
Substrate-Specific Engineering of Amino Acid Dehydrogenase Superfamily for Synthesis of a Variety of Chiral Amines and Amino Acids


Abstract
:1. Introduction
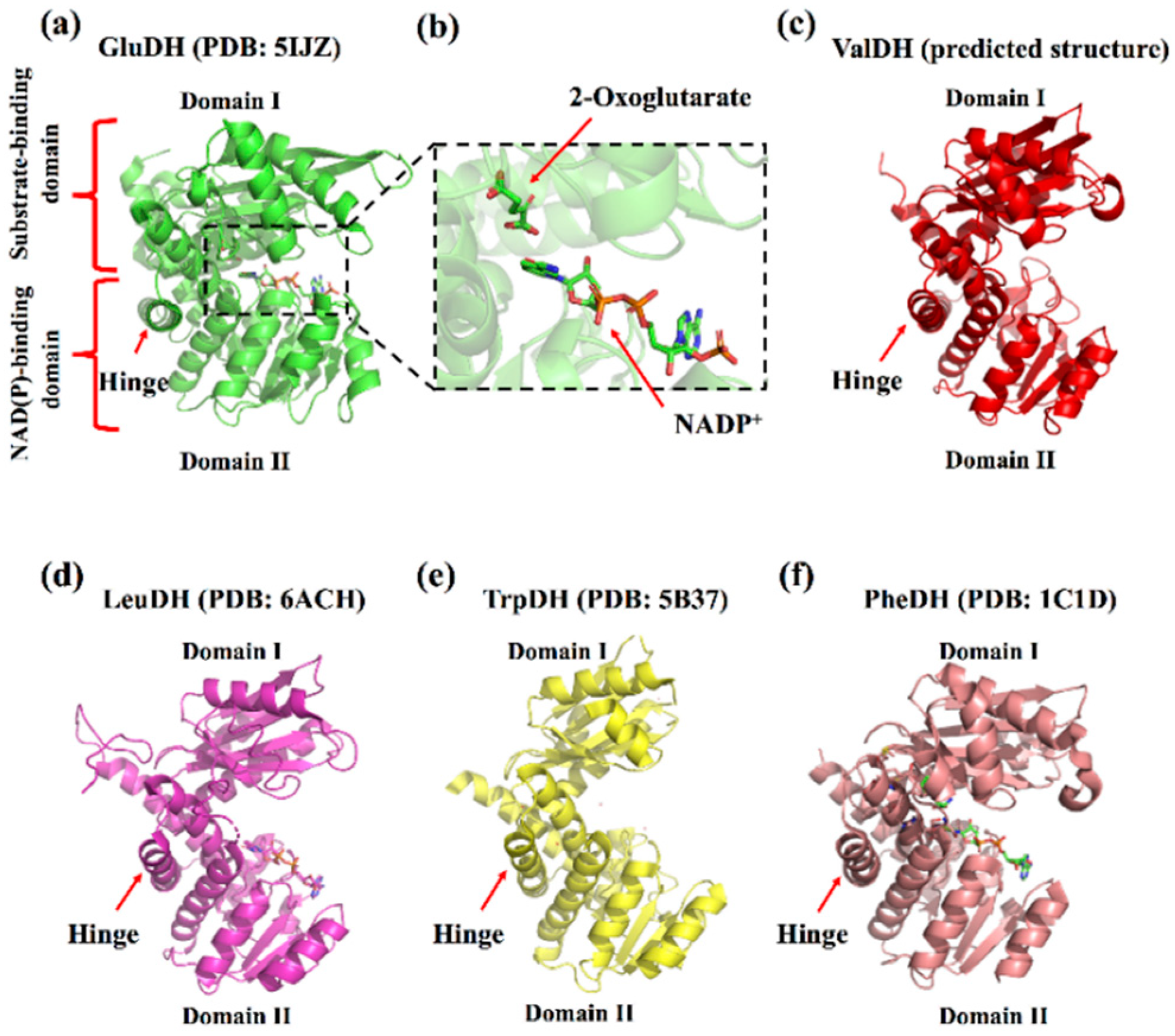
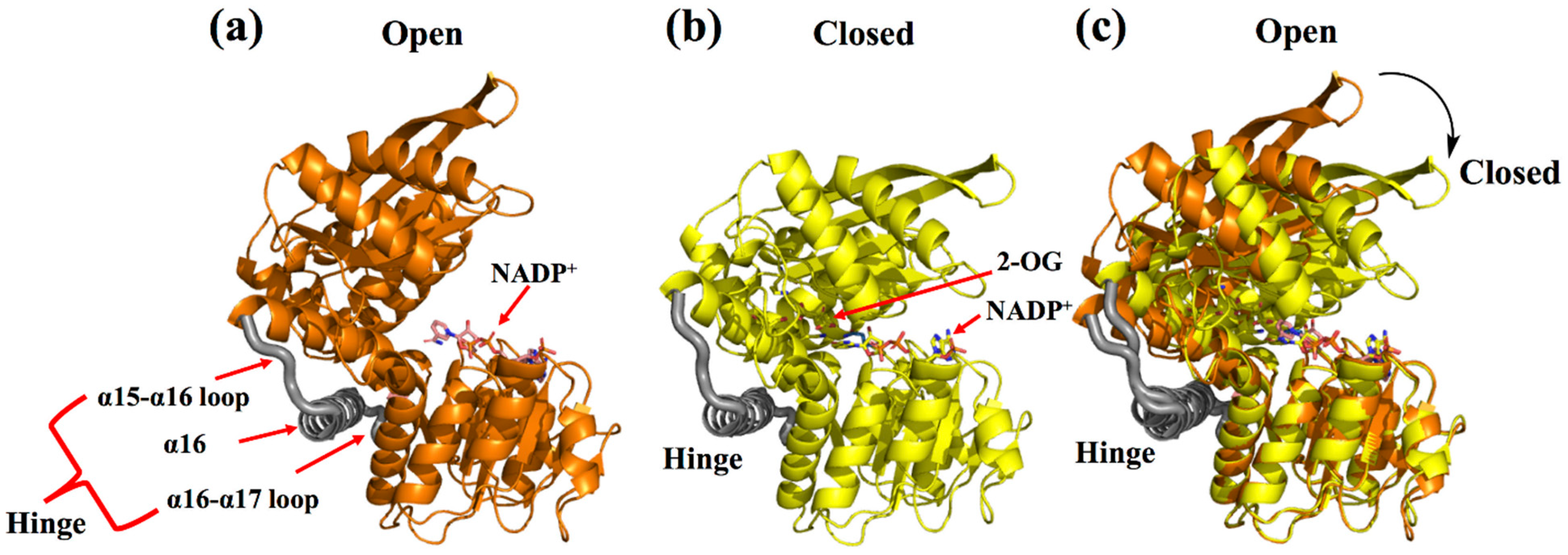
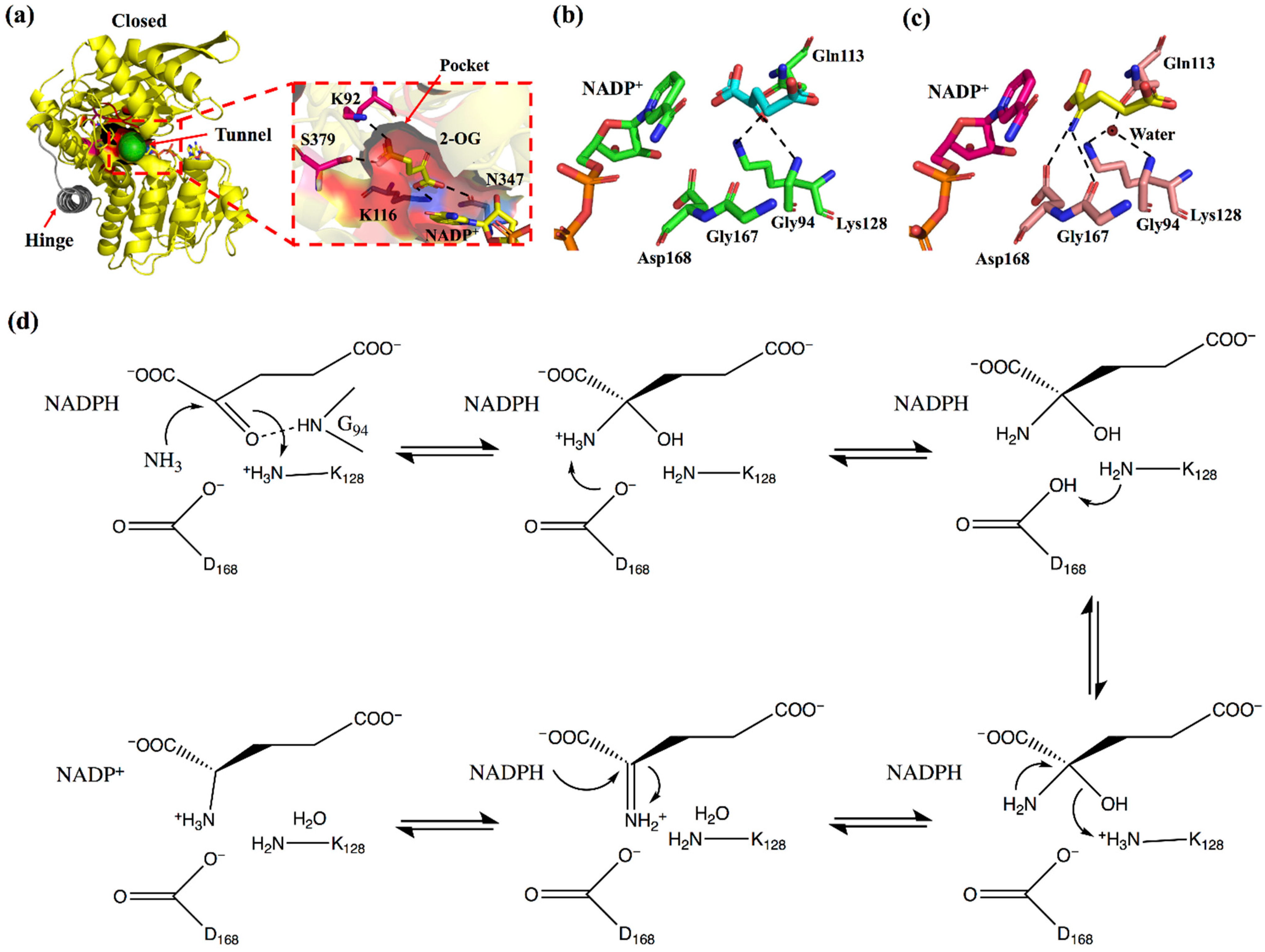
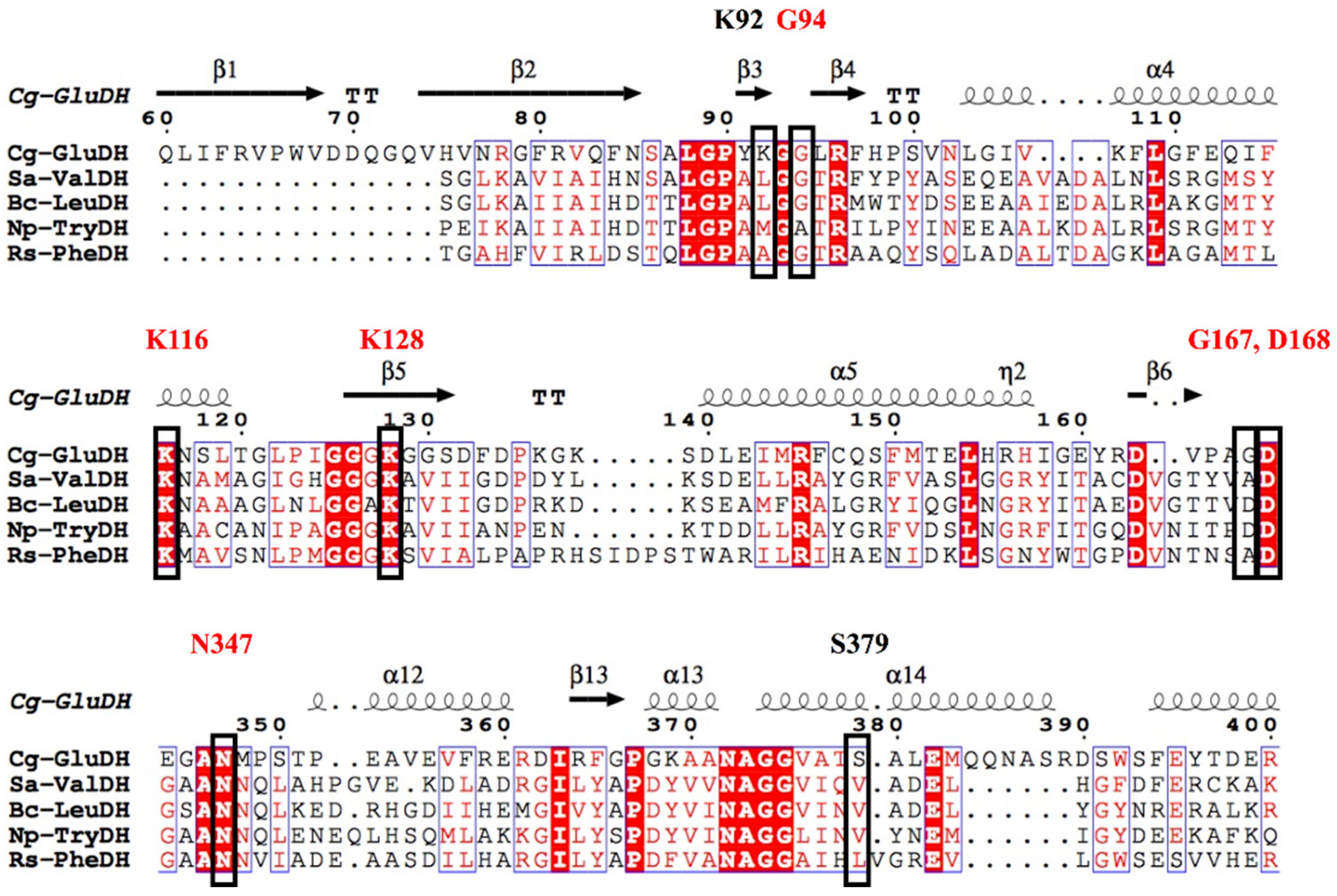
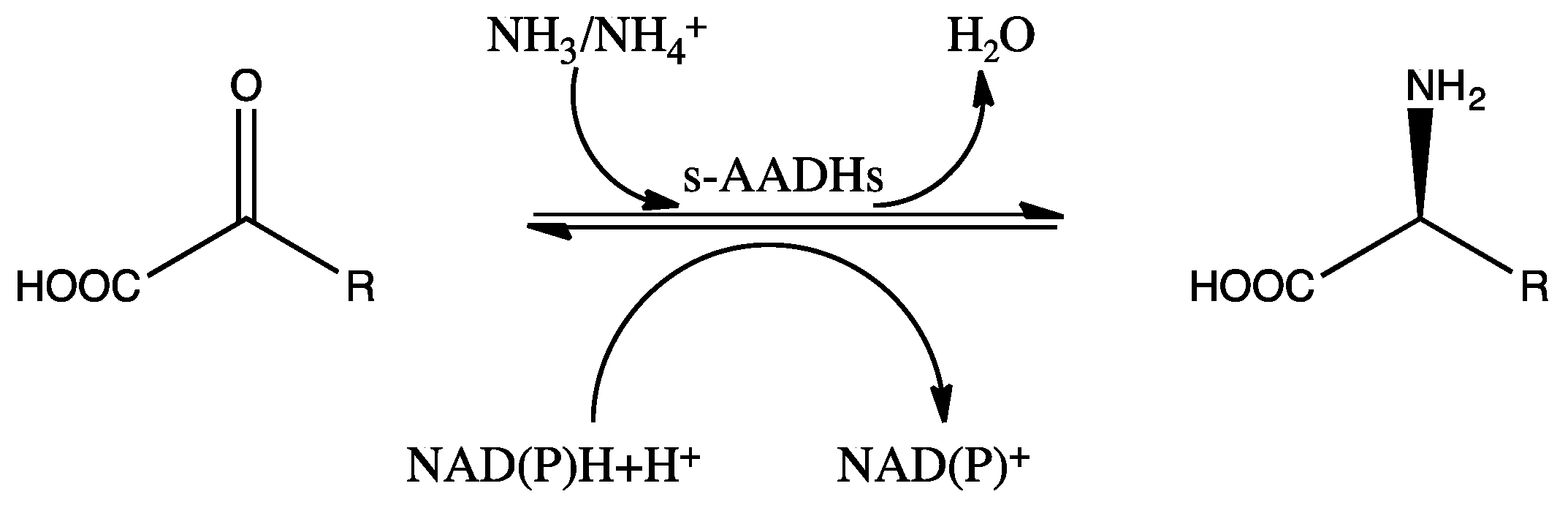
2. Enzyme Structure and Catalytic Mechanism of s-AADHs
3. Substrate-Specific Engineering of s-AADHs
3.1. GluDH
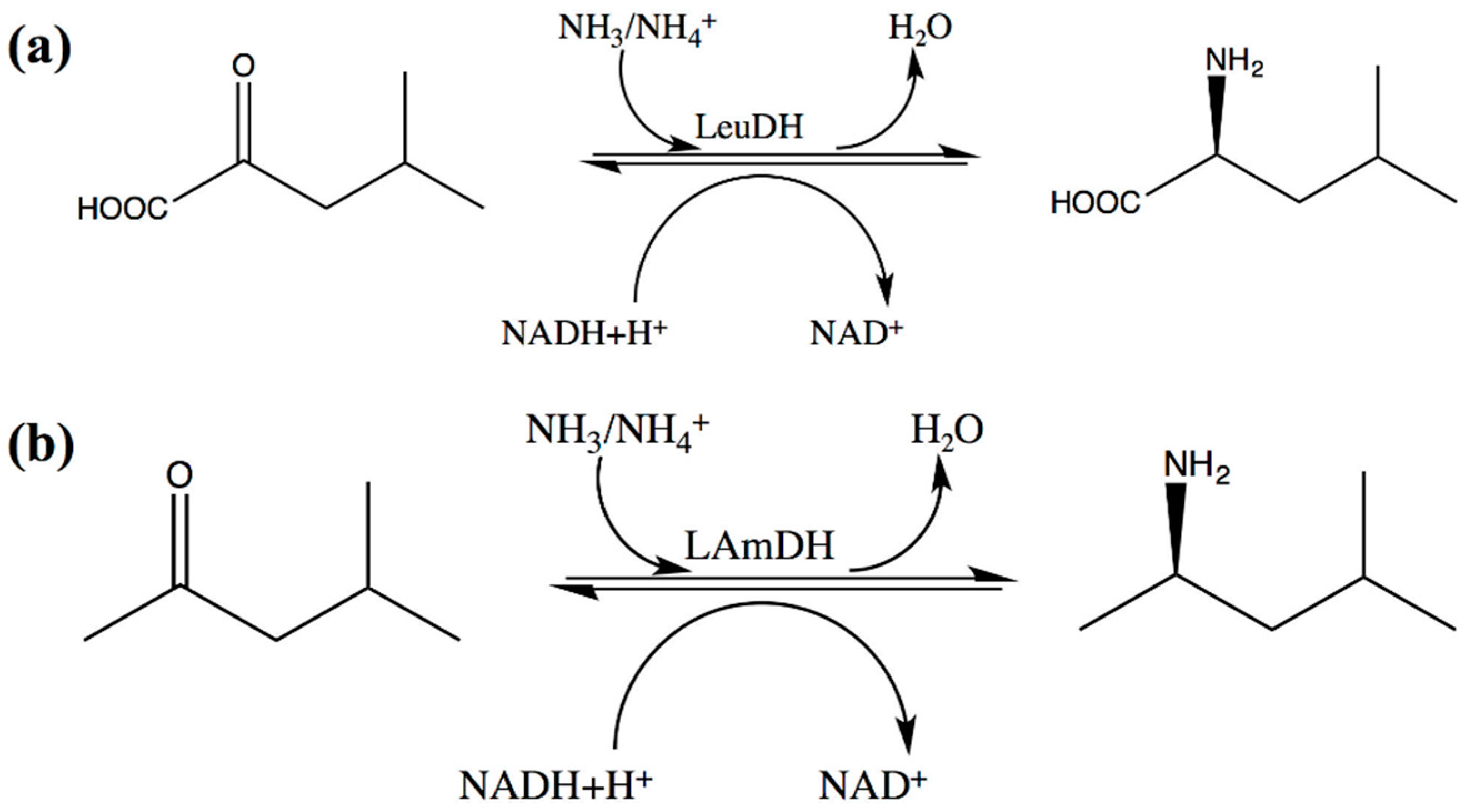
3.2. ValDH and LeuDH
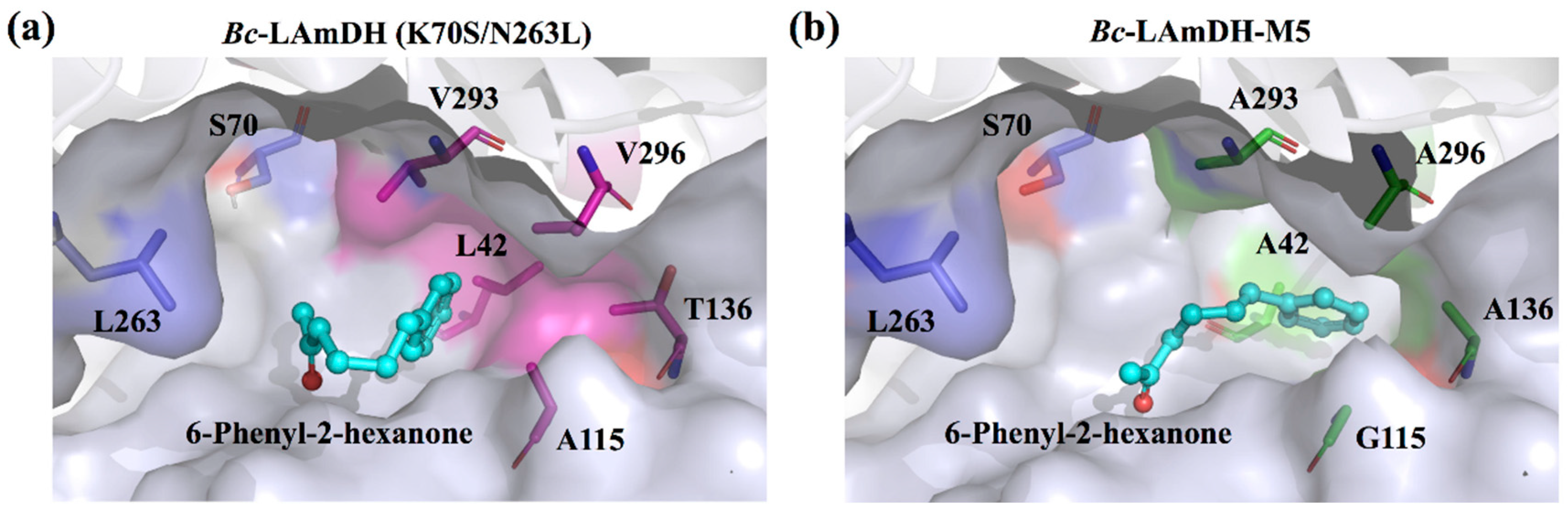
3.2.1. The Substrate-Specific Engineering of LeuDHs and Their Synthesis of L-α-Amino Acids
3.2.2. The Substrate-Specific Engineering of LeuDHs and Their Synthesis of Chiral Amines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gs-LAmDH | K68SO/N261LO/E114VT/V291CI |  | 92.5 | (R), 99.8 | 0.690 | 0.46, 15.1 | [23] |
| 2 | Es-LAmDH | K77SO/N270LO |  | 94 | (R), >99 | 0.796 | 0.84, 9.2 | [47,49] |
| 3 |  | 97 | N.D. b | 0.868 | N.D. | |||
| 4 |  | 21 | (R), >99 | 0.0985 | N.D. | |||
| 5 | Lf-LAmDH | K68SO/N261LO |  | >99 | (R), >99 | 0.930 | 1.7, 30.6 | [49] |
| 6 | K68SO/N261LO/A113GI/T134GI |  (n = 1, 2, 3, 4, 5) | 98, >99, >99, 80, 22 | (R), >99 | - c | N.D. | ||
| 7 | Gs-LAmDH | K68SO/N261LO/E114VT/V291CI/D32A/L39AI/F101S/A112GI/C290VI |  (n = 6) | N.D. | N.D. | 0.1405 | N.D. | [50] |
| 8 | Lf-LAmDH | K68TO/N261LO |  | 91 | (S), >99 | 0.233 | 0.227, 7.2 | [24] |
| 9 | K68TO/N261LO/A113GI/T134GI |  (n = 4) | 99 | (S), >99 | >2 | N.D. | ||
| 10 | Cs- LAmDH | K68SO/D261LO |  | >99 | (S), >99 | N.D. | 1.79, 2.53 | [51] |
| 11 |  | 73 | (S), >99 | N.D. | N.D. | |||
| 12 | Bc-LAmDH | K70SO/N263LO/L42AI/A115GI/T136AI/V293AI/V296AI |  (n = 2) | >99 | (R), >99 | 0.0948 | 0.15, 10.5 | [52] |
| 13 |  (n = 3) | N.D. | N.D. | 0.137 | 0.20, 6.7 | |||
| 14 |  (n = 4) | 93 | (R), >99 | 0.0803 | 0.143, 11.6 |
3.3. TrpDH and PheDH
3.3.1. The Substrate-Specific Engineering of PheDHs and Their Synthesis of L-α-Amino Acids
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) c | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | BsPheDH | wild type |  | N.D. b | N.D. | N.D. | 113, 0.37 | [15] |
| 2 | N145I | N.D. | N.D. | N.D. | 43, 0.012 | |||
| 3 | wild type |  | N.D. | N.D. | N.D. | 90, 0.19 | ||
