
Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective


Department of Chemistry, National Taiwan Normal University, Taipei 11677, Taiwan
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(8), 890; https://doi.org/10.3390/catal12080890
Submission received: 17 July 2022
/
Revised: 7 August 2022
/
Accepted: 9 August 2022
/
Published: 12 August 2022
(This article belongs to the Topic Materials and Catalysts for Pollutants and CO2 Capture and Transformation)
Abstract
:The inclusion of transition metal elements within metal–organic frameworks (MOFs) is considered one of the most promising approaches for enhancing the catalytic capability of MOFs. In this study, MOF-253 containing bipyridine coordination sites is investigated for possible transition metal chelation, and a consequent possible CO2 reduction mechanism in the formation of formic acid. All transition metal elements of the third, fourth and fifth periods except hafnium and the lanthanide series are considered using density functional theory calculations. Two distinct types of CO2 reduction mechanisms are identified: (1) the five-coordination Pd center, which promotes formic acid generation via an intramolecular proton transfer pathway; (2) several four-coordination metal centers, including Mn, Pd, and Pt, which generate formic acid by means of heterolytic hydrogen activation. The MOF-253 environment is found to promote beneficial steric hindrance, and to constrain metal–ligand orientation, which consequently facilitates the formation of formic acid, particularly with the tetrahedral Mn center at high-spin electronic state.
1. Introduction
Metal-organic frameworks (MOFs) are synthetic materials based upon coordination chemistry and supramolecules. MOF structures are periodically constructed, with metal ions or metal oxide clusters as the connectors and organic linkers, extending multidimensionally to form porous crystalline materials. In addition to this, the geometric advantages of MOFs such as large surface area, tunable pore size, and structural design flexibility can provide a basis for applications in gas adsorption and storage, mixture separation, molecule recognition, and drug delivery. The diverse chemical functionality of organic linkers can be also used to advantage in sensing, catalysis, optical and luminescence applications [1].
The pioneer study of MOF-5 synthesis using zinc oxide clusters and 1,4-benzenedicarboxylic acid (bdc) demonstrated the achievement of stable porosity without the presence of guest molecules, thus preventing the structure from collapsing, resulting in the formation of a zeolite-like architecture [2]. In later work, a sub-network of inorganic connectors was formed in one or two dimensions, instead of the three dimensions of MOF-5, by using M(III)-oxide clusters, M = Cr and Al, and a bdc linker, to form MIL-53 [3,4]. The resulting MIL-53 architecture contained an array of one-dimensional pore channels; however, its accompanying breathing phenomena limit the utilization of its surface area [5]. In order to understand the origin of the breathing effect, Senkovska et al. introduced 2,6-naphthalene dicarboxylate (ndc) and 4,4′-biphenyl dicarboxylate (bpdc) linkers to the Al-oxide connectors and produced non-porous DUT-4 and permanent-porous DUT-5, with specific pore volumes of 0.68 and 0.81 g/cm3, respectively [6].
Gotthardt et al. functionalized a bpdc linker by introducing an additional amine, azide, alkyne, or nitro group at the 2-position [7]. The crystalline characters of these functionalized cases were found to remain similar to the parent DUT-5, except in the case of the azide group which decayed to form carbazole. The specific surface area measurements showed a noticeable reduction from 1880 m2/g for the parent materials to 1570, 1270, and 550 m2/g for the amine, alkyne, and nitro substituted versions, respectively. The authors also synthesized mixed linker versions containing a reduced number of amine groups for the post-synthetic modification (PSM), and the dilution of amine site density helped raise the peak surface area after PSM to a level close to that of the parent materials [7].
Bloch et al. introduced 2,2′-bipyridine-5,5′-dicarboxylic acid (bpydc) as linkers to aluminum oxide connectors, resulting in the formation of MOF-253 architecture [8]. The hard and oxophilic Al cations were found to preferentially bind with the hard carboxylate groups and left the soft bipyridine moiety for the subsequent metal ion chelation during the PSM process. PdCl2 was, therefore, successfully chelated to the bpydc binding site—a conclusion validated by extended X-ray adsorption fine structure (EXAFS) spectroscopy—with improved selective CO2(g) adsorption over N2(g).
Considering such a coordination environment as the MOF-253 architecture already mentioned, intuition suggests that it may be worthwhile to investigate possible CO2 reduction reactivity with respect to the various types of transition metal chelation. In a recent review by Wang and Astruc, abundant results from the literature were summarized as evidence for catalysis of organic reactions, electrocatalysis, and photocatalysis, using MOF-based or MOF-derived nanocatalytic materials [9]. However, only non-MOF-253 systems such as Ni/ZIF-8-C [10], Cu/Zn@C-submm [11], Pt/Au@Pd@UIO-66 [12], Ni@UIO-66 [13], 20Ni@MIL-101 [14], PZ8-400 [15], and FeZnK-NC [16] produced positive results for thermal-driven CO2 reduction catalysis. The full potential utilization of the bidentate bipyridine moiety of MOF-253, particularly for the purpose of CO2 reduction, still remains unclear, and it is, therefore, a matter worthy of further computational study. In the case of electrochemical catalysis, CO2 reduction using MOF-based materials is generally considered to be hindered by its insufficient electrical conductivity which results in excessive overpotential. For photocatalytic catalysis, a few instances of incorporation of Re and Ru into MOF-253 architecture were achieved by Li and coworkers [17,18,19].
To assess the diversity of transition metal chelation during the post-synthesis modifications process with MOF-253 architecture, we systematically investigated theoretical CO2 reduction mechanisms for various transition metal centers using density functional theory (DFT) calculations. The current computational study starts with a chlorinated metal site, followed by suitable hydride generation and CO2 hydrogenation, with respect to all transition metals of the periodic table under the MOF-253 architecture. In summary, this comprehensive theoretical analysis surveys the chelation selections and electronic structure profiles, step by step through the mechanistic process, and provides insightful information which may be beneficial for the development of chemical applications using MOF-253 materials.
2. Results and Discussion
2.1. Energetics and Structures of Metal Chloride Coordinated in MOF-253
The pristine MOF-253 supercell containing eight available N, N′–chelation sites is shown in Figure S1a. The energetics describing Pd or Sc coordination to the N, N′–chelation sites were investigated using MCl2 precursor presumptively, and the corresponding results are summarized in Table S1. Scenarios for six-to-eight coordinations are calculated for PdCl2 while four-to-eight-coordination scenarios are calculated for Sc. There are multiple geometric combinations for simulating the double vacancy; notations for labeling these vacancies are shown in Figure S1b and include the aa-type denoting two vacant sites (highlighted by two red circles) with positioning along the lattice a–vector. The choice of Pd or Sc as binding metal centers is based upon the molecular calculations of organometallic complex models, and the corresponding results are summarized in later sections.
As shown in Table S1, the average formation energy (Eavg) of nMCl2 coordination for both Pd and Sc cases appears to remain fairly consistent regardless of the amount of MCl2 chelation. Such a constant level of energetics suggests that the localized characteristic of metal chloride coordination to MOF-253 is well described within the first coordination shell. Consequently, an isolated organometallic complex model can qualitatively describe the electronic structure of metal-center reactivity. A substantial structural difference is identified with these two metal centers; the PdCl2 coordination (singlet d8 center) promotes a planar structure with respect to the bipyridine linker and ScCl2 (doublet d1 center) retains a tetrahedral structure. This structural difference has the potential to affect the CO2RR mechanistics and will be discussed in a later section.
