
Selectivity in the Aliphatic C–H Bonds Oxidation (Hydroxylation) Catalyzed by Heme- and Non-Heme Metal Complexes—Recent Advances


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
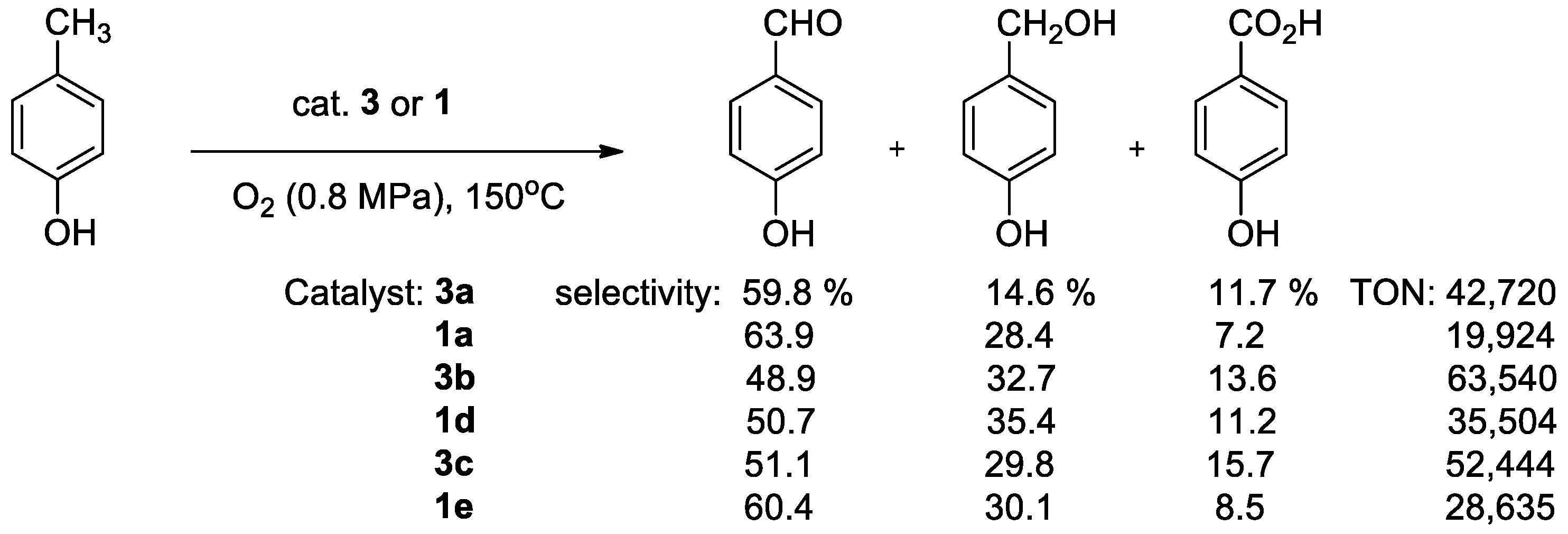
{kind=link}
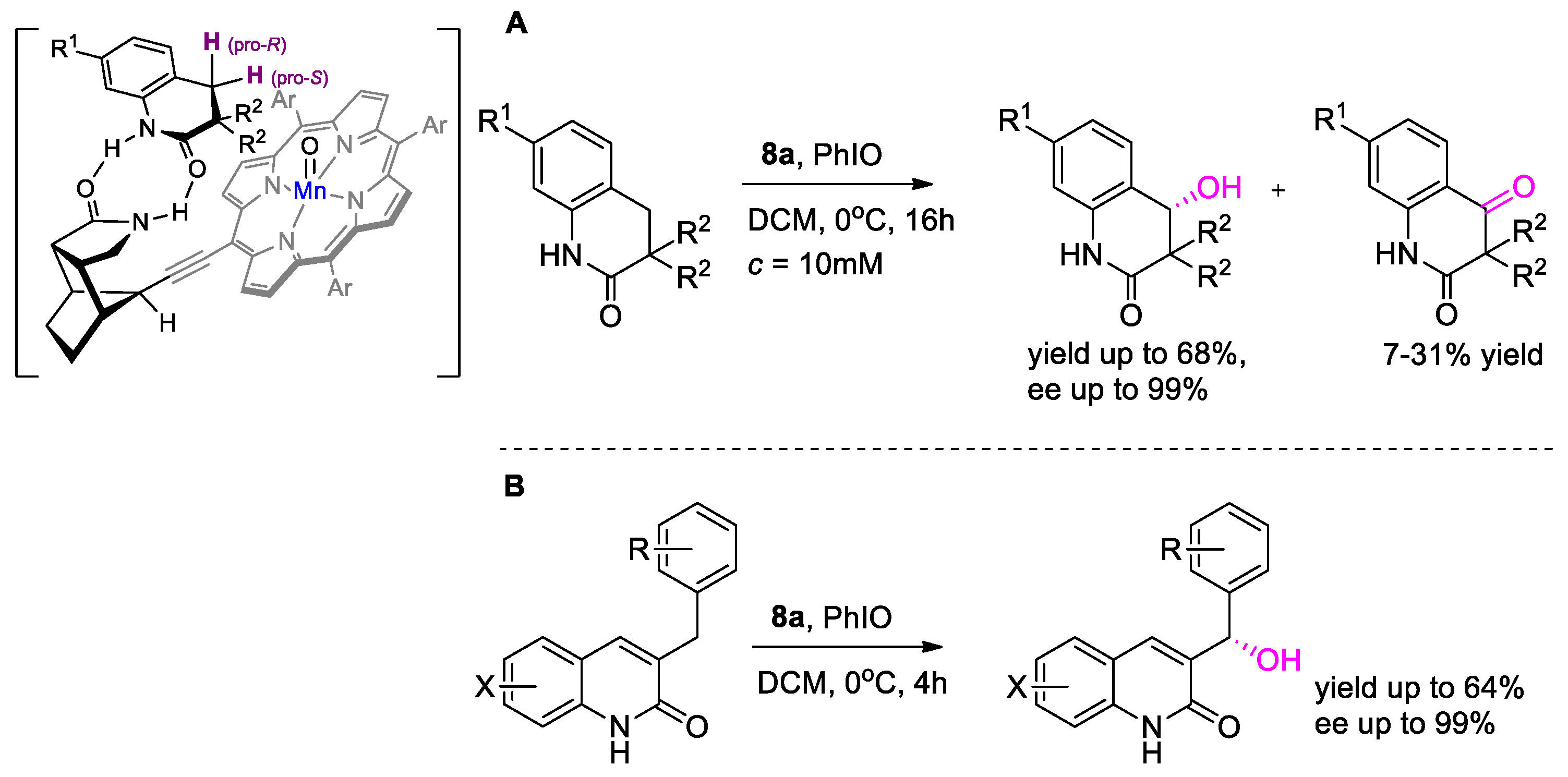
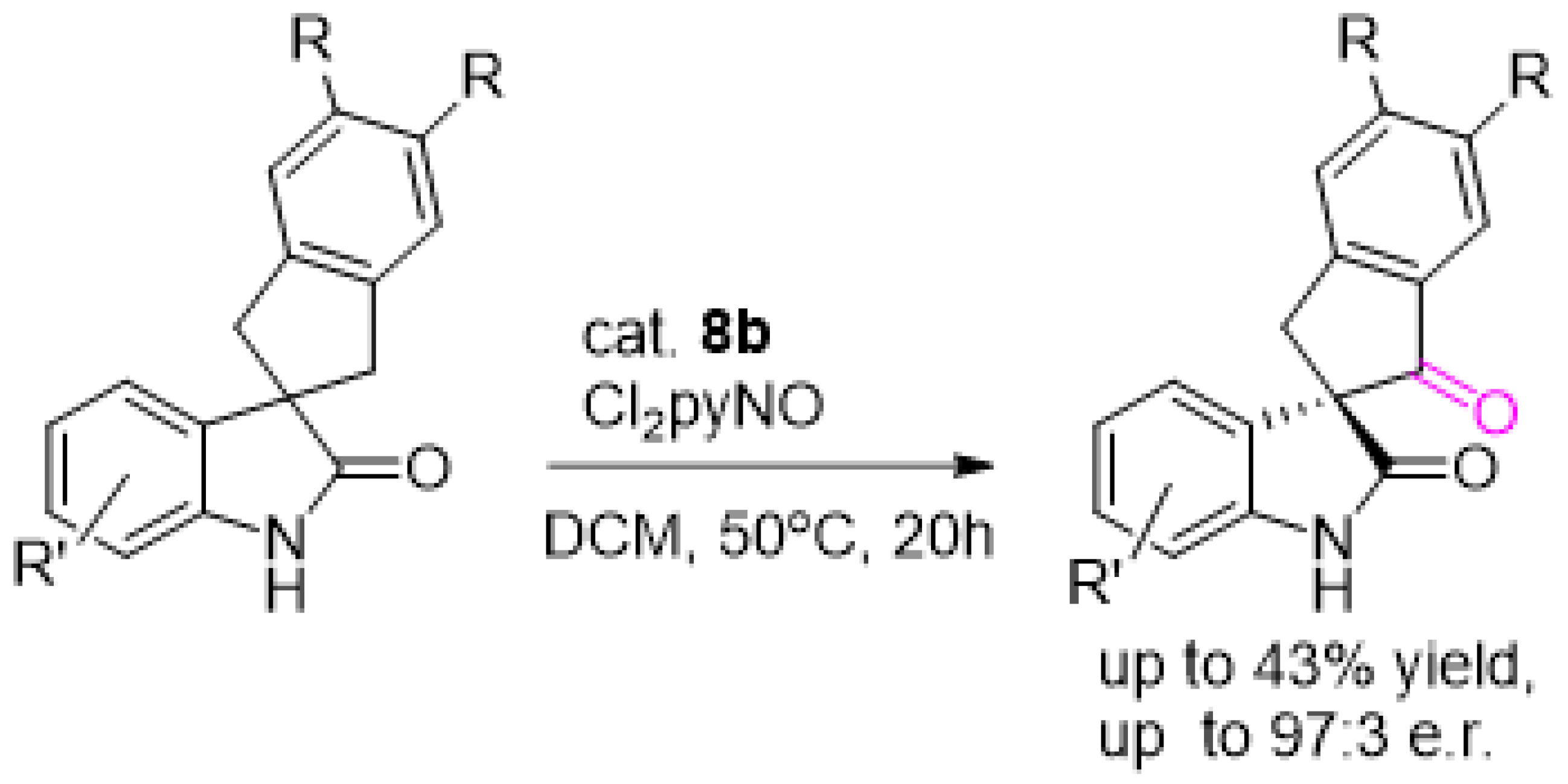
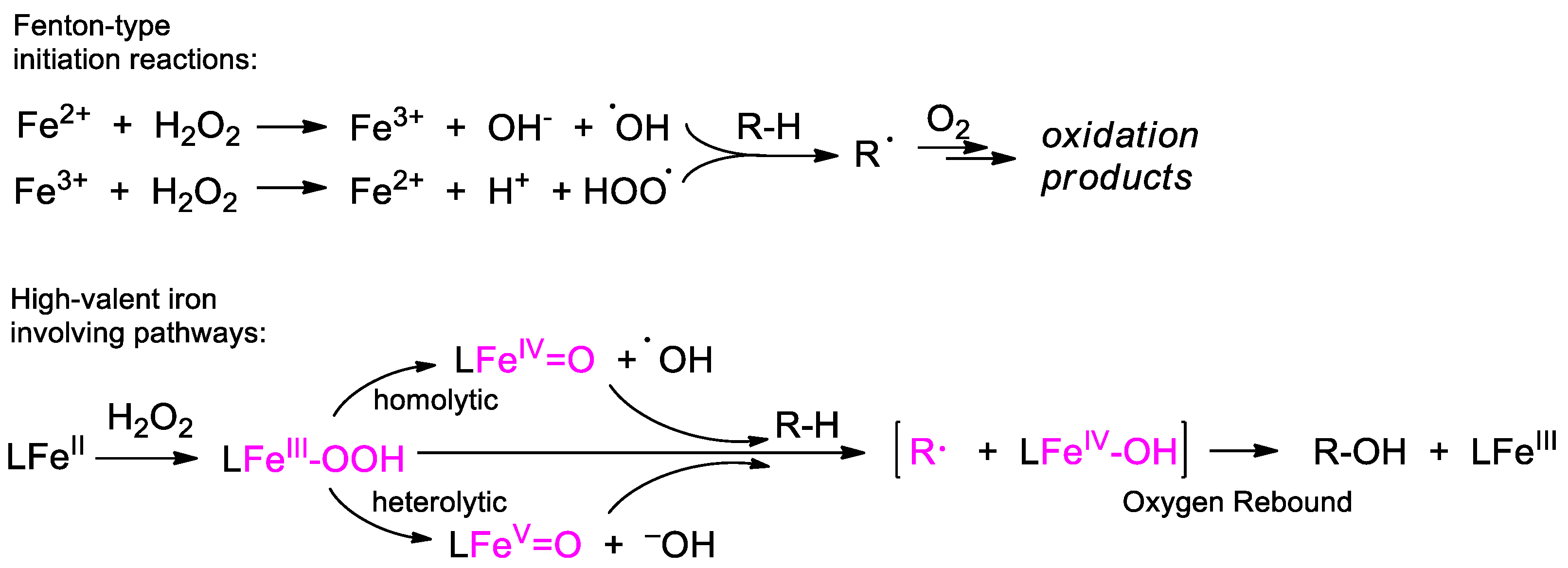{kind=link}
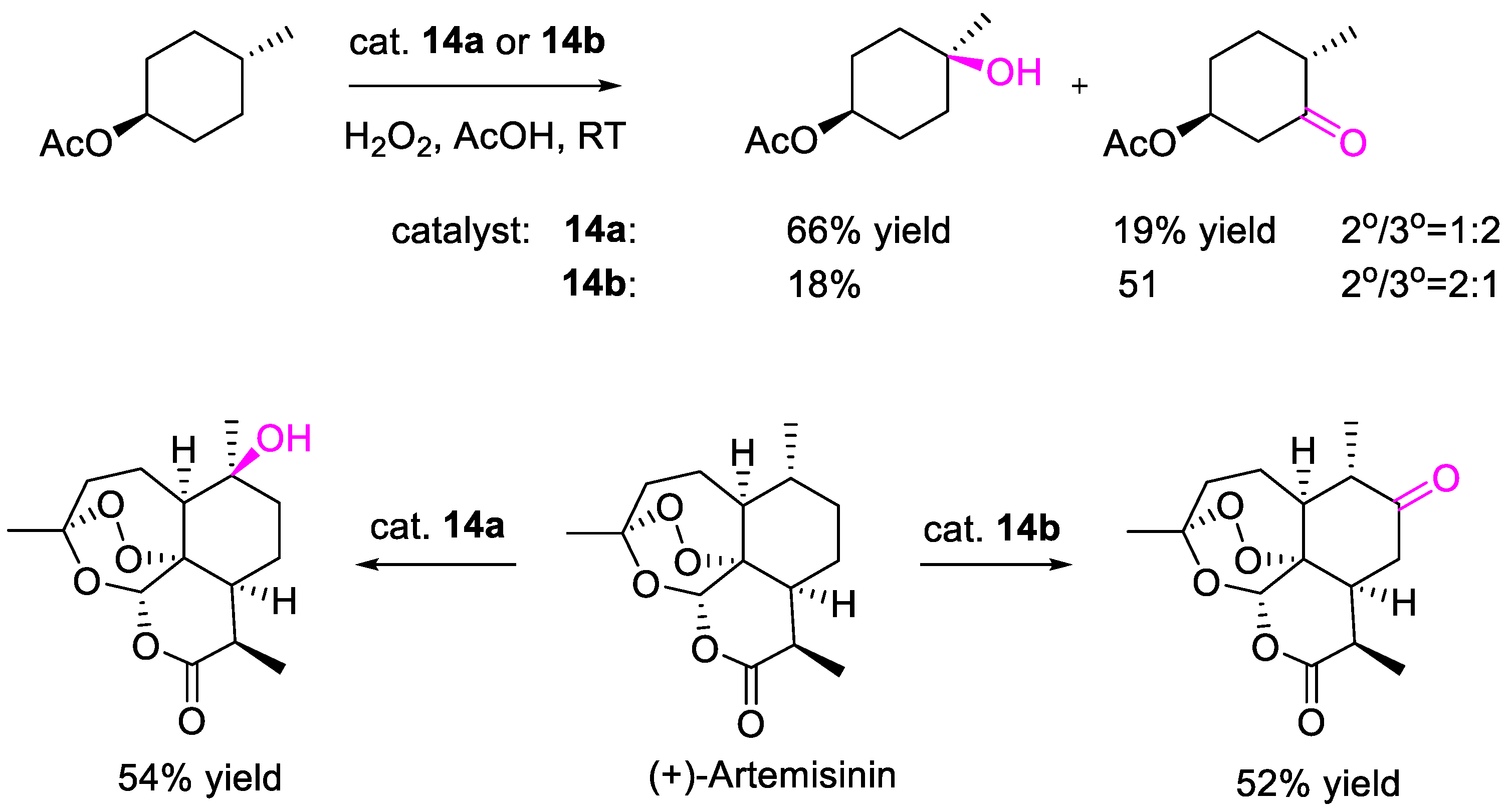{kind=link}
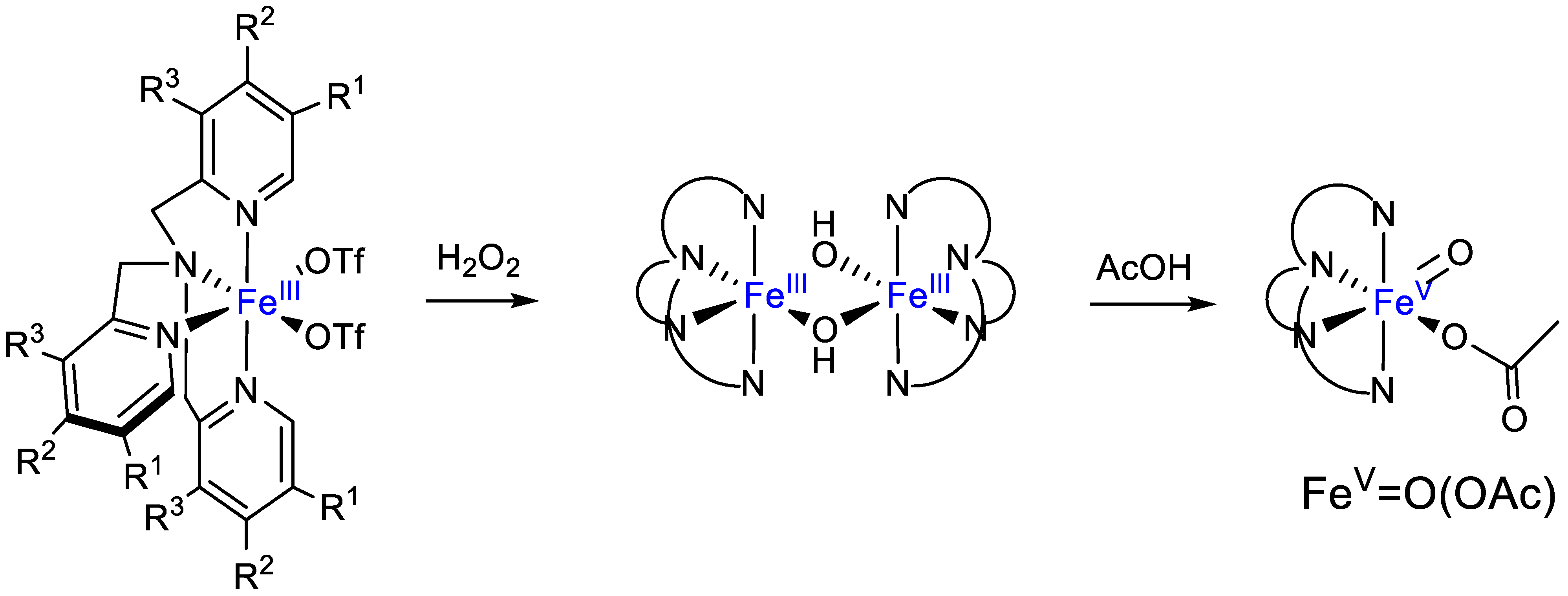{kind=link}
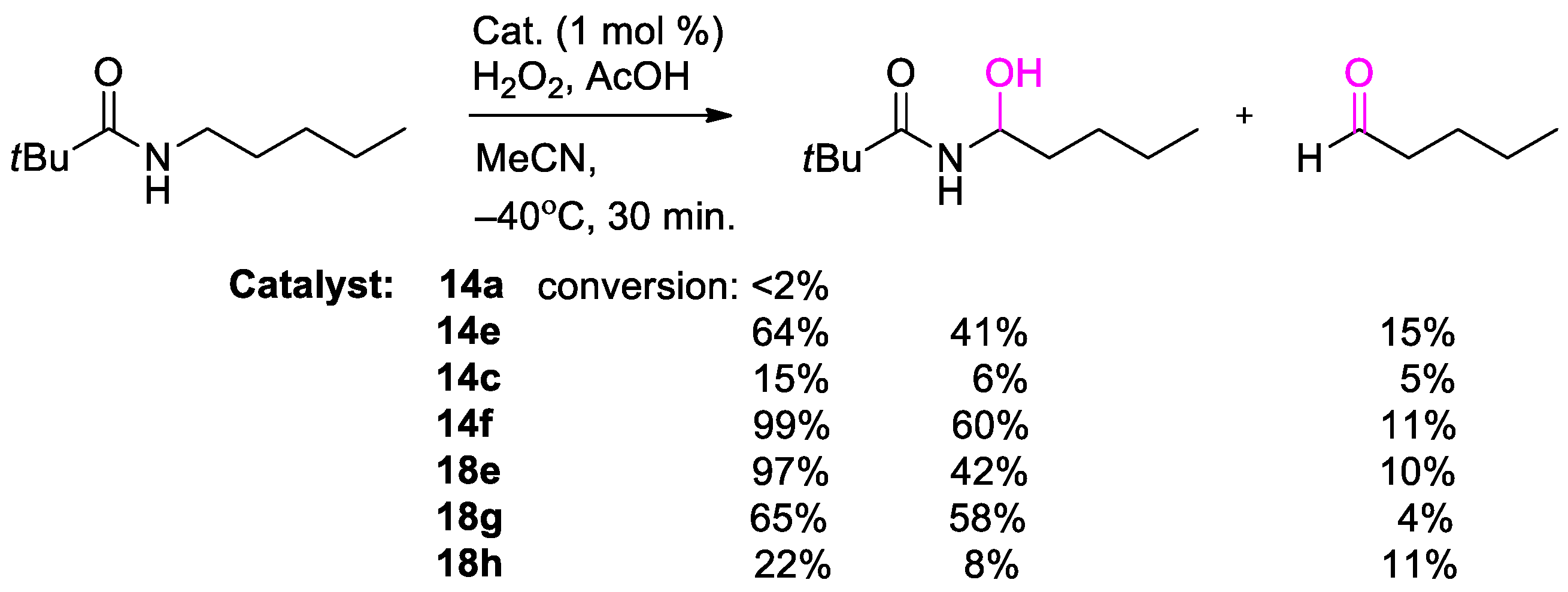{kind=link}
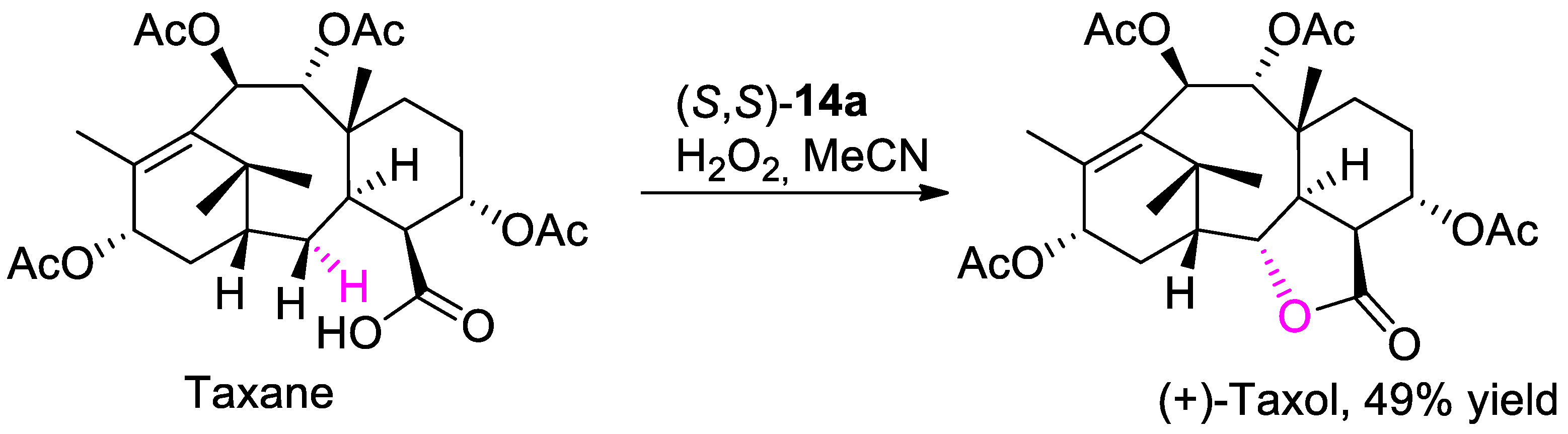{kind=link}
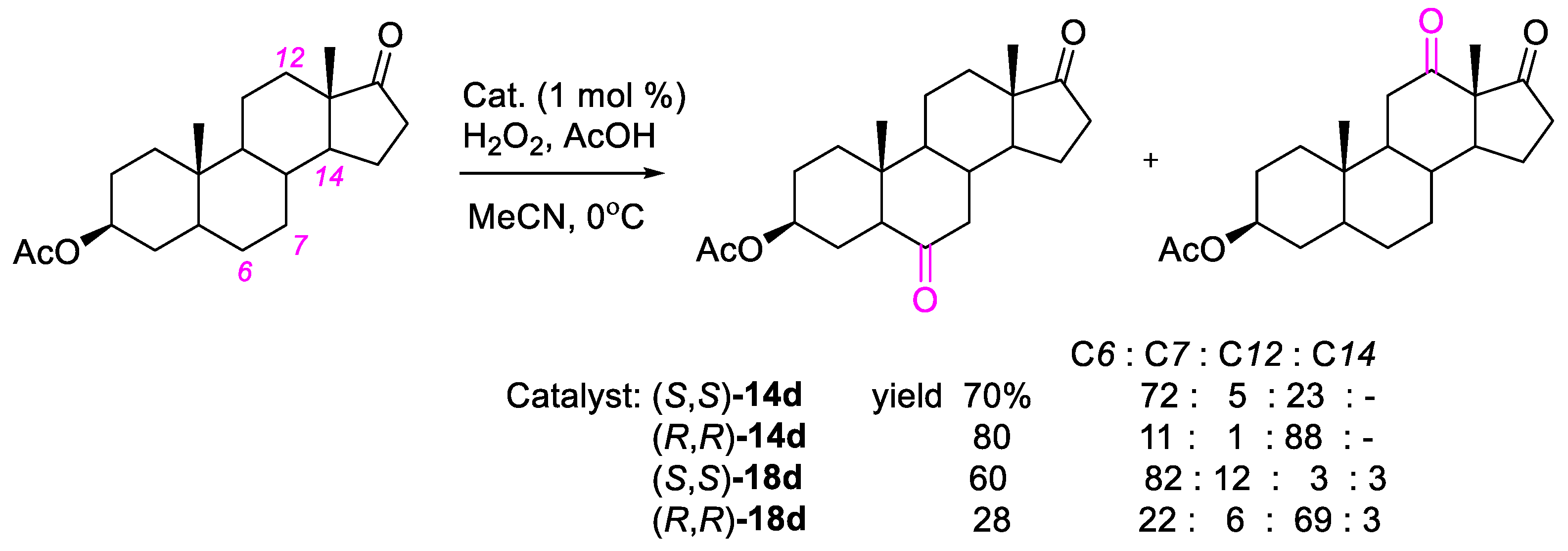{kind=link}
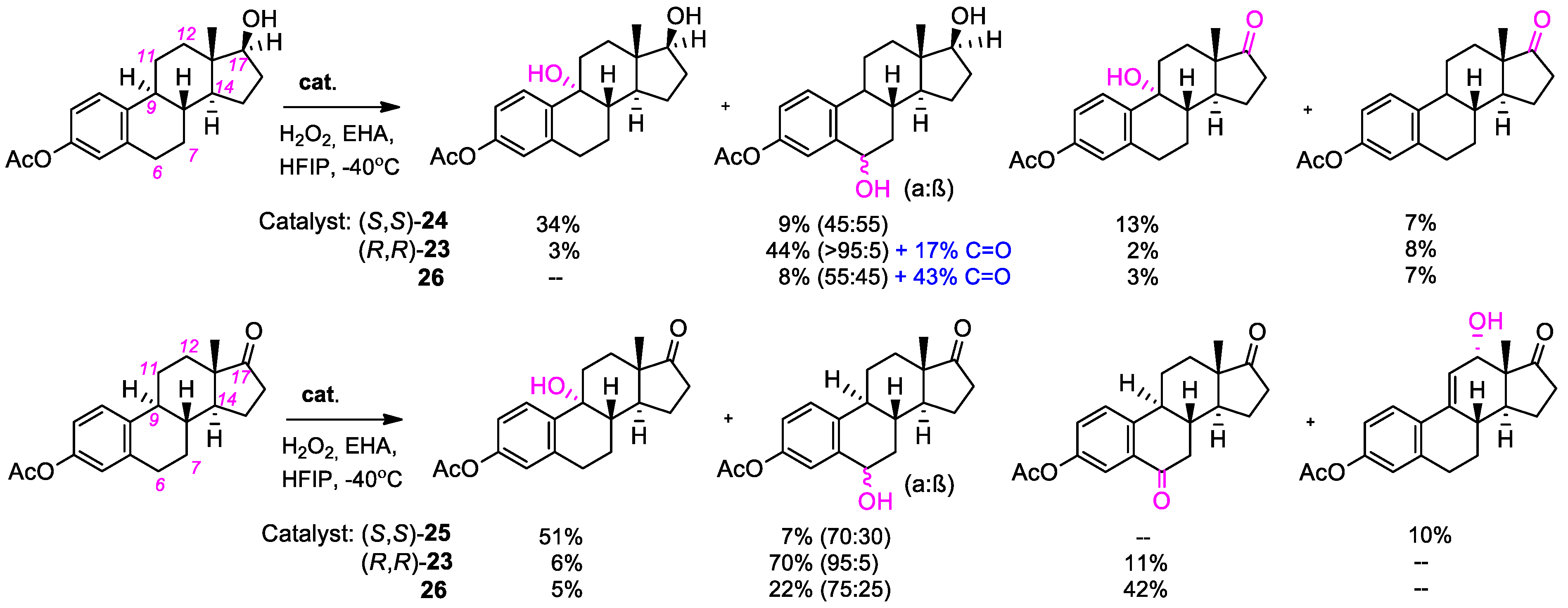{kind=link}
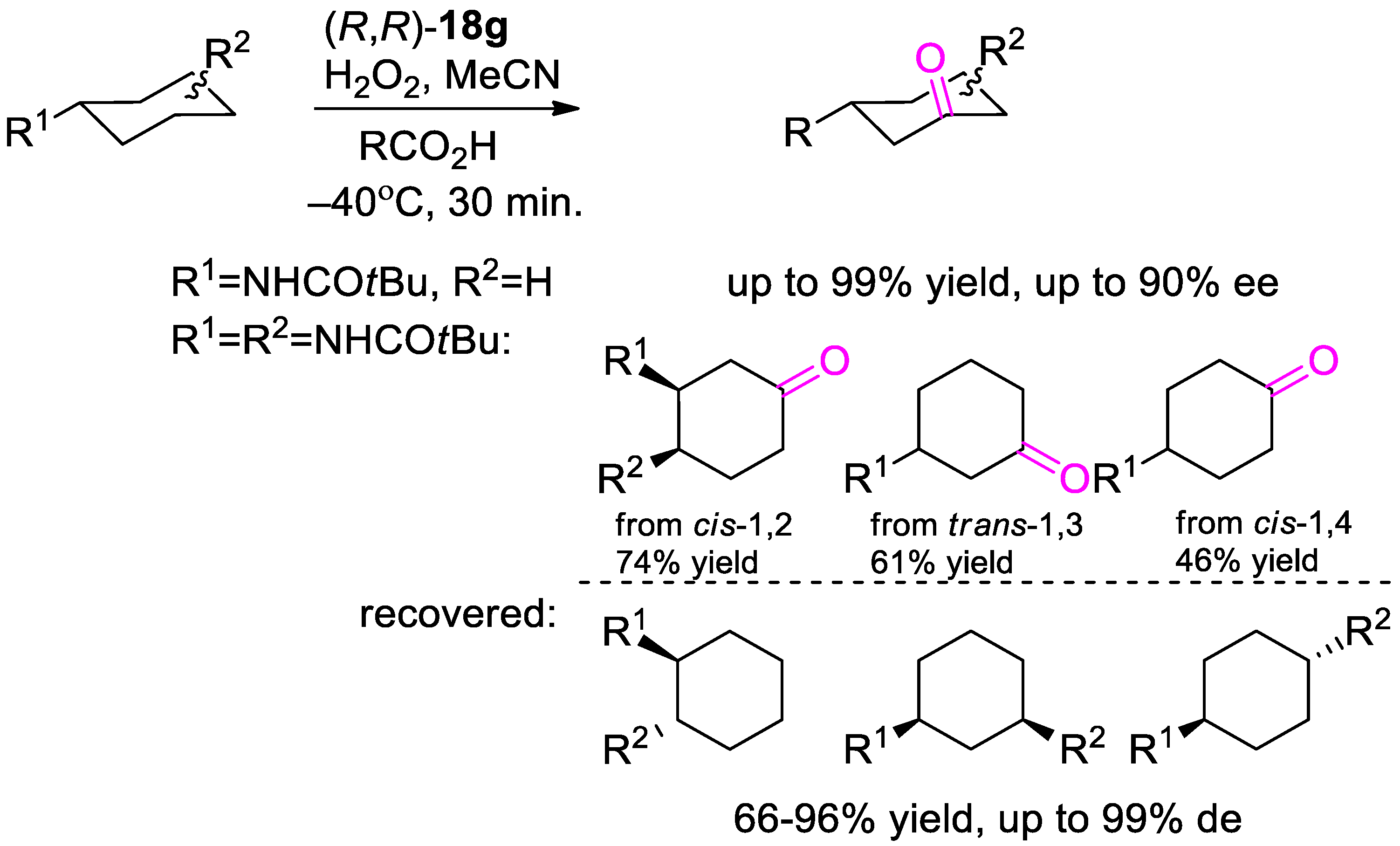{kind=link}
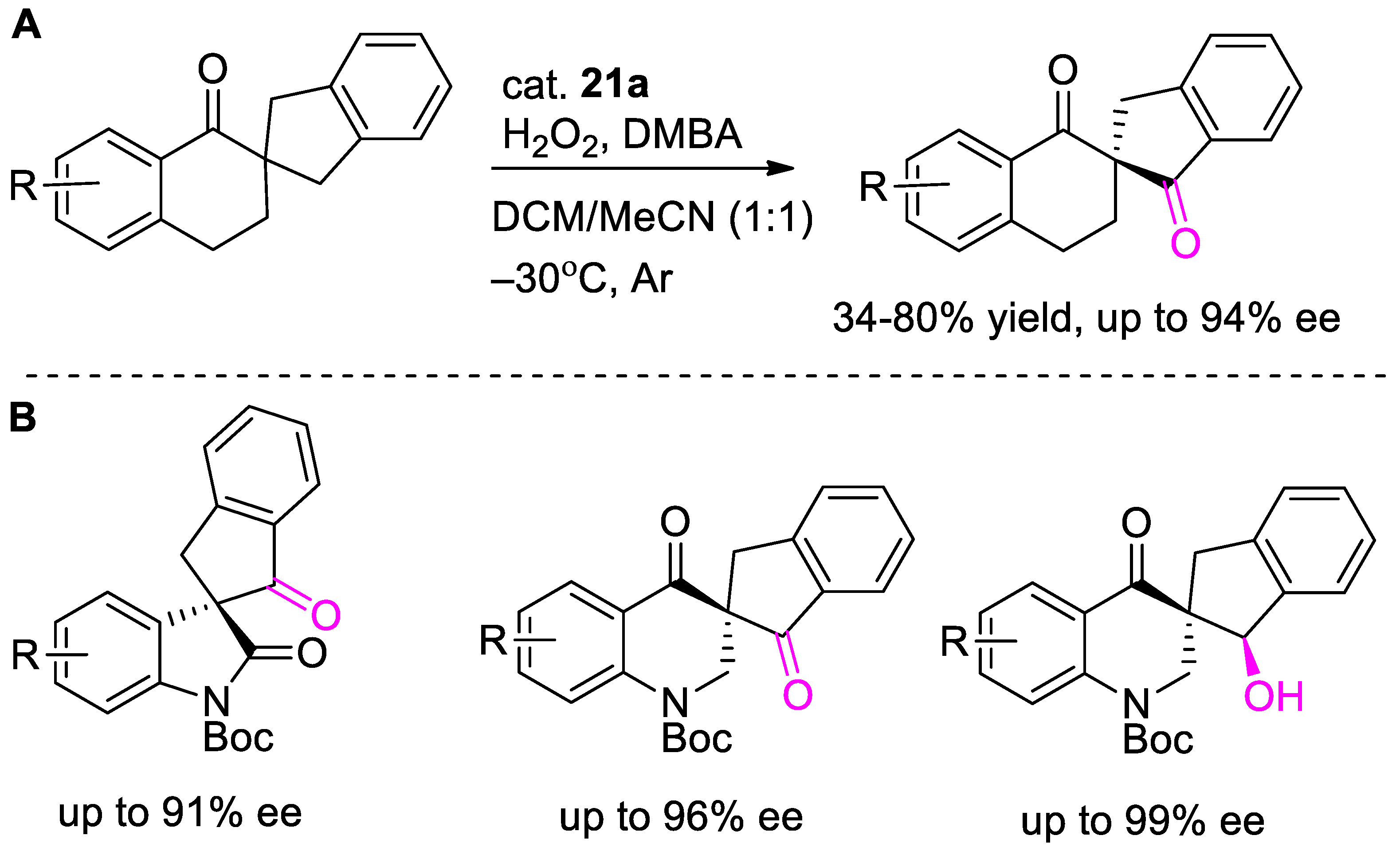{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tools to Evaluate the Selectivity and Usability of Catalytic Systems
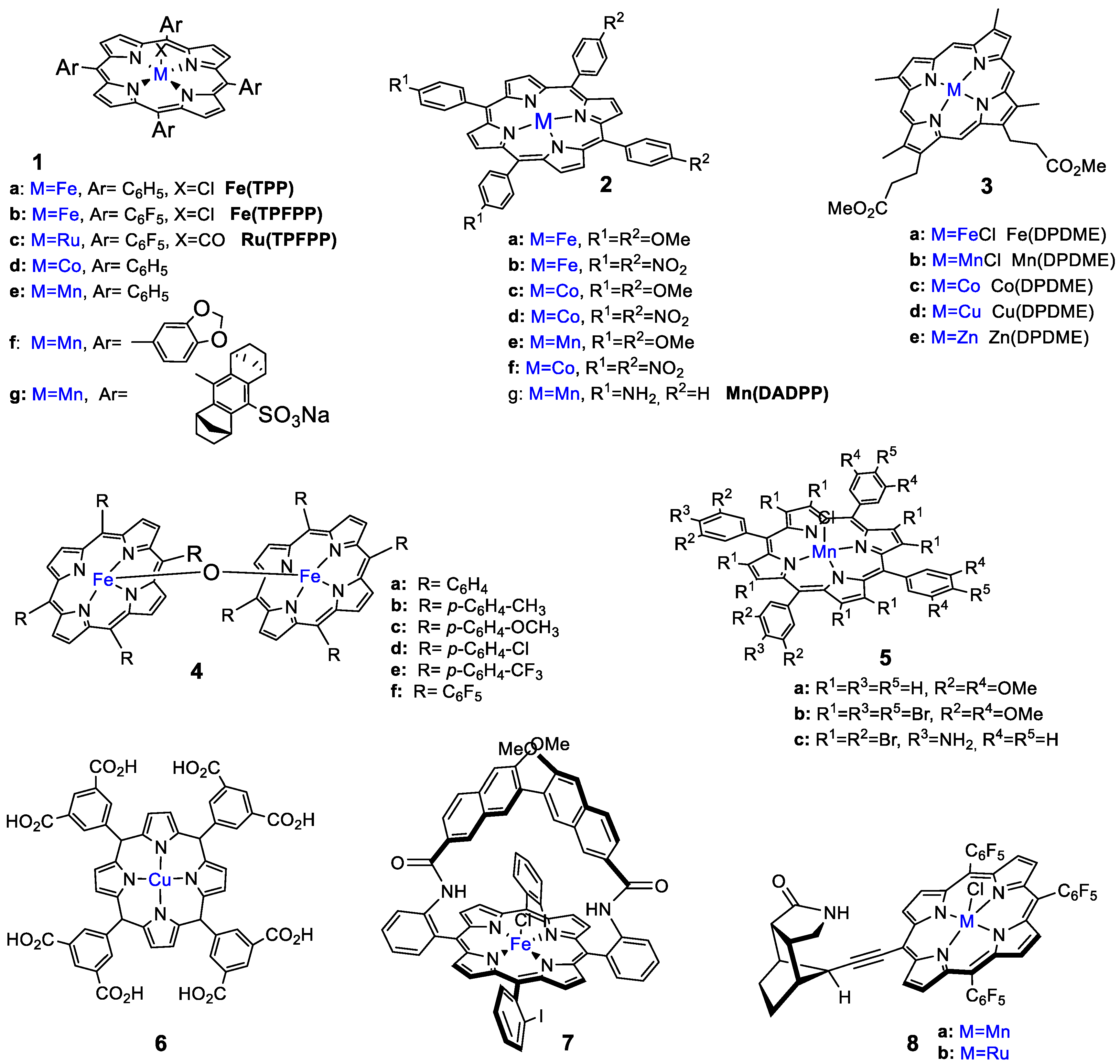
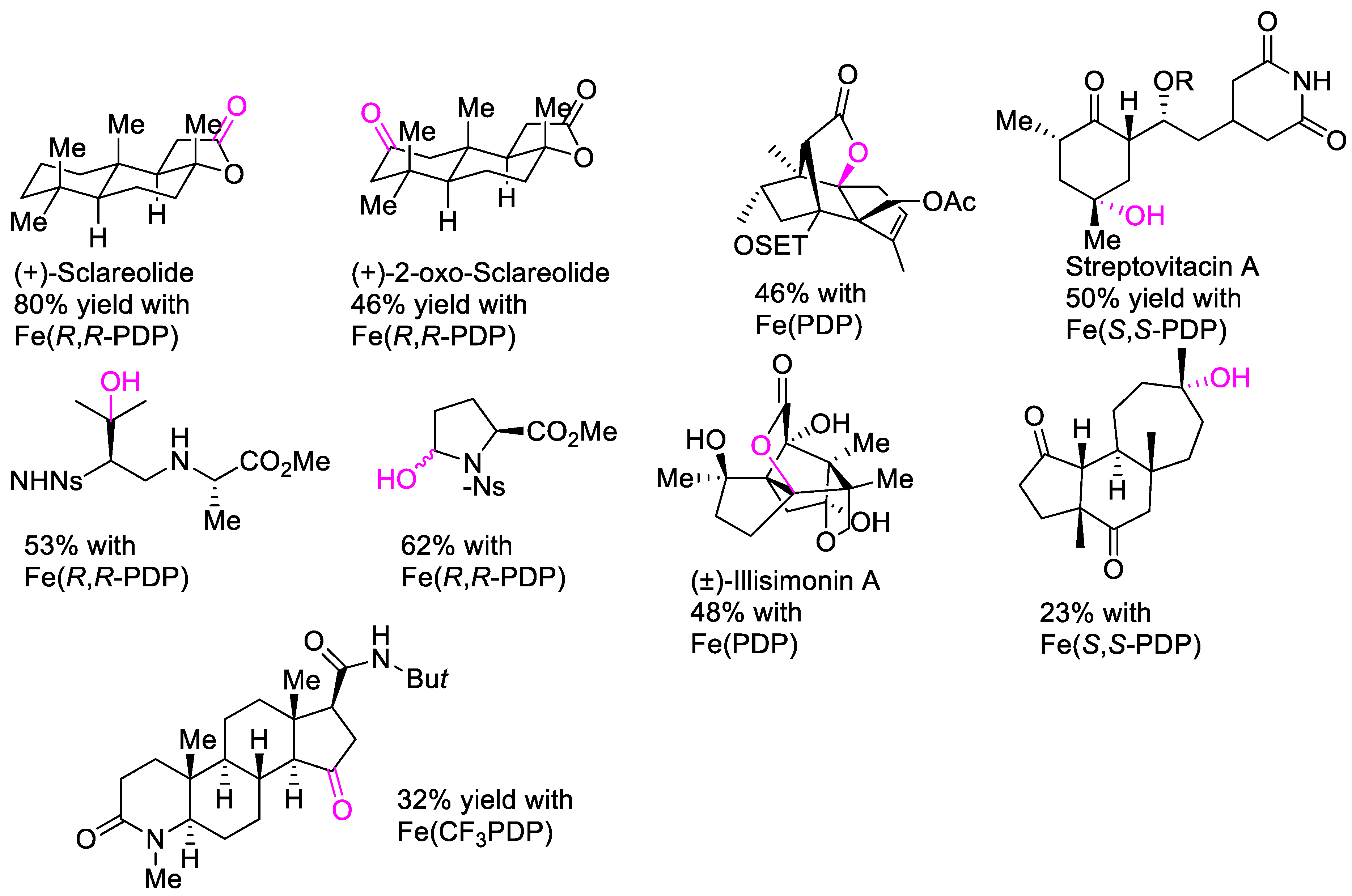
3. Oxidation with Metalloporphyrin Complexes
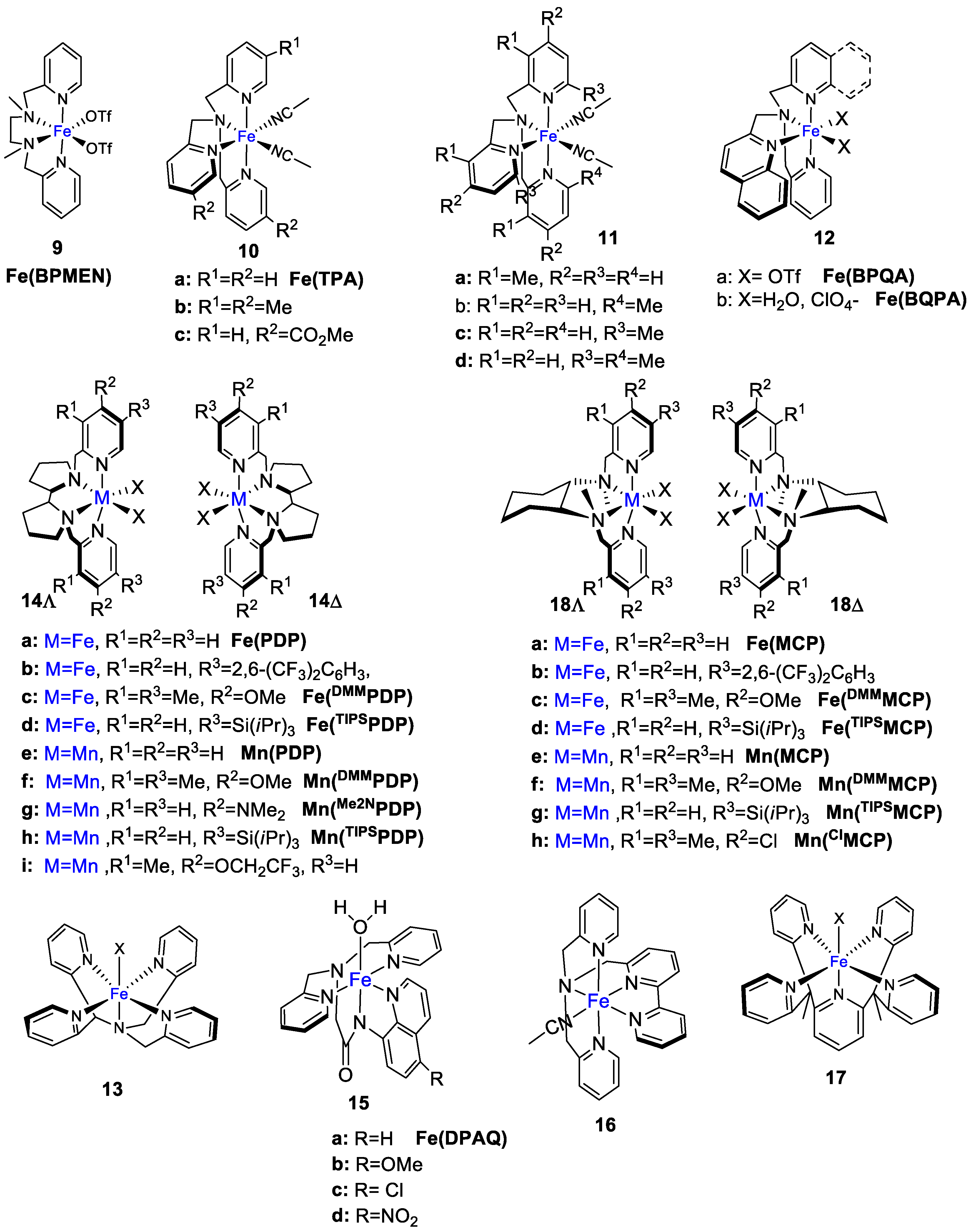
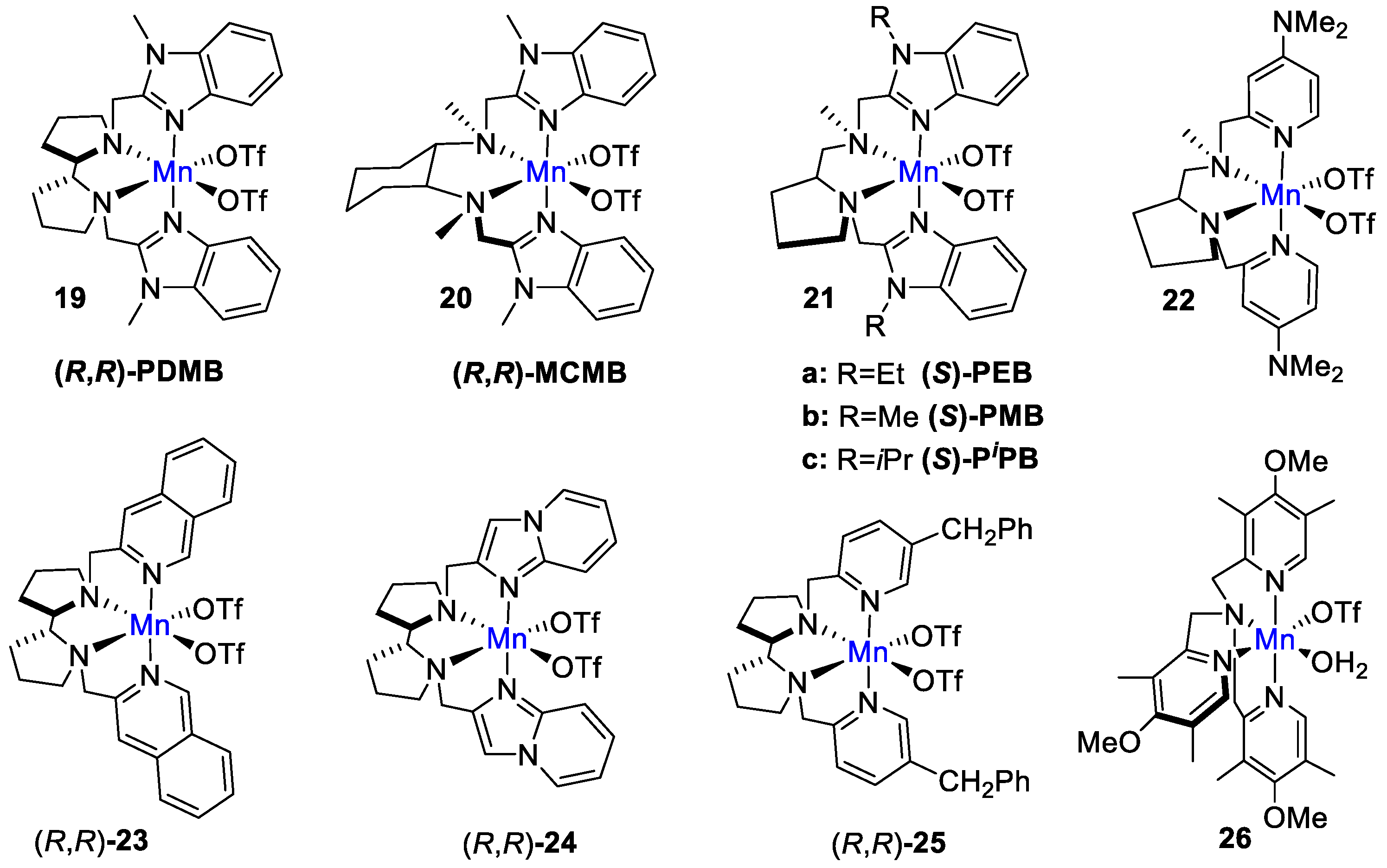
4. Oxidation with Non-Heme Complexes
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Groves, J.T.; Haushalter, R.C.; Nakamura, M.; Nemo, T.E.; Evans, B.J. High-Valent Iron-Porphyrin Complexes Related to Peroxidase and Cytochrome P-450. J. Am. Chem. Soc. 1981, 103, 2884–2886. [Google Scholar] [CrossRef]
- Gonsalves, A.; Pereira, M.M. State of the art in the development of biomimetic oxidation catalysts. J. Mol. Catal. A Chem. 1996, 113, 209–221. [Google Scholar] [CrossRef]
- Riley, D.; Stern, M.; Ebner, J. The Activation of Dioxygen and Homogeneous Catalytic Oxidation; Barton, D.H.R., Martell, A.E., Sawyer, D.T., Eds.; Plenum Press: New York, NY, USA, 1993; p. 31. [Google Scholar]
- Costas, K.; Chen, K.K.; Que, L., Jr. Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 200, 517–544. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shteinman, A.A. Oxygen Atom Transfer into C–H Bond in Biological and Model Chemical Systems. Mechanistic Aspects. Acc. Chem. Res. 1999, 32, 763–771. [Google Scholar] [CrossRef]
- Stavropoulos, P.; Çelenligil-Çetin, R.; Tapper, A.E. The Gif Paradox. Acc. Chem. Res. 2001, 34, 745–752. [Google Scholar] [CrossRef]
- 9 Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen. Chem. Rev. 2005, 105, 2329–2364. [Google Scholar] [CrossRef]
- Kim, C.; Chen, K.; Kim, J.; Que, L., Jr. Stereospecific Alkane Hydroxylation with H2O2 Catalyzed by an Iron(II)–Tris(2-pyridylmethyl)amine Complex. J. Am. Chem. Soc. 1997, 119, 5964–5965. [Google Scholar] [CrossRef]
- Luo, Y.R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press LLC: BocaRaton, FL, USA, 2003; p. 34. [Google Scholar]
- Raynea, S.; Forest, K. Gas phase homolytic bond dissociation enthalpies of common laboratory solvents: A G4 theoretical study. Nat. Prec. 2010. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. Low-Spin and High-Spin Perferryl Intermediates in Non-Heme Iron Catalyzed Oxidations of Aliphatic C–H Groups. Chem. Eur. J. 2021, 27, 7781–7788. [Google Scholar] [CrossRef]
