
CO Oxidation over Alumina-Supported Copper Catalysts

1
College of Chemistry and Chemical Engineering, Xi’an Shiyou University, Xi’an 710065, China
2
Key Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science and Technology, Xi’an 710021, China
3
Shaanxi Collaborative Innovation Center of Industrial Auxiliary Chemistry and Technology, Shaanxi University of Science and Technology, Xi’an 710021, China
4
College of Chemistry and Chemical Engineering, Xianyang Normal University, Xianyang 712000, China
5
Department of Chemical Engineering, Norwegian University of Science and Technology, Sem Sælands vei 4, 7034 Trondheim, Norway
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(9), 1030; https://doi.org/10.3390/catal12091030
Submission received: 12 August 2022
/
Revised: 3 September 2022
/
Accepted: 6 September 2022
/
Published: 10 September 2022
(This article belongs to the Special Issue Advanced Oxidation Catalysts)
Abstract
:CO oxidation, one of the most important chemical reactions, has been commonly studied in both academia and the industry. It is one good probe reaction in the fields of surface science and heterogeneous catalysis, by which we can gain a better understanding and knowledge of the reaction mechanism. Herein, we studied the oxidation state of the Cu species to seek insight into the role of the copper species in the reaction activity. The catalysts were characterized by XRD, N2 adsorption-desorption, X-ray absorption spectroscopy, and temperature-programmed reduction. The obtained results suggested that adding of Fe into the Cu/Al2O3 catalyst can greatly shift the light-off curve of the CO conversion to a much lower temperature, which means the activity was significantly improved by the Fe promoter. From the transient and temperature-programmed reduction experiments, we conclude that oxygen vacancy plays an important role in influencing CO oxidation activity. Adding Fe into the Cu/Al2O3 catalyst can remove part of the oxygen from the Cu species and form more oxygen vacancy. These oxygen vacancy sites are the main active sites for CO oxidation reaction and follow a Mars-van Krevelen-type reaction mechanism.
1. Introduction
The catalytic oxidation of carbon monoxide (CO), a simple and typical heterogeneous catalytic reaction, is a key step in C1 chemistry and has been widely investigated for decades [1]. It has been considered the most studied probe reaction in heterogeneous catalysis, especially in the field of surface science, owing to its simple molecules [2,3,4,5]. With an in-depth understanding of this simple reaction, one can gain more and deeper fundamental new insights or knowledge of the reaction chemistry and mechanism [1,6]. Further, this knowledge is supposed to allow us to make progress in catalyst design and optimization. Therefore, CO oxidation, although seemingly a simple chemical reaction, is still widely under exploration [2,7]. It is not only useful as a model reaction system for fundamental studies for a better understanding of the reaction mechanism and the surface properties of the catalysts, but also imperative for some practical applications such as air cleaning, automotive emission control, and removal of CO impurities from H2 for polymer electrolyte membrane fuel cells [8]. In addition, CO is a poisonous molecule for some catalytic chemical reactions [9,10,11], in which it can strongly be adsorbed on the surface of the working catalysts; it will block or inhibit the other reactants’ adsorption and significantly hamper the catalytic performance. Those noble metals have long been used as the most efficient catalysts for CO oxidation with high activity and stability.
Many types of catalysts have been developed and proposed including noble metals, like Pt, Rh, Au, and Pd, either supported or non-supported catalysts [7,12]. However, owing to the high cost and limited availability of noble metals, the focus has been on transition metals and/or their oxides as the substitute for noble metal catalysts. Transition metals like Cu, Ru, and so on have been reported [13,14,15,16,17,18,19,20]. Among them, copper-based catalysts have been explored widely, and have been recognized as a possible substitute for noble metals for their high activity toward CO oxidation [13,21,22].
Much effort has been devoted to reaction mechanism studies in CO oxidation. The traditional Langmuir-Hinshelwood mechanism has been commonly reported [23,24,25,26], and the O2 adsorption has been reported as the rate-determining step. Besides, Mars–van Krevelen reaction mechanisms were also reported [6,15,27,28], especially for the catalysts with O-vacancy, in which the catalyst participates in the reaction with the reactants. Although, CO oxidation has been studied for decades in both academia and industry and uses multiple techniques. It still has significant meaning to continuously place some more focus on this “simple and classical” chemical reaction.
In the present work, to gain better insights into the relationship between the oxidation state of Cu species and CO oxidation activity, we report the effect of adding Fe as the promoter to the Cu/γ-Al2O3 catalyst to boost the activity of CO oxidation. We found that the Cu/γ-Al2O3 catalyst shows a weak or poor CO oxidation activity in the tested temperature range. Meanwhile, the activity is greatly enhanced by adding Fe into the Cu/γ-Al2O3 catalyst. A full conversion can be obtained at 250 °C. The promoter of Fe can greatly increase the reduction of CuO. The discoveries in this work provide a systematic understanding of the redox dynamics of Cu species under reaction conditions, which has implications for a broad range of catalytic reactions beyond CO oxidation.
2. Results and Discussion
2.1. Catalyst Properties
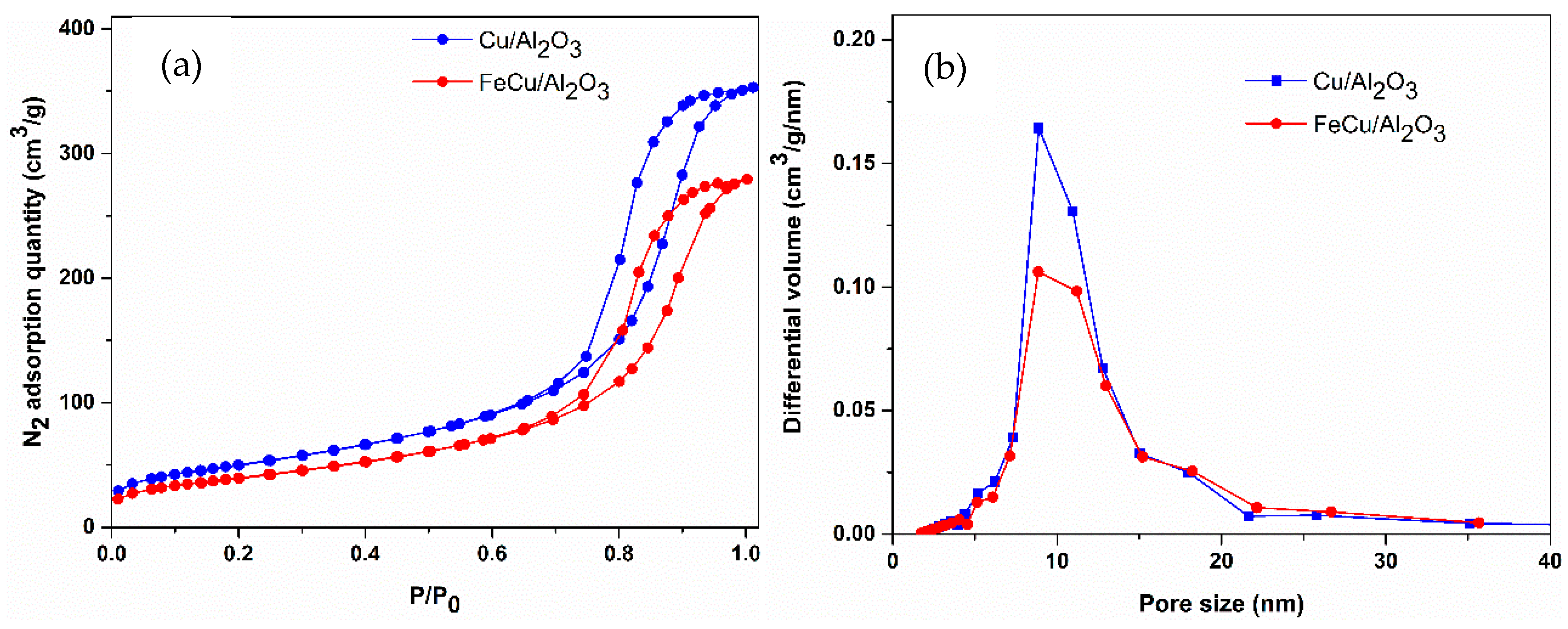
The compositions of the prepared catalysts were analyzed by X-ray fluorescence spectroscopy (XRF). The final metal loadings can be obtained as nominal with preparation. The physical properties of the catalysts, such as specific surface area, pore volume, and pore size, are summarized in Table 1. From the table, we know that, when depositing the metals on the γ-Al2O3, the surface area, pore volume, and pore size decreased slightly compared with the support. The nitrogen adsorption/desorption isotherms of the fresh samples shown in Figure 1a can be categorized as type IV isotherms, the typical character of mesoporous materials [29,30]. The pore size distribution shown in Figure 1b also demonstrates the mesoporous properties of the prepared catalysts.