Table S2 summarizes the structural deviation from the pristine MOF-253 subject to PdCl2 coordination. The ∠Al-O-Al′ bending angle, as depicted in Figure S1c, represents the strain along the one-dimensional aluminum oxide backbone due to the introduction of PdCl2 coordination. The pristine MOF-253 angle is estimated at 131.80 degrees in cases where seven–coordination or eight–coordination slightly deform the Al–O–Al chain. Such a trivial deformation suggests that high catalyst loading to MOF-253 could be enthalpically assessable despite the presence of vacant coordination sites produced by entropy effects. The dihedral angles (ϕ) of the linker–Al–Al′–linker′, as shown schematically in Figure S1d, also suggest a similar deformation effect corresponding to PdCl2 coordination. The rotation angle (τbpy) of bipyridine moiety along the C–C bond to the carboxylate group anchoring on the aluminum oxide backbone is depicted in Figure S1e. The pristine MOF-253 is found to have τbpy = 7.27° while the bpy moiety and carboxylate group retains a fairly coplanar structure to maximize its π conjugation. With the introduction of PdCl2, τbpy increases substantially to values above 20° in order to reduce the steric hindrance between the metal chloride and the neighboring linker. Upon vertical removal of PdCl2, the angles of τbpy for the optimized structures do not fall back to 7.27°, thus indicating the floppiness of bpy rotation.
2.2. Metal-Based CO2 Reduction Mechanism
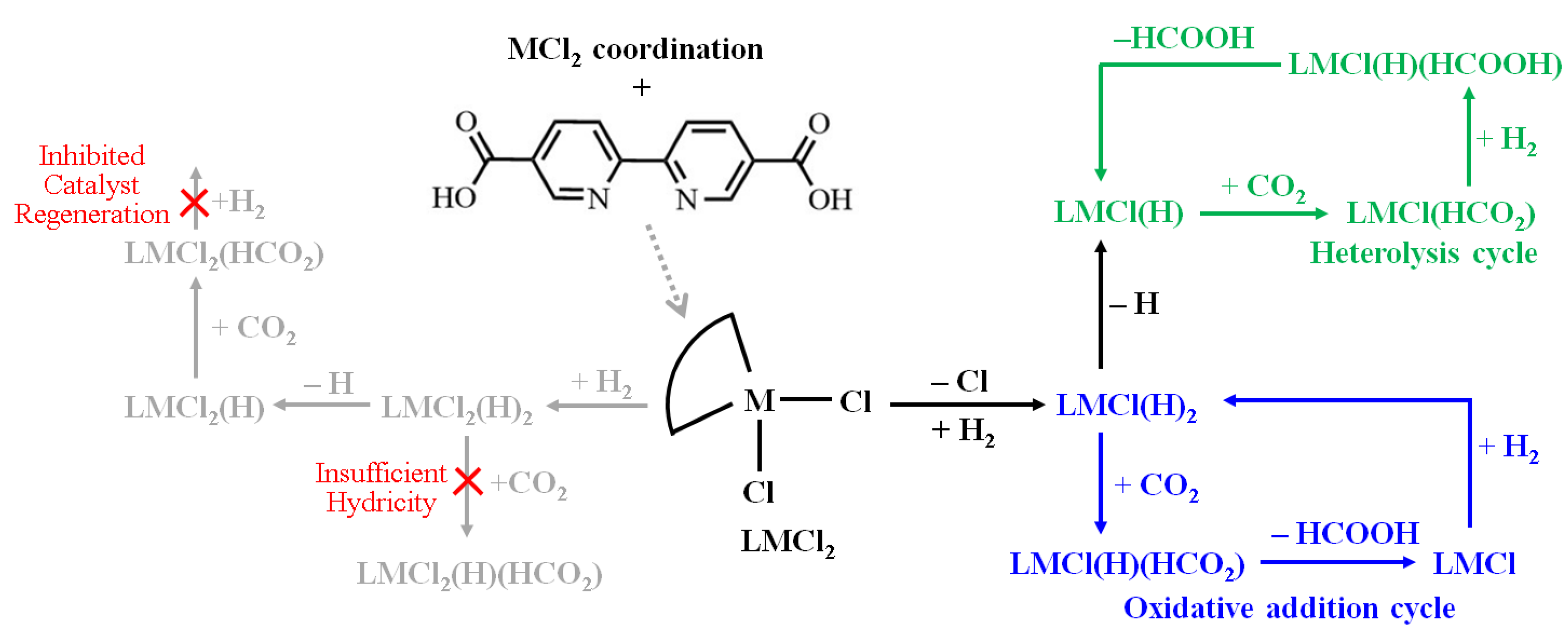
For the remainder of the discussion, the notation for the bpydc ligand is simplified by use of the term L in the expressions of metal complexes. For example, the M(bpydc)Cl2(H)2 complex is expressed as LMCl2(H)2. The mechanistic steps of CO2RR are depicted schematically in Figure 1, starting from the metal chloride coordination, followed by hydrogen activation and the subsequent CO2 hydrogenation.
The proposed catalytic mechanism focuses on the reaction pathways in the gas phase starting from LMCl2 + H2(g) + CO2(g). This assumed gas phase was chosen to avoid the chemical instability of MOFs resulting from an aqueous environment. On the basis of this assumption, four possible pathways are discussed in this study. The proposed four CO2 reduction pathways are: (a) CO2 reacting with six-coordination LMCl2(H)2 resulting from H2 cleavage on metal site; (b) CO2 reacting with five-coordination LMCl2(H) resulting from hydrogen atom transfer (HAT) of LMCl2(H)2; (c) CO2 reacting with five-coordination LMCl(H)2 resulting from H2 cleavage on LMCl; (d) CO2 reacting with four-coordination LMCl(H) resulting from HAT of LMCl(H)2.
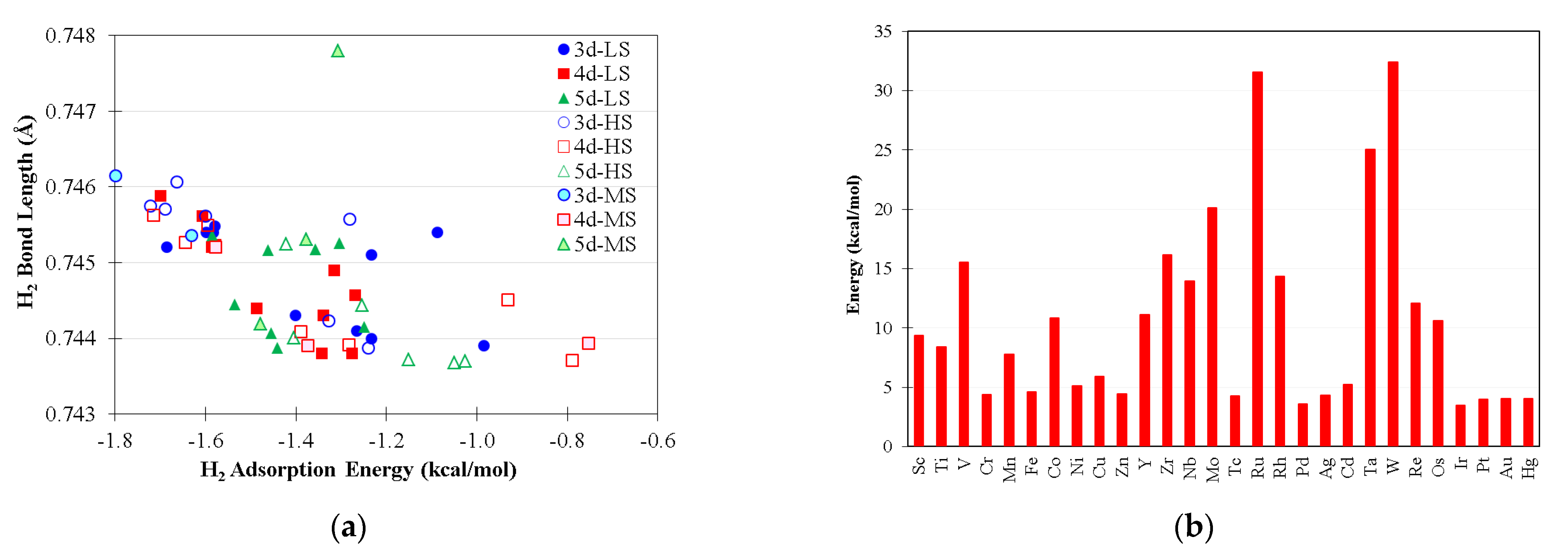
Figure 2a shows the adsorption energy of H2 on LMCl2 complexes, taking into account all possible electronic configurations, and Figure 2b shows the CO2 desorption energies of low-spin LMCl2(CO2) intermediates. The adsorption energies of H2 on LMCl2 are predicted to be noticeably weaker than in the majority of CO2 cases, and this implies that CO2 adsorption could competitively hinder H2 activation on the metal sites. Consequently, as increase in hydrogen gas pressure is important for the experimental design, as this should promote H2 activation. The metal back-donation to the σ* orbital of H2 on LMCl2, leading to H–H bond elongation, is fairly trivial, except in the case of the triplet LCrCl2 complex (s = 1) which binds H2 at −4.32 kcal/mol with rHH at 0.7724Å (not shown in Figure 2a).
