
Redrawing HER Volcano with Interfacial Processes—The Role of Hydrogen Spillover in Boosting H2 Evolution in Alkaline Media
 , , , , and
, , , , and 
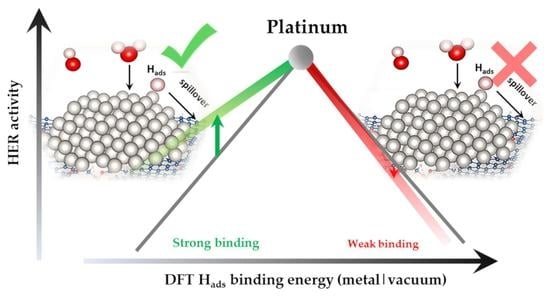
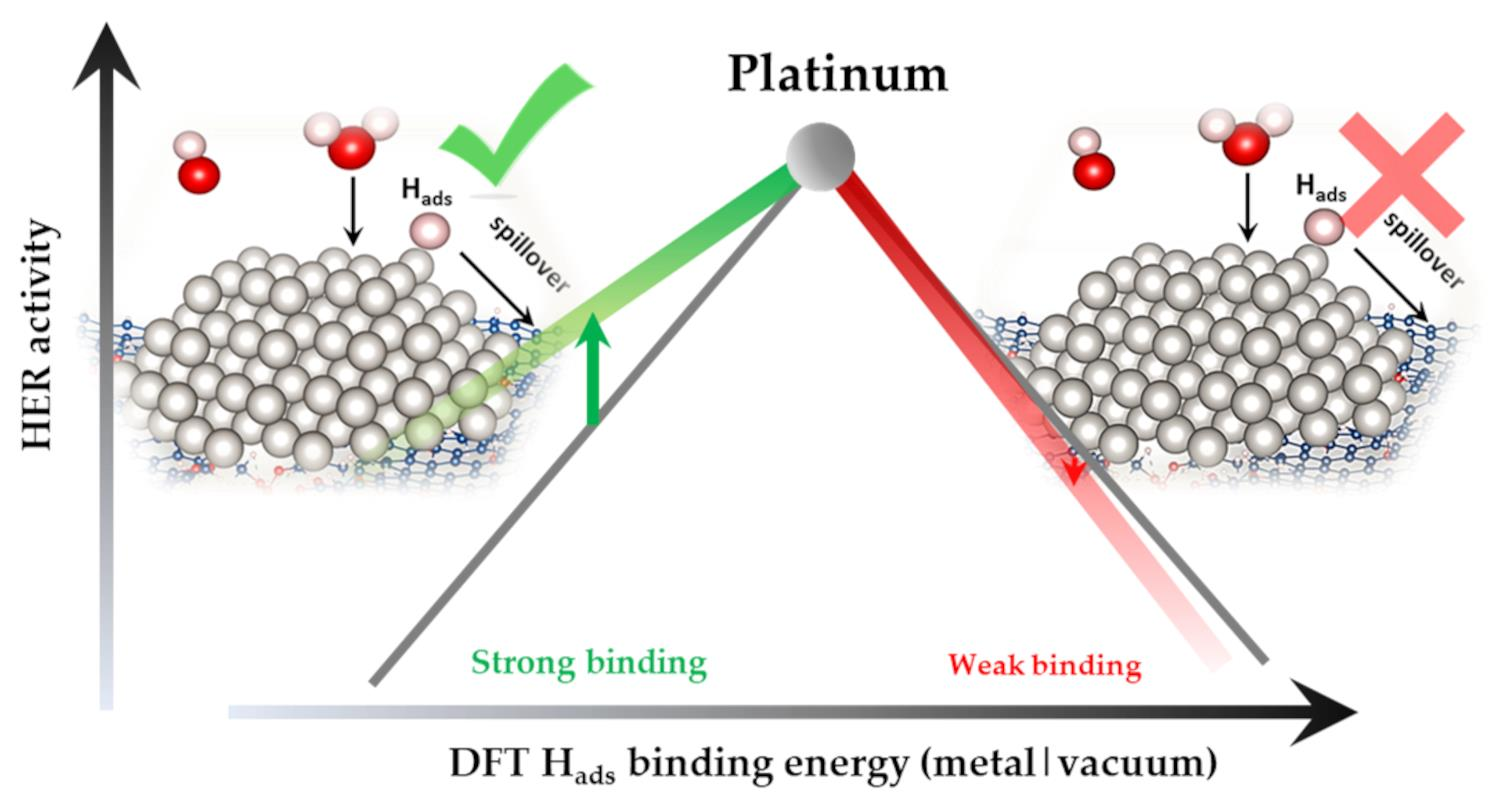
Abstract
:
1. Introduction
2. Results and Discussion
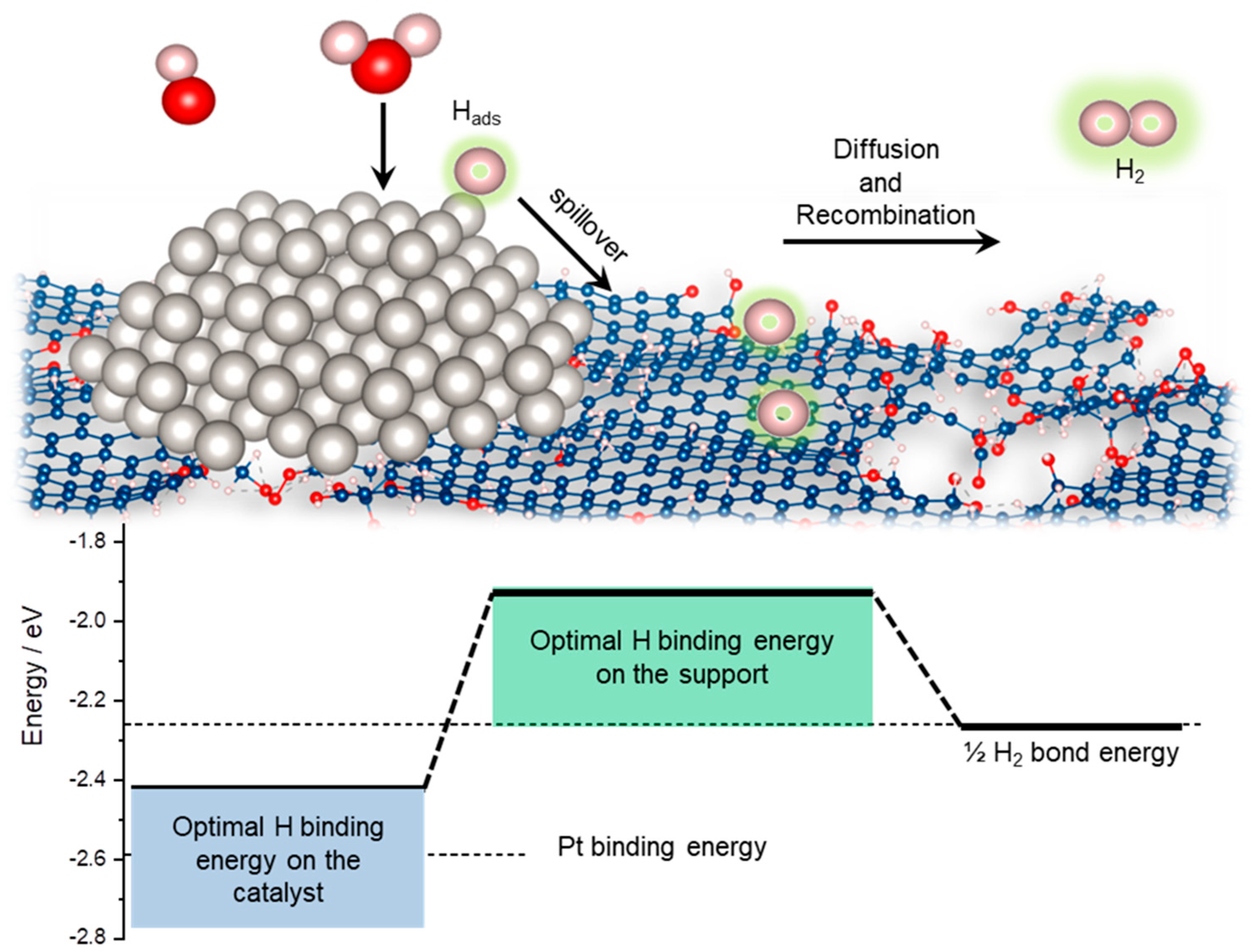
2.1. KMC Simulations—Model
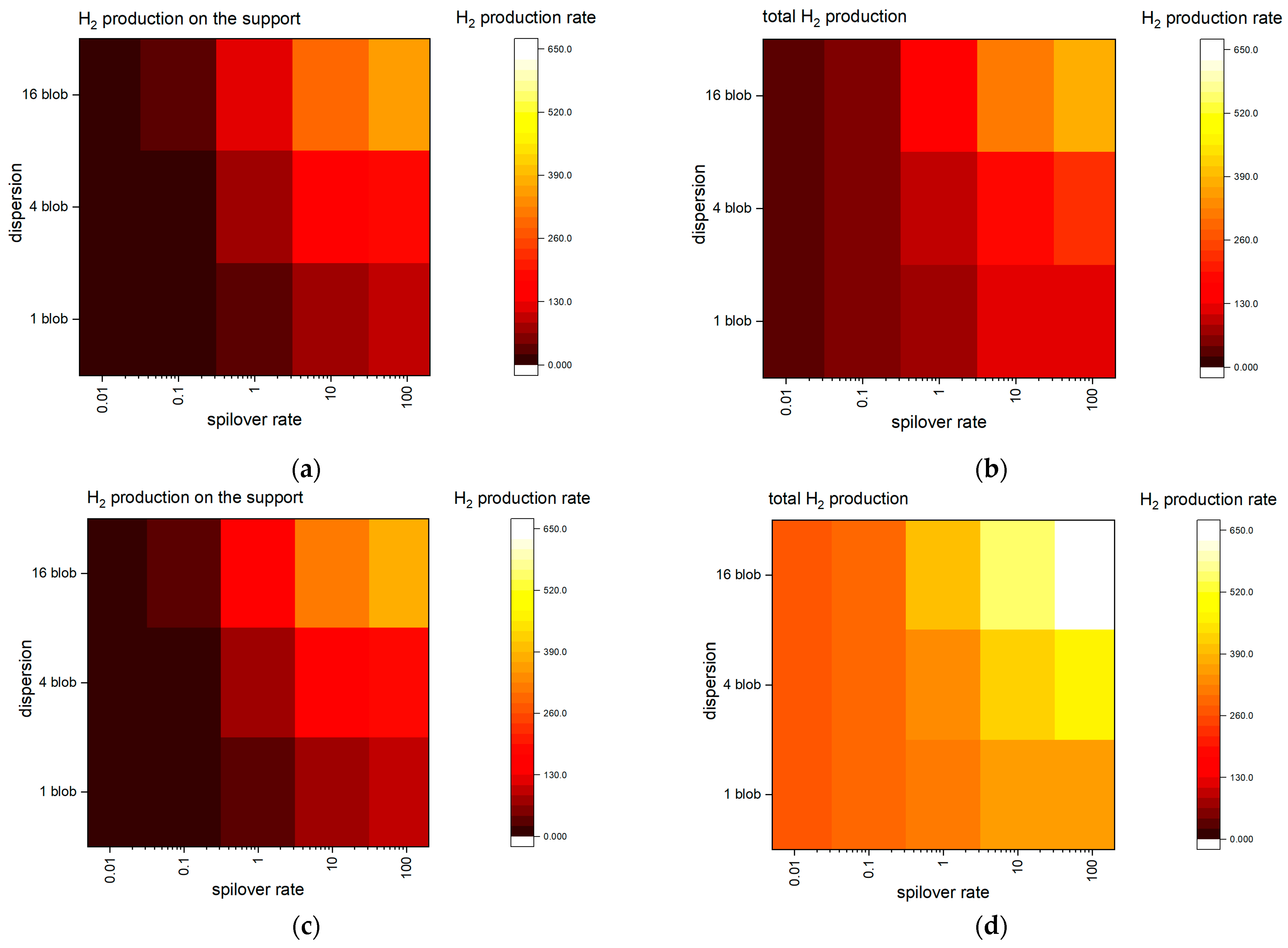
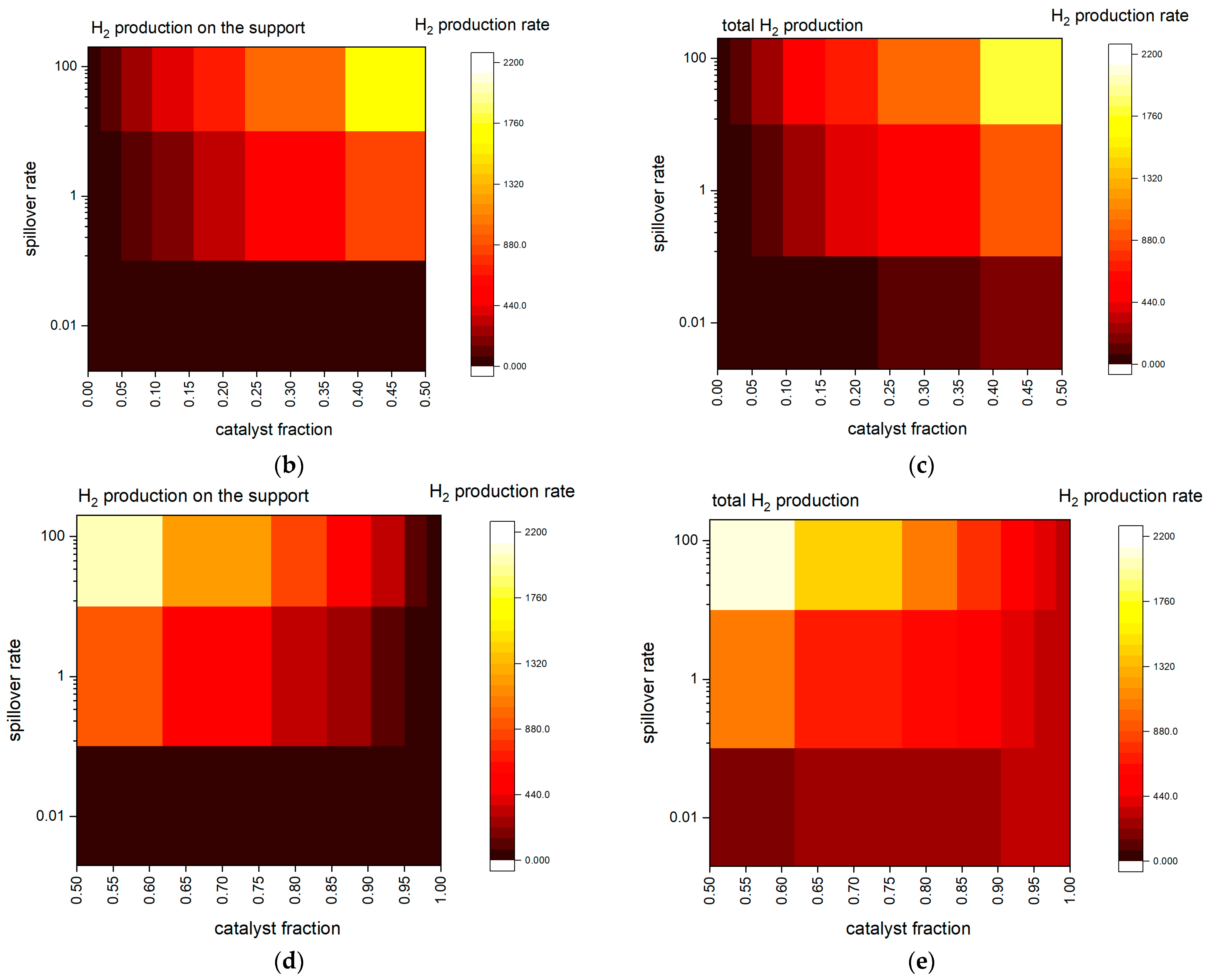
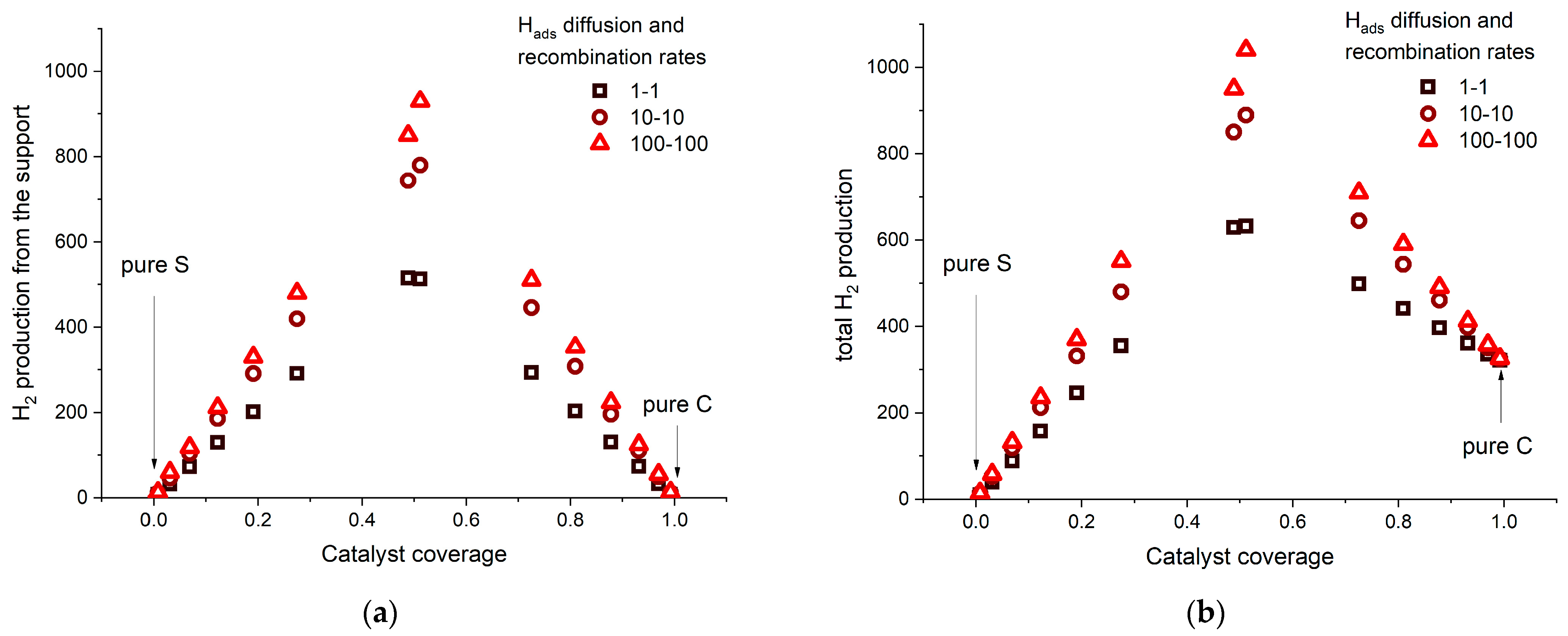
2.2. KMC Simulations—Insights
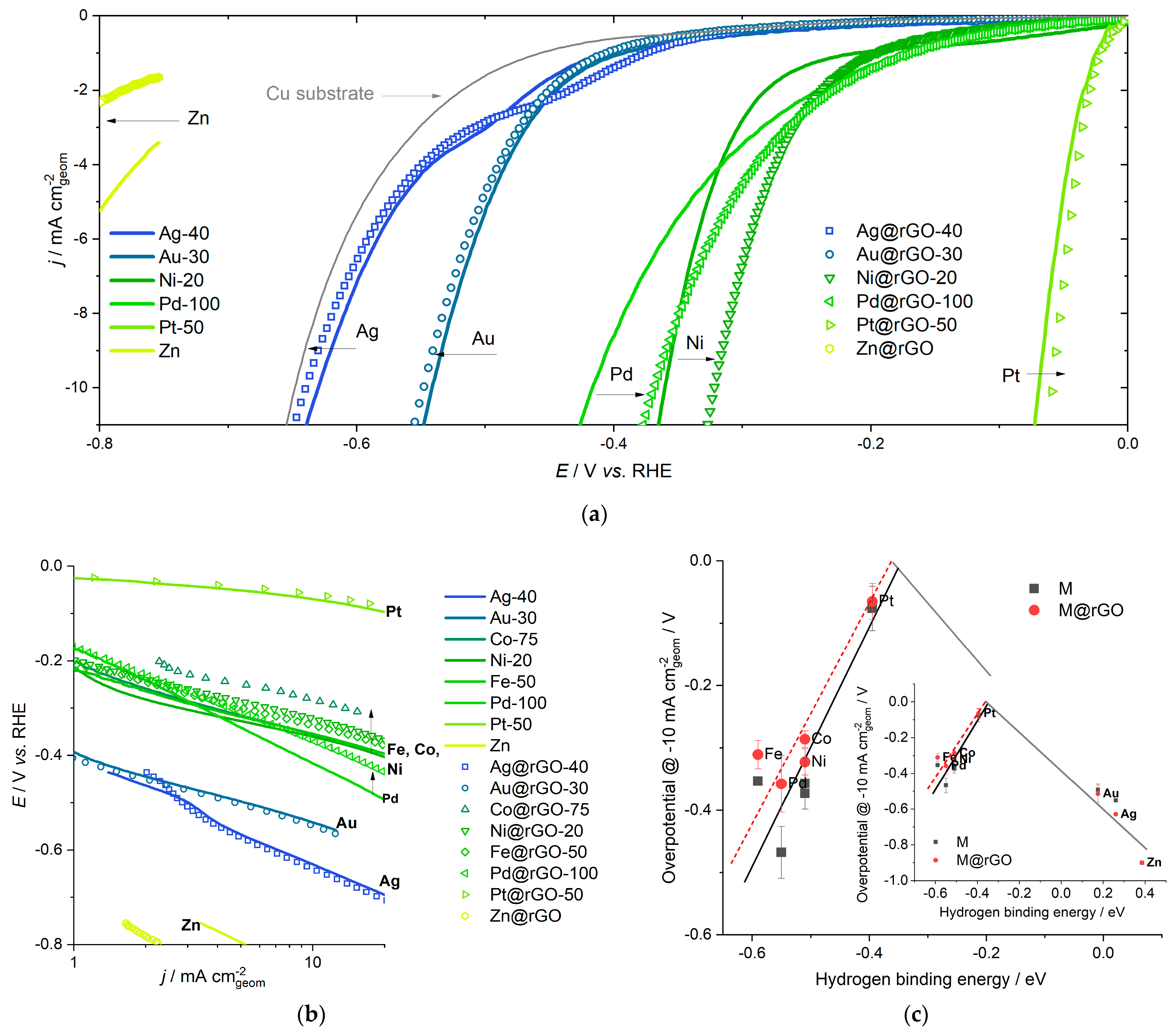
2.3. Choosing the Catalyst—Support Combination
2.4. Trends in Hydrogen Evolution Reaction
3. Discussion
4. Materials and Methods
4.1. KMC Simulations
4.2. Metal Deposition on Drop-Casted-GO Films
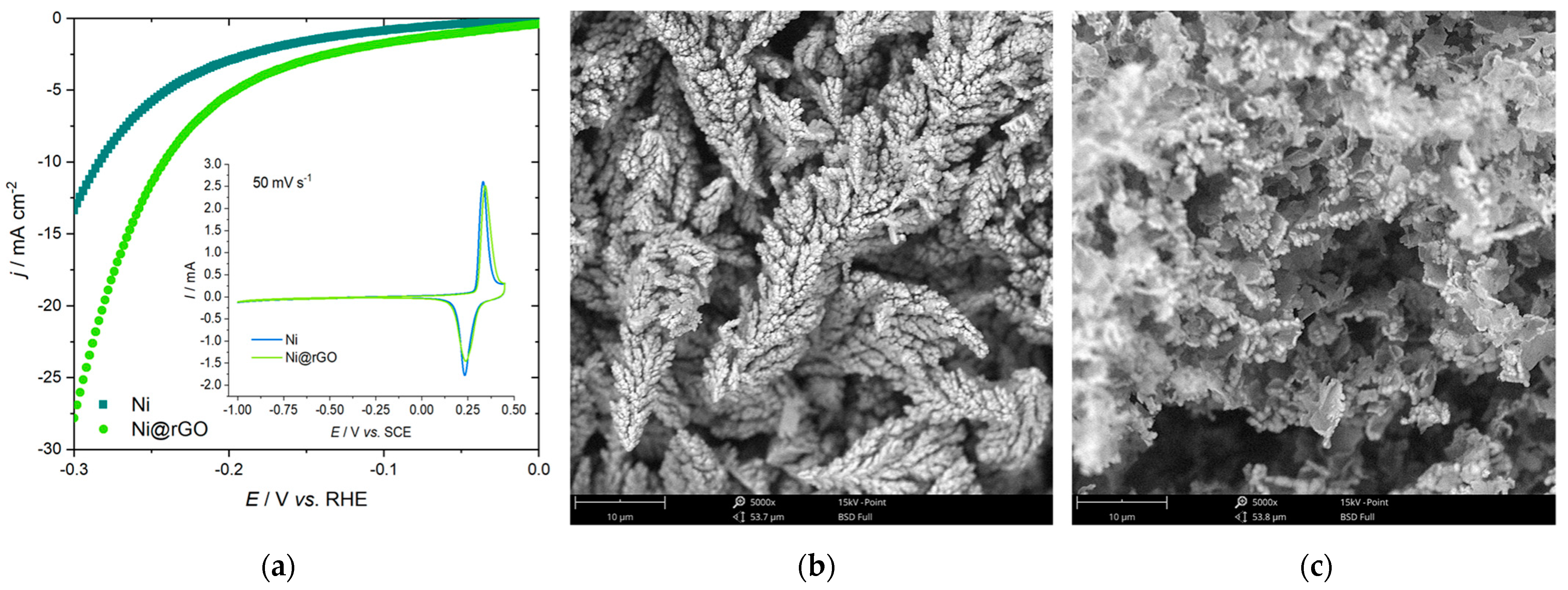
4.3. Co-Deposition of Ni and rGO and the Measurements of HER Activity
4.4. DFT Calculations
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strmcnik, D.; Papa Lopes, P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Markovic, N.M. Interfacing electrochemistry. Nat. Mater. 2013, 12, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Markovic, N.M. The road from animal electricity to green energy: Combining experiment and theory in electrocatalysis. Energy Environ. Sci. 2012, 5, 9246–9256. [Google Scholar] [CrossRef]