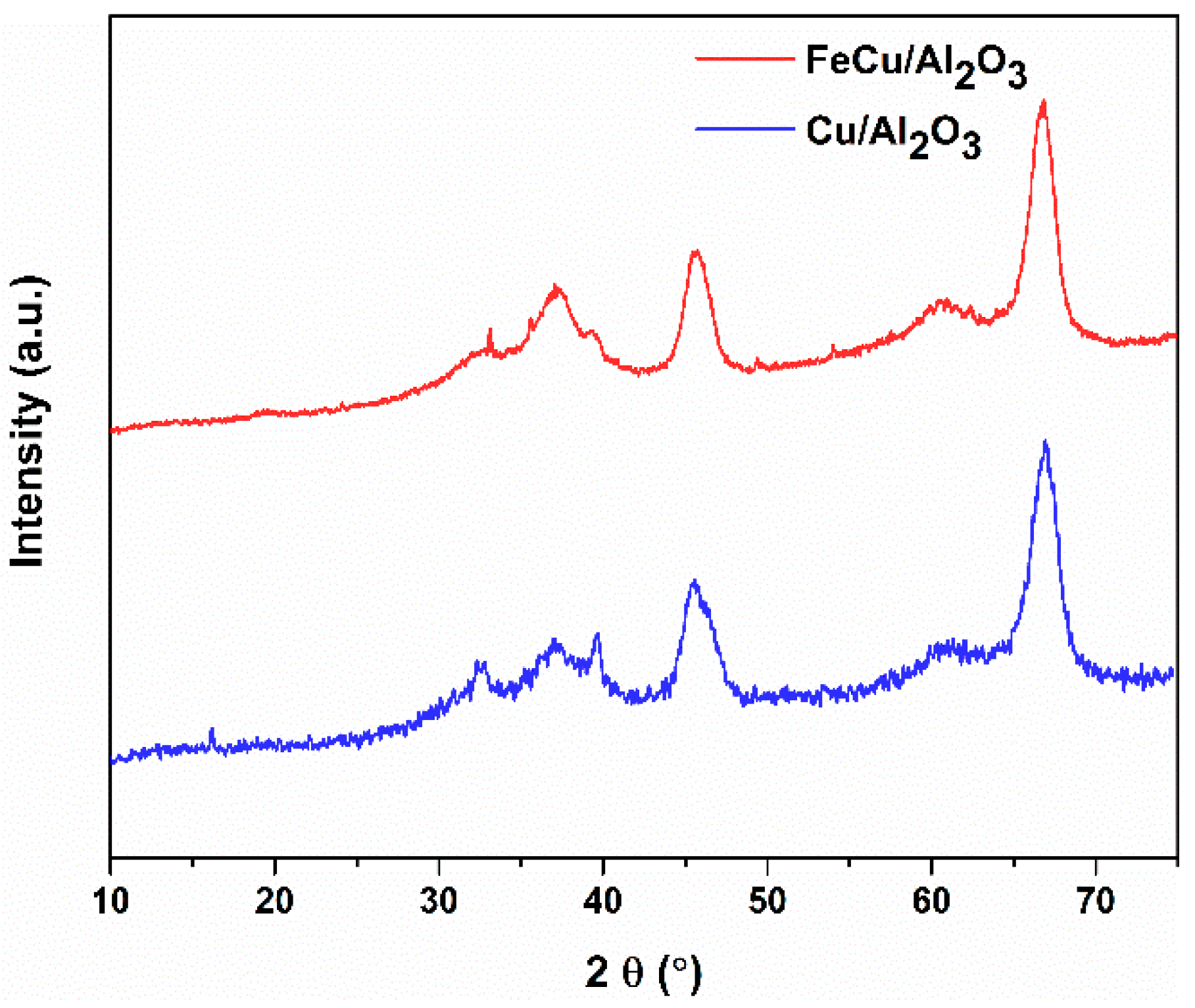
Figure 2 shows the XRD patterns of the fresh calcinated catalysts. The displayed diffraction peaks can be assigned to the phase of γ-Al2O3 [31,32], and no diffraction peaks of Fe and Cu species are observed. This indicates that both Fe and Cu are highly dispersed on the surface of γ-Al2O3. Besides, no mixed metal oxides can be detected on the XRD pattern. It was commonly reported that transition metal salts and oxides can be spontaneously highly dispersed on the surface of the oxide support (like γ-Al2O3 and TiO2) and form a monolayer or even sub-monolayer [31,32,33,34,35,36,37]. This was proven to be a thermodynamic process during the impregnation process. This type of monolayer catalyst has been reported and applied in multiple catalytic systems, forming one imported supported catalyst. Owing to the high dispersion of the Cu species on the support, the influence of other dopants on the Cu species is supposed to be imperative in affecting either the chemical state of the Cu species or the catalytic performance of the total reaction.