None of the optimized LMCl2(H)2 models are able to carry out hydride transfer (HdT) to the incoming CO2 and form a stable LMCl2(H)(HCO2) structure at the low-spin states of all metal centers. The hydrido ligands are found to preferentially coordinate with the metal centers. The presence of two chloro ligands decreases metal center electron density subsequently hinders hydricity. Additionally, some of the metal centers cannot even activate H2 and undergo oxidative addition to form stable dihydrido intermediates, as shown in Table S4.
The insufficient hydricity of the six-coordination LMCl2(H)2 intermediates can be addressed by hydrogen atom transfer (HAT) or chlorine atom transfer (ClAT) steps, leading to the formation of five-coordination LMCl2H or LMCl(H)2 intermediates.. The recommendation of possible reaction reagents for carrying out these atom-transfer processes in the gas phase is beyond the scope of this study, and only the bonding-breaking energetics of HAT or ClAT are reported herein. The free energies of HAT and ClAT processes from LMCl2(H2) are summarized in Figure S2. The HAT step is defined with respect to LMCl2(H2) containing hydrogen in molecular form, and this reference selection provides the equal basis for comparing all metal candidates. The ClAT process appears to be generally more energy-consuming than the corresponding HAT process.
2.3. Metal Screening
2.3.1. Five-Coordination Acting Catalysts
After one ClAT step, the hydricity of LMCl(H)2 is improved with respect to the di-chloro counterparts. Most of the calculated LMCl(H)2 models at the low-spin state, i.e., M = Sc–Zn, Y–Cd, and Hf–Hg, exhibit stable geometries, except in the particular cases of M = Mn, Fe, Ni, Cu, and Ag. These unstable cases were found to favor the molecular hydrogen form as LMCl(H2). Among the stable LMCl(H)2 cases, M = Sc, Ti, Y, Zr, and Pd models were able to react with CO2 and subsequently form the hydride transferred intermediates. Table 1 summarizes the reaction energetics of the successful hydride transfer reactions for M = Sc, Ti, Y, Zr, and Pd cases at the low-spin state. Only three types of HdT intermediates are identified in Table 1, i.e., ScClH(η2-OCOH), TiClH(η1-OCHO), and PdClH(η1-HCO2). The M = Y and Zr cases were found to have similar coordination geometries to the Ti case.
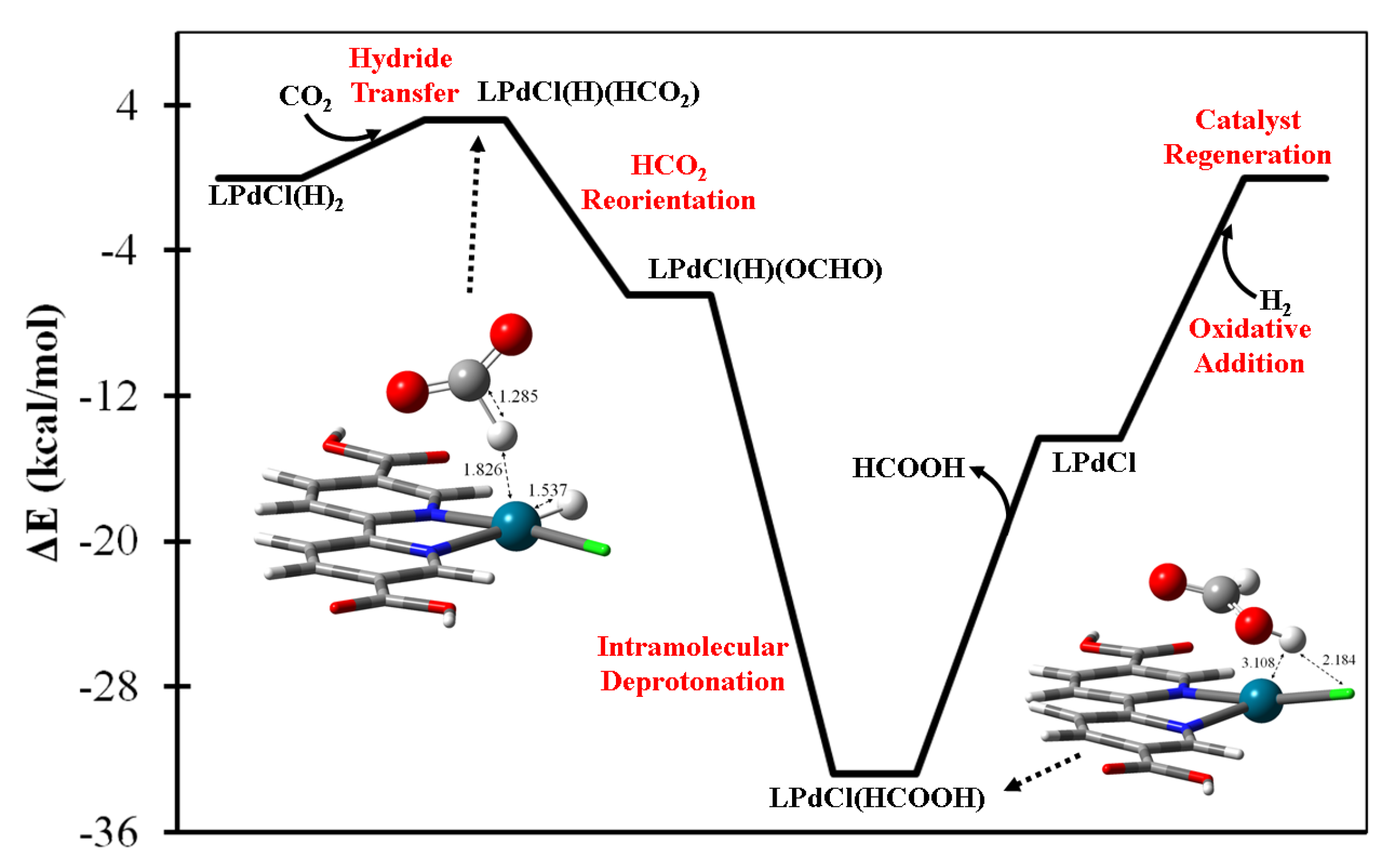
The LPdCl(H)2 complex is predicted to be the acting catalyst for formic acid generation resulting from gaseous H2 and CO2 molecules. The proposed catalytic energetics are summarized in Figure 3 which illustrates how LPd(H)(HCO2) is formed via the HdT step. The thermal rotation could facilitate the reorientation of the HCO2 ligand and result in a more stable oxygen-coordinated LPdCl(H)(OCHO) intermediate, followed by intramolecular deprotonation to form formic acid—LPdCl(HCOOH)—as shown in the legend of Figure 3. Upon the desorption of HCOOH, the bare LPdCl complex is capable of undergoing oxidative addition with the second equivalent H2(g) to regenerate the acting LPdCl(H)2 complex. The most energy-consuming step is predicted to be formic acid desorption, at a level of 18.5 kcal/mol.