- De Giz, M.J.; Tremiliosi-Filho, G.; Gonzalez, E.R.; Srinivasan, S.; Appleby, A.J. The hydrogen evolution reaction on amorphous nickel and cobalt alloys. Int. J. Hydrog. Energy 1995, 20, 423–427. [Google Scholar] [CrossRef]
- Kibsgaard, J.; Tsai, C.; Chan, K.; Benck, J.D.; Nørskov, J.K.; Abild-Pedersen, F.; Jaramillo, T.F. Designing an improved transition metal phosphide catalyst for hydrogen evolution using experimental and theoretical trends. Energy Environ. Sci. 2015, 8, 3022–3029. [Google Scholar] [CrossRef]
- Benck, J.D.; Hellstern, T.R.; Kibsgaard, J.; Chakthranont, P.; Jaramillo, T.F. Catalyzing the Hydrogen Evolution Reaction (HER) with Molybdenum Sulfide Nanomaterials. ACS Catal. 2014, 4, 3957–3971. [Google Scholar] [CrossRef]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous Molybdenum Sulfide Catalysts for Electrochemical Hydrogen Production: Insights into the Origin of their Catalytic Activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Du, H.; Kong, R.-M.; Guo, X.; Qu, F.; Li, J. Recent progress in transition metal phosphides with enhanced electrocatalysis for hydrogen evolution. Nanoscale 2018, 10, 21617–21624. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in hydrogen electrocatalysis-uses and abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, W.; Myint, M.; Chen, J.G.; Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 2013, 6, 1509–1512. [Google Scholar] [CrossRef]