2.2. Catalytic Evaluation of the CO Oxidation
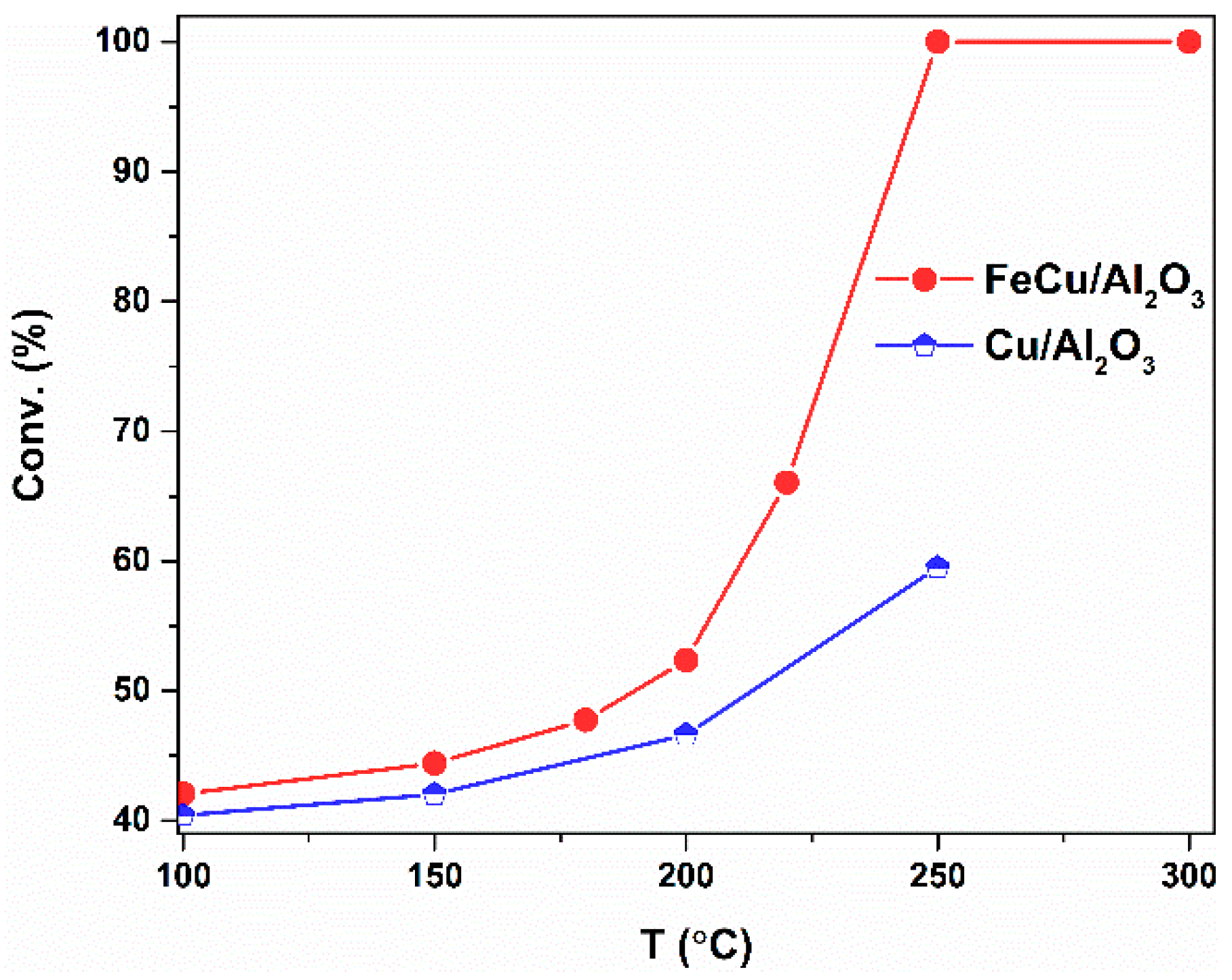
In the following section, the CO oxidation is evaluated over the Cu- and FeCu-supported γ-Al2O3 catalysts. The light-off curve is the conversion–temperature plot of a catalytic reaction; it can directly be used for the criteria for the catalyst comparison and catalyst development. The light-off curve of the FeCu/Al2O3 and Cu/Al2O3 catalysts is shown in Figure 3. The conversion of CO oxidation over the two catalysts increases when the temperature increases. The activity of CO oxidation over the two catalysts is quite different. Cu/Al2O3 shows a rather low CO conversion, in the temperature range from 100 °C to 250 °C, and the conversion can be reached at about 60% at 250 °C. However, when the catalyst is promoted with Fe, the scenarios are different. The light-off curve is shifted to the lower temperature range. The activity of CO oxidation is significantly enhanced over the tested temperature range. A full conversion can be obtained at a temperature of 250 °C.
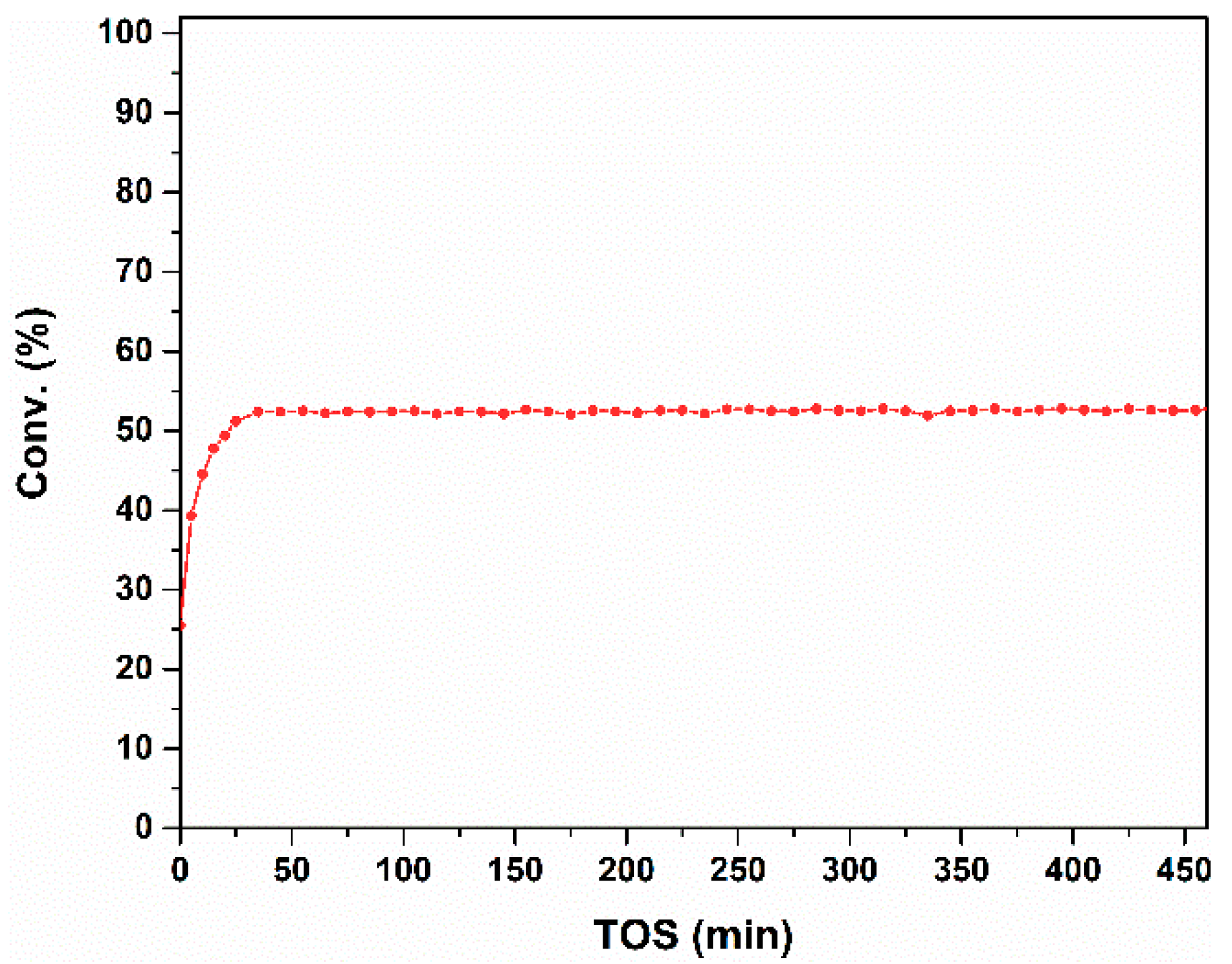
The stability of the FeCu/Al2O3 catalyst is also evaluated at a temperature of 250 °C, and the result is shown in Figure 4. The initial increasing phase was reported as an induction period of the catalyst. After the induction period, the conversion of CO is very stable over the 450 min test. No decreasing tendency is observed in the time-on-stream tests, and a longer reaction time can even be expected. This indicates that FeCu/Al2O3 is a good catalyst for CO oxidation.
