

Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency


1
LAQV-REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal
2
UCIBIO, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal
3
Associate Laboratory i4HB, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal
*
Authors to whom correspondence should be addressed.
†
Current address: ITQB NOVA, Instituto de Tecnologia Química e Biológica António Xavier, Universidade NOVA de Lisboa, Avenida da República, 2780-157 Oeiras, Portugal.
Catalysts 2023, 13(2), 386; https://doi.org/10.3390/catal13020386
Submission received: 23 January 2023
/
Revised: 5 February 2023
/
Accepted: 6 February 2023
/
Published: 10 February 2023
(This article belongs to the Special Issue Feature Papers in Catalysis in Organic and Polymer Chemistry)
Abstract
:A series of nine cationic heteroleptic aryl-BIAN-copper(I) (BIAN = bis-iminoacenaphthene) complexes with the general formula [Cu((E-C6H4)2BIAN)(PPh3)2][X] (E = p-Me, p-iPr, o-iPr; X = BF4, OTf, NO3) 1X–3X were synthesized and fully characterized using several analytical techniques, including NMR spectroscopy and single-crystal X-ray diffraction. Except for complexes 2BF4 and 3BF4, which were already reported in our previous works, all remaining complexes are herein described for the first time. Two different strategies were used for the preparation of the complexes: complexes bearing BF4− or OTf− counter-ions (1BF4, 1OTf, 2OTf, and 3OTf) were obtained using the appropriate copper(I) precursors [Cu(NCMe)4][BF4] or [Cu(NCMe)4][OTf], whereas for derivatives 1NO3–3NO3, [Cu(PPh3)2NO3] was used. Their activity as catalysts for the copper azide-alkyne cycloaddition (CuAAC) was assessed alongside other high activity, previously reported Cu(I) complexes. Comparative studies to determine the influence of the counter-ion and of the aryl substituents were performed. All complexes behaved as active catalysts under neat reaction conditions, at 25 °C and in short reaction times without requiring the use of any additive, with complex 2NO3 being the most efficient derivative, along with other NO3−-bearing complexes.
1. Introduction
1,2,3-triazole derivatives are an important class of compounds with useful applications in various fields such as materials science, polymers, bioconjugation and medicinal chemistry and can be found as the center piece on a large number of therapeutic compounds [1].
The development of innovative new ways to synthesize complex molecular architectures following green chemistry principles is paramount for modern chemists. The copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC), discovered independently in 2002 by Meldal [2] and Sharpless [3] became the gold standard for the synthesis of 1,4-disubstituted 1,2,3-triazoles. The importance of such a breakthrough as click chemistry, in which two building blocks are coupled together quickly and efficiently, was recently highlighted by the awarding of the Nobel Prize in Chemistry in 2022 [4]. However, one downside of the systems developed by Meldal and Sharpless was the amount of copper used and the necessity to use a reductive agent to guarantee the stability of the Cu(I) species in the reaction medium [5]. Afterwards, the use of Cu(I) complexes stabilized by ancillary ligands increased both the stability of the metal center and also led to an increase of their activity [6,7]. Despite many Cu(I) catalysts being efficient in catalyzing this reaction [5,8,9], the conventional Sharpless-Fokin catalyst (CuSO4/sodium ascorbate) is still the most common system used by synthetic chemists, notwithstanding some drawbacks: the large amount of catalyst required, a slow rate, the difficulty of separating copper residues of the products [3,10], and sometimes the detrimental effects on rates and yield observed in the presence of chlorides, bromides and iodides [11].
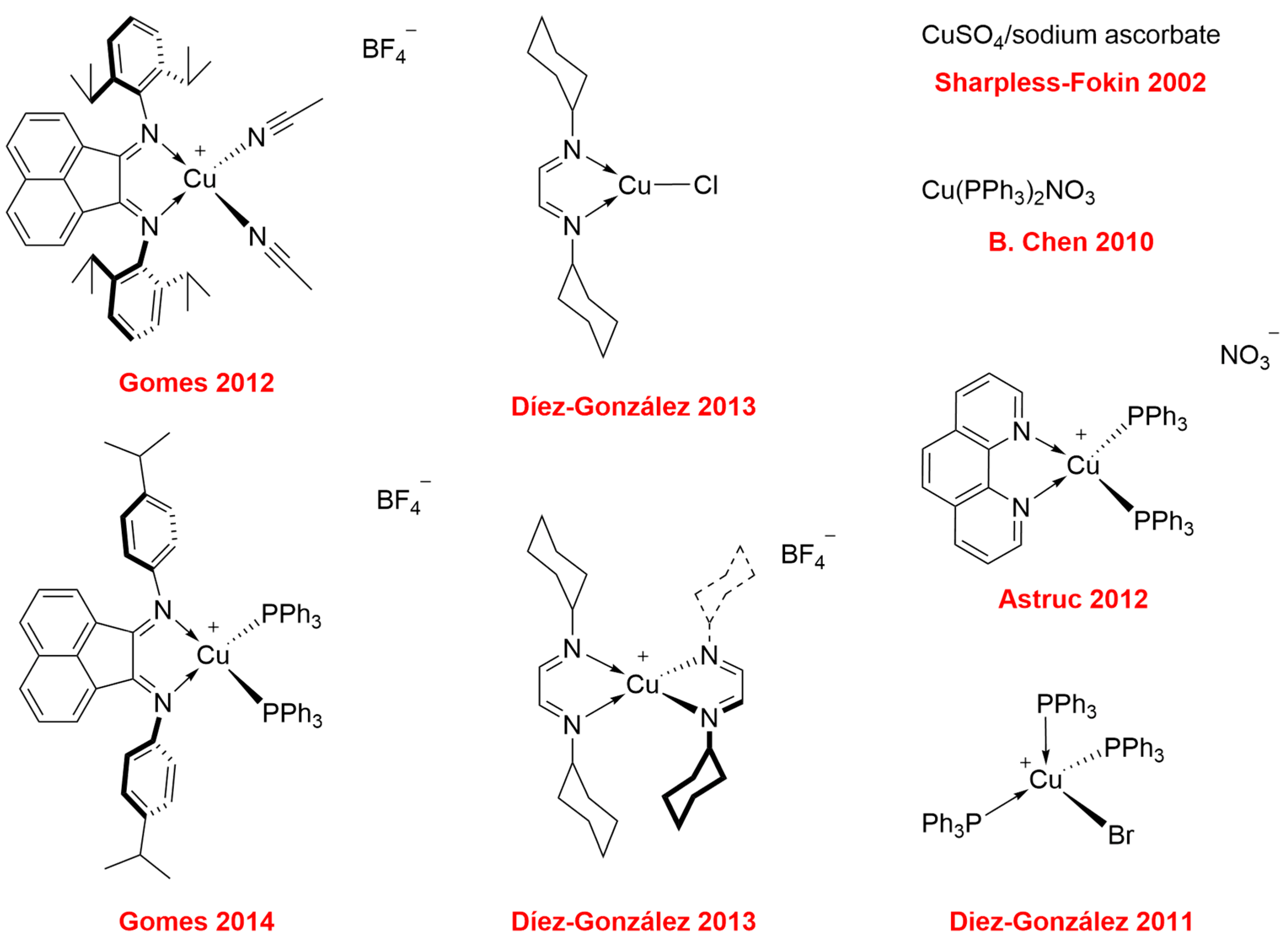
To avoid the use of a large excess of metal ‘to catalyze the reactions, which leads to potential side products and difficulty in its removal after the reaction’s completion, the development of well-defined and versatile catalysts is crucial. It is important that such compounds can exhibit group functionality tolerance, activity in a variety of solvents, do not require the use of additives, and be air and moisture insensitive. Therefore, the pursuit of Cu(I) catalysts that meets these requirements has incessantly occurred in recent decades. Since 2002, a series of very efficient catalysts have been developed and evaluated in the CuAAC reaction (Figure 1) [2,3,12,13,14,15,16,17].
In 2012, Wang et al. showed that complexes [Cu(PPh3)2][NO3] and [Cu(Phen)(PPh3)2][NO3] presented good to excellent yields in reactions carried out under neat conditions [18], at room temperature and in short reaction times. Diez-González and colleagues also showed that [CuBr(PPh3)3] “followed the click principles” at even lower catalyst loadings [15]. In fact, few heteroleptic Cu(I) complexes have been investigated as potential catalysts for the CuAAC reaction.
We have been interested in the chemistry of copper complexes supported by aryl-BIAN (BIAN = bis-iminoacenaphthene) chelating ligands for applications in catalytic processes such as the CuAAC reaction and the reverse atom-transfer radical polymerization (ATRP) of styrene [19,20]. As evidenced in our previous papers, the substituents on the bis-aryl moieties have some impact on the catalytic efficiency of the complexes [19,21]. Moreover, the potential effect of the substitution of the phosphine ligand by an arsine or a stibine analogue did not present any evidence that either As or Sb ligands have improved activity of the complexes as catalysts of the CuAAC reaction. However, an important feature that, so far, has not been investigated is the potential influence of the counter-ions on the activity of the cationic aryl-BIAN-Cu(I) catalysts. Such influence has also been very scarcely studied for other systems.
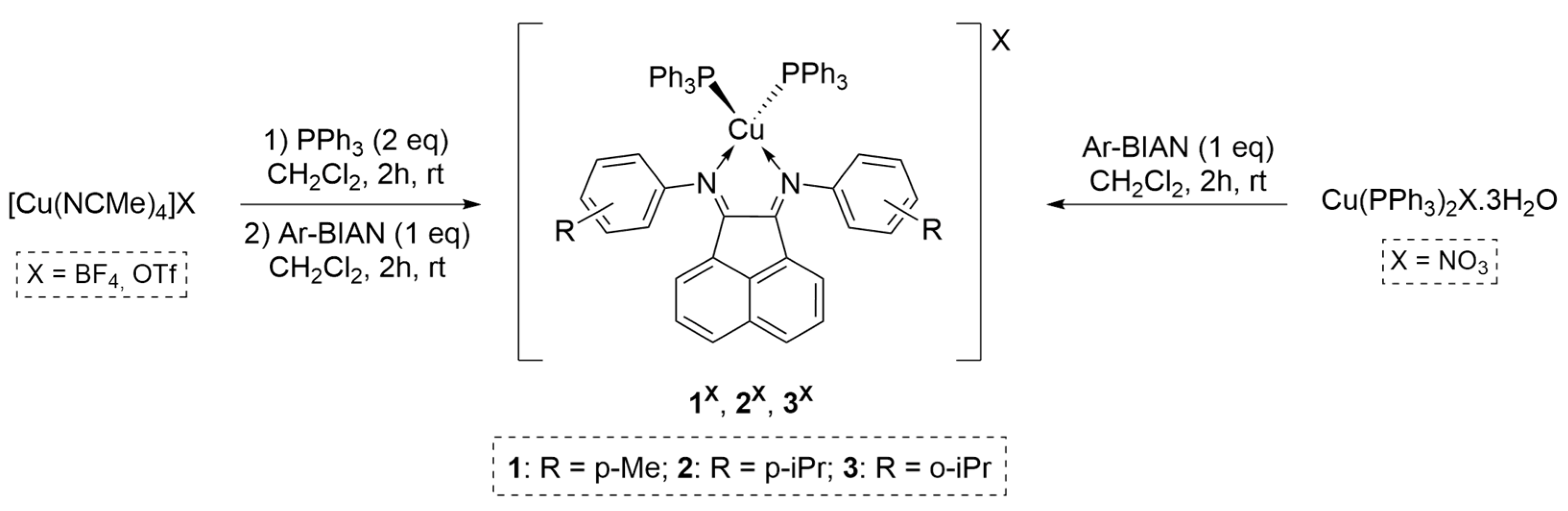
Taking this into account, we decided to design and investigate a new family of cationic heteroleptic copper(I) complexes with the general formula [Cu(aryl-BIAN)(PPh3)2][X], where X = tetrafluoroborate, triflate or nitrate. Herein, we report their synthesis and full characterization, and their evaluation as catalysts for the CuAAC reaction, including the scope of substrates and reaction solvents. The influence of the counter-ion, a subject scarcely investigated so far, was studied to assess if any influence exists on the activity of the catalyst.
