, 273–277. [Google Scholar] [CrossRef]
- Fukuhara, C.; Hayakawa, K.; Suzuki, Y.; Kawasaki, W.; Watanabe, R. A novel nickel-based structured catalyst for CO2 methanation: A honeycomb-type Ni/CeO2 catalyst to transform greenhouse gas into useful resources. Appl. Catal. A Gen. 2017, 532, 12–18. [Google Scholar] [CrossRef]
- Guo, Y.; Mei, S.; Yuan, K.; Wang, D.-J.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Low-Temperature CO2 Methanation over CeO2 -Supported Ru Single Atoms, Nanoclusters, and Nanoparticles Competitively Tuned by Strong Metal−Support Interactions and H-Spillover Effect. ACS Catal. 2018, 8, 6203–6215. [Google Scholar] [CrossRef]
- Solis-Garcia, A.; Louvier-Hernandez, J.F.; Almendarez-Camarillo, A.; Fierro-Gonzalez, J.C. Participation of surface bicarbonate, formate and methoxy species in the carbon dioxide methanation catalyzed by ZrO2-supported Ni. Appl. Catal. B Environ. 2017, 218, 611–620. [Google Scholar] [CrossRef]
- Ashok, J.; Ang, M.L.; Kawi, S. Enhanced activity of CO2 methanation over Ni/CeO2-ZrO2 catalysts: Influence of preparation methods. Catal. Today 2017, 281, 304–311. [Google Scholar] [CrossRef]
- Daturi, M.; Finocchio, E.; Binet, C.; Lavalley, J.; Fally, F.; Perrichon, V.; Vidal, H.; Hickey, N.; Kaspar, J. Reduction of High Surface Area CeO2-ZrO2 Mixed Oxides. J. Phys. Chem. B 2000, 104, 9186–9194. [Google Scholar] [CrossRef]
- Kähler, K.; Holz, M.C.; Rohe, M.; Van Veen, A.C.; Muhler, M. Methanol oxidation as probe reaction for active sites in Au/ZnO and Au/TiO2 catalysts. J. Catal. 2013, 299, 162–170. [Google Scholar] [CrossRef]
- Wu, W.; Chuang, C.; Lin, J. Bonding Geometry and Reactivity of Methoxy and Ethoxy Groups Adsorbed on Powdered TiO2. J. Phys. Chem. B 2000, 104, 8719–8724. [Google Scholar] [CrossRef]
- Manzoli, M.; Chiorino, A.; Boccuzzi, F. Decomposition and combined reforming of methanol to hydrogen: A FTIR and QMS study on Cu and Au catalysts supported on ZnO and TiO2. Appl. Catal. B Environ. 2005, 57, 201–209. [Google Scholar] [CrossRef]
- Chen, T.; Feng, Z.; Wu, G.; Shi, J.; Ma, G.; Ying, P.; Li, C. Mechanistic Studies of Photocatalytic Reaction of Methanol for Hydrogen Production on Pt/TiO2 by in situ Fourier Transform IR and Time-Resolved IR Spectroscopy. J. Phys. Chem. B 2007, 111, 8005–8014. [Google Scholar] [CrossRef]
- Chuang, C.-C.; Chen, C.-C.; Lin, J.-L. Photochemistry of Methanol and Methoxy Groups Adsorbed on Powdered TiO2. J. Phys. Chem. B 1999, 103, 2439–2444. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, J.; Yang, N.; Yu, R.; Wang, D. Core-shell nano/microstructures for heterogeneous tandem catalysis. Mater. Chem. Front. 2021, 5, 1126. [Google Scholar] [CrossRef]
- González-Castaño, M.; González-Arias, J.; Bobadilla, L.F.; Ruíz-López, E.; Odriozola, J.A.; Arellano-García, H. In-Situ Drifts Steady-State Study of CO2 and Co Methanation Over Ni-Promoted Catalysts. Fuel 2023, 338, 127241. [Google Scholar] [CrossRef]
- Gil, A.; Díaz, A.; Gandía, L.M.; Montes, M. Influence of the preparation method and the nature of the support on the stability of nickel catalysts. Appl. Catal. A Gen. 1994, 109, 167–179. [Google Scholar] [CrossRef]
- Gandía, L.M.; Diaz, A.; Montes, M. Selectivity in the High-Temperature Hydrogenation of Acetone with Silica-Supported Nickel and Cobalt catalysts. J. Catal. 1995, 157, 461–471. [Google Scholar] [CrossRef]
- Wuttke, S.; Bazin, P.; Vimont, A.; Serre, C.; Seo, Y.-K.; Hwang, Y.K.; Chang, J.-S.; Férey, G.; Daturi, M. Discovering the Active Sites for C3 Separation in MIL-100(Fe) by Using Operando IR Spectroscopy. Chem. Eur. J. 2012, 18, 11959–11967. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Marie, O.; Bazin, P.; Lietti, L.; Visconti, C.G.; Corbetta, M.; Manenti, F.; Daturi, M. Modelling a reactor cell for operando IR studies: From qualitative to fully quantitative kinetic investigations. Catal. Today 2017, 283, 176–184. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Ni Content (wt.%) | SBET (m2/gcat.) | Metallic Ni Surface Area (SNi, m2/gNi) | Degree of Reduction (DORNi, %) | Nickel Dispersion (DNi, %) | Mean Ni Particle Size (dNi, nm) |
|---|---|---|---|---|---|---|
| Ni/Al | 7.6 | 201 | 41.7 | 68.9 | 6.3 | 7.1 |
| Ni/Ce | 8.2 | 93 | 34.3 | 103 | 5.1 | 13 |
| Ni/ZrCe | 8.9 | 87 | 19.1 | 131 | 2.9 | 23 |
| Ni/Ti | 8.7 | 53 | 8.1 | 120 | 1.2 | 67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Rangulan, V.V.; Reyero, I.; Bimbela, F.; Romero-Sarria, F.; Daturi, M.; Gandía, L.M. CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements. Catalysts 2023, 13, 448. https://doi.org/10.3390/catal13020448
González-Rangulan VV, Reyero I, Bimbela F, Romero-Sarria F, Daturi M, Gandía LM. CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements. Catalysts. 2023; 13(2):448. https://doi.org/10.3390/catal13020448
Chicago/Turabian StyleGonzález-Rangulan, Vigni V., Inés Reyero, Fernando Bimbela, Francisca Romero-Sarria, Marco Daturi, and Luis M. Gandía. 2023. "CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements" Catalysts 13, no. 2: 448. https://doi.org/10.3390/catal13020448