- Sabatier, F. La Catalyse en Chimie Organique; Berauge: Paris, France, 1920. [Google Scholar]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Chang, K.-C.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Enhancing the Alkaline Hydrogen Evolution Reaction Activity through the Bifunctionality of Ni(OH)2/Metal Catalysts. Angew. Chem. Int. Ed. 2012, 51, 12495–12498. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Chang, K.-C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Khoobiar, S. Particle to Particle Migration of Hydrogen Atoms on Platinum—Alumina Catalysts from Particle to Neighboring Particles. J. Phys. Chem. 1964, 68, 411–412. [Google Scholar] [CrossRef]
- Conner, W.C., Jr.; Falconer, J.L. Spillover in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; Van de Vondele, J.; Ekinci, Y.; van Bokhoven, J.A. Catalyst support effects on hydrogen spillover. Nature 2017, 541, 68–71. [Google Scholar] [CrossRef]
- Prins, R. Hydrogen Spillover. Facts and Fiction. Chem. Rev. 2012, 112, 2714–2738. [Google Scholar] [CrossRef]
- Psofogiannakis, G.M.; Froudakis, G.E. Fundamental studies and perceptions on the spillover mechanism for hydrogen storage. Chem. Commun. 2011, 47, 7933–7943. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; van der Vliet, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef]