| 4 | N145I | N.D. | N.D. | N.D. | 80, 0.55 | |||
| 5 | wild type |  (n = 2) | N.D. | N.D. | N.D. | 51, 13 | ||
| 6 | N145A | N.D. | N.D. | N.D. | 66, 0.25 | |||
| 7 | N145L |  (R = -OCH3) | N.D. | N.D. | 114.8 (3.85) | N.D. | [60] | |
| 8 | N145A |  | N.D. | N.D. | 130.5 (4.47) | N.D. | ||
| 9 |  (R = -CF3) | N.D. | N.D. | 29.4 (16.33) | N.D. | |||
| 10 | LsPheDH | N145A |  (R = -Br) | >99 | (S), >99 | >50 (≈1.67) | N.D. | [21] |
| 11 | BbPheDH | V309G/L306V/V144G |  (n = 2) | > 99 | (S), >99 | N.D. | 83, 74 | [22] |
| 12 | BbPheDH | G123S |  | N.D. | N.D. | 2.16 (0.03) | 1.55, 227 | [61] |
| 13 |  | N.D. | N.D. | 12 (5.59) | 1.54, 3.3 | |||
| 14 |  | N.D. | N.D. | 10.68 (74.6) | 1.48, 3.8 | |||
| 15 | BsPheDH | G124A/E313G |  | N.D. | N.D. | N.D. | 0.92, 23.2 | [62] |
3.3.2. The Substrate-Specific Engineering of PheDHs and Their Synthesis of Chiral Amines
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Bb-PAmDH | K77SO/N276LO |  | >90 | (R), >99.8 | N.D.b | 6.85, 7.75 | [27] |
| 2 | Bb-PAmDH | K77SO/N276LO |  | 34 | n.a.c | N.D. | N.D. | [68] |
| 3 | Rs-PAmDH | K66QO/N262CO/S149GI |  | N.D. | (R), >98 | 0.042 | 0.70, 4.0 | [28] |
| 4 |  (n = 2) | 95 | (R), >98 | 0.043 | 0.72, 1.4 | |||
| 5 |  (R = -OCH3, -CF3) | 98, 98 | (R), >99 | N.D. | N.D. | [68] | ||
| 6 |  (R = -OCH3, -CH3) | >99, 98 | (R), >99 | N.D. | N.D. | |||
| 10 |  (R = -CH3, -CH2CH3) (R = -CH3, -CH2CH3) | >99, 71 | (R), >99 | N.D. | N.D. | |||
| 11 |  (n = 2) | 96 | n.a. | N.D. | N.D. | |||
| 12 |  (n = 2; R = -CH3, -CH2CH3) | 99, 87 | (R), >99 | N.D. | N.D. | |||
| 13 |  | 99 | n.a. | N.D. | N.D. | |||
| 14 |  | 91 | (R), >99 | N.D. | N.D. | |||
| 15 |  (n = 3, 4) | 99, 93 | (R), >99 | N.D. | N.D. | |||
| 16 | cFL1-AmDH | N-terminal domain of Bb-PAmDH and C-terminal domain of Bs-LAmDH |  (R = -F) | 93 | (R), >99 | 1.725 | 1.24, 1.1 | [68] |
| 17 |  | 34 | (R), >99 | 0.301 | 0.24, 5.2 | |||
| 18 |  | N.D. | N.D. | 0.107 | N.D. | [65] | ||
| 19 |  | N.D. | N.D. | 0.069 | N.D. | |||
| 20 |  | N.D. | N.D. | 0.133 | N.D. | |||
| 21 |  | >99 | (R), >99 | N.D. | N.D. | [68] | ||
| 22 |  (R = -CH3, -F) | 39, 43 | (R), >99 | N.D. | N.D. | |||
| 23 |  (R = -CH3, -F) | 9, 22 | (R), >99 | N.D. | N.D. | |||
| 24 |  | >99 | n.a. | N.D. | N.D. | |||
| 25 |  | 96 | (R), >99 | N.D. | N.D. | |||
| 26 |  (n = 1, 2, 3, 4) | 75, 92, 98, 50 | (R), >99 | N.D. | N.D. |