- Krusic, P.J.; Meakin, P.; Jesson, J.P. Electron spin resonance studies of conformations and hindered internal rotation in transient free radicals. J. Phys. Chem. 1971, 75, 3438–3453. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2002; pp. 2000–2003. [Google Scholar]
- Meunier, B. Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Pereira, M.M.; Dias, L.D.; Calvete, M.J.F. Metalloporphyrins: Bioinspired Oxidation Catalysts. Acs Catal. 2018, 8, 10784–10808. [Google Scholar] [CrossRef]
- Che, C.-M.; Lo, V.K.-L.; Zhou, C.-Y.; Huang, J.-S. Selective functionalisation of saturated C–H bonds with metallopzorphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Que, L., Jr. Stereospecific Alkane Hydroxylation by Non-Heme Iron Catalysts: Mechanistic Evidence for an FeV=O Active Species. J. Am. Chem. Soc. 2001, 123, 6327–6337. [Google Scholar] [CrossRef]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci. 2019, 5, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Shteinman, A.A. Iron oxygenases: Structure, mechanism and modelling. Russ. Chem. Rev. 2008, 77, 945–966. [Google Scholar] [CrossRef]
- Rana, S.; Biswas, J.P.; Paul, S.; Paik, A.; Maiti, D. Organic Synthesis with the Most Abundant Transition Metal-Iron: From Rust to Multitasking Catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar] [PubMed]
- Nehru, K.; Kim, S.J.; Kim, I.Y.; Seo, M.S.; Kim, Y.; Kim, S.; Kim, J.; Nam, W. A highly efficient non-heme manganese complex in oxygenation reactions. Chem. Commun. 2007, 44, 4623–4625. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, F.; Liu, P.; Hao, F.; Luo, H. Synthesis and catalytic properties of trans-A2B2-type metalloporphyrins in cyclohexane oxidation. Can. J. Chem. 2014, 92, 49–53. [Google Scholar] [CrossRef]
- Liu, Y.; You, T.; Wang, H.-X.; Tang, Z.; Zhou, C.-Y.; Che, C.-M. Iron- and cobalt-catalyzed C(sp3)–H bond functionalization reactions and their application in organic synthesis. Chem. Soc. Rev. 2020, 49, 5310–5358. [Google Scholar] [CrossRef] [PubMed]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D.P. Structure and Spectroscopy of Copper–Dioxygen Complexes. Chem. Rev. 2004, 104, 1013–1046. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Tolman, W.B. Mononuclear Cu-O2 Complexes: Geometries, Spectroscopic Properties, Electronic Structures, and Reactivity. Acc. Chem. Res. 2007, 40, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Matsuo, H.; Matsuda, Y. Catalytic hydrocarbon oxygenation by ruthenium–pyridylamine complexes with alkyl hydroperoxides: A mechanistic insight. Inorg. Chim. Acta 2000, 300–302, 661–667. [Google Scholar] [CrossRef]
- Chen, M.; Pan, Y.; Kwong, H.-K.; Zeng, R.J.; Lau, K.-C.; Lau, T.-C. Catalytic oxidation of alkanes by a (salen)osmium(VI) nitride complex using H2O2 as the terminal oxidant. Chem. Comun. 2015, 51, 13686–13689. [Google Scholar] [CrossRef]
- Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Ucoski, G.M.; Castro, K.A.D.d.F.; Ciuffi, K.J.; Ricci, G.P.; Marques, J.A.; Nunes, F.S.; Nakagaki, S. Use of iron and manganese porphyrins in solution and immobilized on silica obtained by the sol–gel process as catalyst in the oxidation of organic substrates. Appl. Catal. A 2011, 404, 120–128. [Google Scholar] [CrossRef]
- Singh, A.; Agarwala, A.; Kamaraj, K.; Bandyopadhyay, D. The mechanistic aspects of iron(III) porphyrin catalyzed oxidation reactions in mixed solvents. Inorg. Chim. Acta 2011, 372, 295–303. [Google Scholar] [CrossRef]
- Che, C.-M.; Zhang, J.-L.; Zhang, R.; Huang, J.-S.; Lai, T.-S.; Tsui, W.-M.; Zhou, X.-G.; Zhou, Z.-Y.; Zhu, N.; Chang, C.K. Hydrocarbon Oxidation by β-Halogenated Dioxoruthenium (vi) Porphyrin Complexes: Effect of Reduction Potential (RuVI/V) and C–H Bond-Dissociation Energy on Rate Constants. Chem. Eur. J. 2005, 11, 7040–7053. [Google Scholar] [CrossRef]
- Zhang, J.L.; Huang, J.S.; Che, C.M. Oxidation Chemistry of Poly(ethylene glycol)-Supported Carbonylruthenium(II) and Dioxoruthenium(VI) meso-Tetrakis(pentafluorophenyl)porphyrin. Chem. Eur. J. 2006, 12, 3020–3031. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; She, Y.B.; Fu, H.Y.; Zhao, W.B.; Wang, M. Oxidation of Alkylaromatics to Aromatic Ketones Catalyzed by Metalloporphyrins Under the Special Temperature Control Method. Can. J. Chem. 2014, 92, 1059–1065. [Google Scholar] [CrossRef]
- She, Y.B.; Wang, W.J.; Li, G.J. Oxidation of p/o-Cresols to p/o-Hydroxybenzaldehydes Catalyzed by Metalloporphyrins with Molecular Oxygen. Chin. J. Chem. Eng. 2012, 20, 262–266. [Google Scholar] [CrossRef]
- Zhou, W.Y.; Sun, C.G.; Xu, S.C.; Hu, B.C. Metallo-Deuteroporphyrin as a Novel Catalyst for p-Xylene Oxidation Using Molecular Oxygen. Inorg. Chim. Acta 2012, 382, 167–170. [Google Scholar] [CrossRef]
- Tabor, E.; Poltowicz, J.; Pamin, K.; Basag, S.; Kubiak, W. Influence of Substituents in meso-Aryl Groups of Iron mu-Oxo Porphyrins on Their Catalytic Activity in the Oxidation of Cycloalkanes. Polyhedron 2016, 119, 342–349. [Google Scholar] [CrossRef]
- Da Silva, V.S.; Nakagaki, S.; Ucoski, G.M.; Idemori, Y.M.; De Freitas-Silva, G. New highly brominated Mn-porphyrin: A good catalyst for activation of inert C–H bonds. RSC Adv. 2015, 5, 106589–106598. [Google Scholar] [CrossRef]
- Da Silva, V.S.; Idemori, Y.M.; DeFreitas-Silva, G. Biomimetic Alkane Oxidation by Iodosylbenzene and Iodobenzene Diacetate Catalyzed by a New Manganese Porphyrin: Water Effect. Appl. Catal. A 2015, 498, 54–62. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, C.D. Biomimetic Activation of Molecular Oxygen with a Combined Metalloporphyrinic Framework and Cocatalyst Platform. ChemCatChem 2017, 9, 1192–1196. [Google Scholar] [CrossRef]
- Groves, J.T.; Viski, P. Asymmetric Hydroxylation by a Chiral Iron Porphyrin. J. Am. Chem. Soc. 1989, 111, 8537–8538. [Google Scholar] [CrossRef]