2. Results
2.1. Synthesis and Characterization
Aryl-BIAN ligands, (p-iPrC6H4)2BIAN (L2) and (o-iPrC6H4)2BIAN (L3), were synthetized using the traditional methodology, consisting in the acid-catalyzed Schiff base reaction using an alcoholic solvent. On the other hand, in the case of (p-MeC6H4)2BIAN (L1), a more sustainable procedure, i.e., a template reaction using ZnCl2, acenaphthenequinone and toluidine in refluxing acetic acid for 30 min, was employed [22], followed by demetallation with a K2CO3 solution.
The synthesis of heteroleptic cationic complexes 1X−3X (X = BF4, OTf, NO3) proceeded through two distinct strategies using inert atmosphere techniques (Scheme 1). Complexes bearing BF4− or OTf− counterions (1BF4, 1OTf, 2OTf, and 3OTf) were obtained using the appropriate copper(I) precursors [Cu(NCMe)4][BF4] or [Cu(NCMe)4][OTf] in a two-step sequential procedure (Scheme 1, left). The first step consisted in the 2 h reaction between one equivalent (1 eq.) of the suitable metal salt and two equivalents of triphenylphosphine, followed by the second step with the addition of the aryl-BIAN ligand. Due to the very sensitive nature of [Cu(NCMe)4][NO3], which oxidized overnight under an inert atmosphere, a different precursor for the NO3 complexes was chosen, [Cu(PPh3)2][NO3]. This was used in a one-step procedure to synthesize the derivatives 1NO3, 2NO3, and 3NO3 (Scheme 1, right). Compounds 2BF4 and 3BF4 were already reported in previous works [18] and were synthetized to compare their catalytic activity with the newly obtained complexes. All complexes were obtained in high to very high yields (69–98%) and easily crystallized in a mixture of CH2Cl2-pentane.
Complexes 1X−3X (X = BF4, OTf, NO3) were fully characterized using elemental analysis; 1H, 13C{1H}, 31P{1H} and 19F{1H} NMR, UV-Vis and FT-IR spectroscopies; mass spectrometry; and, when possible, single-crystal X-ray diffraction. It was possible to fully assign the 1H and 13C resonances for all derivatives. However, for analogous cations, the counter-ion variation does not influence the 1H NMR chemical shifts (see Section 3.2.1 and the Supplementary Information for detailed NMR assignment). The similarity of every spectrum allows for the assumption that no other electronic or chemical factors will have an effect on the complexes’ catalytic activity, other than ionic influence in solubility, stability and/or coordination to the metal center.
Analysis of the 31P{1H}-NMR spectra of all complexes displayed a single resonance between 2 and −2 ppm (Section 3.2.1 and Supplementary Information), and, similarly to what was observed with the 1H and 13C NMR spectra, no correlation between the different counter-ions could be determined. Nevertheless, a slight difference was noticeable considering the position of the R group in the aryl moiety of the BIAN ligand. For the -para substituted aryl moieties, 31P chemical shifts were ca. 1.3 ppm, and for the -ortho substituted ones, 31P chemical shifts were ca. −1.7 ppm.
The presence of fluorine atoms in the counter-anions BF4− and OTf−, and their inexistence in the NO3−, allowed a differentiation between the three types of derivatives (for example, 19F chemical shift is −154 and −78 ppm for 1BF4 and 1OTf, respectively).
The UV-Vis spectra of the complexes also corroborated the NMR spectroscopy observations, showing no effect of the counter-ion on the shape of the curve (Figure S1 in Supplementary Information), as well as indicating the inexistence of coordination of the NO3 anion to the copper(I) center. All derivatives 1X−3X (X = BF4, OTf, NO3) display bands at ca. 460 and 322 nm, which can be attributed to π–π* transitions of the aryl-BIAN ligands (ILCT). These are also observable in the spectra of the free ligands. However, these have undergone a bathochromic shift, which is indicative of coordination to the metal center [23]. Furthermore, in the case of complexes bearing ligands L1 and L2, it is also possible to observe a shoulder at 372 nm that can be attributed to metal–ligand transitions (MLCT). The absence of coordination was also confirmed by FT ATR-IR (Figure S2 in Supplementary Information). This spectroscopic technique also granted the identification of specific groups, such as C=N, PPh3, BF4, CF3, S=O and NO3. This, along with mass spectrometry, the aforementioned 19F NMR and, when possible, single-crystal X-ray diffraction, were the methods which allowed the distinction between complexes with different counter-ions (Section 2.2 and Section 3.2 and the Supplementary Information for FT ATR-IR spectra, 19F NMR and X-ray diffraction data). The stability of all the synthesized compounds was investigated, showing to be air and moisture stable in their solid form, which is rare in Cu(I) species. However, in solution, although complexes bearing BF4 and OTf counter-anions showed no indication of degradation after a week standing in CDCl3, the NO3 derivatives showed signs of degradation after a couple of days.
2.2. X-ray Diffraction Studies
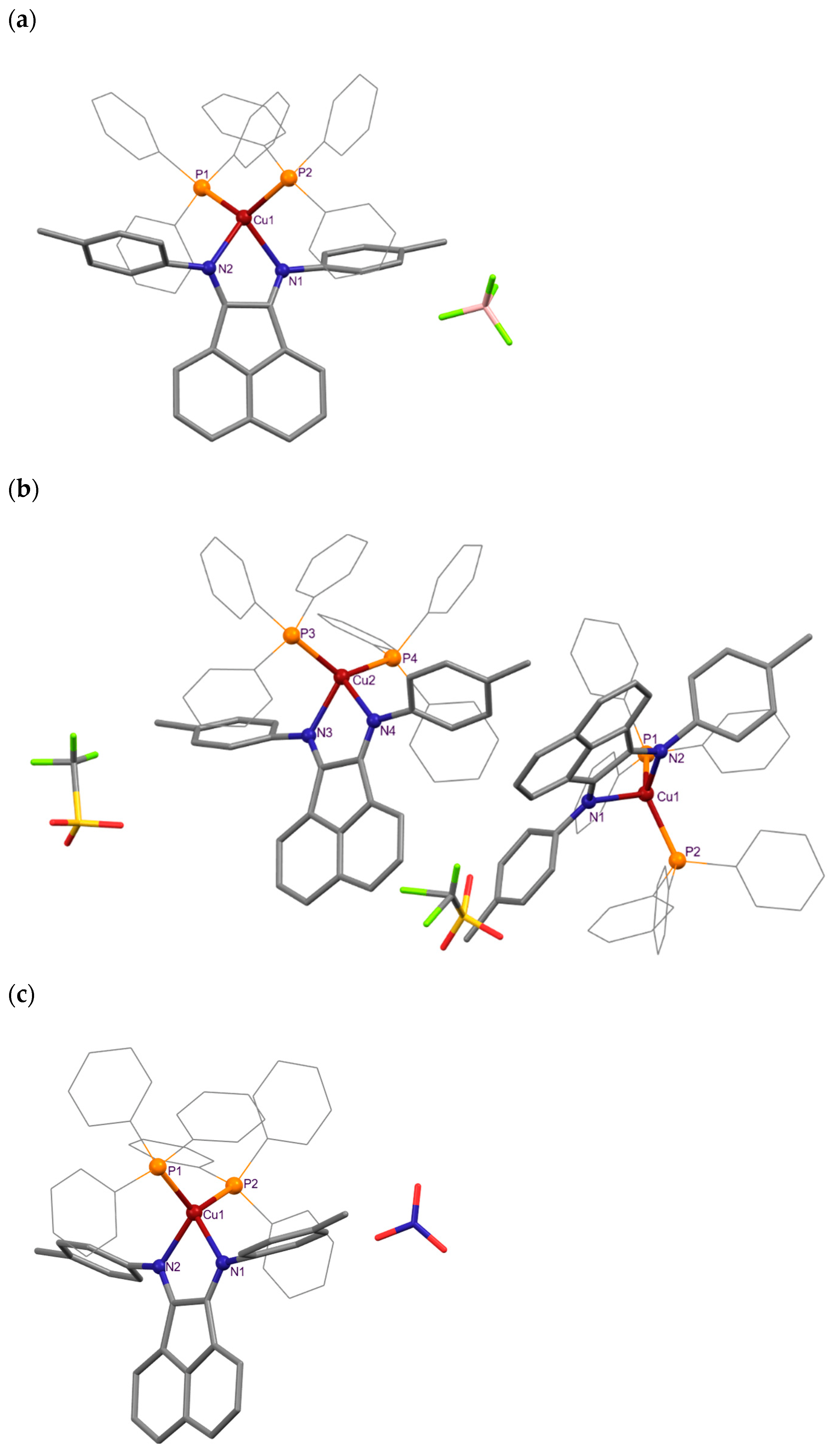
Crystals suitable for single-crystal X-ray diffraction were obtained from solutions of dichloromethane double-layered with pentane for the majority of the compounds reported herein, namely for derivatives 1BF4, 1OTf, 1NO3, 2NO3 and 3NO3, and also for ligand L3 ((o-iPrC6H4)2BIAN). In fact, although the synthesis of ligand L3 was already reported in the literature, to the best of our knowledge, no reports on its crystal structure were found. Their molecular structures are depicted in Figure 2 and Figures S10–S13, while their bond lengths, angles and other relevant structural parameters are displayed in Table 1.
Compound L3, bearing an o-iPr group in the aryl moiety, crystallized in triclinic system, P-1 space group, with one molecule in the asymmetric unit. As expected, and similarly to what is observed in other aryl-BIAN derivatives [24], the acenaphthene backbone is planar with the N-aryl-substituents lying nearly perpendicular to the acenaphthene plane (Figure S10 and Table 1). The presence of a single substituent in one of the ortho positions of the aryl ring makes it asymmetric, allowing the existence of both cis and trans isomers due to the free rotation in the single bond N–Cipso-aryl. In the crystal structure of L3, however, only the cis isomer is observed, i.e., the isomer containing both o-iPr groups on the same side of the acenaphthene plane. All the bond distances and angles are within the expected values for this type of ligands [24].
Complexes 1BF4, 1OTf, 1NO3, 2NO3 and 3NO3 crystallized in a wide variety of space groups and crystal systems: triclinic P-1, 1OTf; monoclinic P21/c, 1NO3; orthorhombic Pbca, 1BF4 and 2NO3; and tetragonal P41, 3NO3. Except for 1OTf, which displays two molecules in the asymmetric unit, all derivatives only exhibit a single metal complex in their asymmetric units.
As can be observed in Table 1, the Cu–N and Cu–P bond distances (ca. 2.1 and ca. 2.26 Å) are constant throughout the derivatives. The same applies to the N–Cu–N bite angle, which is approximately 78°, and to the C=N bond length, which does not suffer modification upon coordination to the metal center. This can be attributed to the rigidity of the BIAN backbone and agrees with previous reports [25,26].