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Obermeier, N.; Poralla, K. Experiments on the role of leucine dehydrogenase in initiation of Bacillus subtilis spore germination. FEMS Microbiol. Lett. 1979, 5, 81–83. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.X.; Hutchinson, C.R. Amino acid catabolism and antibiotic synthesis: Valine is a source of precursors for macrolide biosynthesis in Streptomyces ambofaciens and Streptomyces fradiae. J. Bacteriol. 1994, 176, 6107–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balskus, E.P.; Walsh, C.T. Investigating the initial steps in the biosynthesis of cyanobacterial sunscreen scytonemin. J. Am. Chem. Soc. 2008, 130, 15260–15261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, P.; Punekar, N.S.; Hareshwar, P.V. Structural basis for the catalytic mechanism and α-ketoglutarate cooperativity of glutamate dehydrogenase. J. Biol. Chem. 2018, 293, 6241–6258. [Google Scholar] [CrossRef] [Green Version]
- Britton, K.L.; Baker, P.J.; Engel, P.C.; Rice, D.W.; Stillman, T.J. Evolution of substrate diversity in the superfamily of amino acid dehydrogenases. J. Mol. Biol. 1993, 234, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Waugh, M.L.; Wang, X.-G.; Stillman, T.J.; Turnbull, A.P.; Engel, P.C.; Rice, D.W. Determinants of substrate specificity in the superfamily of amino acid dehydrogenases. Biochemistry 1997, 36, 16109–16115. [Google Scholar] [CrossRef] [PubMed]
- Hyun, C.G.; Kim, S.S.; Park, K.H.; Suh, J.W. Valine dehydrogenase from Streptomyces albus: Gene cloning, heterologous expression and identification of active site by site-directed mutagenesis. FEMS Microbiol. Lett. 2000, 182, 29–34. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Sakuraba, H.; Kitamura, M.; Hakumai, Y.; Fukui, K.; Ohnishi, K.; Ashiuchi, M.; Ohshima, T. Structural insights into L-tryptophan dehydrogenase from a photoautotrophic cyanobacterium, Nostoc punctiforme. Appl. Environ. Microbiol. 2017, 83, e02710-16. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, T.; Soda, K. Stereoselective biocatalysis: Amino acid dehydrogenases and their applications. In Stereoselective Biocatalysis; Patel, R.N., Ed.; Marcel Dekker Inc.: New York, NY, USA, 2000; pp. 877–902. [Google Scholar] [CrossRef]
