
Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective


Abstract
:1. Introduction
2. Results and Discussion
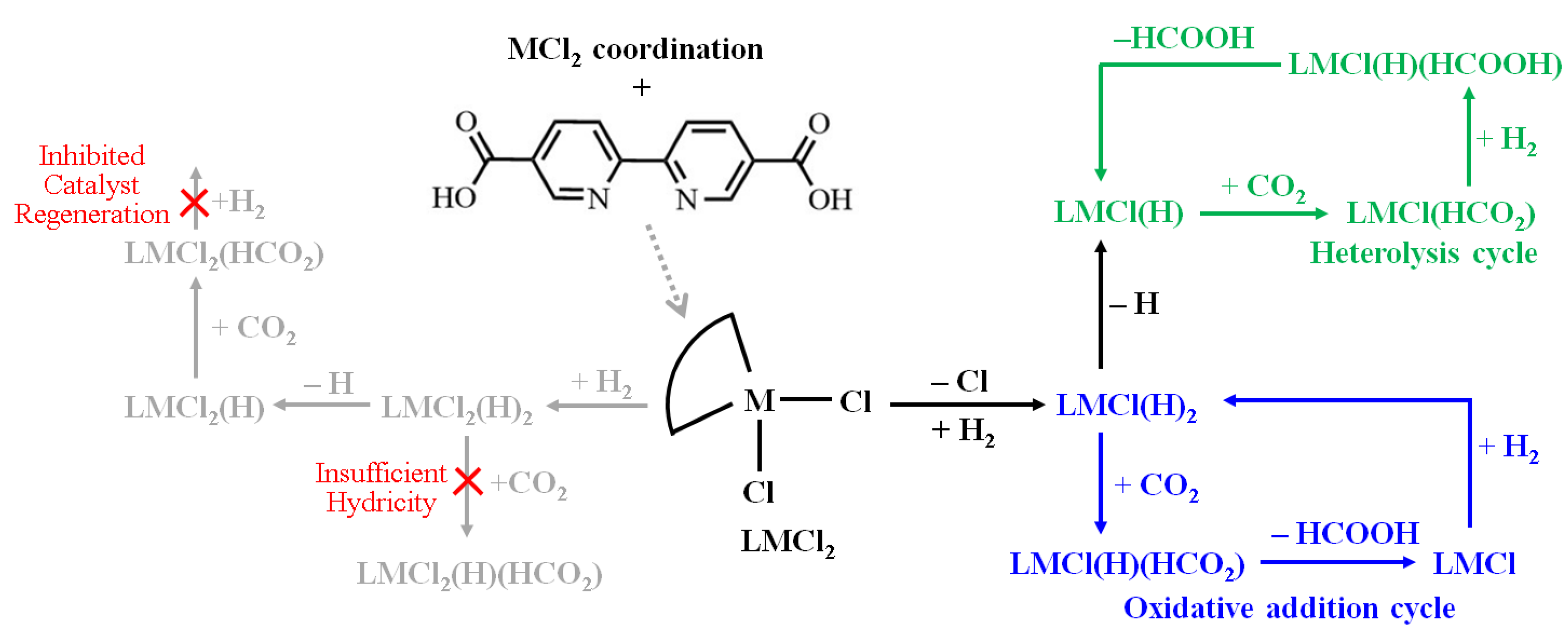
2.1. Energetics and Structures of Metal Chloride Coordinated in MOF-253
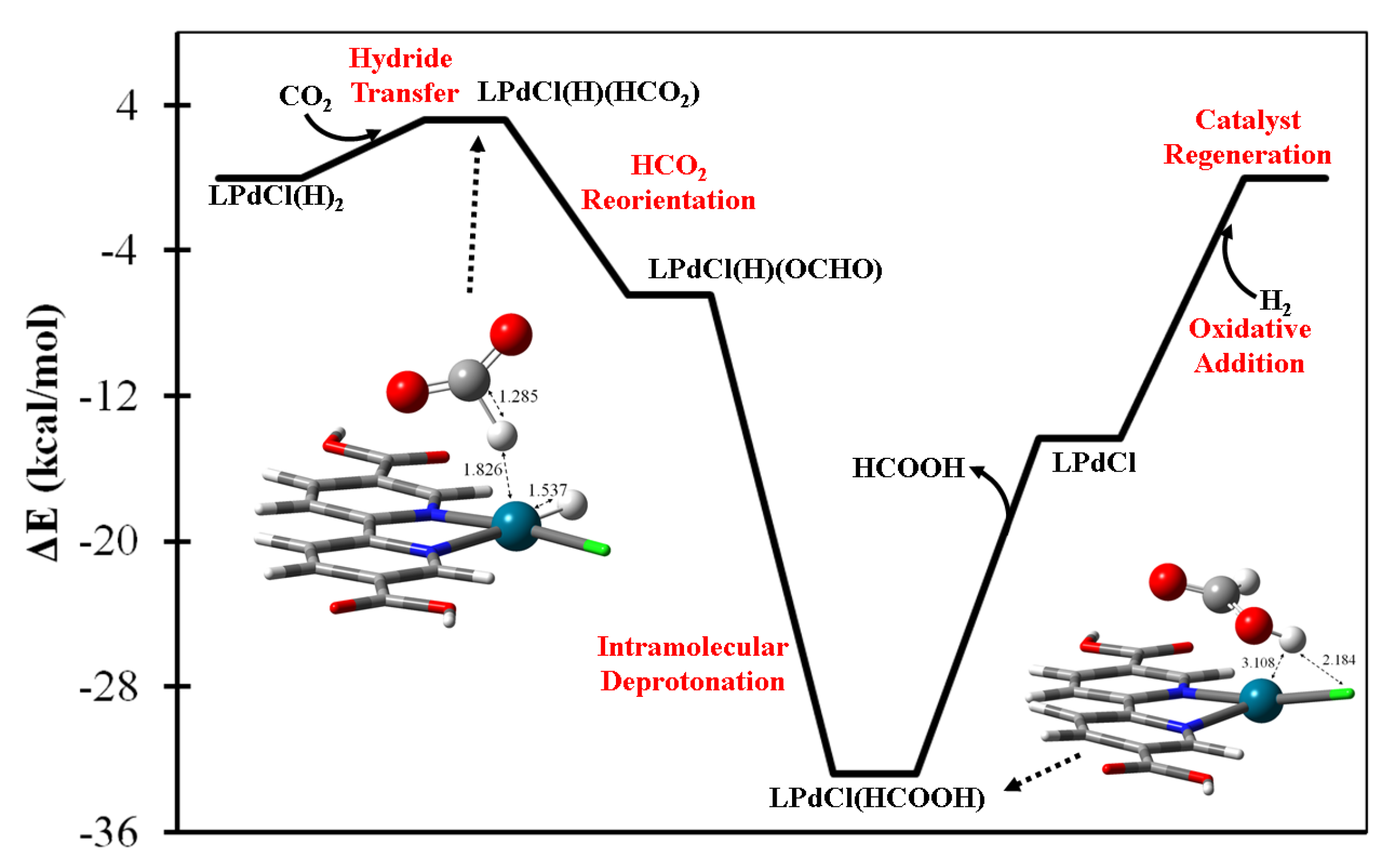
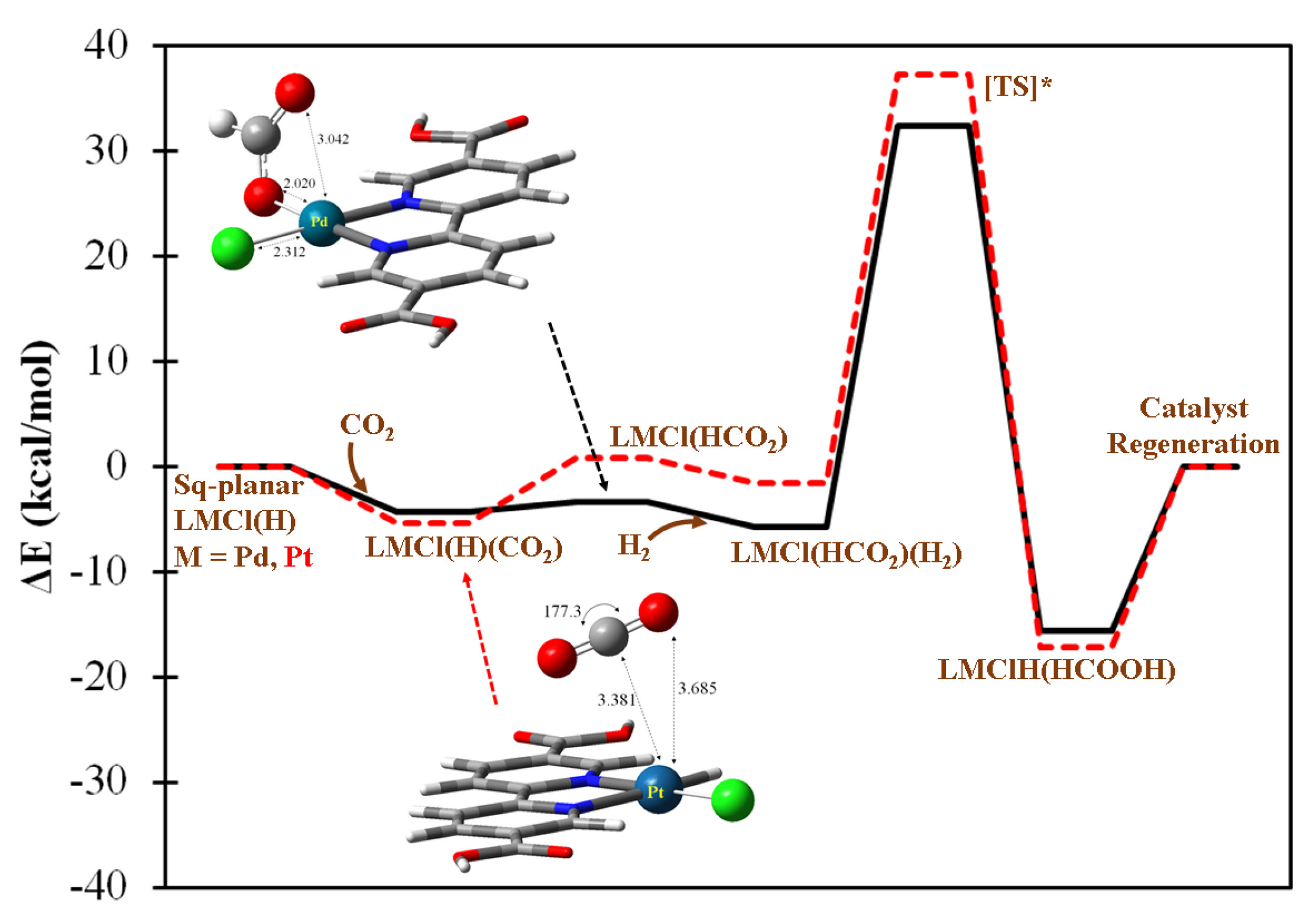
2.2. Metal-Based CO2 Reduction Mechanism
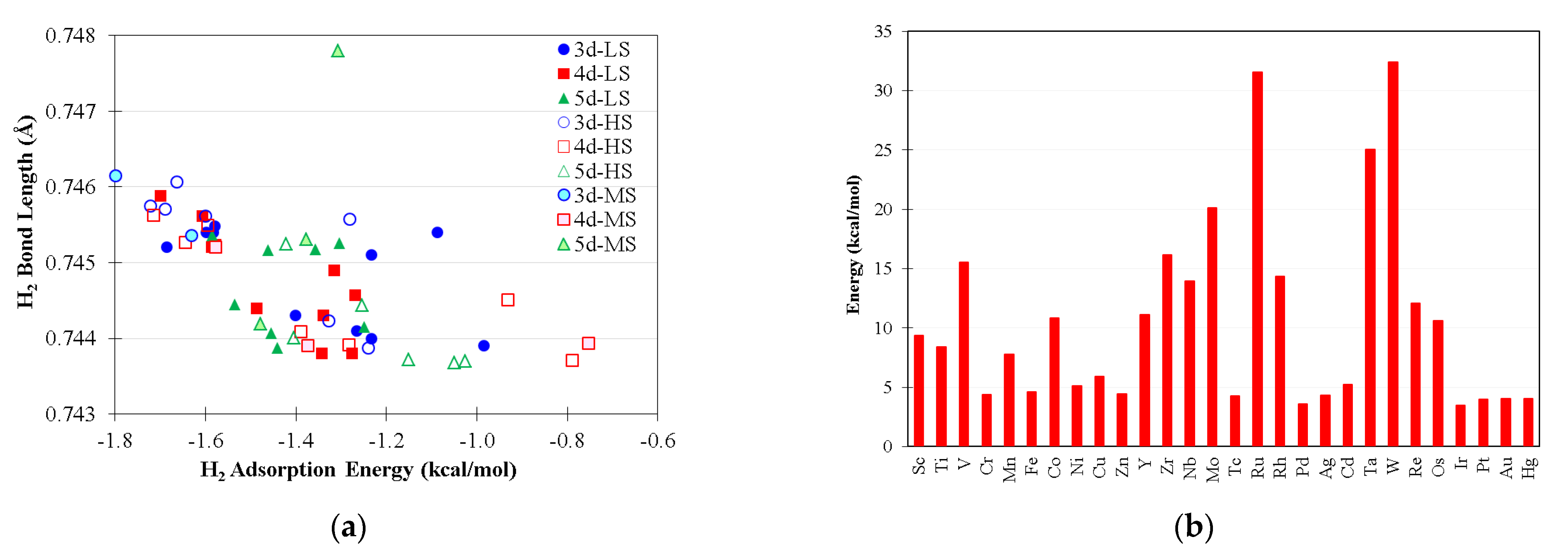
2.3. Metal Screening
2.3.1. Five-Coordination Acting Catalysts
2.3.2. Four-Coordination Acting Catalysts
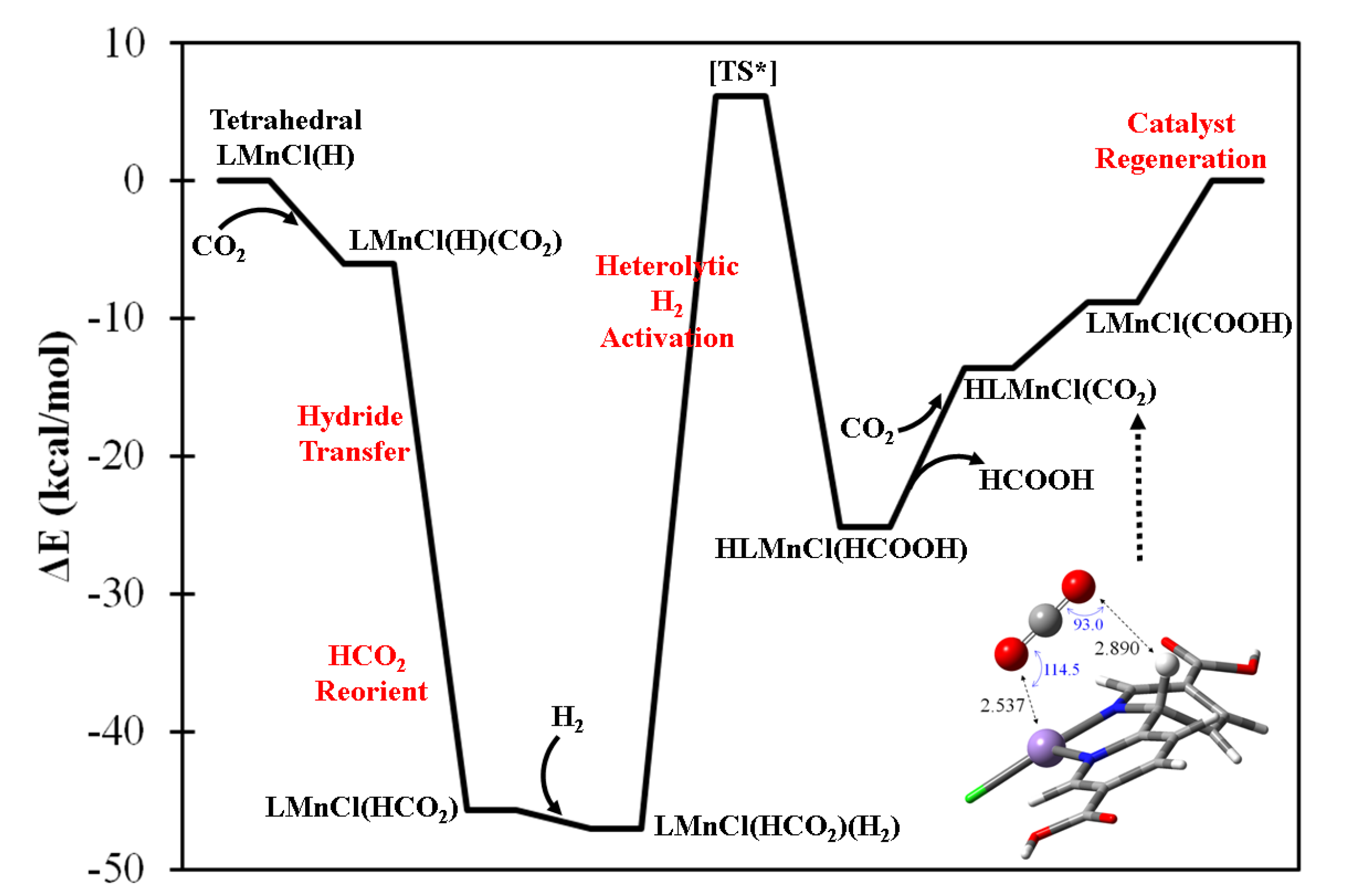
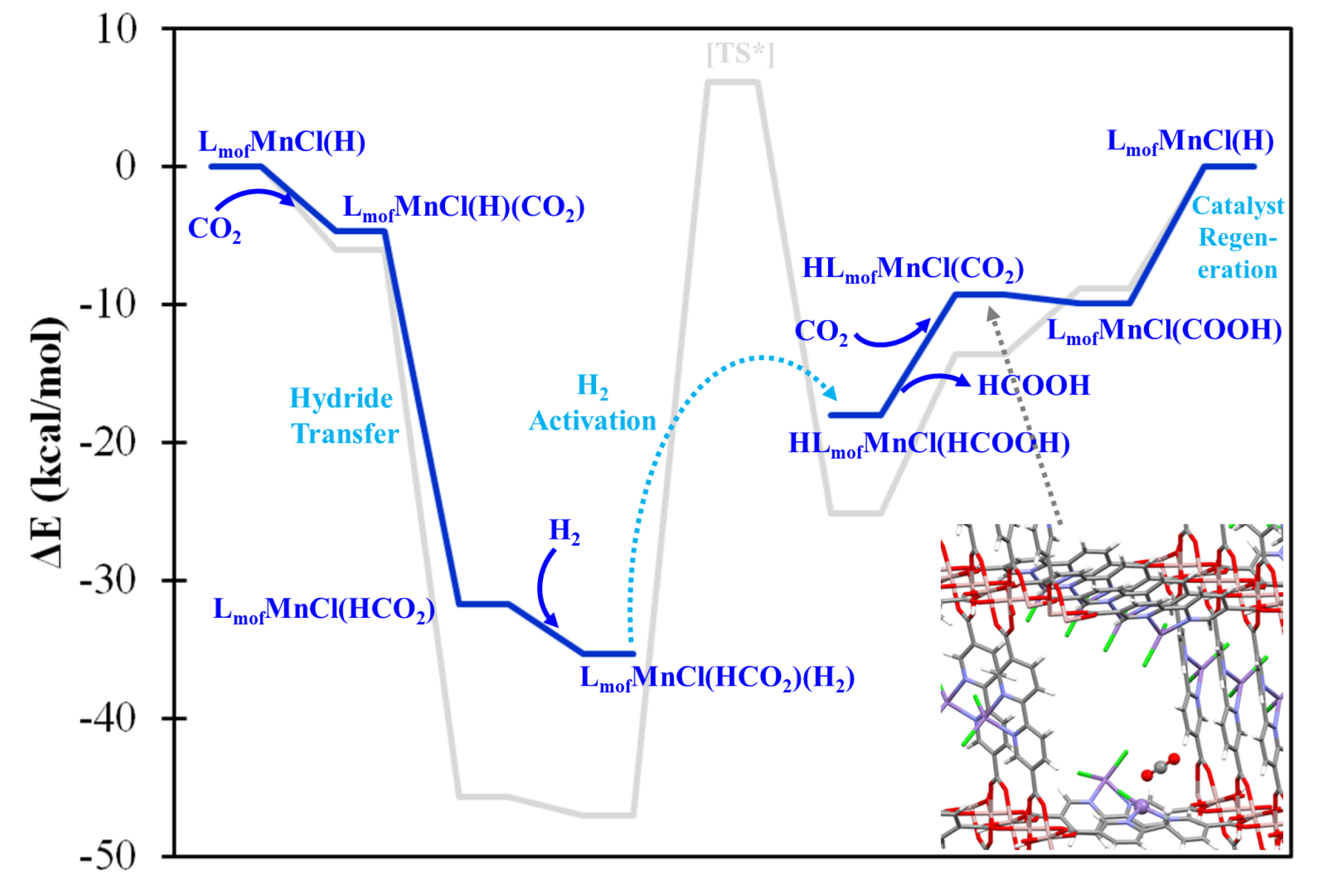
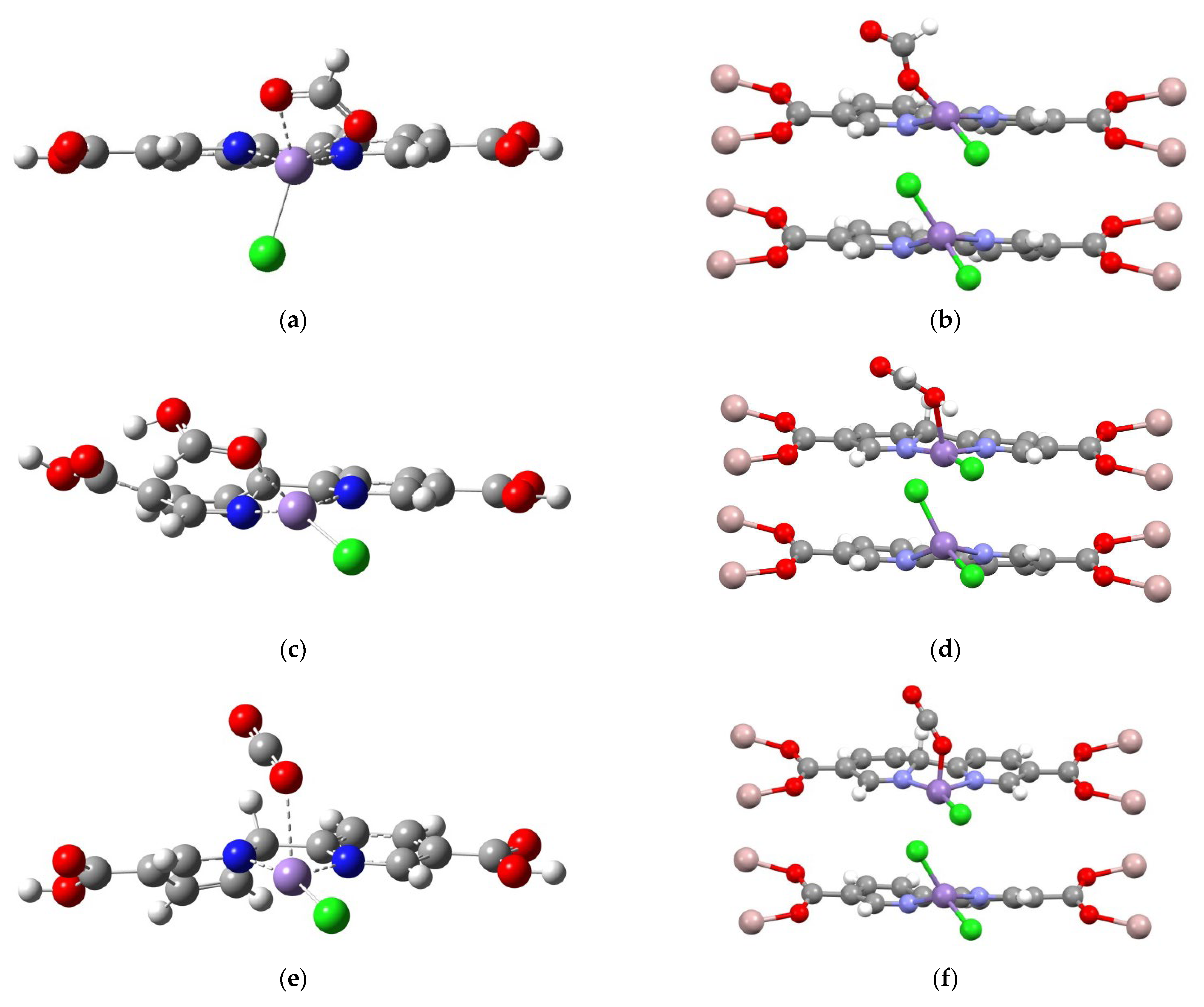
2.4. Confirming the LMnCl(H) Model in MOF-253
3. Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowsell, J.L.C.; Yaghi, O.M. Metal–organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Serre, C.; Millange, F.; Thouvenot, C.; Noguès, M.; Marsolier, G.; Louër, D.; Férey, G. Very Large Breathing Effect in the First Nanoporous Chromium(III)-Based Solids: MIL-53 or