The second type of five-coordination complexes—LMCl2(H)—resulting from the HAT process of LMCl2(H)2 are presumed to be catalytic-inactive for CO2 reduction as noted in Figure 1. Even if the hydride-transferred intermediate—LMCl2(HCO2)—was able to form, such a saturated coordinated metal center would prohibit further H2 activation, and prevent the regeneration of LMCl2(H) and release of HCOOH.
2.3.2. Four-Coordination Acting Catalysts
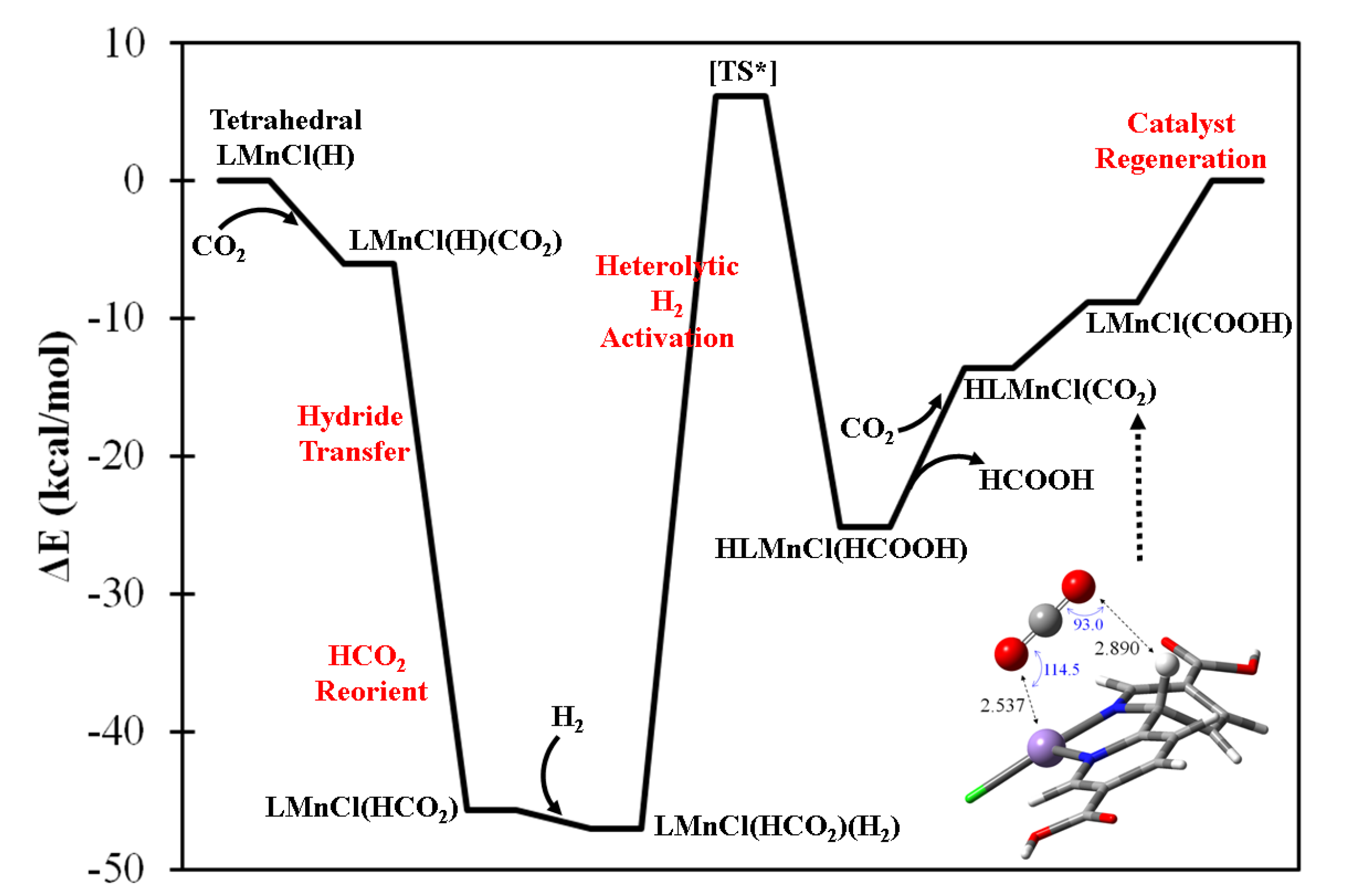
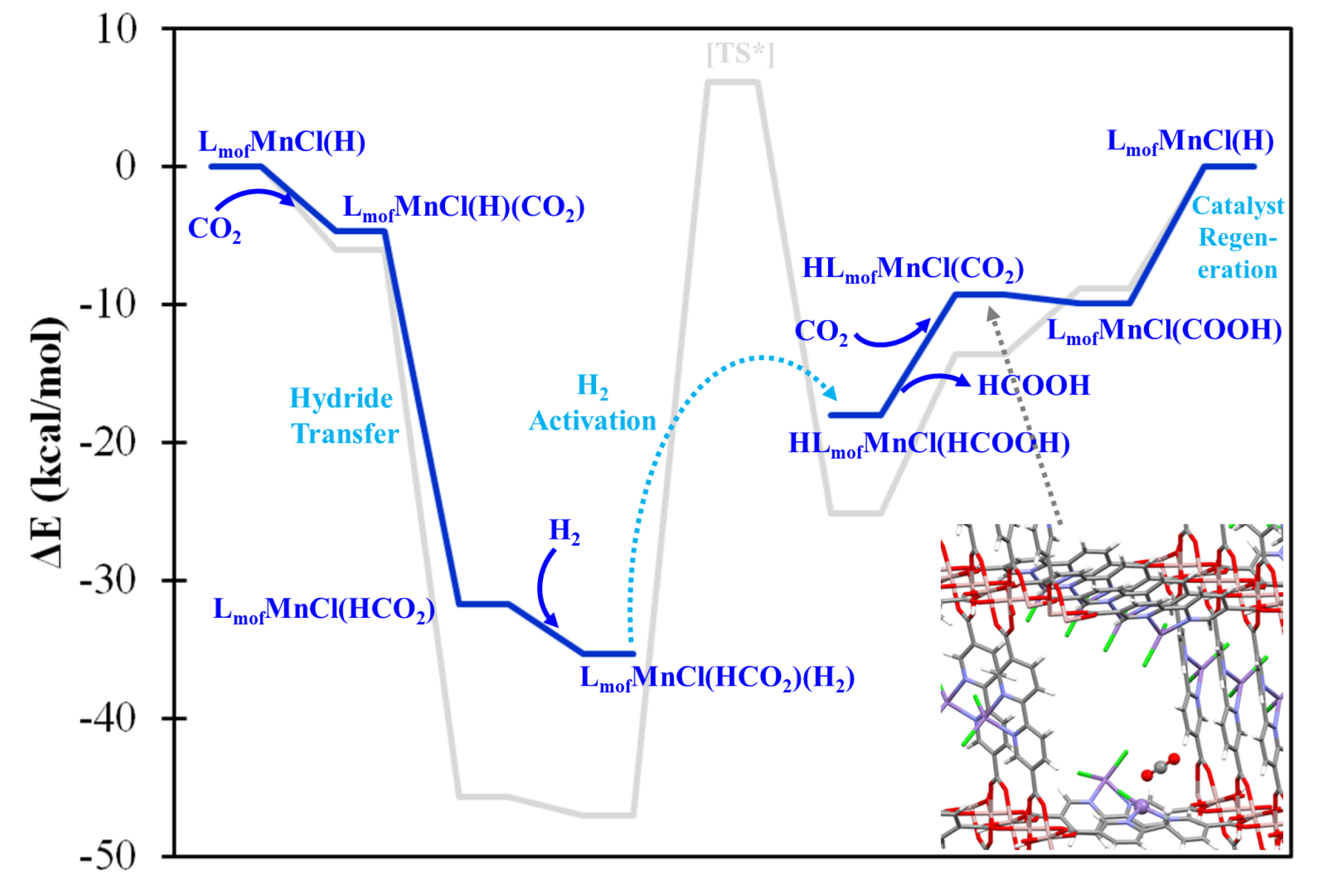
The four-coordination LMCl(H) complexes are generally considered to be hydricity-enhanced forms in comparison with their five-coordination LMCl(H)2 counterparts. The formation of LMCl(H) complexes can be achieved by one step of HAT from the five-coordination LMCl(H)2 complexes. Table S5 summarizes the calculated HAT energetics with respect to the lowest-energy electronic configuration of LMCl(H) and LMCl(H)2 complexes. Several metal centers appear to possess sizable and exothermic HAT energies, i.e., M = V, Cr, Mn, Zn, Pd, Cd, Pt, and Hg, indicating the possible stability of these acting catalytic forms. As an example, Figure 4 shows the predicted catalytic mechanism of formic acid formation using tetrahedral LMnCl(H) complex in which the oxidation state of metal center is determined as Mn2+ with s = 5/2. The hydrido ligand is able to react with the incoming CO2 molecule exothermically, and subsequently form LMnCl(HCO2) complex. The HCO2− ligand could further reorient to form a bidentate coordination, and facilate the H2 heterolytic cleavage process, being cooperatively influenced by the cationic Mn center. The atomic charges of H2 of the approximated transition state (see Figure S3) indicate the presence of a strongly polarized inter-hydrogen bond. However, the back-donation effect from Mn center is subtle, and this is supported by the calculated net charge of H2 of almost zero. The energy for the heterolytic H2 activation is approximated as 53.1 kcal/mol with respect to the stable H2-adsorbed intermediate—LMnCl(HCO2)(H2)—as shown in Figure S3. Because the bond dissociation energy of a gaseous H2 molecule is estimated to be 109.9 kcal/mol at the current DFT level, the