In all derivatives, the cationic Cu(I) metal center is surrounded by four atoms, two N belonging to the BIAN chelating ligand and two phosphines, giving rise to a geometry of a distorted tetrahedron. This degree of distortion can be assessed by the dihedral angle ω (Table 1), which consists of the dihedral angle between C=N1–Cu–N2=C and P1–Cu–P2. For the cationic copper(I) complexes described in this work, ω varies between 80.97(9) and 105.99(14)°. A pure tetrahedral geometry would present a ω dihedral angle of 90°. In the case of complex 1OTf, which shows two independent molecules in the asymmetric unit, ω varies considerably with values of 103.36(11)° for molecule 1 and 97.96(10)° for molecule 2, the latter being the less distorted derivative and displaying a value similar to that of 1NO3, 97.64(7)° (Table 1). Moreover, the angle φ (dihedral angle between the acenaphthene backbone and the aryl rings) is also a relevant parameter that gives important information regarding the relative conformation of the aryl substituents in relation to the rigid BIAN moiety. The observed values in Table 1 are somewhat irregular (58.41(16)° to 104.59(17)°), showing that the aryl rings have some degree of distortion from perpendicularity. These values are within the observed for the free ligand L3, not showing significative variation upon coordination. Curiously, the main difference between the free ligand L3 and the coordinated ligand in 3NO3 is the fact that L3 presents the o-iPr groups in a cis conformation, while in 3NO3 these groups are located at opposite sides of the acenaphthene plane, i.e., in a trans geometry. Finally, the dihedral angle χ is related to the relative position of the two aryl substituents. Except for molecule 2 of complex 1OTf, which shows a greater tendency for coplanarity of the aryl rings, the remaining structures present dihedral angles of ca. 50°.
Interestingly, π...π stacking between acenaphthene moieties of two independent molecules, in a tail-to-tail fashion, at a distance of 3.373 Å, can be observed in the structure of complex 1OTf (Figure S13 in Supplementary Information).
2.3. Catalytic Studies
2.3.1. Catalysts’ Screening
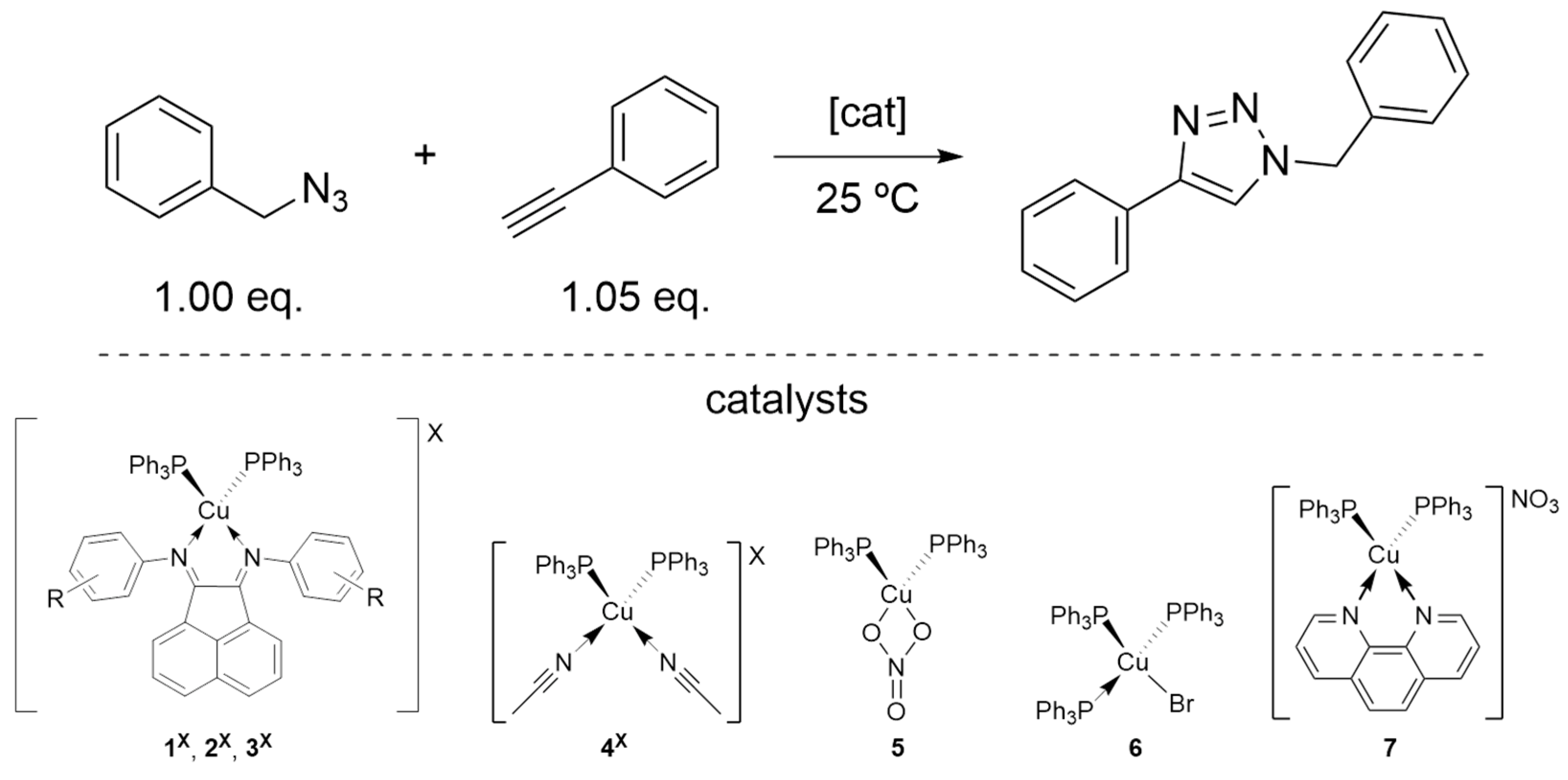
The newly synthesized heteroleptic cationic copper(I) complexes 1X–3X (X = BF4, OTf, NO3) were tested as catalysts for the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC). Benzyl azide and phenylacetylene, which are standard reagents for this type of reaction, were chosen as model substrates for this study. The catalytic activity of the Cu(I) complexes was investigated under neat conditions, i.e., solventless, at room temperature with an initial loading of 0.5 mol% relative to substrate loading, specifically azide (1.00 eq.). Alkyne was added with a slight excess (1.05 eq.). The reaction was performed at the temperature of 25 °C, with the reaction time set to 30 min (see Table S1 for complete data). In this primary screening, some complexes showed little catalytic activity, which led us to increase the time to 1h when necessary. On the other hand, for the complexes that showed good catalytic activity, the catalyst loading was decreased, for optimal minimal conditions for all complexes 1X−3X (X = BF4, OTf, NO3). The obtained results are summarized in Table 2. Conversions were calculated by 1H NMR spectroscopy, and η represents the isolated yield.
For comparison, the copper precursors of each counter-ion bearing phosphine ligands, 4X and 5 (see Scheme 2 and Table 2), were also studied to verify the role of the aryl-BIAN ligands in this reaction. Furthermore, we prepared and studied, under the same reaction conditions, efficient copper(I) catalysts previously reported by Diez et al. [Cu(PPh3)3Br] (6) [15], and by Wang et al. [Cu(Phen)(PPh3)2][NO3] (7) [27].
Within complexes 1X−3X, complexes bearing NO3− as counter-ion showed better catalytic activity. It was possible to decrease the catalyst loading down to 0.1 mol%, while maintaining high conversion and isolated yields, in only 30-min reaction time. Moreover, in the case of 2NO3, attempts to decrease the catalyst loading down to 500 ppm were investigated. After 2 h, even at such low catalyst loading, conversion of 88% and an isolated yield of 76% were obtained. However, it was difficult to reproduce with precision due to the low measured amount of catalyst. Also, complex 2OTf kept high conversion and yield with decreased cat. loading, although only down to 0.3 mol%. The remaining complexes needed longer reaction times even at 0.5 mol% catalyst loading. This is the first indication that the NO3− counter-ion has a great positive effect on the catalytic efficiency of these copper(I) complexes. Further studies to gain a better grasp of those effects are currently being undertaken and will be published as soon as possible.
As expected, the isolated yields were slightly lower than the conversion, due to some product losses during the purification process. No indication of side products was found after analysis of the crude reaction product.
Copper precursors 4X (X = BF4, OTf) and 5 showed no activity in the tested conditions. Complexes 6 and 7, however, obtained very high conversions and yields, comparable to the ones reported here for the first time. However, they are less favorable. Some difficulty was observed in storage of these species under solid form. The copper(I) complexes easily oxidized, especially [Cu(Phen)(PPh3)2][NO3] (7) after only one week.
2.3.2. Solvents’ Screening
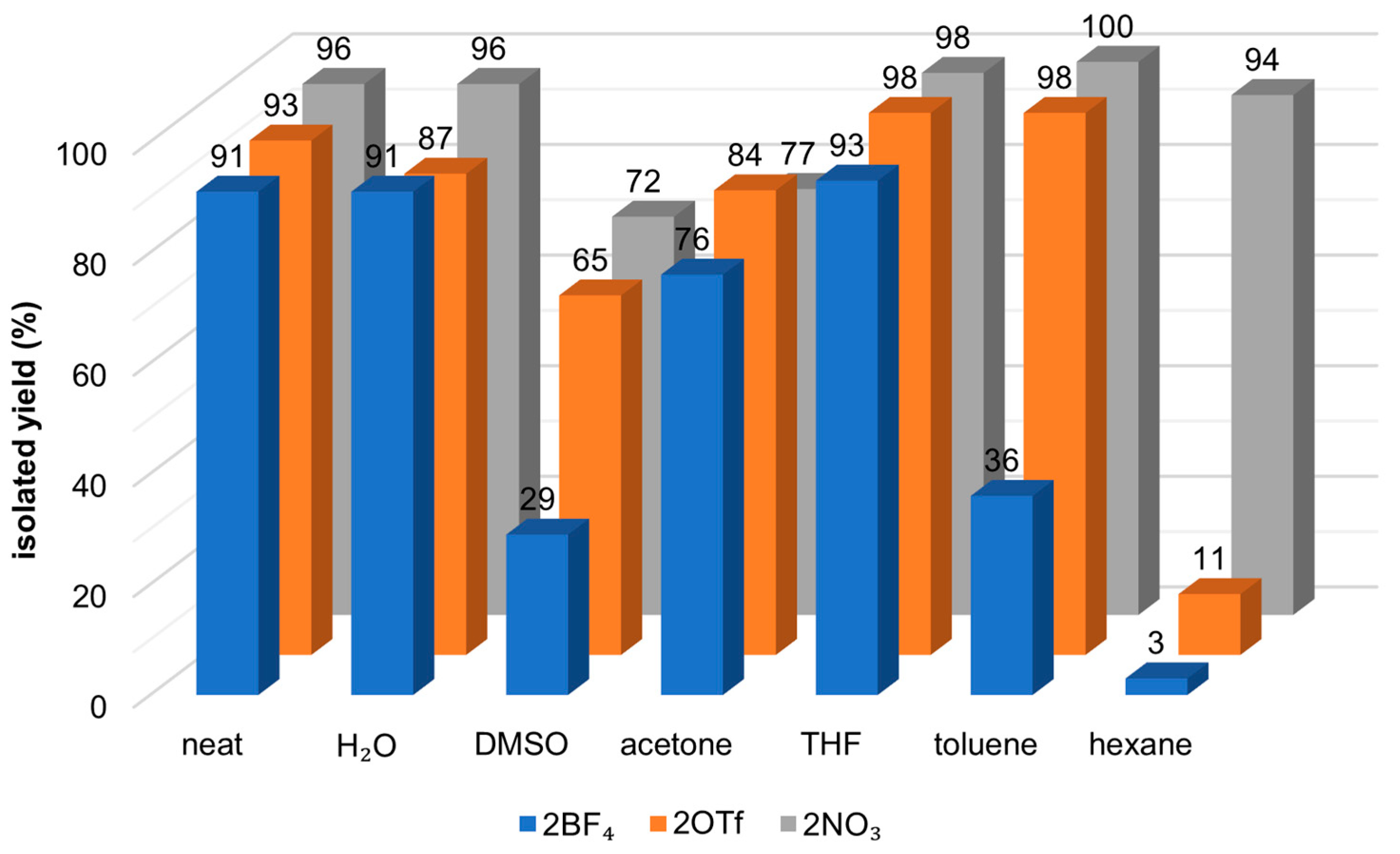
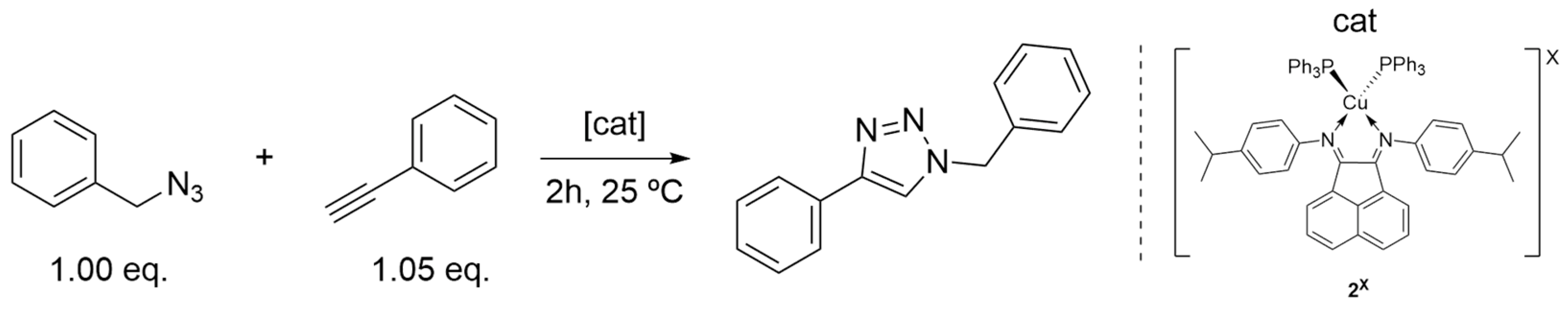
The solvent assays were carried out with only complexes 2X (see Scheme 3). Six different solvents were chosen based on their wide range of polarity and usage in synthetic laboratories worldwide: water, DMSO, acetone, THF, toluene and hexane. The obtained results are depicted in Figure 3. Only isolated yields were calculated due to the nature of the different solvents and consequent reaction work-up.