CrIII(OH)·{O2C−C6H4−CO2}·{HO2C−C6H4−CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, T.; Serre, C.; Huguenard, C.; Fink, G.; Taulelle, F.; Henry, M.; Bataille, T.; Férey, G. A Rationale for the Large Breathing of the Porous Aluminum Terephthalate (MIL-53) Upon Hydration. Chem. Eur. J. 2004, 10, 1373–1382. [Google Scholar] [CrossRef]
- Férey, G.; Serre, C. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar] [CrossRef]
- Senkovska, I.; Hoffmann, F.; Fröba, M.; Getzschmann, J.; Böhlmann, W.; Kaskel, S. New highly porous aluminium based metal-organic frameworks: Al(OH)(ndc) (ndc=2,6-naphthalene dicarboxylate) and Al(OH)(bpdc) (bpdc=4,4′-biphenyl dicarboxylate). Microporous Mesoporous Mater. 2009, 122, 93–98. [Google Scholar] [CrossRef]
- Gotthardt, M.A.; Grosjean, S.; Brunner, T.S.; Kotzel, J.; Gänzler, A.M.; Wolf, S.; Bräse, S.; Kleist, W. Synthesis and post-synthetic modification of amine-, alkyne-, azide- and nitro-functionalized metal–organic frameworks based on DUT-5. Dalton Trans. 2015, 44, 16802–16809. [Google Scholar] [CrossRef]