LMnCl(HCO2) intermediate is considered to substantially activate the H–H bond in a heterolytic manner. Once H2 is successfully activated, one proton is predicted to bond with the HCO2– ligand, forming HCOOH, and the hydride is predicted to react with the bpydc ligand. With the desorption of HCOOH, the Mn center becomes spatially available to interact with another CO2 molecule and form a four-coordination CO2-containing (carbon-on) intermediate. The hydrogenated bpydc ligand of HLMnCl(CO2) intermediate could presumably undergo a proton-coupled electron transfer process to form the LMnCl(COOH) intermediate and require only 4.8 kcal/mol reaction energy. As shown in Figure S4, two vibrational modes of LMnCl(COOH) are identified which initiate the regeneration of the acting form of LMnCl(H) through COOH ligand reorientation and the consequent CO2 desorption.
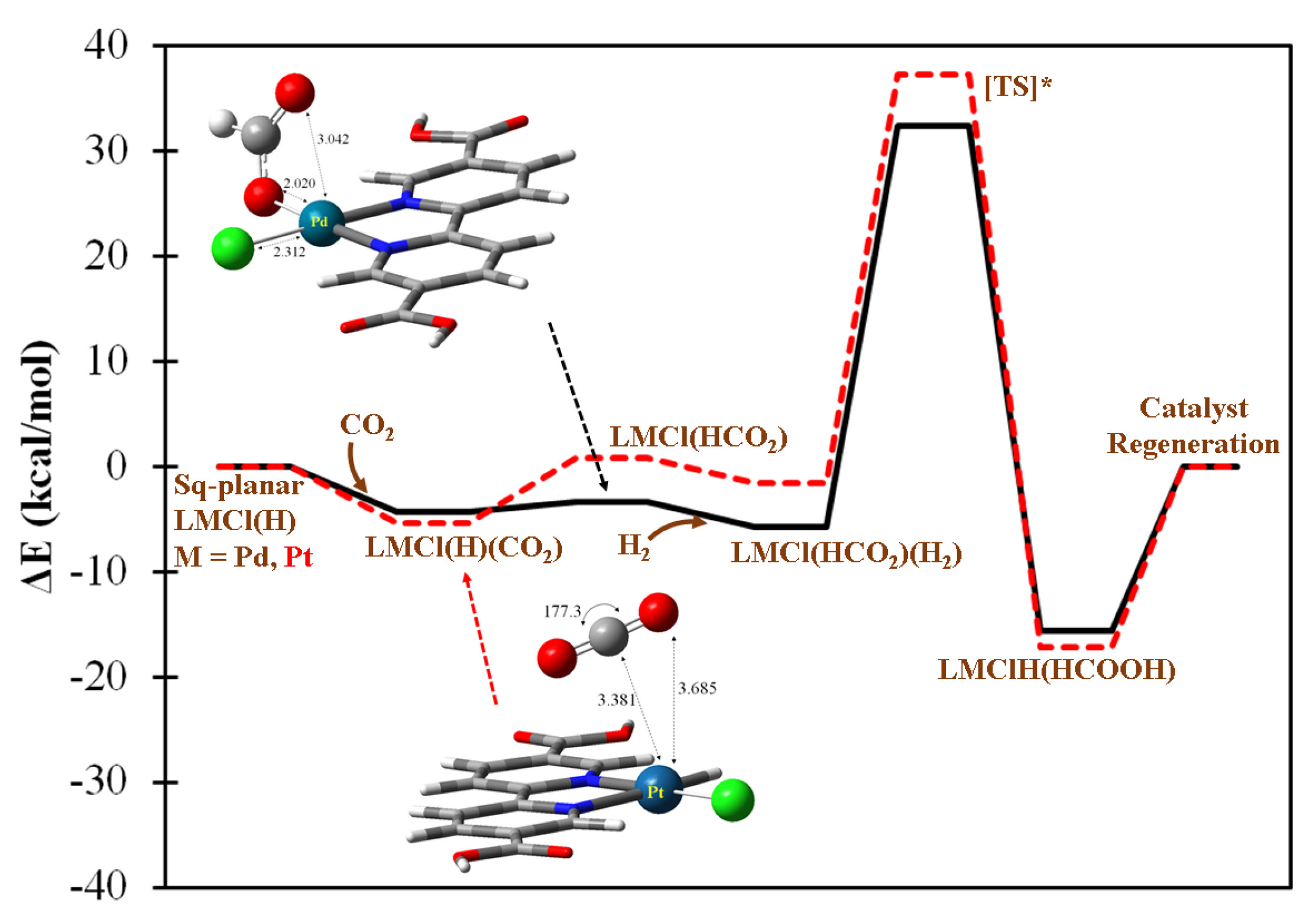
Two square planar LMCl(H), M = Pd and Pt, complexes are also identified as catalytically active for the purposes of formic acid generation. The predicted mechanistic profiles for Pd and Pt cases are fairly similar, as can be seen in Figure 5 which shows how CO2 can be adsorbed by the metal centers exothermically, this being followed by the highly accessible hydride transfer step to form LMCl(HCO2) intermediates. The cleavage of H2 by both metal centers is found to be more substantially accessible (~38 kcal/mol) than in the aforementioned Mn case due to the presence of a noticeable back-donation effect from these late-transition metal centers. In Figure S5, the values of the net charge balance of H2 for the approximated transition state structures are shown to deviate substantially from zero. This could imply the electronic density transferred from the complex moiety is nontrivial, subsequently facilitating the H2 bond breaking step.
The schematic H2 activation profiles for other four-coordination LMCl(H) complexes are summarized in Figure S6. The V, Zn, Cd, and Hg cases appear to follow heterolytic H2 activation pathways with energy demands close to the Mn model; the remaining metal centers are not considered as actively catalytic on account of their low stability, or non-physical H2 activation profiles.