As was the case within neat conditions, 2NO3 showed the best activity overall. Yet, it is noteworthy to point out that 2OTf shows as good or better activities in more polar solvents, when compared to 2NO3. On the other hand, 2BF4 was consistently the worst catalyst on the various solvents studied, indicating that the coordination capability of the triflate and the nitrate counter-ions provides an advantage to these catalysts. On the two conditions where the catalysts were insoluble, acting in heterogenous conditions, water and hexane, two different outcomes arose. In an aqueous solution, all three catalysts showed yields superior to 87%. In hexane, however, 2NO3 was the only catalyst to show good results, with 94% yield.
2.3.3. Catalysis Scope
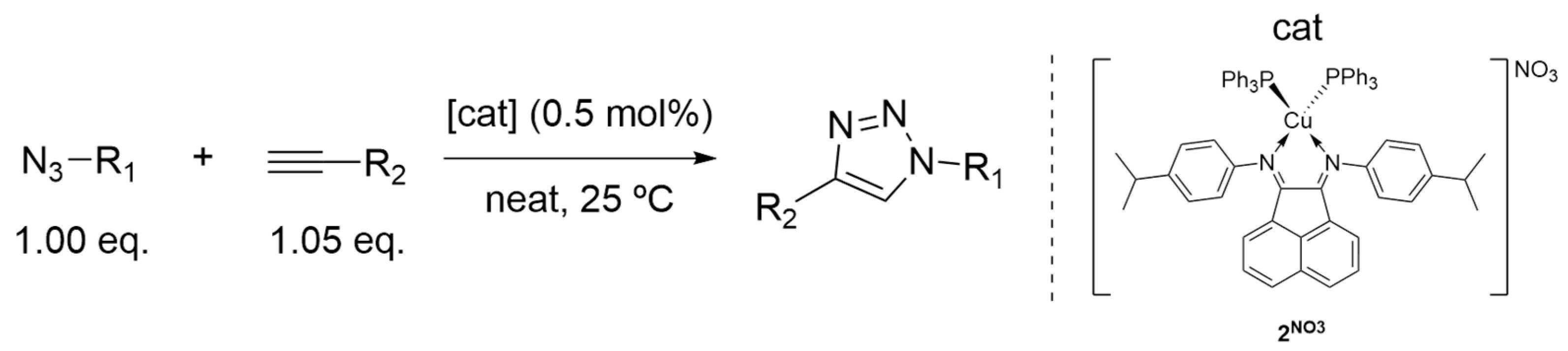
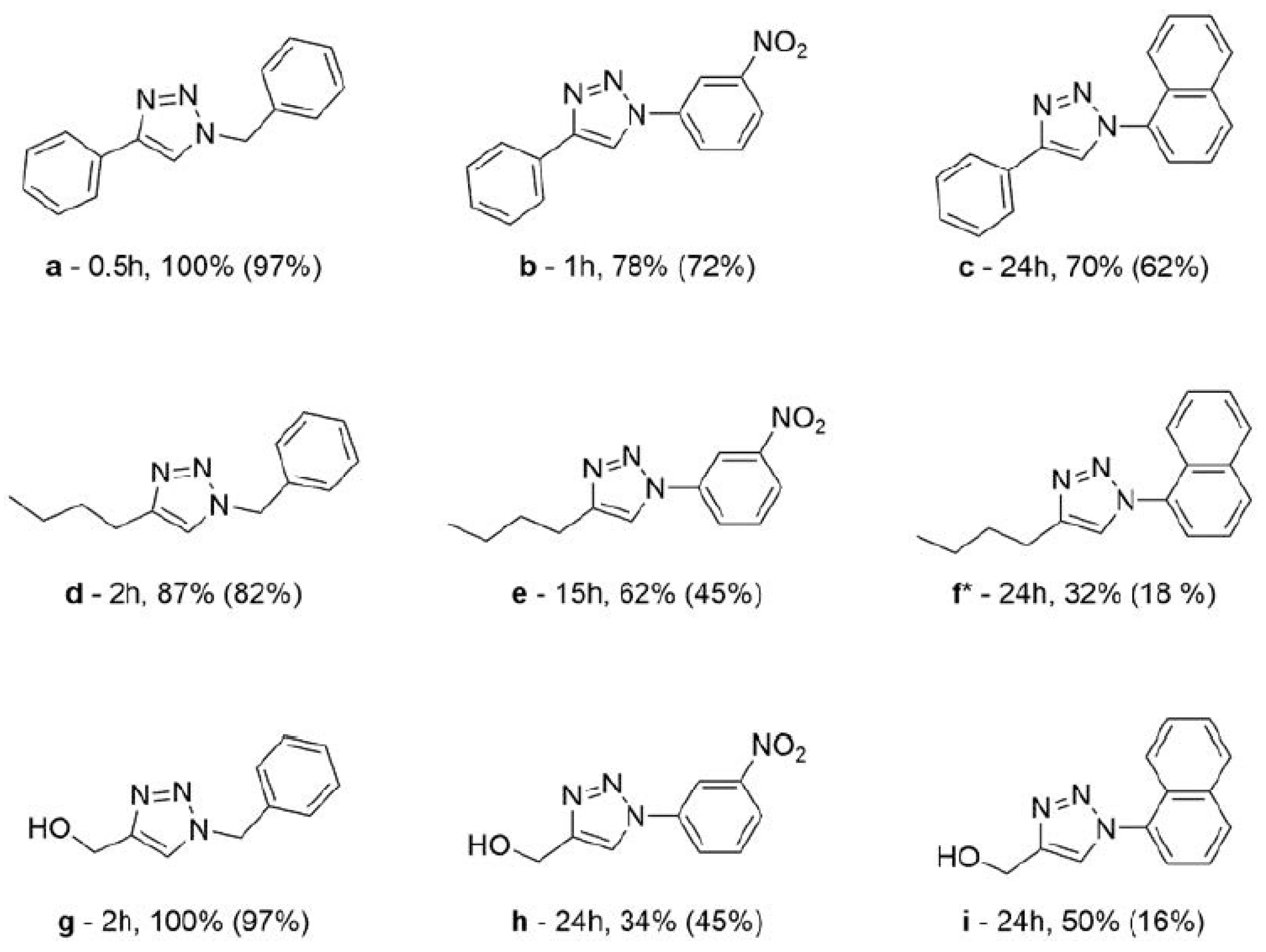
The solvent and catalyst screenings allowed for selection of the best reaction conditions and catalyst for the determination of the system scope. Complex 2NO3 was chosen as the catalyst for further studies, since it showed the best results across the board under different conditions. A catalyst loading of 0.5 mol% was applied in neat conditions to keep the system robust. Different azide and alkyne derivatives were employed; 1-hexyne and propargyl alcohol, in addition to phenylacetylene, were chosen as alkyne substrates, and as azide substrates, 1-azidonaphthalene, 3-nitrophenyl azide and benzyl azide (see Scheme 4 and Scheme 5).
Two trends can be determined by evaluating the results. The limitation of the neat system is evident when one of the substrates is solid. When both substrates are liquid at 25 °C, the best results were obtained. The use of benzyl azide yielded the best results with the three different alkynes; the presence of electron-withdrawing groups on the alkyne led to better yields.
3. Materials and Methods
3.1. General Considerations
All reactions were carried out using Schlenk techniques under argon atmosphere with the use of dry solvents unless otherwise specified. Diethyl ether and pentane were dried over metallic sodium prior to distillation under argon. Dichloromethane and acetonitrile were dried over calcium hydride prior to distillation.
Alkynes were purchased at Sigma-Aldrich (Madrid, Spain), Alfa Aesar (Madrid, Spain) and TCI (Zwijndrecht, Belgium) and used as received.
Ligands (p-MeC6H4)2BIAN (L1) [28], (p-iPrC6H4)2BIAN (L2) and (o-iPrC6H4)2BIAN (L3) were synthesized following the reported methodology, and characterization corresponded to the described data. Complexes [Cu(NCMe)4][BF4] [29], [Cu(NCMe)4][OTf] (OTf = CF3SO3), [Cu(NCMe)2(PPh3)2][BF4] [30], [Cu(NCMe)2(PPh3)2][OTf], [Cu(PPh3)3Br] [31], [Cu(PPh3)2][NO3] [32], [Cu(Phen)(PPh3)2][NO3] [33], [Cu((p-iPrC6H4)2BIAN)(PPh3)2][BF4] (2BF4) [19] and [Cu((o-iPr-MeC6H4)2BIAN)(PPh3)2][BF4] (3BF4) [19] were prepared according to the literature. 3-Nitrophenyl azide was prepared according to the literature [34].
UV-Vis spectra were acquired in a Varian Cary 100 Bio spectrophotometer (Varian Spectrophotometer Cary-100 Bio, Agilent Technologies, Santa Clara, CA, USA). Samples were prepared in dichloromethane previously filtered through basic alumina, with a 5 × 10 M−1 concentration.
IR spectra were acquired in a FT-IR Spectrum Two (Perkin-Elmer Waltham, MA, EUA) in ATR mode (FT-IR, Perkin-Elmer Waltham, MA, EUA).