- Bloch, E.D.; Britt, D.; Lee, C.; Doonan, C.J.; Uribe-Romo, F.J.; Furukawa, H.; Long, J.R.; Yaghi, O.M. Metal Insertion in a Microporous Metal−Organic Framework Lined with 2,2′-Bipyridine. J. Am. Chem. Soc. 2010, 132, 14382–14384. [Google Scholar] [CrossRef]
- Wang, Q.; Astruc, D. State of the Art and Prospects in Metal-Organic Framework (MOF)-Based and MOF-Derived Nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar] [CrossRef]
- Li, Y.; Cai, X.; Chen, S.; Zhang, H.; Zhang, K.H.L.; Hong, J.; Chen, B.; Kuo, D.-H.; Wang, W. Highly Dispersed Metal Carbide on ZIF-Derived Pyridinic-N-Doped Carbon for CO2 Enrichment and Selective Hydrogenation. ChemSusChem 2018, 11, 1040–1047. [Google Scholar] [CrossRef]
- Zhang, J.; An, B.; Hong, Y.; Meng, Y.; Hu, X.; Wang, C.; Lin, J.; Lin, W.; Wang, Y. Pyrolysis of metal–organic frameworks to hierarchical porous Cu/Zn-nanoparticle@carbon materials for efficient CO2 hydrogenation. Mater. Chem. Front. 2017, 1, 2405–2409. [Google Scholar] [CrossRef]