- Amiri, N.; Le Maux, P.; Srour, H.; Nasri, H.; Simonneaux, G. Nitration of Halterman porphyrin: A new route for fine tuning chiral iron and manganese porphyrins with application in epoxidation and hydroxylation reactions using hydrogen peroxide as oxidant. Tetrahedron 2014, 70, 8836–8842. [Google Scholar] [CrossRef]
- Srour, H.; Le Maux, P.; Simonneaux, G. Enantioselective Manganese-Porphyrin-Catalyzed Epoxidation and C–H Hydroxylation with Hydrogen Peroxide in Water/Methanol Solutions. Inorg. Chem. 2012, 51, 5850–5856. [Google Scholar] [CrossRef]
- Burg, F.; Gicquel, M.; Breitenlechner, S.; Pöthig, A.; Bach, T. Site- and Enantioselective C-H Oxygenation Catalyzed by a Chiral Manganese Porphyrin Complex with a Remote Binding Site. Angew. Chem. Int. Ed. 2018, 57, 2953–2957. [Google Scholar] [CrossRef] [PubMed]
- Burg, F.; Breitenlechner, S.; Jandl, C.; Bach, T. Enantioselective oxygenation of exocyclic methylene groups by a manganese porphyrin catalyst with a chiral recognition site. Chem. Sci. 2020, 11, 2121–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.R.; Huber, S.M.; Breitenlechner, S.; Bannwarth, C.; Bach, T. Enantiotopos-Selective CH Oxygenation Catalyzed by a Supramolecular Ruthenium Complex. Angew. Chem. Int. Ed. 2015, 54, 691–695. [Google Scholar]
- Oloo, W.N.; Que, L., Jr. Bioinspired Nonheme Iron Catalysts for C–H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Chen, K.; Que, L., Jr. Evidence for the participation of a high-valent iron–oxo species in stereospecific alkane hydroxylation by a non-heme iron catalyst. Chem. Commun. 1999, 15, 1375–1376. [Google Scholar] [CrossRef]
- Okuno, T.; Ito, S.; Ohba, S.; Nishida, Y. µ-Oxo bridged diiron (III) complexes and hydrogen peroxide: Oxygenation and catalase-like activities. J. Chem. Soc. Dalton Trans. 1997, 19, 3547–3551. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; England, J.; White, A.J.P. Non-heme Iron(II) Complexes Containing Tripodal Tetradentate Nitrogen Ligands and Their Application in Alkane Oxidation Catalysis. Inorg. Chem. 2005, 44, 8125–8134. [Google Scholar] [CrossRef]
- England, J.; Gondhia, R.; Bigorra-Lopez, L.; Petersen, A.R.; White, A.J.P.; Britovsek, G.J.P. Towards robust alkane oxidation catalysts: Electronic variations in non-heme iron(II) complexes and their effect in catalytic alkane oxidation. Dalton Trans. 2009, 27, 5319–5334. [Google Scholar] [CrossRef] [Green Version]
- Lyakin, O.Y.; Bryliakov, K.P.; Britovsek, G.J.P.; Talsi, E.P. EPR Spectroscopic Trapping of the Active Species of Nonheme Iron-Catalyzed Oxidation. J. Am. Chem. Soc. 2009, 131, 10798–10799. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science 2010, 327, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Gómez, L.; Garcia-Bosch, I.; Company, A.; Benet-Buchholz, J.; Polo, A.; Sala, X.; Ribas, X.; Costas, M. Stereospecific C-H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed. 2009, 48, 5720–5723. [Google Scholar] [CrossRef] [PubMed]