2.4. Confirming the LMnCl(H) Model in MOF-253
In order to assess the viability of the formic acid generation mechanism predicted by the molecular models, the high-spin Mn complexes were explicitly incorporated in MOF-253, being represented by a periodic boundary condition model with experimental lattice constants. The high-spin LMnCl(H) model would favor tetrahedral coordination, and such a structure would be significantly repelled by the neighboring linker in the MOF environment. Conversely, the five-coordination LMCl(H)2 and four-coordination square planar LMCl(H) complexes would be more spatially feasible in the MOF-253 environment. Figure 6 shows the energetic profile (blue line) of LmofMnCl(H) where Lmof denotes the bpy binding site of MOF-253. The predicted profile of MOF-253 is consistent with that of the molecular model (gray line), though with a more moderate energy requirement. Despite this difference in profile between the molecular model and periodic model, which might be attributed to the use of different DFT functionals, at least in part, the steric hindrance introduced by the MOF environment appears to be the dominant factor involved, and thus facilitates formic acid generation.
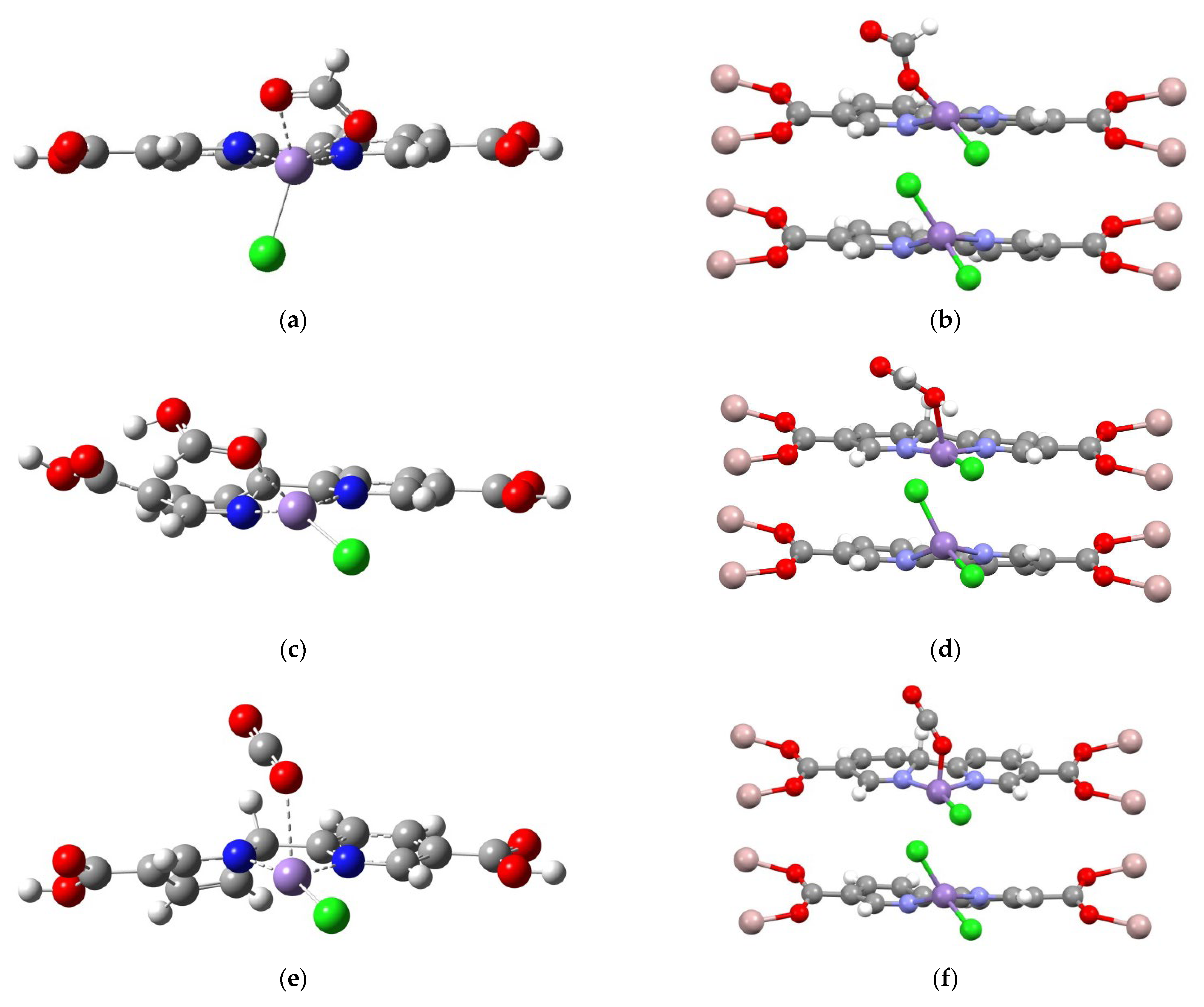
In Figure 7, three characteristic structural features of the MOF environment which benefit the catalytic mechanism are identified. A comparison of Figure 7a–d shows that the LmofMnCl(HCO2) model retains a distorted square planar geometry, while the corresponding molecular LMnCl(HCO2) model favors tetrahedral-like coordination (the η2-HCO2 ligand is qualitatively accounted as monodentate). Such a distorted square planar geometry could subsequently enhance H2 adsorption. In Figure 7b, the HLMnCl(HCOOH) intermediate described by the molecular model appears to have substantial out-of-plane distortion at the carboxylate groups, due to the intrinsic quality of negative-charged Hbpy moiety and the hydrogen bond interaction between the HCOOH ligand and the carboxylate group. Such an unfavorable distortion is suppressed by the carboxylated–Al coordination of MOF, consequently constraining the bpydc ligand to a coplanar geometry and enhancing HCOOH desorption. In Figure 7c, the molecular model of HLMnCl(CO2) intermediate contains the out-of-plane distortion of the Hbpy moiety. However, the constrained planar geometry of Hbpy moiety in MOF (see Figure 7f) can improve hydride transfer back to the CO2 ligand, subsequently forming the LmofMnCl(COOH) intermediate.
3. Computational Methodology
The CO2 reduction mechanism was assessed using a series of single M(bpydc)Cl2 complex models in the gaseous condition without including the aluminum secondary building unit (SBU) computationally in the models. The transition metal elements, including Sc–Zn, Y–Cd, and Hf–Hg, were investigated. The energetics of possible intermediates during the reduction mechanism process were optimized with the hybrid B3LYP functional [20,21,22], in which elements H, C, N, and O were described by basis set of 6–31 g with diffuse functions [23], with the addition of a polarized function [24] to the heavy atoms. This hybrid function has been commonly adopted for theoretical characterizations of the formic acid generation mechanism by organometallic complexes [25,26,27,28,29,30,31,32] The transition metals were described by triple-zeta quality LANL2TZ pseudopotential basis set except Hf [33]. The van der Waals interactions were taken into account using the D3 version of Grimme’s dispersion with Becke–Johnson damping [34,35]. The minimum structures were determined by frequency calculations. All the DFT calculations using the complex models were carried out by the Gaussian 16 package [36].
Several of the selected transition metal centers, which were recommended by the complex modeling and will be discussed in later sections, were subsequently investigated for the presence of SBU. The unit cell was taken from the experimental orthorhombic morphology with corresponding lattice constants (a = 23.59 Å, b = 6.91 Å, c = 19.84 Å) [8]. The periodic boundary condition (PBC) models were a 1 × 2 × 1 supercell with extension along the Al-O-Al chain in order to take into account eight binding sites in the models. The periodic models were calculated with generalized gradient approximation (GGA) using Perdew–Burke–Ernzerh (PBE) [37] exchange–correlation functional and projector augmented-wave (PAW) [38,39] methods. The plane wave basis set was expanded to a cutoff energy level of 400 eV. The van der Waals interactions of the solid materials were also described by the D3 version of Grimme’s dispersion Becke–Johnson damping. The convergence criteria were determined as 10−5 eV for total energy change and 0.04 eV/Å for all forces for optimization, with the lattice constants remaining frozen. All periodic simulations were carried out by Vienna Ab initio Simulation Package (VASP 5.3.5) [40,41,42].