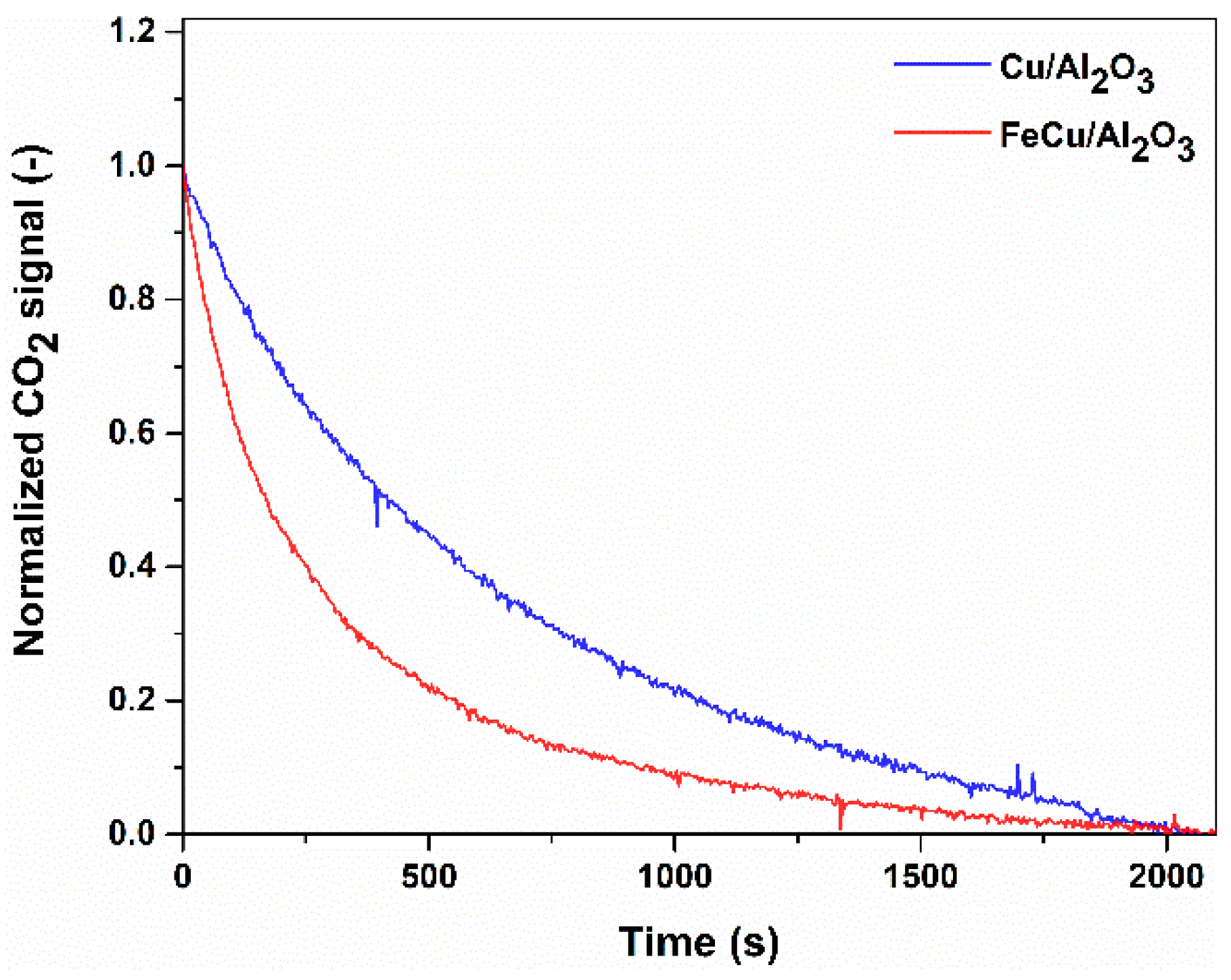
It has been commonly reported that CO oxidation follows the Mars-van Krevelen reaction mechanism over the Cu-based catalyst [13], in which Cu undergoes oxidation and reduction reactions via the oxygen vacancy. To gain a better understanding of the reaction, we also performed the transient experiment. The catalysts were first treated by the O2 atmosphere so that the catalyst is in the highest oxidation state. Then, by introducing CO into the catalyst, the catalyst will be reduced with the production of CO2, which can be traced by the online mass spectra. The normalized CO2 formation signal over the catalysts is shown in Figure 5 for qualitative comparison. To make the comparison more reasonable and to eliminate the influence caused by the baseline of the mass spectra, the signal was normalized in the same time range. The relative difference between the two curves will be discussed. We can see that the production rate over the FeCu/Al2O3 catalyst is much faster than that over Cu/Al2O3, as the slope of the curve is much higher for the FeCu/Al2O3 catalyst, which is also confirmed by the activity test in Figure 3. The most likely reason is that adding Fe into the catalyst can enhance the reduction of CuO with CO to a lower Cu oxidation state and CO2; therefore, the activity of CO oxidation is much higher on the FeCu/Al2O3 catalyst. Another parameter we should mention is the peak areas, which are related to the produced amount of CO2 on the two pre-oxidized catalysts. We can know that more oxygen can be removed from the Cu/Al2O3 catalyst because more CO2 is produced. As reported, the oxygen vacancy participates in the MvK type reaction cycle. Herein, we can know greater oxygen vacancy is produced on the FeCu/Al2O3 catalyst.