- Xue, Y.-P.; Cao, C.-H.; Zheng, Y.-G. Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 2018, 47, 1516–1561. [Google Scholar] [CrossRef] [PubMed]
- Hummel, W.; Gröger, H. Reductive Amination of Keto Acids. In Enzyme Catalysis in Organic Synthesis; John Wiley & Sons, Ltd.: Weinheim, Germany, 2012; pp. 1165–1203. [Google Scholar] [CrossRef]
- Wang, X.G.; Britton, K.L.; Stillman, T.J.; Rice, D.W.; Engel, P.C. Conversion of a glutamate dehydrogenase into methionine/norleucine dehydrogenase by site-directed mutagenesis. Eur. J. Biochem. 2001, 268, 5791–5799. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.H.; Kim, H.; Ashida, H.; Ishikawa, T.; Shibata, H.; Sawa, Y. Altering the substrate specificity of glutamate dehydrogenase from Bacillus subtilis by site-directed mutagenesis. Biosci. Biotechnol. Biochem. 2005, 69, 1802–1805. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.P.; Baker, P.J.; Rice, D.W. Analysis of the quaternary structure, substrate specificity, and catalytic mechanism of valine dehydrogenase. J. Biol. Chem. 1997, 272, 25105–25111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seah, S.Y.K.; Britton, K.L.; Rice, D.W.; Asano, Y.; Engel, P.C. Single amino acid substitution in Bacillus sphaericus phenylalanine dehydrogenase dramatically increases its discrimination between phenylalanine and tyrosine substrates. Biochemistry 2002, 41, 11390–11397. [Google Scholar] [CrossRef] [Green Version]
- Hyun, C.-G.; Suk Kim, S.; Lee, I.H.; Suh, J.-W. Alteration of substrate specificity of valine dehydrogenase from Streptomyces albus. Antonie Van Leeuwenhoek 2000, 78, 237–242. [Google Scholar] [CrossRef]
- Kataoka, K.; Tanizawa, K. Alteration of substrate specificity of leucine dehydrogenase by site-directed mutagenesis. J. Mol. Catal. B Enzym. 2003, 23, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Diao, S.; Sun, Y.; Jiang, S.; Liu, Y.; Wang, H.; Wei, D. Rational engineering of Acinetobacter tandoii glutamate dehydrogenase for asymmetric synthesis of L-homoalanine through biocatalytic cascades. Catal. Sci. Technol. 2021, 11, 4208–4215. [Google Scholar] [CrossRef]
- Zhang, K.; Li, H.; Cho, K.M.; Liao, J.C. Expanding metabolism for total biosynthesis of the nonnatural amino acid L-homoalanine. Proc. Natl. Acad. Sci. USA 2010, 107, 6234–6239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Mu, X.; Nie, Y.; Xu, Y. Enhanced catalytic efficiency and coenzyme affinity of leucine dehydrogenase by comprehensive screening strategy for L-tert-leucine synthesis. Appl. Microbiol. Biotechnol. 2021, 105, 3625–3634. [Google Scholar] [CrossRef]