- Tan, Y.; Zhu, Y.; Cao, X.; Liu, Y.; Li, J.; Chen, Z.; Hu, J. Discovery of Hydrogen Spillover-Based Binary Electrocatalysts for Hydrogen Evolution: From Theory to Experiment. ACS Catal. 2022, 12, 11821–11829. [Google Scholar] [CrossRef]
- Chanda, D.; Hnát, J.; Dobrota, A.S.; Pašti, I.A.; Paidar, M.; Bouzek, K. The effect of surface modification by reduced graphene oxide on the electrocatalytic activity of nickel towards the hydrogen evolution reaction. Phys. Chem. Chem. Phys. 2015, 17, 26864–26874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutić, S.J.; Jovanović, A.Z.; Dobrota, A.S.; Metarapi, D.; Rafailović, L.D.; Pašti, I.A.; Mentus, S.V. Simple routes for the improvement of hydrogen evolution activity of Ni-Mo catalysts: From sol-gel derived powder catalysts to graphene supported co-electrodeposits. Int. J. Hydrog. Energy 2018, 43, 16846–16858. [Google Scholar] [CrossRef]
- Wolfschmidt, H.; Paschos, O.; Stimming, U. Hydrogen Reactions on Nanostructured Surfaces. In Fuel Cell Science: Theory, Fundamentals, and Biocatalysis; Wieckowski, A., Nørskov, J.K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Pašti, I.A.; Leetmaa, M.; Skorodumova, N.V. General principles for designing supported catalysts for hydrogen evolution reaction based on conceptual Kinetic Monte Carlo modeling. Int. J. Hydrog. Energy 2016, 41, 2526–2538. [Google Scholar] [CrossRef]