4. Conclusions
A series of M(bpydc)Cl2, M = Sc–Zn, Y–Cd, and Ta–Hg complexes were computationally investigated for formic acid generation resulting from CO2 and H2. Two types of catalytic pathways were proposed using five-coordination LMCl(H)2 and four-coordination LMCl(H) complexes. LPdCl(H)2 was selected to undergo an intramolecular proton transfer mechanism with an energy consumption level of below 20 kcal/mol. LMCl(H), M = Mn (high spin), Pd, and Pt were the representative four-coordination acting catalysts with characteristic heterolytic hydrogen activation steps where the most energy-consuming steps are greater than 40 kcal/mol. With the metal coordination to MOF-253 materials, the electronic structure of the bpydc ligand appeared to be localized to a single metal site, as suggested by the energetics of the periodic model simulations. However, the steric hindrance of linker in MOF was found to enhance the catalytic energetics of the tetrahedral-type four-coordination Mn center, particularly in the case of the hydride-transferred LMnCl(HCO2), HLMnCl(HCOOH), and HLMnCl(CO2) intermediates. For the square pyramidal five-coordination and square planar four-coordination types of metal centers, the beneficial role resulting from the steric effect of linker is likely trivial. The current computational study provides new insights into catalytic mechanisms involved in formic acid generation in MOF materials, as well as establishing a helpful modeling protocol for the post-synthesis of transition metal coordination to MOF materials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12080890/s1. Figure S1: the geometric label definition used in MOF-253 model; Figure S2: energetic comparison between H atom transfer and Cl atom transfer for low-spin LMH2Cl2 complexes; Figure S3: the calculated energetics along H–H interatomic distance of LMnCl(HCO2)(H2) complex; Figure S4: the identified vibrational modes of LMnCl(COOH) minimum structures; Figure S5: the calculated energetics along H–H interatomic distance of LPdCl(HCO2)(H2) and LPtCl(HCO2)(H2) complexes; Figure S6: the H2 activation profiles of MCl(HCO2), M = V, Zn, Cd, and Hg complexes; Table S1: various MCl2, M = Pd and Sc, formation energetics in MOF-253; Table S2: calculated structural information of MOF-253 upon PdCl2 coordination; Table S3: the labels of the spin states of LMCl2(H2) intermediates; Table S4: the H-H bond lengths of the optimized 6-coordinated LMCl2(H)2 intermediates; Table S5: the H-H bond lengths of the optimized 5-coordinated LMCl(H)2 intermediates.
Author Contributions
Methodology and data curation: M.-C.H. Draft writing—review and editing: M.-C.H., R.K., and M.-K.T. Conceptualization and supervision: M.-K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This study is supported by the Ministry of Science and Technology of Taiwan (110-2124-M-003-001 and 110-2113-M-003-015). R.K. extends his gratitude to MOST for a postdoctoral fellowship (MOST 110-2811-M-003-522). M.C.H. extends his gratitude to MOST-NTNU for award of outstanding doctoral student scholarship.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful for the computational resources provided by the National Center for High-Performance Computing of Taiwan and the Center for Cloud Computing at National Taiwan Normal University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rowsell, J.L.C.; Yaghi, O.M. Metal–organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Serre, C.; Millange, F.; Thouvenot, C.; Noguès, M.; Marsolier, G.; Louër, D.; Férey, G. Very Large Breathing Effect in the First Nanoporous Chromium(III)-Based Solids: MIL-53 or CrIII(OH)·{O2C−C6H4−CO2}·{HO2C−C6H4−CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, T.; Serre, C.; Huguenard, C.; Fink, G.; Taulelle, F.; Henry, M.; Bataille, T.; Férey, G. A Rationale for the Large Breathing of the Porous Aluminum Terephthalate (MIL-53) Upon Hydration. Chem. Eur. J. 2004, 10, 1373–1382. [Google Scholar] [CrossRef]
- Férey, G.; Serre, C. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar] [CrossRef]
- Senkovska, I.; Hoffmann, F.; Fröba, M.; Getzschmann, J.; Böhlmann, W.; Kaskel, S. New highly porous aluminium based metal-organic frameworks: Al(OH)(ndc) (ndc=2,6-naphthalene dicarboxylate) and Al(OH)(bpdc) (bpdc=4,4′-biphenyl dicarboxylate). Microporous Mesoporous Mater. 2009, 122, 93–98. [Google Scholar] [CrossRef]
- Gotthardt, M.A.; Grosjean, S.; Brunner, T.S.; Kotzel, J.; Gänzler, A.M.; Wolf, S.; Bräse, S.; Kleist, W. Synthesis and post-synthetic modification of amine-, alkyne-, azide- and nitro-functionalized metal–organic frameworks based on DUT-5. Dalton Trans. 2015, 44, 16802–16809. [Google Scholar] [CrossRef]
- Bloch, E.D.; Britt, D.; Lee, C.; Doonan, C.J.; Uribe-Romo, F.J.; Furukawa, H.; Long, J.R.; Yaghi, O.M. Metal Insertion in a Microporous Metal−Organic Framework Lined with 2,2′-Bipyridine. J. Am. Chem. Soc. 2010, 132, 14382–14384. [Google Scholar] [CrossRef]
- Wang, Q.; Astruc, D. State of the Art and Prospects in Metal-Organic Framework (MOF)-Based and MOF-Derived Nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar] [CrossRef]
- Li, Y.; Cai, X.; Chen, S.; Zhang, H.; Zhang, K.H.L.; Hong, J.; Chen, B.; Kuo, D.-H.; Wang, W. Highly Dispersed Metal Carbide on ZIF-Derived Pyridinic-N-Doped Carbon for CO2 Enrichment and Selective Hydrogenation. ChemSusChem 2018, 11, 1040–1047. [Google Scholar] [CrossRef]
- Zhang, J.; An, B.; Hong, Y.; Meng, Y.; Hu, X.; Wang, C.; Lin, J.; Lin, W.; Wang, Y. Pyrolysis of metal–organic frameworks to hierarchical porous Cu/Zn-nanoparticle@carbon materials for efficient CO2 hydrogenation. Mater. Chem. Front. 2017, 1, 2405–2409. [Google Scholar] [CrossRef]
- Zheng, Z.; Xu, H.; Xu, Z.; Ge, J. A Monodispersed Spherical Zr-Based Metal–Organic Framework Catalyst, Pt/Au@Pd@UIO-66, Comprising an Au@Pd Core–Shell Encapsulated in a UIO-66 Center and Its Highly Selective CO2 Hydrogenation to Produce CO. Small 2018, 14, 1702812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-W.; Zhou, X.; Liu, Y.-N.; Shen, C.-C.; Yuan, C.-Z.; Jiang, Y.-F.; Zhao, S.-J.; Ma, L.-B.; Cheang, T.-Y.; Xu, A.-W. Ultrasmall Ni nanoparticles embedded in Zr-based MOFs provide high selectivity for CO2 hydrogenation to methane at low temperatures. Catal. Sci. Technol. 2018, 8, 3160–3165. [Google Scholar] [CrossRef]
- Zhen, W.; Gao, F.; Tian, B.; Ding, P.; Deng, Y.; Li, Z.; Gao, H.; Lu, G. Enhancing activity for carbon dioxide methanation by encapsulating (111) facet Ni particle in metal–organic frameworks at low temperature. J. Catal. 2017, 348, 200–211. [Google Scholar] [CrossRef]
- Yin, Y.; Hu, B.; Li, X.; Zhou, X.; Hong, X.; Liu, G. Pd@zeolitic imidazolate framework-8 derived PdZn alloy catalysts for efficient hydrogenation of CO2 to methanol. Appl. Catal. B 2018, 234, 143–152. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Y.; Jiang, X.; Zhang, A.; Song, C.; Guo, X. Pyrolyzing ZIF-8 to N-doped porous carbon facilitated by iron and potassium for CO2 hydrogenation to value-added hydrocarbons. J. CO2 Util. 2018, 25, 120–127. [Google Scholar] [CrossRef]