Elemental analysis was done in a Thermo Finnigan-CE Instruments Flash EA 1112 CHNS series and mass spectrometry in a LC Agilent 1200 Series with a binary pump/MS Agilent 6130B Single Quadrupole with an ESI source (LC-ESI, Agilent, Santa Clara, CA, EUA) at LAQV-REQUIMTE (DQ) Laboratório de Análises.
NMR spectra were recorded using a Bruker Avance III 400 MHz or 500 MHz spectrometer, with probe QNP 100 MHZ S1 with Z-gradient (NMR, Bruker, Billerica, MA, EUA), and processed with the TopSpin software (version 3.2, Bruker BioSpin GmbH, Ettlingen, Germany). Spectra were referenced internally using the residual protio solvent resonance relative to tetramethylsilane (δ = 0). Multiplicities were abbreviated as follows: singlet (s), broad (br), doublet (d), triplet (t), sextet (sx), septet (sept) and multiplet (m).
Purifications by column chromatography were made using silica gel 60 (0.040–0.063 mm), 230–400 mesh.
3.2. Synthetic Procedures
3.2.1. Syntheses of Complexes 1BF4, 1OTf, 2OTf and 3OTf
A typical synthetic procedure is described as follows: to a suspension of copper tetrakis acetonitrile BF4 or OTf, triphenylphosphine (2.00 eq.) was added in dichloromethane (10 mL). The mixture was left under stirring at room temperature. After two hours, a solution (10 mL) of the respective bis-aryl-BIAN (1.00 eq.), in dichloromethane, was added. The resulting solution was left stirring for two additional hours. After completion of the reaction, the solution was filtered, concentrated under reduced pressure, and layered with pentane to precipitate the desired compound.
Data for [Cu((p-MeC6H4)2BIAN)(PPh3)2][BF4] (1BF4): following the general procedure, [Cu(NCMe)4][BF4] (0.15 g, 0.20 mmol), PPh3 (0.11 g, 0.42 mmol) and L1 (0.07 g, 0.20 mmol), complex 1BF4 was obtained as red crystals (0.18 g, η = 88%). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.21 (d, J = 8.3 Hz, 2H), 7.59 (t, J = 7.8 Hz, 2H), 7.36 (t, J = 7.5 Hz, 6H), 7.33 (d, J = 7.2 Hz, 2H), 7.12 (t, J = 7.5 Hz, 12H), 7.00 (d, J = 7.9 Hz, 4H), 6.92–6.80 (br, 12H), 6.31 (d, J = 8.0 Hz, 4H), 2.45 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ (ppm): 162.8, 144.6, 143.7, 137.9, 133.3, 132.0, 131.52, 131.49 (t, 1JCP = 17.4 Hz), 130.6, 130.4, 129.1, 128.7, 126.0, 125.1, 120.8, 21.3. 19F{1H} NMR (376 MHz, CDCl3) δ (ppm): −154.6. 31P{1H} NMR (162 MHz, CDCl3) δ (ppm): 1.1. UV-Vis: (CH2Cl2) λ (nm) ε (×104 M−1·cm−1): 322 nm (1.40), 372sh (0.60), 460 nm (0.77). IR ATR ν max. (cm−1): 1636 (w, C=N), 1434 (m, PPh3), 1050 (s, BF4), 1035 (s, BF4), 694 (s, C conj.). Elemental analysis calc. for C62H50CuBN2F4P2·1/2CH2Cl2: C 69.65, H 4.77, N 2.60, exp.: C 69.35, H 4.75, N 2.57. LC/MS (NCMe/H2O) m/z: Positive mode: 783.2 [Cu(L1)2]+, 685.2 (100%) [Cu(L1)(PPh3)]+, 587.2 [Cu(PPh3)2]+, 361.1 [L1 + H]+, 263.1 [PPh3 + H]+. Negative mode: 197.1 [(BF4)2 + Na]−, 87.1 (100%) [BF4]−.
Data for [Cu((p-MeC6H4)2BIAN)(PPh3)2][OTf] (1OTf): following the general procedure, [Cu(NCMe)4][OTf] (0.15 g, 0.40 mmol), PPh3 (0.22 g, 0.83 mmol) and L1 (0.15 g, 0.42 mmol), complex 1OTf was obtained as red crystals (0.37 g, η = 85%). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.20 (d, J = 8.0 Hz, 2H), 7.59 (t, J = 7.8 Hz, 2H), 7.36 (t, J = 7.5 Hz, 6H), 7.33 (d, J = 7.3 Hz, 2H), 7.12 (t, J = 7.4 Hz, 12H), 7.00 (d, J = 8.0 Hz, 4H), 6.92–6.81 (br, 12H), 6.31 (d, J = 7.8 Hz, 4H), 2.45 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ (ppm): 162.8, 144.6, 143.7, 137.9, 133.3, 131.9, 131.5 (overlap: t, 1JCP = 17,4 Hz; s), 130.6, 130.4, 129.1, 128.7, 126.0, 125.1, 120.8, 21.3. 19F{1H} NMR (376 MHz, CDCl3) δ (ppm): −78.0. 31P{1H} NMR (162 MHz, CDCl3) δ (ppm): 1.3. UV-Vis (CH2Cl2) λmáx. (nm) [ε (×104 M−1·cm−1)]: 322 (1.36), 372sh (0.58), 459 (0.75). IR ATR ν max. (cm−1): 1635 (w, C=N), 1435 (m, PPh3), 1267 (s, CF3), 1155 (m, S=O), 1032 (s, C-F), 695 (s, C conj.), 636 (s, CF3). Elemental analysis calc. for C63H50CuN2O3F3P2S·1CH2Cl2 (%): C 65.00, H 4.43, N 2.37, S 2.71; exp.: C 65.00, H 4.45, N 2.39, S 2,59. LC/MS (NCMe/H2O) m/z: Positive mode: 783.2 [Cu(L1)2]+, 685.2 (100%) [Cu(L1)(PPh3)]+, 587.1 [Cu(PPh3)2]+, 361.1 [L1 + H]+, 263.1 [PPh3 + H]+. Negative mode: 321.0 [(OTf)2 + Na]−, 149.0 (100%) [OTf]−.
3.2.2. Syntheses of Complexes 1NO3, 2NO3 and 3NO3
A typical synthetic procedure is described as follows: to a solution of [Cu(PPh3)2][NO3] (1.00 eq.) complex in dichloromethane (10 mL), a solution (10 mL) of the respective bisaryl-BIAN (1.00 eq.) in dichloromethane was added. The resulting mixture was left stirring for two hours. After completion of the reaction, the solution was filtered, concentrated under reduced pressure, and layered with pentane to precipitate the desired compound.
Data for [Cu((p-MeC6H4)2BIAN)(PPh3)2][NO3] (1NO3): following the general procedure, [Cu(PPh3)2(NO3)] (0.15 g, 0.24 mmol) and L1 (0.09 g, 0.24 mmol), complex 1NO3 was obtained as a red powder (0.22 g η = 92%) and subsequently recrystallized by slow diffusion of hexane in a chloroform solution. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.21 (d, J = 8.2 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.39–7,31 (m, 8H), 7.12 (t, J = 7.4 Hz, 12H), 7.00 (d, J = 7.8 Hz, 4H), 6.96–6.75 (br., 12H), 6.42 – 6.21 (br., 4H), 2.45 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ (ppm): 162.8, 144.6, 143.6, 137.9, 133.3, 131.9, 131.5 (overlap.: t, J = 17.5 Hz; s), 130.6, 130.4, 129.0, 128.7, 126.0, 125.0, 120.8, 21.3. 31P{1H} NMR (162 MHz, CDCl3) δ (ppm): 1.2. UV-Vis (CH2Cl2) λmáx. (nm) [ε (× 104 M−1.cm−1)]: 321 (1.34), 372sh (0.58), 460 (0.72). IR ATR ν máx (cm−1): 1641 (w, C=N), 1434 (m, PPh3), 1365, 1332 (m, NO3), 693 (s, C conj.). Elemental analysis calc. For C62H51CuN3O3P2·½CH2Cl2 (%): C 71.29, H 4.88, N 3.99; exp.: C 71.42, H 4.93, N 3.85. LC/MS (NCMe/H2O) m/z: positive mode: 783.2 [Cu(L1)2]+, 685.1 (100%) [Cu(L1)(PPh3)]+, 587.1 [Cu(PPh3)2]+, 361.2 [L1 + H]+, 263.1 [PPh3 + H]+. Negative mode: 186.8 [Cu(NO3)2]−, 62.1 (100%) [NO3]−.
Data for [Cu((p-iPrC6H4)2BIAN)(PPh3)2][NO3] (2NO3): following the general procedure, [Cu(PPh3)2(NO3)] (0.41 g, 0.63 mmol) and L2 (0.26 g, 0.63 mmol), complex 2NO3 was obtained as a red powder (0.22 g η = 92%). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.23 (d, J = 8.2 Hz, 2H), 7.59 (t, J = 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 6H), 7.29 (d, J = 7.2 Hz, 2H), 7.12 (t, J = 7.5 Hz, 12H), 7.05 (d, J = 7.6 Hz, 4H), 6.99–6.79 (br, 12H), 6.45–6.27 (br., 4H), 2.99 (sept, J = 7.0 Hz, 2H), 1.34 (d, J = 7.0 Hz, 12H). 13C{1H} NMR (126 MHz, CDCl3) δ (ppm): 162.9, 148.9, 144.8, 143.7, 133.3, 132.0, 131.5 (overlap: t, JCP = 17.2 Hz; s), 130.6, 129.0, 128.7, 127.8, 126.0, 125.0, 120.8, 34.0, 24.3. 31P{1H} NMR (162 MHz, CDCl3) δ (ppm): 1.3. UV-Vis (CH2Cl2) λmáx. (nm) [ε (×104 M−1·cm−1)]: 322 (1.41), 372sh (0.63), 460 (0.76). IR ATR ν máx (cm−1): 1635 (w, C=N), 1434 (m, PPh3), 1345, 1339 (m, NO3), 695 (s, C conj.). Elemental analysis calc. for C66H58CuN3O3P2 (%): C 74.32, H 5.48, N 3.94; exp.: C 74.53, H 5.12, N 3.89. LC/MS (NCMe/H2O) m/z: positive mode: 895.3 [Cu(L2)2]+, 741.2 (100%) [Cu(L2)(PPh3)]+, 587.1 [Cu(PPh3)2]+, 417.2 [L2 + H]+, 263.1 [PPh3 + H]+. Negative mode: 186.9 [Cu(NO3)2]−, 62.0 (100%) [NO3]−.
Data for [Cu((o-iPrC6H4)2BIAN)(PPh3)2][NO3] (3NO3): following the general procedure, [Cu(PPh3)2(NO3)] (0.15 g, 0.23 mmol) and L3 (0.10 g, 0.23 mmol), complex 3NO3 was obtained as a red powder (0.17 g η = 69%). 1H NMR (400 MHz, CDCl3, 0 °C) δ (ppm): 8.22 (d, J = 8.3 Hz, 2H), 7.54 (t, J = 7.8 Hz, 2H), 7.46 (t, J = 7.8 Hz, 2H), 7.41–7.29 (overlap: m, 6H), 7.33 (t, J = 7.6 Hz, 2H), 7.22 (overlap: br, 14H), 6.90–6.34 (overlap: br, 10H; 6.79 (d, J = 7.5 Hz, 2 H); 6.70 (t, J = 7.6 Hz, 2H)), 5.58 (d, J = 7.6 Hz, 2H), 3.32 (sept, J = 6.8 Hz, 2H), 0.87–0.82 (m, 7H), 0.42 (d, J = 6.7 Hz, 5H). 13C{1H} NMR (101 MHz, DMSO-d6) δ (ppm): 165.2, 145.9, 142.8, 140.0, 133.1, 131.8, 131.0, 130.6 (overlap), 129.0, 128.8, 128.1, 127.1, 126.6, 126.4, 124.7, 121.0, 28.9, 25.6, 25.2, 20.1. 31P{1H} NMR (162 MHz, CDCl3) δ (ppm): −1.7. UV-Vis (CH2Cl2) λmáx. (nm) [ε (×104 M−1·cm−1)]: 319 (1.31), 456 (0.36). IR ATR ν máx (cm−1): 1634 (w, C=N), 1434 (m, PPh3), 1357, 1326 (m, NO3), 693 (s, C conj.). Elemental analysis calc. for C66H58CuN3O3P2·⅖CH2Cl2 (%): C 72.46, H 5.38, N 3.82; exp.: C 72.52, H 5.52, N 3.69. LC/MS (NCMe/H2O) m/z: positive mode: 895.2 [Cu(L3)2]+, 741.2 (100%) [Cu(L3)(PPh3)]+, 587.0 [Cu(PPh3)2]+, 417.2 [L3 + H]+, 263.1 [PPh3 + H]+. Negative mode: 62.0 (100%) [NO3]−.