- Khorsand, F.; Murphy, C.D.; Whitehead, A.J.; Engel, P.C. Biocatalytic stereoinversion of D-para-bromophenylalanine in a one-pot three-enzyme reaction. Green Chem. 2017, 19, 503–510. [Google Scholar] [CrossRef]
- Wu, T.; Mu, X.; Xue, Y.; Xu, Y.; Nie, Y. Structure-guided steric hindrance engineering of Bacillus badius phenylalanine dehydrogenase for efficient L-homophenylalanine synthesis. Biotechnol. Biofuels 2021, 14, 207–213. [Google Scholar] [CrossRef]
- Abrahamson, M.J.; Vázquez-Figueroa, E.; Woodall, N.B.; Moore, J.C.; Bommarius, A.S. Development of an amine dehydrogenase for synthesis of chiral amines. Angew. Chem. Int. Ed. Engl. 2012, 51, 3969–3972. [Google Scholar] [CrossRef]
- Chen, F.-F.; Cosgrove, S.C.; Birmingham, W.R.; Mangas-Sanchez, J.; Citoler, J.; Thompson, M.P.; Zheng, G.-W.; Xu, J.-H.; Turner, N.J. Enantioselective synthesis of chiral vicinal amino alcohols using amine dehydrogenases. ACS Catal. 2019, 9, 11813–11818. [Google Scholar] [CrossRef]
- Liu, L.; Wang, D.-H.; Chen, F.-F.; Zhang, Z.-J.; Chen, Q.; Xu, J.-H.; Wang, Z.-L.; Zheng, G.-W. Development of an engineered thermostable amine dehydrogenase for the synthesis of structurally diverse chiral amines. Catal. Sci. Technol. 2020, 10, 2353–2358. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, Y.; Mu, X.; Nie, Y. A sustainable approach for synthesizing (R)-4-aminopentanoic acid from levulinic acid catalyzed by structure-guided tailored glutamate dehydrogenase. Front. Bioeng. Biotechnol. 2022, 9, 770302. [Google Scholar] [CrossRef]
- Abrahamson, M.J.; Wong, J.W.; Bommarius, A.S. The evolution of an amine dehydrogenase biocatalyst for the asymmetric production of chiral amines. Adv. Synth. Catal. 2013, 355, 1780–1786. [Google Scholar] [CrossRef]