- Tierney, H.L.; Baber, A.E.; Kitchin, J.R.; Sykes, E.C. H Hydrogen Dissociation and Spillover on Individual Isolated Palladium Atoms. Phys. Rev. Lett. 2009, 103, 246102. [Google Scholar] [CrossRef]
- Nilekar, A.U.; Greeley, J.; Mavrikakis, M. A Simple Rule of Thumb for Diffusion on Transition-Metal Surfaces. Angew. Chem. Int. Ed. 2006, 45, 7046–7049. [Google Scholar] [CrossRef]
- Jakšić, J.M.; Vojnović, M.V.; Krstajić, N.V. Kinetic analysis of hydrogen evolution at Ni–Mo alloy electrodes. Electrochim. Acta 2000, 45, 25–26. [Google Scholar] [CrossRef]
- Krstajić, N.; Popović, M.; Grgur, B.; Vojnović, M.; Šepa, D. On the kinetics of the hydrogen evolution reaction on nickel in alkaline solution: Part I. The mechanism. J. Electroanal. Chem. 2001, 512, 16–26. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. Large-scale, density functional theory-based screening of alloys for hydrogen evolution. Surf. Sci. 2007, 601, 1590–1598. [Google Scholar] [CrossRef]
- Singh, A.K.; Ribas, M.A.; Yakobson, B.I. H-Spillover through the Catalyst Saturation: An Ab Initio Thermodynamics Study. ACS Nano 2009, 3, 1657–1662. [Google Scholar] [CrossRef]
- Gutić, S.J.; Dobrota, A.S.; Leetmaa, M.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. Improved catalysts for hydrogen evolution reaction in alkaline solutions through the electrochemical formation of nickel-reduced graphene oxide interface. Phys. Chem. Chem. Phys. 2017, 19, 13281–13293. [Google Scholar] [CrossRef] [PubMed]