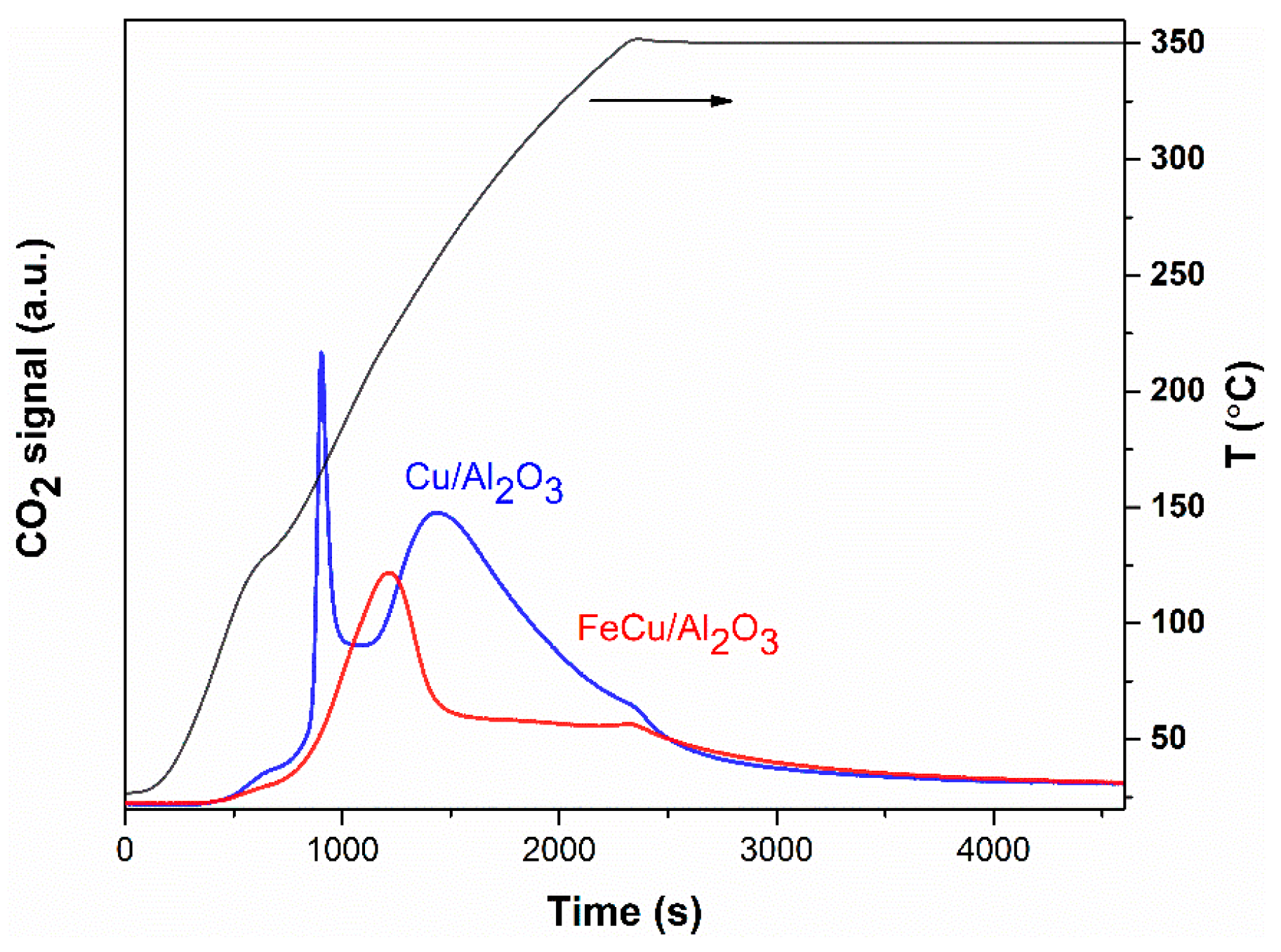
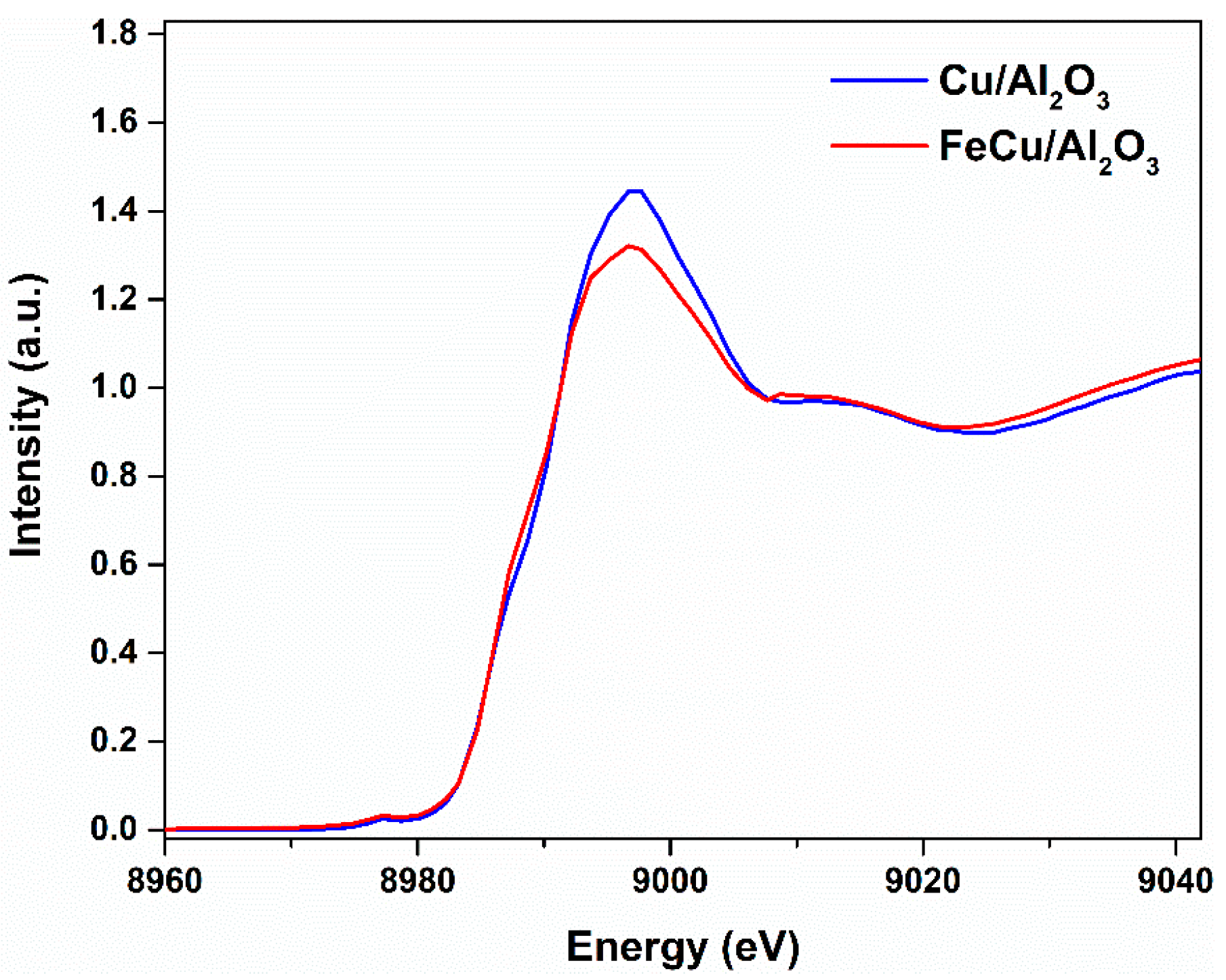
To further verify the discovery in the transient CO experiments, the CO temperature-programmed reduction (CO-TPR) was also performed to have an overview of the oxidation state and the reduction ability of the catalysts. The fresh catalyst was pre-treated in Ar at 100 °C to remove the adsorbed H2O caused by the storage in the atmosphere. Then, the catalyst was reduced by introducing CO, and the temperature was increased to 350 °C and maintained for a certain period until a stable baseline was observed on the mass spectra, as shown in Figure 6. CO2 is produced during the heating process; it indicates that oxygen is removed from the catalyst surface, and the catalyst is undergoing a reduction reaction with CO. This means the Cu or part of the Cu on the catalyst was in the oxidized state. While comparing the two catalysts, we can see that more CO2 is produced on the Cu/Al2O3 catalyst than that on the FeCu/Al2O3 catalyst. This shows that more Cu is in the oxidized state on the Cu/Al2O3 catalyst, and more oxygen species are accessible. Adding Fe as the promoter to the Cu/Al2O3 catalyst can enhance the reduction of CuO during the synthesis process. Greater oxygen vacancy is formed on the FeCu/Al2O3 catalyst. Furthermore, this oxygen vacancy is supposed to influence the activity of CO oxidation. This can also be concluded from the X-ray adsorption near-edge spectroscopy (XANES), as shown in Figure 7. The white line intensity of the FeCu/Al2O3 catalyst is much lower than that of Cu/Al2O3, indicating that the oxidation state of Cu in Cu/Al2O3 is much higher than the FeCu/Al2O3 catalyst. This is consistent with the transient and CO-TPR experiments stating that more oxygen vacancy is formed on the FeCu/Al2O3 catalyst.