- Ye, L.J.; Toh, H.H.; Yang, Y.; Adams, J.P.; Snajdrova, R.; Li, Z. Engineering of amine dehydrogenase for asymmetric reductive amination of ketone by evolving Rhodococcus phenylalanine dehydrogenase. ACS Catal. 2015, 5, 1119–1122. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Chen, J.; Xu, M.; Yang, T.; Zheng, J.; Zhang, X.; Rao, Z. Rational engineering of Bacillus cereus leucine dehydrogenase towards α-keto acid reduction for improving unnatural amino acid production. Biotechnol. J. 2018, 14, 1800253. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Liu, Y.; Meng, L.; Zhou, H.; Wu, J.; Yang, L. Semi-rational hinge engineering: Modulating the conformational transformation of glutamate dehydrogenase for enhanced reductive amination activity towards non-natural substrates. Catal. Sci. Technol. 2020, 10, 3376–3386. [Google Scholar] [CrossRef]
- Son, H.F.; Kim, I.-K.; Kim, K.-J. Structural insights into domain movement and cofactor specificity of glutamate dehydrogenase from Corynebacterium glutamicum. Biochem. Biophys. Res. Commun. 2015, 459, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Yin, L.; Nakamura, S.; Kosono, S.; Kuzuyama, T.; Nishiyama, M. Crystal structure of the 2-iminoglutarate-bound complex of glutamate dehydrogenase from Corynebacterium glutamicum. FEBS Lett. 2017, 591, 1611–1622. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Brunhuber, N.M.; Thoden, J.B.; Blanchard, J.S.; Vanhooke, J.L. Rhodococcus L-phenylalanine dehydrogenase: kinetics, mechanism, and structural basis for catalytic specifity. Biochemistry 2000, 39, 9174–9187. [Google Scholar] [CrossRef]
- Sekimoto, T.; Matsuyama, T.; Fukui, T.; Tanizawa, K. Evidence for lysine 80 as general base catalyst of leucine dehydrogenase. J. Biol. Chem. 1993, 268, 27039–27045. [Google Scholar] [CrossRef]
- Yin, X.; Liu, Y.; Meng, L.; Zhou, H.; Wu, J.; Yang, L. Rational molecular engineering of glutamate dehydrogenases for enhancing asymmetric reductive amination of bulky α-keto acids. Adv. Synth. Catal. 2018, 361, 803–812. [Google Scholar] [CrossRef]
- Engel, P.C. Glutamate dehydrogenases: The why and how of coenzyme specificity. Neurochem. Res. 2014, 39, 426–432. [Google Scholar] [CrossRef]
- Fang, J.-M.; Lin, C.-H.; Bradshaw, C.W.; Wong, C.-H. Enzymes in organic synthesis: Oxidoreductions. J. Chem. Soc. Perkin Trans. 1 1995, 967–978. [Google Scholar] [CrossRef]
- Hanson, R.L.; Schwinden, M.D.; Banerjee, A.; Brzozowski, D.B.; Chen, B.C.; Patel, B.P.; McNamee, C.G.; Kodersha, G.A.; Kronenthal, D.R.; Patel, R.N.; et al. Enzymatic synthesis of L-6-hydroxynorleucine. Bioorg. Med. Chem. 1999, 7, 2247–2252. [Google Scholar] [CrossRef]