- Gutić, S.J.; Šabanović, M.; Metarapi, D.; Pašti, I.A.; Korać, F.; Mentus, S.V. Electrochemically Synthesized Ni@reduced Graphene Oxide Composite Catalysts for Hydrogen Evolution in Alkaline Media—The Effects of Graphene Oxide Support. Int. J. Electrochem. Sci. 2019, 14, 8532–8543. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konda, S.K.; Chen, A. Palladium based nanomaterials for enhanced hydrogen spillover and storage. Mater. Today 2016, 19, 100–108. [Google Scholar] [CrossRef]
- Parambhath, V.B.; Nagar, R.; Sethupathi, K.; Ramaprabhu, S. Investigation of Spillover Mechanism in Palladium Decorated Hydrogen Exfoliated Functionalized Graphene. J. Phys. Chem. C 2011, 115, 15679–15685. [Google Scholar] [CrossRef]
- Tsao, C.-S.; Liu, Y.; Chuang, H.-Y.; Tseng, H.-H.; Chen, T.-Y.; Chen, C.-H.; Yu, M.-S.; Li, Q.; Lueking, A.; Chen, S.-H. Hydrogen Spillover Effect of Pt-Doped Activated Carbon Studied by Inelastic Neutron Scattering. J. Phys. Chem. Lett. 2011, 2, 2322–2325. [Google Scholar] [CrossRef]
- Blanco-Rey, M.; Juaristi, J.I.; Alducin, M.; López, M.J.; Alonso, J.A. Is Spillover Relevant for Hydrogen Adsorption and Storage in Porous Carbons Doped with Palladium Nanoparticles? J. Phys. Chem. C 2016, 120, 17357–17364. [Google Scholar] [CrossRef] [Green Version]