As mentioned above, CO oxidation follows a Mars van-Krevelen reaction mechanism. The catalyst undergoes reduction and oxidation via the oxygen vacancy, as summarized in the following equations.
where OL is surface lattice oxygen and is the surface oxygen vacancy.
In this reaction mechanism, re-oxidation of the catalytic surface is usually faster than the step of withdrawal of oxygen from the copper oxide, so Equation 1 can be recognized as the rate-determining step [13,38]. From the CO-TPR, we know that, by adding Fe into the Cu/Al2O3 catalyst, the ability to withdraw oxygen from the catalyst surface becomes easier. It was reported that variations in copper valence during CO oxidation over CuO and Cu2O cycled between 2 and 0 for CuO, but between 1 and 2 for Cu2O [39]. The activity of CO oxidation over copper oxide species can be explained in terms of species transformation and changes in the amount of surface lattice oxygen. It seems the intermediate Cu oxidation state or non-stoichiometric metastable copper oxide species shows a good ability to transport surface lattice oxygen. Herein, it can be derived from the TPR that, in transient experiments, adding Fe into the Cu/Al2O3 catalyst increased the oxygen vacancy on the catalyst. Thus, the conversion of the FeCu/Al2O3 catalyst is much higher than that on Cu/Al2O3.
3. Materials and Methods
3.1. Catalyst Preparation
All of the catalysts were prepared by wetness impregnation methods. The precursor was CuCl2·2H2O (Sigma-Aldrich, St. Louis, MO, USA, ≥99%) and Fe(NO3)3·9H2O (Sigma-Aldrich, St. Louis, MO, USA, ≥99%), which were impregnated on the γ-Al2O3 (Sasol Germany GmbH, Hamburg, Germany) and mixed well by stirring. The metal loadings for Cu and Fe are 5 wt% and 1 wt%, respectively. Then, the resultant mixture was placed into the oven and dried at 100 °C for 10 h, followed by calcination at a temperature of 500 °C for 2 h with a ramping rate of 5 °C/min. The obtained samples were represented as Cu/Al2O3 and FeCu/Al2O3 in the following context, and the fresh samples were directly used for characterizations and catalytic evaluation.
3.2. Catalyst Characterization
The specific surface areas of the two γ-Al2O3 were measured on a TriStar 3000 instrument at liquid nitrogen temperature using N2 adsorption isotherms. BET and BJH analysis methods were used for the specific surface area and pore volume calculations. Samples were degassed under a vacuum condition at 200 °C overnight before measurements. XRD profiles were recorded with a Bruker D8 Davinci X-ray diffractometer (Bruker Nano GmbH, Berlin, Germany), using a Cu Ka1 (0.154 nm) wavelength.
The sample compositions were analyzed by X-ray fluorescence spectroscopy (XRF, Rigaku Supermini 200, Tokyo, Japan). The samples were dried and prepared in the form of pressed powder pellets.
3.3. X-Ray Absorption Spectroscopy Measurement
The X-ray absorption spectroscopy of the Cu K-edge was collected at BM 31 beamline station in ESRF (Swiss-Norwegian Beamline, European Synchrotron Radiation Facility, Grenoble, France) using the transmission mode with the use of a water-cooled flat Si [1 1 1] double-crystal monochromator. The Cu foil was used as the reference to conduct the energy calibrations. The spectra were normalized to the unity edge jump using Athena software (Demeter version 0.9.26, Bruce Ravel, BNL, NY, USA).
3.4. Catalytic Evaluation of CO Oxidation
The CO oxidation reaction was performed in a fixed bed reactor at 1 bar combined with an online mass spectrum (Omnistar GSD 3010, Asslar, Germany). The reactant gases were introduced into the reactor with specific mass flow controllers. Before the reaction, the catalysts were heated to the target temperature in Ar with a ramping rate of 5 °C/min. In one typical experiment, 0.3 g of catalyst was used at a total flow rate of 100 mL/min. The MS was used to perform the conversion calculation, in which Helium gas was used as the reference.
3.5. Carbon Monoxide Temperature-Programmed Reduction (CO-TPR)
CO-TPR was performed on the same setup with a fixed bed reactor, combined with an online MS recording the effluence gas. Before the TPR tests, the catalyst (0.3 g) was treated in Ar at 100 °C for 1 h to purge out the adsorbed water. Then, the samples were cooled down to room temperature in Ar. When a stable MS baseline was obtained, the samples were heated to 350 °C in 100 mL/min CO/Ar with a ramping rate of 10 °C/min. The final temperature was maintained until a stable MS baseline was obtained.
4. Conclusions
In summary, the γ-Al2O3 supported Cu with and without adding Fe as the promoter, and catalysts were prepared, characterized, and evaluated for CO oxidation reaction. Both Cu and Fe are highly dispersed on the surface of γ-Al2O3. The catalyst with Fe as the promoter shows much better activity than the neat Cu/Al2O3 catalyst over CO oxidation. Both the XAS results and transition experiments demonstrate that, by adding Fe inside the Cu/Al2O3 catalyst, the reduction of CuOx is greatly enhanced, which benefits the CO oxidation reaction. Adding Fe into the base Cu/Al2O3 catalyst, part of the oxygen can be removed, leaving greater oxygen vacancy on the catalyst. We demonstrate the role of oxygen vacancy in influencing the activity of CO oxidation, which follows a Mars-van Krevelen-type reaction mechanism. From the transient experiment and temperature-programmed reduction, we know this oxygen vacancy will further contribute to the activity of CO oxidation, which makes the FeCu/Al2O3 catalyst highly active for CO oxidation. This work also demonstrates the relationship between the activity and the Cu oxidation state.
Author Contributions
Conceptualization, G.M. and H.M.; Methodology, G.M. and H.M.; Writing—Original Draft Preparation, G.M. and H.M.; Writing—Review and Editing, G.M., L.W., X.W., L.L., and H.M.; Funding Acquisition, G.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Open Foundation of Key Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science and Technology (No. KFKT2021-07); the Shaanxi Collaborative Innovation Center of Industrial Auxiliary Chemistry and Technology, Shaanxi University of Science and Technology (No. KFKT2021-07); and the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2020JQ-765). And the APC was funded by the Norwegian University of Science and Technology.