- Zheng, Z.; Xu, H.; Xu, Z.; Ge, J. A Monodispersed Spherical Zr-Based Metal–Organic Framework Catalyst, Pt/Au@Pd@UIO-66, Comprising an Au@Pd Core–Shell Encapsulated in a UIO-66 Center and Its Highly Selective CO2 Hydrogenation to Produce CO. Small 2018, 14, 1702812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-W.; Zhou, X.; Liu, Y.-N.; Shen, C.-C.; Yuan, C.-Z.; Jiang, Y.-F.; Zhao, S.-J.; Ma, L.-B.; Cheang, T.-Y.; Xu, A.-W. Ultrasmall Ni nanoparticles embedded in Zr-based MOFs provide high selectivity for CO2 hydrogenation to methane at low temperatures. Catal. Sci. Technol. 2018, 8, 3160–3165. [Google Scholar] [CrossRef]
- Zhen, W.; Gao, F.; Tian, B.; Ding, P.; Deng, Y.; Li, Z.; Gao, H.; Lu, G. Enhancing activity for carbon dioxide methanation by encapsulating (111) facet Ni particle in metal–organic frameworks at low temperature. J. Catal. 2017, 348, 200–211. [Google Scholar] [CrossRef]
- Yin, Y.; Hu, B.; Li, X.; Zhou, X.; Hong, X.; Liu, G. Pd@zeolitic imidazolate framework-8 derived PdZn alloy catalysts for efficient hydrogenation of CO2 to methanol. Appl. Catal. B 2018, 234, 143–152. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Y.; Jiang, X.; Zhang, A.; Song, C.; Guo, X. Pyrolyzing ZIF-8 to N-doped porous carbon facilitated by iron and potassium for CO2 hydrogenation to value-added hydrocarbons. J. CO2 Util. 2018, 25, 120–127. [Google Scholar] [CrossRef]
- Sun, D.; Gao, Y.; Fu, J.; Zeng, X.; Chen, Z.; Li, Z. Construction of a supported Ru complex on bifunctional MOF-253 for photocatalytic CO2 reduction under visible light. Chem. Commun. 2015, 51, 2645–2648. [Google Scholar] [CrossRef]
- Deng, X.; Albero, J.; Xu, L.; García, H.; Li, Z. Construction of a Stable Ru-Re Hybrid System Based on Multifunctional MOF-253 for Efficient Photocatalytic CO2 Reduction. Inorg. Chem. 2018, 57, 8276–8286. [Google Scholar] [CrossRef]