- Granja-DelRío, A.; Alducin, M.; Juaristi, J.I.; López, M.J.; Alonso, J.A. Absence of spillover of hydrogen adsorbed on small palladium clusters anchored to graphene vacancies. Appl. Surf. Sci. 2021, 559, 149835. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Skorodumova, N.V. A general view on the reactivity of the oxygen-functionalized graphene basal plane. Phys. Chem. Chem. Phys. 2016, 18, 6580–6586. [Google Scholar] [CrossRef]
- Leetmaa, M.; Skorodumova, N.V. KMCLib 1.1: Extended random number support and technical updates to the KMCLib general framework for kinetic Monte-Carlo simulations. Comput. Phys. Commun. 2015, 196, 611–613. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzibum, C.; Ceresoli, D.; Chiarotti, G.L.; Coccoccioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Electrodeposition Bath | Deposition Conditions | Deposition Time/s |
|---|---|---|---|
| Ag | 5 × 10−3 M AgI + 2 M KI in water (AgI is dissolved in excess KI), pH ≈ 7 | −0.5 A dm−2 30 °C | 40, 250 |
| Au | 0.05 M HAuCl4 + 0.42 M Na2SO3 + 0.42 M Na2S2O3 + 0.3 M Na2HPO4, pH ≈ 6 | −0.5 A dm−2 60 °C | 30, 60, 100, 300 |
| Co | 59.4 g dm−3 CoSO4 × 7H2O + 12 g dm−3 Na2SO4 + 12 g dm−3 H3BO3 | −1.2 V vs. Ag/AgCl/KCl, room temperature | 50, 75, 100, 300 |
| Fe | 300 g dm−3 FeSO4 × 7H2O + 6 g dm−3 FeCl2 × H2O + 2.8 g dm−3 H3BO3; pH adjusted to 2.5 with H2SO4; | −5 A dm−2, room temperature | 50, 100, 300 |