Data Availability Statement
All relevant data are included in the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Freund, H.-J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, M.A.; Frenken, J.W.M.; Groot, I.M.N. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef] [PubMed]
- Stamenković, V.; Arenz, M.; Blizanac, B.; Mayrhofer, K.; Ross, P.; Marković, N. In situ CO oxidation on well characterized Pt3Sn (hkl) surfaces: A selective review. Surf. Sci. 2005, 576, 145–157. [Google Scholar] [CrossRef]
- Cui, H.; Liu, Z.; Jia, P. Pd-doped C3N monolayer: A promising low-temperature and high-activity single-atom catalyst for CO oxidation. Appl. Surf. Sci. 2020, 537, 147881. [Google Scholar] [CrossRef]
- Meunier, F.C.; Cardenas, L.; Kaper, H.; Šmíd, B.; Vorokhta, M.; Grosjean, R.; Aubert, D.; Dembélé, K.; Lunkenbein, T. Synergy between metallic and oxidized Pt sites unravelled during room temperature CO oxidation on Pt/ceria. Angew. Chem. Int. Ed. 2021, 60, 3799–3805. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R.J. Activation of Molecular Oxygen and the Nature of the Active Oxygen Species for CO Oxidation on Oxide Supported Au Catalysts. Accounts Chem. Res. 2014, 47, 740–749. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature CO Oxidation and Propene Oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef]
- Lin, J.; Wang, X.; Zhang, T. Recent progress in CO oxidation over Pt-group-metal catalysts at low temperatures. Chin. J. Catal. 2016, 37, 1805–1813. [Google Scholar] [CrossRef]
- Xu, S.; Chansai, S.; Xu, S.; Stere, C.E.; Jiao, Y.; Yang, S.; Hardacre, C.; Fan, X. CO poisoning of Ru catalysts in CO2 hydrogenation under thermal and plasma conditions: A combined kinetic and diffuse reflectance infrared fourier transform spectroscopy–mass spectrometry study. ACS Catal. 2020, 10, 12828–12840. [Google Scholar] [CrossRef]
- Knudsen, J.; Nilekar, A.U.; Vang, R.T.; Schnadt, J.; Kunkes, E.L.; Dumesic, J.A.; Mavrikakis, M.; Besenbacher, F. A Cu/Pt Near-Surface Alloy for Water-Gas Shift Catalysis. J. Am. Chem. Soc. 2007, 129, 6485–6490. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Wang, X.; Mei, B.; Luo, E.; Li, Y.; Meng, Q.; Jin, Z.; Jiang, Z.; Liu, C.; et al. CO-Tolerant PEMFC Anodes Enabled by Synergistic Catalysis between Iridium Single-Atom Sites and Nanoparticles. Angew. Chem. 2021, 133, 26381–26387. [Google Scholar] [CrossRef]
- Mosrati, J.; Abdel-Mageed, A.M.; Vuong, T.H.; Grauke, R.; Bartling, S.; Rockstroh, N.; Atia, H.; Armbruster, U.; Wohlrab, S.; Rabeah, J.; et al. Tiny species with big impact: High activity of Cu single atoms on CeO2–TiO2 deciphered by operando spectroscopy. ACS Catal. 2021, 11, 10933–10949. [Google Scholar] [CrossRef]
- Huang, T.-J.; Tsai, D.-H. CO oxidation behavior of copper and copper oxides. Catal. Lett. 2003, 87, 173–178. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the Catalytic CO Oxidation Activity of Nanoparticles. Angew. Chem. 2008, 120, 4913–4917. [Google Scholar] [CrossRef]
- Jia, A.-P.; Hu, G.-S.; Meng, L.; Xie, Y.-L.; Lu, J.-Q.; Luo, M.-F. CO oxidation over CuO/Ce1−xCuxO2−δ and Ce1−xCuxO2−δ catalysts: Synergetic effects and kinetic study. J. Catal. 2012, 289, 199–209. [Google Scholar] [CrossRef]
- Zhang, X.-m.; Tian, P.; Tu, W.; Zhang, Z.; Xu, J.; Han, Y.-F. Tuning the dynamic interfacial structure of copper–ceria catalysts by indium oxide during CO oxidation. ACS Catal. 2018, 8, 5261–5275. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Hu, P.; Alavi, A. Mechanism for the high reactivity of CO oxidation on a ruthenium–oxide. J. Chem. Phys. 2001, 114, 5956–5957. [Google Scholar] [CrossRef]
- Huang, B.; Kobayashi, H.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Nishida, Y.; Sato, K.; Nagaoka, K.; Haneda, M.; Xie, W.; et al. A CO adsorption site change induced by copper substitution in a ruthenium catalyst for enhanced CO oxidation activity. Angew. Chem. Int. Ed. 2019, 131, 2252–2257. [Google Scholar] [CrossRef]
- Kim, Y.; Over, H.; Krabbes, G.; Ertl, G. Identification of RuO2 as the active phase in CO oxidation on oxygen-rich ruthenium surfaces. Top. Catal. 2000, 14, 95–100. [Google Scholar] [CrossRef]
- Joo, S.H.; Park, J.Y.; Renzas, J.; Butcher, D.R.; Huang, W.; Somorjai, G.A. Size Effect of Ruthenium Nanoparticles in Catalytic Carbon Monoxide Oxidation. Nano Lett. 2010, 10, 2709–2713. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, A.; Saraev, A.; Kremneva, A.; Selivanova, A.; Vorokhta, M.; Šmíd, B.; Bulavchenko, O.; Yakovlev, V.; Kaichev, V. Kinetic and mechanistic study of CO oxidation over nanocomposite Cu-Fe-Al oxide catalysts. ChemCatChem 2020, 12, 4911–4921. [Google Scholar] [CrossRef]
- Fedorov, A.V.; Tsapina, A.M.; Bulavchenko, O.A.; Saraev, A.A.; Odegova, G.V.; Ermakov, D.Y.; Zubavichus, Y.V.; Yakovlev, V.A.; Kaichev, V.V. Structure and chemistry of Cu–Fe–Al nanocomposite catalysts for CO oxidation. Catal. Lett. 2018, 148, 3715–3722. [Google Scholar] [CrossRef]
- Engel, T.; Ertl, G. Surface residence times and reaction mechanism in the catalytic oxidation of CO on Pd(111). Chem. Phys. Lett. 1978, 54, 95–98. [Google Scholar] [CrossRef]
- Cisternas, J.; Holmes, P.; Kevrekidis, I.G.; Li, X. CO oxidation on thin Pt crystals: Temperature slaving and the derivation of lumped models. J. Chem. Phys. 2003, 118, 3312–3328. [Google Scholar] [CrossRef]
- Baxter, R.; Hu, P. Insight into why the Langmuir–Hinshelwood mechanism is generally preferred. J. Chem. Phys. 2002, 116, 4379–4381. [Google Scholar] [CrossRef]
- Hopstaken, M.J.P.; Niemantsverdriet, J.W. Structure sensitivity in the CO oxidation on rhodium: Effect of adsorbate coverages on oxidation kinetics on Rh(100) and Rh(111). J. Chem. Phys. 2000, 113, 5457. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R. Dynamic surface composition in a Mars-van Krevelen type reaction: CO oxidation on Au/TiO2. J. Catal. 2018, 357, 263–273. [Google Scholar] [CrossRef]