- Prat, I.; Gómez, L.; Canta, M.; Ribas, X.; Costas, M. An Iron Catalyst for Oxidation of Alkyl C-H Bonds Showing Enhanced Selectivity for Methylenic Sites. Chem. Eur. J. 2013, 19, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Gormisky, P.E.; White, M.C. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. [Google Scholar] [CrossRef]
- Hung, K.; Condakes, M.L.; Morikawa, T.; Maimone, T.J. Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Synthesis of (+)-Pseudoanisatin. J. Am. Chem. Soc. 2016, 138, 16616–16619. [Google Scholar] [CrossRef] [Green Version]
- Osberger, T.J.; Rogness, D.C.; Kohrt, J.T.; Stephan, A.F.; White, M.C. Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Qu, P.; Snyder, S.A. Total Syntheses of Scaparvins B, C, and D Enabled by a Key C–H Functionalization. J. Am. Chem. Soc. 2017, 139, 18428–18431. [Google Scholar] [CrossRef]
- Kim, K.E.; Adams, A.M.; Chiappini, N.D.; Du Bois, J.; Stolz, B.M. Cyanthiwigin natural product core as a complex molecular scaffold for comparative late-stage C–H functionalization studies. J. Org. Chem. 2018, 83, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.; Condakes, M.L.; Novaes, L.F.T.; Harwood, S.J.; Morikawa, T.; Yang, Z.; Maimone, T.J. Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes. J. Am. Chem. Soc. 2019, 141, 3083–3099. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.S.; Rychnovsky, S.D. Total Synthesis and Structure Revision of (−)-Illisimonin A, a Neuroprotective Sesquiterpenoid from the Fruits of Illicium simonsii. J. Am. Chem. Soc. 2019, 141, 13295–13300. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.M.; Feng, K.; Clark, J.R.; Trzepkowski, L.J.; White, M.C. Remote Oxidation of Aliphatic C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2015, 137, 14590–14593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjo, T.; de Lucca, E.C., Jr.; White, M.C. Remote, Late-Stage Oxidation of Aliphatic C–H Bonds in Amide-Containing Molecules. J. Am. Chem. Soc. 2017, 139, 14586–14591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Bryliakov, K.P.; Talsi, E.P. EPR Spectroscopic Detection of the Elusive FeV=O Intermediates in Selective Catalytic Oxofunctionalizations of Hydrocarbons Mediated by Biomimetic Ferric Complexes. ACS Catal. 2015, 5, 2702–2707. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; García-España, E.; Basallote, M.G.; Münck, E.; Que, L., Jr.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C-H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef]
- Fan, R.; Serrano-Plana, J.; Oloo, W.N.; Draksharapu, A.; Delgado-Pinar, E.; Company, A.; Martin-Diaconescu, V.; Borrell, M.; Lloret-Fillol, J.; García-España, E.; et al. Spectroscopic and DFT Characterization of a Highly Reactive Nonheme FeV–Oxo Intermediate. J. Am. Chem. Soc. 2018, 140, 3916–3928. [Google Scholar] [CrossRef]