| Ni | 76 g dm−3 NiSO4 × 6H2O + 12 g dm−3 H3BO3 | −1.2 V vs. Ag/AgCl/KCl, room temperature | 10, 30, 60, 100, 150, 200 |
| Pd | 100 g dm−3 PdCl2 × 4H2O + NH3 in excess + NH4Cl to pH ≈ 9 | −0.3 A dm−2 room temperature | 60, 100, 200, 300 |
| Pt | 20 g dm−3 H2 [Pt(OH)6] + 15 g dm−3 KOH | −0.75 A dm−2 75 °C | 10, 50, 100 |
| Zn | 300 g dm−3 ZnSO4 + 100 g dm−3 Na2SO4 + 8 g dm−3 H2SO4 | −15 A dm−2 room temperature | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutić, S.J.; Metarapi, D.; Jovanović, A.Z.; Gebremariam, G.K.; Dobrota, A.S.; Nedić Vasiljević, B.; Pašti, I.A. Redrawing HER Volcano with Interfacial Processes—The Role of Hydrogen Spillover in Boosting H2 Evolution in Alkaline Media. Catalysts 2023, 13, 89. https://doi.org/10.3390/catal13010089
Gutić SJ, Metarapi D, Jovanović AZ, Gebremariam GK, Dobrota AS, Nedić Vasiljević B, Pašti IA. Redrawing HER Volcano with Interfacial Processes—The Role of Hydrogen Spillover in Boosting H2 Evolution in Alkaline Media. Catalysts. 2023; 13(1):89. https://doi.org/10.3390/catal13010089
Chicago/Turabian StyleGutić, Sanjin J., Dino Metarapi, Aleksandar Z. Jovanović, Goitom K. Gebremariam, Ana S. Dobrota, Bojana Nedić Vasiljević, and Igor A. Pašti. 2023. "Redrawing HER Volcano with Interfacial Processes—The Role of Hydrogen Spillover in Boosting H2 Evolution in Alkaline Media" Catalysts 13, no. 1: 89. https://doi.org/10.3390/catal13010089