- Sun, D.; Gao, Y.; Fu, J.; Zeng, X.; Chen, Z.; Li, Z. Construction of a supported Ru complex on bifunctional MOF-253 for photocatalytic CO2 reduction under visible light. Chem. Commun. 2015, 51, 2645–2648. [Google Scholar] [CrossRef]
- Deng, X.; Albero, J.; Xu, L.; García, H.; Li, Z. Construction of a Stable Ru-Re Hybrid System Based on Multifunctional MOF-253 for Efficient Photocatalytic CO2 Reduction. Inorg. Chem. 2018, 57, 8276–8286. [Google Scholar] [CrossRef]
- Deng, X.; Qin, Y.; Hao, M.; Li, Z. MOF-253-Supported Ru Complex for Photocatalytic CO2 Reduction by Coupling with Semidehydrogenation of 1,2,3,4-Tetrahydroisoquinoline (THIQ). Inorg. Chem. 2019, 58, 16574–16580. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Ashvar, C.S.; Chabalowski, C.F.; Frisch, M.J. Theoretical Calculation of Vibrational Circular-Dichroism Spectra. Faraday Discuss. 1994, 99, 103–119. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular-Orbital Methods 25. Supplementary Functions for Gaussian-Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Madsen, M.R.; Rønne, M.H.; Heuschen, M.; Golo, D.; Ahlquist, M.S.G.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)3Br as Electrocatalyst and Triethylamine/Isopropanol as Additives. J. Am. Chem. Soc. 2021, 143, 20491–20500. [Google Scholar] [CrossRef] [PubMed]
- Cramer, H.H.; Ye, S.; Neese, F.; Werlé, C.; Leitner, W. Cobalt-Catalyzed Hydrosilylation of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Level—How to Control the Catalytic Network? J. Am. Chem. Soc. Au 2021, 1, 2058–2069. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Song, Y.; Zhang, J.; Zeng, L.; Wang, C.; Lin, W. Cooperative Copper Centres in a Metal–Organic Framework for Selective Conversion of CO2 to Ethanol. Nat. Catal. 2019, 2, 709–717. [Google Scholar] [CrossRef]
- Pascher, T.F.; Ončák, M.; van der Linde, C.; Beyer, M.K. Release of Formic Acid from Copper Formate: Hydride, Proton-Coupled Electron and Hydrogen Atom Transfer All Play their Role. ChemPhysChem 2019, 20, 1420–1424. [Google Scholar] [CrossRef]
- Hameed, Y.; Rao, G.K.; Ovens, J.S.; Gabidullin, B.; Richeson, D. Visible-Light Photocatalytic Reduction of CO2 to Formic Acid with a Ru Catalyst Supported by N,N′-Bis(diphenylphosphino)-2,6-diaminopyridine Ligands. ChemSusChem 2019, 12, 3453–3457. [Google Scholar] [CrossRef]
- Johnson, S.I.; Nielsen, R.J.; Goddard, W.A. Selectivity for HCO2− over H2 in the Electrochemical Catalytic Reduction of CO2 by (POCOP)IrH2. ACS Catal. 2016, 6, 6362–6371. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Neese, F.; Ye, S. Toward Rational Design of 3d Transition Metal Catalysts for CO2 Hydrogenation Based on Insights into Hydricity-Controlled Rate-Determining Steps. Inorg. Chem. 2016, 55, 5438–5444. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations-Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Abinitio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed reaction pathways of CO2 reduction by LMCl2 complexes. The gray pathways are unfavorable cases; green and blue pathways are accessible cases.
Figure 1.
Proposed reaction pathways of CO2 reduction by LMCl2 complexes. The gray pathways are unfavorable cases; green and blue pathways are accessible cases.

Figure 2.
(a) The H2 adsorption energies and bond lengths of LMCl2(H2) intermediates where basis set superposition errors are corrected; LS, MS, and HS denote the low-spin, high-spin, and medium spin states of complexes, respectively (see Table S3); (b) The CO2 desorption energies of low-spin LMCl2(CO2) intermediates.
Figure 2.
(a) The H2 adsorption energies and bond lengths of LMCl2(H2) intermediates where basis set superposition errors are corrected; LS, MS, and HS denote the low-spin, high-spin, and medium spin states of complexes, respectively (see Table S3); (b) The CO2 desorption energies of low-spin LMCl2(CO2) intermediates.

Figure 3.
The predicted catalytic mechanism of HCOOH(g) generation by the (bpydc)PdCl(H)2 complex with H2(g) + CO2(g).
Figure 3.
The predicted catalytic mechanism of HCOOH(g) generation by the (bpydc)PdCl(H)2 complex with H2(g) + CO2(g).

Figure 4.
The predicted catalytic mechanism of formic acid generation by high-spin (s = 5/2) (bpydc)MnCl(H) complex with H2(g) + CO2(g). [TS*] denotes the approximate transition state.
Figure 4.
The predicted catalytic mechanism of formic acid generation by high-spin (s = 5/2) (bpydc)MnCl(H) complex with H2(g) + CO2(g). [TS*] denotes the approximate transition state.

Figure 5.
The predicted catalytic mechanism of formic acid generation by singlet (bpydc)MCl(H), M = Pd or Pt complex with H2(g) + CO2(g). The solid black and dashed red lines denote the catalytic pathways conducted by Pd and Pt complexes, respectively, and share the same text labels as the predicted intermediates. [TS*] denotes the approximate transition state.
Figure 5.
The predicted catalytic mechanism of formic acid generation by singlet (bpydc)MCl(H), M = Pd or Pt complex with H2(g) + CO2(g). The solid black and dashed red lines denote the catalytic pathways conducted by Pd and Pt complexes, respectively, and share the same text labels as the predicted intermediates. [TS*] denotes the approximate transition state.

Figure 6.
The predicted catalytic mechanism (blue lines) of formic acid generation by LmofMnCl(H) catalytic site with H2(g) + CO2(g). The gray lines denote the mechanism of the molecular model. [TS*] denotes the approximate transition state.
Figure 6.
The predicted catalytic mechanism (blue lines) of formic acid generation by LmofMnCl(H) catalytic site with H2(g) + CO2(g). The gray lines denote the mechanism of the molecular model. [TS*] denotes the approximate transition state.

Figure 7.
Schematic comparisons of LMnCl(HCO2), HLMnCl(HCOOH), and HLMnCl(CO2) intermediates using the molecular (a–c) and periodic (d–f) models, respectively.
Figure 7.
Schematic comparisons of LMnCl(HCO2), HLMnCl(HCOOH), and HLMnCl(CO2) intermediates using the molecular (a–c) and periodic (d–f) models, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The hydride transfer results of MCl(H)2 + CO2 → MCl(H)(HCO2) step 1,3.
| Metal | Multi 4 | ΔE 2 |  |
| Sc | 1 | −18.0 | |
| Ti | 2 | −43.4 | |
| Y | 1 | −53.3 | |
| Zr | 2 | −48.7 | |
| Pd | 2 | +3.2 |
1 All possible multiplicities of the neutral MCl(H)(HCO2) intermediates are calculated. See SI for details of the energetics. 2 The reaction energy (in kcal/mol) is estimated under the same multiplicity conditions. 3 Schematic representations of ScClH(OCOH), TiClH(OCHO), and PdClH(HCO2), respectively, with the notation of the important interatomic distances (Å) and bond angles. 4 Multi stands for multiplicity.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hsieh, M.-C.; Krishnan, R.; Tsai, M.-K. Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective. Catalysts 2022, 12, 890. https://doi.org/10.3390/catal12080890
AMA Style
Hsieh M-C, Krishnan R, Tsai M-K. Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective. Catalysts. 2022; 12(8):890. https://doi.org/10.3390/catal12080890
Chicago/Turabian StyleHsieh, Meng-Chi, Ranganathan Krishnan, and Ming-Kang Tsai. 2022. "Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective" Catalysts 12, no. 8: 890. https://doi.org/10.3390/catal12080890
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.