- Hitomi, Y.; Arakawa, K.; Funabiki, T.; Kodera, M. An Iron(III)–Monoamidate Complex Catalyst for Selective Hydroxylation of Alkane CH Bonds with Hydrogen Peroxide. Angew. Chem. Int. Ed. 2012, 51, 3448–3452. [Google Scholar] [CrossRef]
- Hitomi, Y.; Arakawa, K.; Kodera, M. Electron Tuning of Iron-Oxo-Mediated C-H Activation: Effect of Electron-Donating Ligand on Selectivity. Chem. Eur. J. 2013, 19, 14697–14701. [Google Scholar] [CrossRef]
- Wong, E.; Jeck, J.; Grau, M.; White, A.J.P.; Britovsek, G.J.P. A strong-field pentadentate ligand in iron-based alkane oxidation catalysis and implications for iron(IV)oxo intermediates. Catal. Sci. Technol. 2013, 3, 1116–1122. [Google Scholar] [CrossRef]
- Xiang, J.; Li, H.; Wu, J.-S. Synthesis, Characterization, and Catalytic Activity of Iron(II) and Nickel(II) Complexes Containing the Rigid Pentadentate Ligand PY5Me2. Z. Anorg. Allg. Chem. 2014, 640, 1670–1674. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Efficient, Regioselective, and Stereospecific Oxidation of Aliphatic C-H Groups with H2O2, Catalyzed by Aminopyridine Manganese Complexes. Org. Lett. 2012, 14, 4310–4313. [Google Scholar] [CrossRef] [PubMed]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective C–H oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar] [CrossRef]
- Milan, M.; Bietti, M.; Costas, M. Highly Enantioselective Oxidation of Nonactivated Aliphatic C–H Bonds with Hydrogen Peroxide Catalyzed by Manganese Complexes. ACS Cent. Sci. 2017, 3, 196–204. [Google Scholar] [CrossRef]
- Milan, M.; Carboni, G.; Salamone, M.; Costas, M.; Bietti, M. Tuning Selectivity in Aliphatic C–H Bond Oxidation of N-Alkylamides and Phthalimides Catalyzed by Manganese Complexes. ACS Catal. 2017, 7, 5903–5911. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts. Molecules 2016, 21, 1454. [Google Scholar] [CrossRef] [Green Version]
- Milan, M.; Salamone, M.; Costas, M.; Bietti, M. The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C–H Bond Oxygenation. Acc. Chem. Res. 2018, 51, 1984–1995. [Google Scholar] [CrossRef]
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity, Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Borrell, M.; Gil-Caballero, S.; Bietti, M.; Costas, M. Site-Selective and Product Chemoselective Aliphatic C–H Bond Hydroxylation of Polyhydroxylated Substrates. ACS Catal. 2020, 10, 4702–4709. [Google Scholar] [CrossRef]
- Cianfanelli, M.; Olivo, G.; Milan, M.; Klein Gebbink, R.J.M.; Ribas, X.; Bietti, M.; Costas, M. Enantioselective C−H Lactonization of Unactivated Methylenes Directed by Carboxylic Acids. J. Am. Chem. Soc. 2020, 142, 1584–1593. [Google Scholar] [CrossRef]
- Vicens, L.; Bietti, M.; Costas, M. General Access to Modified a-Amino Acids by Bioinspired Stereoselective γ-C–H Bond Lactonization. Angew. Chem. Int. Ed. 2021, 60, 4740–4746. [Google Scholar] [CrossRef] [PubMed]