- Qi, L.; Yu, Q.; Dai, Y.; Tang, C.; Liu, L.; Zhang, H.; Gao, F.; Dong, L.; Chen, Y. Influence of cerium precursors on the structure and reducibility of mesoporous CuO-CeO2 catalysts for CO oxidation. Appl. Catal. B 2012, 119, 308–320. [Google Scholar] [CrossRef]
- Niu, J.; Liland, S.E.; Yang, J.; Rout, K.R.; Ran, J.; Chen, D. Effect of oxide additives on the hydrotalcite derived Ni catalysts for CO2 reforming of methane. Chem. Eng. J. 2018, 377, 119763. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Ma, H.; Sollund, E.S.; Zhang, W.; Fenes, E.; Qi, Y.; Wang, Y.; Rout, K.R.; Fuglerud, T.; Piccinini, M.; Chen, D. Kinetic modeling of dynamic changing active sites in a Mars-van Krevelen type reaction: Ethylene oxychlorination on K-doped CuCl2/Al2O3. Chem. Eng. J. 2021, 407, 128013. [Google Scholar] [CrossRef]
- Ma, H.; Wang, Y.; Zhang, H.; Ma, G.; Zhang, W.; Qi, Y.; Fuglerud, T.; Jiang, Z.; Ding, W.; Chen, D. Facet-Induced Strong Metal Chloride-Support Interaction over CuCl2/γ-Al2O3 Catalyst to Enhance Ethylene Oxychlorination Performance. ACS Catal. 2022, 12, 8027–8037. [Google Scholar] [CrossRef]
- Ma, H.; Wang, Y.; Qi, Y.; Rout, K.R.; Chen, D. Critical Review of Catalysis for Ethylene Oxychlorination. ACS Catal. 2020, 10, 9299–9319. [Google Scholar] [CrossRef]
- Xie, Y.-C.; Tang, Y.-Q. Spontaneous monolayer dispersion of oxides and salts onto surfaces of supports: Applications to heterogeneous catalysis. Adv. Catal. 1990, 37, 1–43. [Google Scholar] [CrossRef]
- Ma, H.; Fenes, E.; Qi, Y.; Wang, Y.; Rout, K.R.; Fuglerud, T.; Chen, D. Understanding of K and Mg co-promoter effect in ethylene oxychlorination by operando UV–vis-NIR spectroscopy. Catal. Today 2020, 369, 227–234. [Google Scholar] [CrossRef]
- Yang, G.; Haibo, Z.; Biying, Z. Monolayer dispersion of oxide additives on SnO2 and their promoting effects on thermal stability of SnO2 ultrafine particles. J. Mater. Sci. 2000, 35, 917–923. [Google Scholar] [CrossRef]
- Yu, X.-F.; Wu, N.-Z.; Xie, Y.-C.; Tang, Y.-Q. A monolayer dispersion study of titania-supported copper oxide. J. Mater. Chem. 2000, 10, 1629–1634. [Google Scholar] [CrossRef]
- Jernigan, G.G.; Somorjai, G.A. Carbon monoxide oxidation over three different oxidation states of copper: Metallic copper, copper (I) oxide, and copper (II) oxide-a surface science and kinetic study. J. Catal. 1994, 147, 567–577. [Google Scholar] [CrossRef]
- Nagase, K.; Zheng, Y.; Kodama, Y.; Kakuta, J. Dynamic Study of the Oxidation State of Copper in the Course of Carbon Monoxide Oxidation over Powdered CuO and Cu2O. J. Catal. 1999, 187, 123–130. [Google Scholar] [CrossRef]
Figure 1.
(a) N2 adsorption/desorption isotherms and (b) pore size distribution of all of the catalysts (calculated by the BJH method).
Figure 1.
(a) N2 adsorption/desorption isotherms and (b) pore size distribution of all of the catalysts (calculated by the BJH method).

Figure 2.
XRD patterns of the γ-Al2O3-supported catalysts.

Figure 3.
The temperature-dependent curve of the CO oxidation over the Al2O3-based catalysts. Reaction conditions: Wcat = 0.3 g, Ftot = 100 mL/min, PCO = 0.02 bar, O2/CO = 10/1, Ptot = 1 bar.
Figure 3.
The temperature-dependent curve of the CO oxidation over the Al2O3-based catalysts. Reaction conditions: Wcat = 0.3 g, Ftot = 100 mL/min, PCO = 0.02 bar, O2/CO = 10/1, Ptot = 1 bar.

Figure 4.
Stability test of the CO oxidation over the FeCu/Al2O3 catalyst. Reaction conditions: Wcat = 0.3 g, Ftot = 150 mL/min, PCO = 0.02 bar, O2/CO = 10/1, T = 250 °C, Ptot = 1 bar.
Figure 4.
Stability test of the CO oxidation over the FeCu/Al2O3 catalyst. Reaction conditions: Wcat = 0.3 g, Ftot = 150 mL/min, PCO = 0.02 bar, O2/CO = 10/1, T = 250 °C, Ptot = 1 bar.

Figure 5.
Transient CO reduction of the oxidized catalyst after treatment in O2. Reaction conditions: Wcat = 0.3 g, Ftot = 100 mL/min, PCO = 0.02 bar, T = 250 °C.
Figure 5.
Transient CO reduction of the oxidized catalyst after treatment in O2. Reaction conditions: Wcat = 0.3 g, Ftot = 100 mL/min, PCO = 0.02 bar, T = 250 °C.

Figure 6.
CO-TPR profile of the Cu/Al2O3 and FeCu/Al2O3 catalyst. Reaction conditions: Wcat = 0.3 g, FCO/Ar = 100 mL/min, ramping rate: 10 °C/min.
Figure 6.
CO-TPR profile of the Cu/Al2O3 and FeCu/Al2O3 catalyst. Reaction conditions: Wcat = 0.3 g, FCO/Ar = 100 mL/min, ramping rate: 10 °C/min.

Figure 7.
Normalized Cu K-edge XANES spectra of Cu/Al2O3 and FeCu/Al2O3 catalysts.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
BET surface area, pore volume, and average pore size for the samples. Pore volume and diameter calculations were determined using the BJH model on desorption data.
Table 1.
BET surface area, pore volume, and average pore size for the samples. Pore volume and diameter calculations were determined using the BJH model on desorption data.
| Catalyst | Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) | Metal Loading (wt%) |
|---|---|---|---|---|
| Cu/γ-Al2O3 | 181 | 0.55 | 9.07 | 5 |
| FeCu/γ-Al2O3 | 144 | 0.43 | 9.10 | 1/5 (Fe/Cu) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ma, G.; Wang, L.; Wang, X.; Li, L.; Ma, H. CO Oxidation over Alumina-Supported Copper Catalysts. Catalysts 2022, 12, 1030. https://doi.org/10.3390/catal12091030
AMA Style
Ma G, Wang L, Wang X, Li L, Ma H. CO Oxidation over Alumina-Supported Copper Catalysts. Catalysts. 2022; 12(9):1030. https://doi.org/10.3390/catal12091030
Chicago/Turabian StyleMa, Guoyan, Le Wang, Xiaorong Wang, Lu Li, and Hongfei Ma. 2022. "CO Oxidation over Alumina-Supported Copper Catalysts" Catalysts 12, no. 9: 1030. https://doi.org/10.3390/catal12091030
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.