- Yin, X.; Wu, J.; Yang, L. Efficient reductive amination process for enantioselective synthesis of L-phosphinothricin applying engineered glutamate dehydrogenase. Appl. Microbiol. Biotechnol. 2018, 102, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Ma, C.-W.; Zeng, A.-P. Reengineering substrate specificity of E. coli glutamate dehydrogenase using a position-based prediction method. Biotechnol. Lett. 2017, 39, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Munawar, N.; Engel, P.C. Prospects for robust biocatalysis: Engineering of novel specificity in a halophilic amino acid dehydrogenase. Extremophiles 2013, 17, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liao, J.C. Development of an NADPH-dependent homophenylalanine dehydrogenase by protein engineering. ACS Synth. Biol. 2014, 3, 13–20. [Google Scholar] [CrossRef]
- Oikawa, T.; Yamanaka, K.; Kazuoka, T.; Kanzawa, N.; Soda, K. Psychrophilic valine dehydrogenase of the antarctic psychrophile, Cytophaga sp. KUC-1. Eur. J. Biochem. 2001, 268, 4375–4383. [Google Scholar] [CrossRef] [PubMed]
- Vancurová, I.; Vancura, A.; Volc, J.; Neuzil, J.; Flieger, M.; Basarová, G.; Bĕhal, V. Isolation and characterization of valine dehydrogenase from Streptomyces aureofaciens. J. Bacteriol. 1988, 170, 5192–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, T.; Misono, H.; Soda, K. Properties of crystalline leucine dehydrogenase from Bacillus sphaericus. J. Biol. Chem. 1978, 253, 5719–5725. [Google Scholar] [CrossRef]
- Chen, F.-F.; Liu, Y.-Y.; Zheng, G.-W.; Xu, J.-H. Asymmetric amination of secondary alcohols by using a redox-neutral two-enzyme Cascade. ChemCatChem 2015, 7, 3838–3841. [Google Scholar] [CrossRef]
- Löwe, J.; Ingram, A.A.; Gröger, H. Enantioselective synthesis of amines via reductive amination with a dehydrogenase mutant from Exigobacterium sibiricum: Substrate scope, co-solvent tolerance and biocatalyst immobilization. Bioorg. Med. Chem. 2017, 26, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-F.; Zheng, G.-W.; Liu, L.; Li, H.; Chen, Q.; Li, F.-L.; Li, C.-X.; Xu, J.-H. Reshaping the active pocket of amine dehydrogenases for asymmetric synthesis of bulky aliphatic amines. ACS Catal. 2018, 8, 2622–2628. [Google Scholar] [CrossRef]