3.2.3. Synthesis of Azides
Benzylazide was prepared according to a modified literature procedure [35]: to a solution of required alkyl bromide (1.00 eq., 1.5 mL, 12.61 mmol) in acetone:water (30:15 mL (v/v)) sodium azide (1.50 eq., 1.23 g, 18.92 mmol) was added in portions. The resulting mixture was left stirring at room temperature for 24 h. After completion of the reaction, ethyl acetate was added (15 mL), and the product was extracted with more ethyl acetate (3 × 10 mL). The organic phase was washed with a brine solution (3 × 8 mL), dried over MgSO4, filtered, concentrated and dried under vacuum.
Data: benzylazide was obtained as a colorless oil (1.62 g η = 97%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.43–7.29 (m, 5H, H-1,2,3); 4,35 (s, 2H, H-5).
1-Azidonaphthalene was prepared according to an adapted literature procedure [34]: to a solution of required aniline (1.00 eq., 1.99 g, 13.62 mmol) in HCl (6 M, 30 mL) in an ice bath, a solution of NaNO2 (1.50 eq., 1.43 g, 20.73 mmol) in water (40 mL) was added dropwise. After 30 min, a solution of sodium azide (3.00 eq., 2.69 mg, 41.38 mmol) in water (50 mL) was added dropwise. The resulting solution was left stirring for two additional hours at room temperature. After completion of the reaction, ethyl acetate was added (15 mL), and the product was extracted with more ethyl acetate (4 × 15 mL). The organic phase was washed with a saturated solution of NaHCO3 (10 mL) and a brine solution (3 × 8 mL). It was dried over MgSO4, filtered, concentrated and dried under vacuum.
Data: 1-azidonaphthalene was obtained as a brown oil (2.15 g η = 93%). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.14–8.07 (m, 1H), 7.85–7.78 (m, 1H), 7.63 (d, J = 8.3 Hz, 1H), 7.55–7.42 (m, 3H), 7.25 (m, 1H).
3.3. X-ray Diffraction
Single crystals of ligand L3 and of complexes 1BF4, 1OTf, 1NO3, 2NO3 and 3NO3 were selected, covered with Fomblin (polyfluoro ether oil) and mounted on a nylon loop. Experimental details are presented in Table 3 and Table 4. The data was collected at 296 K (1OTf, 1NO3 and 3NO3) or at 110 K (L3, 1BF4 and 2NO3) on a Bruker D8 Venture diffractometer equipped with a Photon 100 CMOS detector, using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The data was processed using the APEX3 suite software package, which includes integration and scaling (SAINT), absorption corrections (SADABS) [36] and space group determination (XPREP). Structure solution and refinement were performed using direct methods with the programs SIR2019 [37] or SHELXT 2014/5 and SHELXL (version 2018/3) [38] inbuilt in APEX, and WinGX-Version 2020.1 [39] software packages. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were inserted in idealized positions and allowed to refine, riding on the parent carbon with fixed 1.2 Ueq of their parent carbon atom or 1.5 Ueq for the methyl group. The crystals of 1Otf were of poorer quality and showed low diffracting power, exhibiting dynamic motion or thermal disorder that was impossible to solve. Nevertheless, all atoms were correctly assigned and in agreement with the results obtained by other characterization techniques. The molecular diagrams were drawn with Mercury [40]. The data were deposited in the CCDC under deposit numbers 2,237,417 for L3, 2,237,418 for 1BF4, 2,237,419 for 1OTf, 2,237,420 for 1NO3, 2,237,421 for 2NO3, 2,237,421 for 3NO3.
3.4. General Procedures for the CuAAC Reaction
In a typical neat/solventless reaction, the appropriate Cu(I) complex (0.01–1 mol%) was added to a 15 mL vial, followed by the addition of the required azide (1.00 eq.) and alkyne (1.05 eq). The resulting mixture was stirred for 0.5–24 h, at 25 °C. An aliquot of the final crude mixture was characterized by 1H NMR spectroscopy in CDCl3. The conversion was calculated using 1H NMR spectroscopy, through the integration of specific resonances characteristic of the reagents and product. The crude mixture was washed with diethyl ether and petroleum ether and dried in vacuum to obtain the isolated product.
In a typical reaction employing a solvent, the appropriate Cu(I) complex (0.5 mol%) was dissolved in a selected solvent and added to a 15 mL vial. In a subsequent step, the required azide (1 eq.) and alkyne (1.05–1.10 eq.) were added, and the resulting mixture stirred for 2 h, at 25 °C. In order to isolate the reaction product, depending on the employed solvent, different procedures were used. For water and hexane, the solvent was decanted and the resulting solid washed with diethyl ether and dried in vacuum. For DMSO, water was added to the reaction mixture, followed by extraction with diethyl ether and washing with brine. The organic phase was dried over anhydrous MgSO4, followed by filtration and evaporation under vacuum. Finally, for acetone, THF and toluene, the product was precipitated with petroleum ether, and the resulting solid dried under vacuum. The final product was characterized using 1H NMR spectroscopy.
The triazole products a–i were identified by their 1H NMR spectra, which were consistent with the data reported in the literature [41,42,43,44,45]. In the case of the new triazole derivative f, further purification through column chromatography was required prior to NMR spectroscopy characterization.
Data for 4-butyl-1-(naphthalen-1-yl)-1H-1,2,3 (f) obtained as a brown oil. The crude mixture was subsequently purified by column chromatography (petroleum ether:ethyl acetate 9:1 (v/v)) and yielded a brown-orange solid (0.05 g η = 18%). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.02–7.95 (m, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.67–7.59 (overlap: m, 1H; 7.65, s, 1H), 7.59–7.48 (m, 4H), 2.87 (t, J = 7.7 Hz, 2H), 1.78 (qt, J = 7.6 Hz, 2H); 1.47 (sx, J = 7.4 Hz, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 134.3, 130.4, 128.8, 128.4, 128.0, 127.2, 125.1, 123.7, 122.6, 31.7, 25.5, 22.6, 14.0.
4. Conclusions
A series of cationic aryl-BIAN-copper(I) complexes containing ancillary PPh3 ligands were synthesized and studied as catalysts for the CuAAC reaction. The effectiveness of the application as catalysts for this catalytic reaction in different solvents, water included, was demonstrated. All complexes reached almost full conversion in neat conditions, using catalyst loadings ranging from 0.05 to 0.5 mol%, and reaction times varying between 30 min and 1 h. It is noteworthy mentioning that complex 2NO3 presented the best results, both in neat and in solution conditions, being inclusively able to catalyze the cycloaddition of phenylacetylene to benzyl azide utilizing a catalyst loading of only 500 ppm. This complex also behaved efficiently in a wide scope of substrates.
It was demonstrated that counter-ions play a major role in the reactivity of the complex towards the CuAAC reaction. Moreover, the lack of activity of 2BF4 on solvents, such as water and hexane, demonstrates that scientists should be careful regarding the choice of the counter-ion when designing new catalysts.
These systems showed, due to their practicability, great stability in several solvents and in storage, as well as an easy reaction work-up, low loadings of copper, which limits potential contamination of the final triazoles; they should provide an excellent methodology to be widely applicable in laboratories.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13020386/s1, Figure S1: UV-Vis spectra of complexes 1X–3X and L1–L3; Figure S2: Overlay of all FT ATR-IR spectra of complexes 1X–3X; Figure S3. FT ATR-IR spectrum of complex 1BF4; Figure S4. FT ATR-IR spectrum of complex 1OTf; Figure S5. FT ATR-IR spectrum of complex 1NO3; Figure S6. FT ATR-IR spectrum of complex 2OTf; Figure S7. FT ATR-IR spectrum of complex 2NO3; Figure S8. FT ATR-IR spectrum of complex 3OTf; Figure S9. FT ATR-IR spectrum of complex 3NO3; Figure S10. Mercury representations of the asymmetric unit of ligand L3. All hydrogen atoms were omitted for clarity; Figure S11. Mercury representations of the asymmetric unit of complex 2NO3. All hydrogen atoms and co-crystallized CHCl3 solvent molecules atoms were omitted for clarity; Figure S12. Mercury representations of the asymmetric unit of complex 3NO3. All hydrogen atoms and co-crystallized CHCl3 solvent molecules atoms were omitted for clarity; Figure S13. Mercury representation displaying the tail-to-tail π…π interactions observed between tow acenaphthene moieties; Figure S14. 1H NMR spectrum of complex 1BF4 in CDCl3; Figure S15. 13C NMR spectrum of complex 1BF4 in CDCl3; Figure S16. 19F NMR spectrum of complex 1BF4 in CDCl3; Figure S17. 31P NMR spectrum of complex 1BF4 in CDCl3; Figure S18. 1H NMR spectrum of complex 1OTf in CDCl3; Figure S19. 13C NMR spectrum of complex 1OTf in CDCl3; Figure S20. 19F NMR spectrum of complex 1OTf in CDCl3; Figure S21. 31P NMR spectrum of complex 1OTf in CDCl3; Figure S22. 1H NMR spectrum of complex 1NO3 in CDCl3; Figure S23. 13C NMR spectrum of complex 1NO3 in CDCl3; Figure S24. 31P NMR spectrum of complex 1NO3 in CDCl3; Figure S25. 1H NMR spectrum of complex 2OTf in CDCl3; Figure S26. 13C NMR spectrum of complex 2OTf in CDCl3; Figure S27. 19F NMR spectrum of complex 2OTf in CDCl3; Figure S28. 31P NMR spectrum of complex 2OTf in CDCl3; Figure S29. 1H NMR spectrum of complex 2NO3 in CDCl3; Figure S30. 13C NMR spectrum of complex 2NO3 in CDCl3; Figure S31. 31P NMR spectrum of complex 2NO3 in CDCl3; Figure S33. 13C NMR spectrum of complex 3OTf in CDCl3; Figure S34. 19F NMR spectrum of complex 3OTf in CDCl3; Figure S35. 31P NMR spectrum of complex 3OTf in CDCl3; Figure S36. 1H NMR spectrum of complex 3NO3 in CDCl3 at T = 0 °C; Figure S37. 13C NMR spectrum of complex 3NO3 in DMSO-d6; Figure S38. 31P NMR spectrum of complex 3NO3 in CDCl3; Figure S39. 1H NMR spectrum of benzylazide in CDCl3; Figure S40. 1H NMR spectrum of 1-azidonaphthalene in CDCl3; Figure S41. 1H NMR spectrum of triazole f in CDCl3; Figure S42. 13C NMR spectrum of triazole f in CDCl3. and Table S1. Initial screening and different condition testing of catalytic activities of complexes 1X–3X (X = BF4, OTf, NO3) in the CuAAC reaction of phenylacetylene and benzyl azide.