- Bigi, M.A.; Reed, S.A.; White, M.C. Directed Metal (Oxo) Aliphatic C-H Hydroxylations: Overriding Substrate Bias. J. Am. Chem. Soc. 2012, 134, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Font, D.; Canta, M.; Milan, M.; Cussó, O.; Ribas, X.; Klein Gebbink, R.J.M.; Costas, M. Readily Accessible Bulky Iron Catalysts exhibiting Site Selectivity in the Oxidation of Steroidal Substrates. Angew. Chem. Int. Ed. 2016, 55, 5776–5779. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Samsonenko, D.G.; Nefedov, A.A.; Bryliakov, K.P. Direct regio- and stereoselective mono- and polyoxyfunctionalization of estrone derivatives at C(sp3)-H bonds. J. Catal. 2022, 415, 12–18. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Nefedov, A.A.; Talsi, E.P.; Bryliakov, K.P. Mn aminopyridine oxidase mimics: Switching between biosynthetic-like and xenobiotic regioselectivity in CAH oxidation of (-)-ambroxide. J. Catal. 2021, 399, 224–229. [Google Scholar] [CrossRef]
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [CrossRef]
- Vicens, L.; Olivo, G.; Costas, M. Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar] [CrossRef]
- Milan, M.; Bietti, M.; Costas, M. Aliphatic C–H Bond Oxidation with Hydrogen Peroxide Catalyzed by Manganese Complexes: Directing Selectivity through Torsional Effects. Org. Lett. 2018, 20, 2720–2723. [Google Scholar] [CrossRef]
- Talsi, E.P.; Samsonenko, D.G.; Ottenbacher, R.V.; Bryliakov, K.P. Highly Enantioselective C-H Oxidation of Arylalkanes with H2O2 in the Presence of Chiral Mn-Aminopyridine Complexes. ChemCatChem 2017, 9, 4580–4586. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Highly enantioselective undirected catalytic hydroxylation of benzylic CH2 groups with H2O2. J. Catal. 2020, 390, 170–177. [Google Scholar] [CrossRef]
- Chen, J.; Yao, J.; Li, X.-X.; Wang, Y.; Song, W.; Cho, K.-B.; Lee, Y.-M.; Nam, W.; Wang, B. Bromoacetic Acid-Promoted Nonheme Manganese-Catalyzed Alkane Hydroxylation Inspired by α-Ketoglutarate-Dependent Oxygenases. ACS Catal. 2022, 12, 6756–6769. [Google Scholar] [CrossRef]
- Qiu, B.; Xu, D.; Sun, Q.; Miao, C.; Lee, Y.-M.; Li, X.-X.; Nam, W.; Sun, W. Highly enantioselective oxidation of spirocyclic hydrocarbons by bioinspired manganese catalysts and hydrogen peroxide. ACS Catal. 2018, 8, 2479–2487. [Google Scholar] [CrossRef]
- Qiu, B.; Xu, D.; Sun, Q.; Lin, J.; Sun, W. Manganese-Catalyzed Asymmetric Oxidation of Methylene C–H of Spirocyclic Oxindoles and Dihydroquinolinones with Hydrogen Peroxide. Org. Lett. 2019, 21, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, W. Catalytic Enantioselective Methylene C(sp3)–H Hydroxylation Using a Chiral Manganese Complex/Carboxylic Acid System. Org. Lett. 2020, 22, 9529–9533. [Google Scholar] [CrossRef]