- Deng, X.; Qin, Y.; Hao, M.; Li, Z. MOF-253-Supported Ru Complex for Photocatalytic CO2 Reduction by Coupling with Semidehydrogenation of 1,2,3,4-Tetrahydroisoquinoline (THIQ). Inorg. Chem. 2019, 58, 16574–16580. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Ashvar, C.S.; Chabalowski, C.F.; Frisch, M.J. Theoretical Calculation of Vibrational Circular-Dichroism Spectra. Faraday Discuss. 1994, 99, 103–119. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular-Orbital Methods 25. Supplementary Functions for Gaussian-Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Madsen, M.R.; Rønne, M.H.; Heuschen, M.; Golo, D.; Ahlquist, M.S.G.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)3Br as Electrocatalyst and Triethylamine/Isopropanol as Additives. J. Am. Chem. Soc. 2021, 143, 20491–20500. [Google Scholar] [CrossRef] [PubMed]
- Cramer, H.H.; Ye, S.; Neese, F.; Werlé, C.; Leitner, W. Cobalt-Catalyzed Hydrosilylation of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Level—How to Control the Catalytic Network? J. Am. Chem. Soc. Au 2021, 1, 2058–2069. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Song, Y.; Zhang, J.; Zeng, L.; Wang, C.; Lin, W. Cooperative Copper Centres in a Metal–Organic Framework for Selective Conversion of CO2 to Ethanol. Nat. Catal. 2019, 2, 709–717. [Google Scholar] [CrossRef]
- Pascher, T.F.; Ončák, M.; van der Linde, C.; Beyer, M.K. Release of Formic Acid from Copper Formate: Hydride, Proton-Coupled Electron and Hydrogen Atom Transfer All Play their Role. ChemPhysChem 2019, 20, 1420–1424. [Google Scholar] [CrossRef]
- Hameed, Y.; Rao, G.K.; Ovens, J.S.; Gabidullin, B.; Richeson, D. Visible-Light Photocatalytic Reduction of CO2 to Formic Acid with a Ru Catalyst Supported by N,N′-Bis(diphenylphosphino)-2,6-diaminopyridine Ligands. ChemSusChem 2019, 12, 3453–3457. [Google Scholar] [CrossRef]
- Johnson, S.I.; Nielsen, R.J.; Goddard, W.A. Selectivity for HCO2− over H2 in the Electrochemical Catalytic Reduction of CO2 by (POCOP)IrH2. ACS Catal. 2016, 6, 6362–6371. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Neese, F.; Ye, S. Toward Rational Design of 3d Transition Metal Catalysts for CO2 Hydrogenation Based on Insights into Hydricity-Controlled Rate-Determining Steps. Inorg. Chem. 2016, 55, 5438–5444. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations-Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Abinitio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Multi 4 | ΔE 2 |  |
| Sc | 1 | −18.0 | |
| Ti | 2 | −43.4 | |
| Y | 1 | −53.3 | |
| Zr | 2 | −48.7 | |
| Pd | 2 | +3.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.-C.; Krishnan, R.; Tsai, M.-K. Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective. Catalysts 2022, 12, 890. https://doi.org/10.3390/catal12080890
Hsieh M-C, Krishnan R, Tsai M-K. Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective. Catalysts. 2022; 12(8):890. https://doi.org/10.3390/catal12080890
Chicago/Turabian StyleHsieh, Meng-Chi, Ranganathan Krishnan, and Ming-Kang Tsai. 2022. "Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective" Catalysts 12, no. 8: 890. https://doi.org/10.3390/catal12080890