Author Contributions
M.S.V. developed the synthesis of all complexes and the CuAAC studies; X-ray diffraction studies, C.S.B.G.; Conceptualization, V.R. and C.S.B.G.; writing—original draft preparation, V.R., C.S.B.G. and M.S.V.; writing—review and editing, M.S.V., V.R. and C.S.B.G.; supervision, V.R. and C.S.B.G.; project administration, V.R.; funding acquisition, V.R. and C.S.B.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fundação para a Ciência e Tecnologia FCT/MCTES through projects UIDB/50006/2020, UIDP/50006/2020 and LA/P/0008/2020 of the Associate Laboratory for Green Chemistry—LAQV, UIDB/04378/2020, UIDP/04378/2020 and LA/P/0140/2020 of UCIBIO and Associate Laboratory i4HB, respectively.
Data Availability Statement
The data presented in this study are available from the authors.
Acknowledgments
The authors acknowledge LabRMN at FCT-UNL and Rede Nacional de RMN (RNRMN) for access to the facilities. The NMR spectrometers are part of The National NMR Facility, supported by FCT (ROTEIRO/0031/2013—PINFRA/22161/2016) (co-financed by FEDER through COMPETE 2020, POCI, and PORL and FCT through PIDDAC). Data Mass Spectrometry obtained by the Laboratório de Análises/LAQV REQUIMTE—Chemistry department FCT University NOVA of Lisbon. The SCXR determinations were performed in house (equipment financed by national funds through project RECI/BBBBEP/0124/2012 from FCT/MCTES).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Meldal, M.; Diness, F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew. Chemie Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- The Royal Swedish Academy of Sciences. The Nobel Prize in Chemistry 2022; The Royal Swedish Academy of Sciences: Stockholm, Sweden, 2022. [Google Scholar]
- Meldal, M.; Tomøe, C.W. Cu-catalyzed azide-Alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Wang, C.; Ikhlef, D.; Kahlal, S.; Saillard, J.Y.; Astruc, D. Metal-catalyzed azide-alkyne ‘click’ reactions: Mechanistic overview and recent trends. Coord. Chem. Rev. 2016, 316, 1–20. [Google Scholar] [CrossRef]
- Rodionov, V.O.; Presolski, S.I.; Díaz, D.D.; Fokin, V.V.; Finn, M.G. Ligand-accelerated Cu-catalyzed azide-alkyne cycloaddition: A mechanistic report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) ‘click’ reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(i) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Fokin, V.V.; Matyjaszewski, K. Organic Chemistry-Breakthroughs, Perspectives; Wiley: Hoboken, NJ, USA, 2012; pp. 247–277. [Google Scholar]
- Moorman, R.M.; Collier, M.B.; Frohock, B.H.; Womble, M.D.; Chalker, J.M. Halide inhibition of the copper-catalysed azide-alkyne cycloaddition. Org. Biomol. Chem. 2014, 13, 1974–1978. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. [(NHC)2Cu]X complexes as efficient catalysts for azide-alkyne click chemistry at low catalyst loadings. Angew. Chem. Int. Ed. 2008, 47, 8881–8884. [Google Scholar] [CrossRef]
- Barta, J.M.; Díez-González, S. Well-Defined Diimine Copper(I) Complexes as Catalysts in Click Azide-Alkyne Cycloaddition Reactions. Molecules 2013, 18, 8919–8928. [Google Scholar] [CrossRef]
- Lal, S.; Díez-González, S. [CuBr(PPh3)3] for azide-alkyne cycloaddition reactions under strict click conditions. J. Org. Chem. 2011, 76, 2367–2373. [Google Scholar] [CrossRef]
- Lazreg, F.; Slawin, A.M.Z.; Cazin, C.S.J. Heteroleptic bis(N-heterocyclic carbene)copper(I) complexes: Highly efficient systems for the [3+2] cycloaddition of azides and alkynes. Organometallics 2012, 31, 7969–7975. [Google Scholar] [CrossRef]
- Sau, S.C.; Roy, S.R.; Sen, T.K.; Mullangi, D.; Mandal, S.K. An abnormal N-heterocyclic carbene-copper(I) complex in click chemistry. Adv. Synth. Catal. 2013, 355, 2982–2991. [Google Scholar] [CrossRef]
- Wang, D.; Li, N.; Zhao, M.; Shi, W.; Ma, C.; Chen, B. Solvent-free synthesis of 1,4-disubstituted 1,2,3-triazoles using a low amount of Cu(PPh3)2NO3 complex. Green Chem. 2010, 12, 2120–2123. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Rosa, V.; Figueira, C.A.; Lemos, M.A.N.D.A.; Duarte, M.T.; Avilés, T.; Gomes, P.T. Synthesis and structural characterisation of (aryl-BIAN)copper(i) complexes and their application as catalysts for the cycloaddition of azides and alkynes. Dalton Trans. 2012, 41, 5144–5154. [Google Scholar] [CrossRef]
- Fliedel, C.; Rosa, V.; Santos, C.I.M.; Gonzalez, P.J.; Almeida, R.M.; Gomes, C.S.B.; Gomes, P.T.; Lemos, M.A.N.D.A.; Aullón, G.; Welter, R.; et al. Copper(ii) complexes of bis(aryl-imino)acenaphthene ligands: Synthesis, structure, DFT studies and evaluation in reverse ATRP of styrene. Dalton Trans. 2014, 43, 13041–13054. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Figueira, C.A.; Gomes, C.S.B.; Duarte, M.T.; Rosa, V.; Fliedel, C.; Avilés, T.; Gomes, P.T. Cationic and Neutral (Ar-BIAN)Copper(I) Complexes Containing Phosphane and Arsane Ancillary Ligands: Synthesis, Molecular Structure and Catalytic Behaviour in Cycloaddition Reactions of Azides and Alkynes. Eur. J. Inorg. Chem. 2013, 2013, 1404–1417. [Google Scholar] [CrossRef]
- van Asselt, R.; Elsevier, C.J.; Smeets, W.J.J.; Spek, A.L.; Benedix, R. Synthesis and characterization of rigid bidentate nitrogen ligands and some examples of coordination to divalent palladium. X-ray crystal structures of bis (p-tolylimino) acenaphthene and methylchloro [bis(o,o′-diisopropylphenyl-imino) acenaphthene] palla. Recl. Trav. Chim. Pays-Bas 1994, 113, 88–98. [Google Scholar] [CrossRef]
- Rosa, V.; Santos, C.I.M.; Welter, R.; Aullón, G.; Lodeiro, C.; Avilés, T. Comparison of the structure and stability of new α-diimine complexes of copper(I) and silver(I): Density functional theory versus experimental. Inorg. Chem. 2010, 49, 8699–8708. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- McCormick, T.; Jia, W.-L.; Wang, S. Phosphorescent Cu(I) complexes of 2-(2′-pyridylbenzimidazolyl) benzene: Impact of phosphine ancillary ligands on electronic and photophysical properties of the Cu(I) complexes. Inorg. Chem. 2005, 45, 147–155. [Google Scholar] [CrossRef]
- Saravanabharathi, D.; Monika; Venugopalan, P.; Samuelson, A.G. Solid state structural and solution studies on the formation of a flexible cavity for anions by copper(I) and 1,2-bis(diphenylphosphino)ethane. Polyhedron 2002, 21, 2433–2443. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, M.; Liu, X.; Chen, Y.; Li, N.; Chen, B. Quick and highly efficient copper-catalyzed cycloaddition of organic azides with terminal alkynes. Org. Biomol. Chem. 2011, 10, 229–231. [Google Scholar] [CrossRef]
- Ferretti, F.; Rota, L.; Ragaini, F. Unexpected coordination behavior of ruthenium to a polymeric α-diimine containing the poly[bis(arylimino)acenaphthene] fragment. Inorg. Chim. Acta 2021, 518, 120257. [Google Scholar] [CrossRef]
- Angelic, R.J. Inorganic Syntheses. In Reagents for Transition Metal Complex and Organometallic Syntheses; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; Volume 28, pp. 68–70. [Google Scholar] [CrossRef]
- Knight, D.A.; Keller, S.W. Solvent dependant ligand displacement and anion coordination in selected copper(I) coordination compounds. J. Chem. Crystallogr. 2006, 36, 531–542. [Google Scholar] [CrossRef]
- Gujadhur, R.; Venkataraman, D.; Kintigh, J.T. Formation of aryl-nitrogen bonds using a soluble copper(I) catalyst. Tetrahedron Lett. 2001, 42, 4791–4793. [Google Scholar] [CrossRef]
- Gysling, H.J.; Kubas, G.J. Coordination Complexes of Copper(I) Nitrate. Inorg. Synth. 1979, 19, 92–97. [Google Scholar] [CrossRef]
- Villarreal, W.; Colina-Vegas, L.; Visbal, G.; Corona, O.; Corrêa, R.S.; Ellena, J.; Cominetti, M.R.; Batista, A.A.; Navarro, M. Copper(I)−Phosphine Polypyridyl Complexes: Synthesis, Characterization, DNA/HSA Binding Study, and Antiproliferative Activity. Inorg. Chem. 2017, 56, 3781–3793. [Google Scholar] [CrossRef]
- Zhao, J.; Guo, J.; Huang, M.; You, Y.; Wu, Z. Design, synthesis and biological evaluation of new steroidal β -triazoly enones as potent antiproliferative agents. Steroids 2019, 150, 108431. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.; Elsinga, P.H.; Mirfeizi, L.; Dierckx, R.A.; Feringa, B.L. Copper-free ‘click’: 1,3-dipolar cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Beltrán, Á.; Gata, I.; Maya, C.; Avó, J.; Lima, J.C.; Laia, C.A.T.; Peloso, R.; Outis, M.; Nicasio, M.C. Dinuclear Cu(I) Halides with Terphenyl Phosphines: Synthesis, Photophysical Studies, and Catalytic Applications in CuAAC Reactions. Inorg. Chem. 2020, 59, 10894–10906. [Google Scholar] [CrossRef]
- Wang, S.; Jia, K.; Cheng, J.; Chen, Y.; Yuan, Y. Dual roles of substituted thiourea as reductant and ligand in CuAAC reaction. Tetrahedron Lett. 2017, 58, 3717–3721. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; Cruz, E.H.G.; Rodrigues, B.L.; Ramos, D.F.; Oliveira, R.N.; Silva, P.E.A. On the Search for Potential Antimycobacterial Drugs: Synthesis of Naphthoquinoidal, Phenazinic and 1,2,3-Triazolic Compounds and Evaluation Against Mycobacterium tuberculosis. J. Braz. Chem. Soc. 2015, 26, 1013–1027. [Google Scholar] [CrossRef]
- Gannarapu, M.R.; Vasamsetti, S.B.; Punna, N.; Kotamraju, S.; Banda, N. Synthesis of novel 1-substituted triazole linked 1,2-benzothiazine 1,1-dioxido propenone derivatives as potent anti-inflammatory agents and inhibitors of monocyte-to-macrophage differentiation. MedChemComm 2015, 6, 1494–1500. [Google Scholar] [CrossRef]
- Dutta, S.; Gupta, S.J.; Sen, A.K. Silver trifluoromethanesulfonate and metallic copper mediated syntheses of 1,2,3-triazole-O- and triazolyl glycoconjugates: Consecutive glycosylation and cyclization under one-pot condition. Tetrahedron Lett. 2016, 57, 3086–3090. [Google Scholar] [CrossRef]
Figure 1.
Examples of CuAAC’s copper(I) catalysts reported in the literature [2,3,12,13,14,15,16,17].

Scheme 1.
Syntheses of heteroleptic copper(I) complexes 1X–3X (X = BF4, OTf, NO3).