- Olivo, G.; Farinelli, G.; Barbieri, A.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Supramolecular recognition allows remote, site-selective C-H oxidation of methylenic sites in linear amines. Angew. Chem. Int. Ed. 2017, 56, 16347–16351. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Enzyme-like substrate-selectivity in C–H oxidation enabled by recognition. Chem. Commun. 2019, 55, 917–920. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Ticconi, B.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Predictable selectivity in remote C-H Oxidation of steroids: Analysis of substrate binding mode. Angew. Chem. Int. Ed. 2020, 59, 12703–12711. [Google Scholar] [CrossRef]
- Kodera, M.; Ishiga, S.; Tsuji, T.; Sakurai, K.; Hitomi, Y.; Shiota, Y.; Sajith, P.K.; Yoshizawa, K.; Mieda, K.; Ogura, T. Formation and High Reactivity of the anti-Dioxo Form of High-Spin μ-Oxodioxodiiron(IV) as the Active Species That Cleaves Strong C-H Bonds. Chem. Eur. J. 2016, 22, 5924–5936. [Google Scholar] [CrossRef]
- Balamurugan, M.; Suresh, E.; Palaniandavar, M. μ-Oxo-bridged diiron(III) complexes of tripodal 4N ligands as catalysts for alkane hydroxylation reaction using m-CPBA as an oxidant: Substrate vs. self-hydroxylation. RSC Adv. 2021, 11, 21514–21526. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; England, J.; Spitzmesser, S.K.; White, A.J.P.; Williams, D.J. Synthesis of iron(II), manganese(II), cobalt(II) and ruthenium(II) complexes containing tridentate nitrogen ligands and their application in the catalytic oxidation of alkanes. Dalton Trans. 2005, 5, 945–955. [Google Scholar] [CrossRef]
- Shejwalkar, P.; Rath, N.P.; Bauer, E.B. New iron(II) α-iminopyridine complexes and their catalytic activity in the oxidation of activated methylene groups and secondary alcohols to ketones. Dalton Trans. 2011, 40, 7617–7631. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Arancio, G.; Mandolini, L.; Lanzalunga, O.; Di Stefano, S. Hydrocarbon oxidation catalyzed by a cheap nonheme imine-based iron(II) complex. Catal. Sci. Technol. 2014, 4, 2900–2903. [Google Scholar] [CrossRef]
- Olivo, G.; Nardi, M.; Diego Vìdal, D.; Barbieri, A.; Lapi, A.; Gómez, L.; Lanzalunga, O.; Costas, M.; Di Stefano, S. C–H Bond Oxidation Catalyzed by an Imine-Based Iron Complex: A Mechanistic Insight. Inorg. Chem. 2015, 54, 10141–10152. [Google Scholar] [CrossRef]
- Silva, R.; Mourao, T.; Rocha, J. Oxidation of cyclohexane by transition-metal complexes with biomimetic ligands. Catal. Today 2013, 203, 81–86. [Google Scholar] [CrossRef]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siedlecka, R. Selectivity in the Aliphatic C–H Bonds Oxidation (Hydroxylation) Catalyzed by Heme- and Non-Heme Metal Complexes—Recent Advances. Catalysts 2023, 13, 121. https://doi.org/10.3390/catal13010121
Siedlecka R. Selectivity in the Aliphatic C–H Bonds Oxidation (Hydroxylation) Catalyzed by Heme- and Non-Heme Metal Complexes—Recent Advances. Catalysts. 2023; 13(1):121. https://doi.org/10.3390/catal13010121
Chicago/Turabian StyleSiedlecka, Renata. 2023. "Selectivity in the Aliphatic C–H Bonds Oxidation (Hydroxylation) Catalyzed by Heme- and Non-Heme Metal Complexes—Recent Advances" Catalysts 13, no. 1: 121. https://doi.org/10.3390/catal13010121