Figure 2.
Mercury representations of the asymmetric units of complexes (a) 1BF4, (b) 1OTf and (c) 1NO3. All hydrogen atoms and co-crystallized CH2Cl2 (in 1BF4) and CHCl3 (in 1OTf) solvent molecules were omitted for clarity.
Figure 2.
Mercury representations of the asymmetric units of complexes (a) 1BF4, (b) 1OTf and (c) 1NO3. All hydrogen atoms and co-crystallized CH2Cl2 (in 1BF4) and CHCl3 (in 1OTf) solvent molecules were omitted for clarity.

Scheme 2.
Reaction conditions of catalyst screening using model substrates, benzyl azide and phenylacetylene.
Scheme 2.
Reaction conditions of catalyst screening using model substrates, benzyl azide and phenylacetylene.

Scheme 3.
Reaction conditions of solvent screening using model substrates, benzyl azide and phenylacetylene, and catalysts 2X.
Scheme 3.
Reaction conditions of solvent screening using model substrates, benzyl azide and phenylacetylene, and catalysts 2X.

Figure 3.
Representation of isolated yields of 1-benzyl-4-phenyl-1,2,3-triazole in different solvents (1 mL) utilizing complexes 2X as catalysts. Isolated yields are the average of at least two independent runs.
Figure 3.
Representation of isolated yields of 1-benzyl-4-phenyl-1,2,3-triazole in different solvents (1 mL) utilizing complexes 2X as catalysts. Isolated yields are the average of at least two independent runs.

Scheme 4.
Reaction conditions of catalysis scope using the chosen catalyst 2NO3.

Scheme 5.
Screening of the scope of the catalytic system. Conditions: [2NO3] = 0.5 mol% (entry f: 1 mol% 2NO3) neat, 25 °C. Conversion and isolated yields, in parenthesis, are the average of at least two independent runs. * 1 mol% used in this assay. The purification of the different products was not optimized. (a–i) = 1,4-substituted 1,2,3-triazoles prepared under the screening of the scope of the catalytic system.
Scheme 5.
Screening of the scope of the catalytic system. Conditions: [2NO3] = 0.5 mol% (entry f: 1 mol% 2NO3) neat, 25 °C. Conversion and isolated yields, in parenthesis, are the average of at least two independent runs. * 1 mol% used in this assay. The purification of the different products was not optimized. (a–i) = 1,4-substituted 1,2,3-triazoles prepared under the screening of the scope of the catalytic system.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond distances (Å) and angles (°), and other relevant structural parameters for compounds L3, 1 BF4, 1OTf, 1NO3, 2NO3 and 3NO3.
Table 1.
Selected bond distances (Å) and angles (°), and other relevant structural parameters for compounds L3, 1 BF4, 1OTf, 1NO3, 2NO3 and 3NO3.
| L3 | 1 BF4 | 1OTf | 1NO3 | 2NO3 | 3NO3 | ||
|---|---|---|---|---|---|---|---|
| Molecule 1 | Molecule 2 | ||||||
| Cu1–N1 | 2.121(3) | 2.161(4) | 2.109(4) | 2.089(3) | 2.122(3) | 2.165(5) | |
| Cu1–N2 | 2.100(3) | 2.091(4) | 2.125(4) | 2.092(3) | 2.104(3) | 2.157(5) | |
| Cu1–P1 | 2.2571(10) | 2.2580(14) | 2.2522(14) | 2.2551(11) | 2.2606(11) | 2.2800(19) | |
| Cu1–P2 | 2.2656(10) | 2.2709(14) | 2.2573(14) | 2.2608(11) | 2.2763(10) | 2.2788(19) | |
| C1–N1 | 1.277(3) | 1.272(5) | 1.282(6) | 1.271(6) | 1.273(5) | 1.280(4) | 1.274(9) |
| C11–N2 | 1.275(3) | 1.281(5) | 1.286(6) | 1.266(6) | 1.288(5) | 1.278(4) | 1.263(8) |
| N1–Cu1–N2 | 78.84(11) | 78.63(15) | 78.76(15) | 79.44(12) | 78.90(11) | 78.79(18) | |
| P1–Cu1–P2 | 122.10(4) | 118.37(5) | 130.45(6) | 120.46(4) | 124.34(4) | 123.21(7) | |
| ω a | 104.73(8) | 103.36(11) | 97.96(10) | 97.64(7) | 80.97(9) | 105.99(14) | |
| φ1 b | 111.65(7) | 70.45(11) | 58.41(16) | 60.56(16) | 77.02(12) | 61.30(13) | 72.4(2) |
| φ2 b | 65.41(6) | 108.28(9) | 104.59(17) | 62.25(17) | 71.63(14) | 109.70(12) | 72.2(2) |
| χ c | 47.57(9) | 43.21(13) | 49.3(2) | 115.5(2) | 141.63(16) | 51.86(16) | 142.3(3) |
a Dihedral angle between C=N1–Cu–N2=C and P1–Cu–P2. b Dihedral angle between the acenaphthene and the aryl rings. c Dihedral angle between the aryl rings.
Table 2.
Catalytic activities of complexes 1X–3X (X = BF4, Otf, NO3) in the CuAAC reaction of phenylacetylene and benzyl azide.
Table 2.
Catalytic activities of complexes 1X–3X (X = BF4, Otf, NO3) in the CuAAC reaction of phenylacetylene and benzyl azide.
| Entry a | Cat. | Cat. Loading (mol%) | Time (h) | Conversion (%) b | η (%) c |
|---|---|---|---|---|---|
| 1 | 1BF4 | 0.5 | 1 | 98 | 89 |
| 2 | 1Otf | 0.5 | 1 | 99 | 94 |
| 3 | 1NO3 | 0.1 | 0.5 | 98 | 92 |
| 4 | 2BF4 | 0.5 | 1 | 99 | 91 |
| 5 | 2Otf | 0.3 | 0.5 | 99 | 93 |
| 6 | 2NO3 | 0.1 | 0.5 | 97 | 96 |
| 7 | 3BF4 | 0.5 | 1 | 98 | 89 |
| 8 | 3Otf | 0.5 | 1 | 100 | 92 |
| 9 | 3NO3 | 0.1 | 0.5 | 98 | 92 |
| 10 | 4BF4 | 0.5 | 0.5 | 2 | / |
| 11 | 4Otf | 0.5 | 0.5 | 2 | / |
| 12 | 5 | 0.5 | 0.5 | 8 | / |
| 13 | 6 | 0.5 | 0.5 | 97 | 94 |
| 14 | 7 | 0.5 | 0.5 | 100 | 87 |
| 15 | / | / | 0.5 | 0 | 0 |
a General reaction conditions: benzyl azide (1.00 eq.), phenylacetylene (1.05 eq.), T = 25 °C. b Determined by 1H NMR spectroscopy from starting material and product proportion. c Isolated yield. Conversion and isolated yields are the average of at least two independent runs.
Table 3.
Crystallographic data and details about refinement for structures L3, 1BF4 and 1OTf.
| L3 | 1BF4 | 1OTf | |
|---|---|---|---|
| Formula | C30H28N2 | C63H52BCl2Cu2F4N2P2 | C63H50CuF3N2O3P2S |
| M | 416.54 | 1120.25 | 1097.59 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| T (K) | 110(2) | 110(2) | 296(2) |
| Crystal system | Triclinic | Orthorhombic | Triclinic |
| Space group | P-1 | Pbca | P-1 |
| a (Å) | 8.9160(7) | 17.4954(15) | 15.2564(10) |
| b (Å) | 11.0941(10) | 23.7716(18) | 16.0253(11) |
| c (Å) | 12.5663(10) | 26.188(2) | 23.0757(15) |
| α (°) | 94.783(3) | 90 | 90.355(3) |
| β (°) | 103.724(3) | 90 | 93.137(3) |
| γ (°) | 107.064(3) | 90 | 98.235(3) |
| V (Å3) | 1138.58(17) | 10,891.2(15) | 5574.6(6) |
| Z | 2 | 8 | 4 |
| ρcalc (g·cm−3) | 1.215 | 1.366 | 1.308 |
| µ (mm−1) | 0.071 | 0.614 | 0.544 |
| Crystal size | 0.40 × 0.20 × 0.20 | 0.40 × 0.20 × 0.20 | 0.30 × 0.20 × 0.20 |
| Crystal color | Yellow | Red | Red |
| Crystal description | Prism | Block | Prism |
| θmax (°) | 25.680 | 25.740 | 25.350 |
| Total data | 25,850 | 112,589 | 189,596 |
| Unique data | 4323 | 10,365 | 20,386 |
| Rint | 0.1400 | 0.1721 | 0.1505 |
| R [I > 2σ(I)] | 0.0609 | 0.0581 | 0.0788 |
| Rw | 0.1266 | 0.1464 | 0.2088 |
| Goodness of fit | 1.002 | 1.048 | 1.047 |
| ρmin ρmax | −0.371 0.230 | −1.240 0.888 | −0.698 0.954 |
Table 4.
Crystallographic data and details about refinement for structures 1NO3, 2NO3 and 3NO3.
| 1NO3 | 2NO3 | 3NO3 | |
|---|---|---|---|
| Formula | C63H51Cl3CuN3O3P2 | C67H59Cl3CuN3O3P2 | C69H61Cl9CuN3O3P2 |
| M | 1129.89 | 1186.00 | 1097.59 |
| λ (Å) | 0.71073 | 0.71073 | 1424.73 |
| T (K) | 296(2) | 110(2) | 296(2) |
| Crystal system | Monoclinic | Orthorhombic | Tetragonal |
| Space group | P21/c | Pbca | P41 |
| a (Å) | 16.7053(12) | 17.979(3) | 13.2253(4) |
| b (Å) | 14.7608(11) | 24.990(4) | 13.2253(4) |
| c (Å) | 22.8229(17) | 27.167(4) | 40.3250(19) |
| α (°) | 90 | 90 | 90 |
| β (°) | 95.361(2) | 90 | 90 |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 5603.1(7) | 12,206(3) | 7053.2(5) |
| Z | 4 | 8 | 4 |
| ρcalc (g·cm−3) | 1.339 | 1.291 | 1.342 |
| µ (mm−1) | 0.639 | 0.590 | 0.742 |
| Crystal size | 0.40 × 0.26 × 0.14 | 0.30 × 0.24 × 0.20 | 0.26 × 0.20 × 0.16 |
| Crystal color | Red | Red | Red |
| Crystal description | Prism | Prism | Prism |
| θmax (°) | 26.815 | 25.772 | 25.749 |
| Total data | 104,765 | 216,199 | 21,487 |
| Unique data | 11,970 | 11,645 | 10,328 |
| Rint | 0.1603 | 0.1057 | 0.0503 |
| R [I > 2σ(I)] | 0.0728 | 0.0638 | 0.0611 |
| Rw | 0.1987 | 0.1818 | 0.1616 |
| Goodness of fit | 1.093 | 1.056 | 1.047 |
| ρmin ρmax | −0.732 1.527 | −0.615 0.777 | −0.652 0.579 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Viana, M.S.; Gomes, C.S.B.; Rosa, V. Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency. Catalysts 2023, 13, 386. https://doi.org/10.3390/catal13020386
AMA Style
Viana MS, Gomes CSB, Rosa V. Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency. Catalysts. 2023; 13(2):386. https://doi.org/10.3390/catal13020386
Chicago/Turabian StyleViana, Maria S., Clara S. B. Gomes, and Vitor Rosa. 2023. "Heteroleptic Copper Complexes as Catalysts for the CuAAC Reaction: Counter-Ion Influence in Catalyst Efficiency" Catalysts 13, no. 2: 386. https://doi.org/10.3390/catal13020386
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.