- Franklin, R.D.; Mount, C.J.; Bommarius, B.R.; Bommarius, A.S. Separate sets of mutations enhance activity and substrate scope of amine dehydrogenase. ChemCatChem 2020, 12, 2436–2439. [Google Scholar] [CrossRef]
- Wang, H.; Qu, G.; Li, J.K.; Ma, J.A.; Guo, J.; Miao, Y.; Sun, Z. Data mining of amine dehydrogenases for the synthesis of enantiopure amino alcohols. Catal. Sci. Technol. 2020, 10, 5945–5952. [Google Scholar] [CrossRef]
- Mu, X.; Wu, T.; Mao, Y.; Zhao, Y.; Xu, Y.; Nie, Y. Iterative alanine scanning mutagenesis confers aromatic ketone specificity and activity of L-amine dehydrogenases. ChemCatChem 2021, 13, 5243–5253. [Google Scholar] [CrossRef]
- Minh, T.H.; Kutáček, M.; Šebánek, J. Growth-correlative effect of the root on the apical part of the epicotyl in pea seedlings regarding the IAA content and L-tryptophan aminotransferase and L-tryptophan dehydrogenase activities. Biol. Plant. 1984, 26, 342–348. [Google Scholar] [CrossRef]
- El Bahr, M.K.; Kutáček, M.; Opatrný, Z. L-Tryptophan aminotransferase and L-tryptophan dehydrogenase, enzymes of IAA synthesis, in normal and tumorous tobacco tissue cultures. Biochem. Physiol. Pflanz. 1987, 182, 213–222. [Google Scholar] [CrossRef]
- Ogura, R.; Wakamatsu, T.; Mutaguchi, Y.; Doi, K.; Ohshima, T. Biochemical characterization of an L-tryptophan dehydrogenase from the photoautotrophic cyanobacterium Nostoc punctiforme. Enzym. Microb. Technol. 2014, 60, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Matsui, D.; Okazaki, S.; Matsuda, M.; Asano, Y. Enhancement of stability of L-tryptophan dehydrogenase from Nostoc punctiforme ATCC29133 and its application to L-tryptophan assay. J. Biotechnol. 2015, 196–197, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Seah, S.Y.; Britton, K.L.; Baker, P.J.; Rice, D.W.; Asano, Y.; Engel, P.C. Alteration in relative activities of phenylalanine dehydrogenase towards different substrates by site-directed mutagenesis. FEBS Lett. 1995, 370, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Seah, S.Y.K.; Britton, K.L.; Rice, D.W.; Asano, Y.; Engel, P.C. Kinetic analysis of phenylalanine dehydrogenase mutants designed for aliphatic amino acid dehydrogenase activity with guidance from homology-based modelling. Eur. J. Biochem. 2003, 270, 4628–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, F.; Ataei, F.; Arab, S.S.; Hosseinkhani, S. Increase of Bacillus badius phenylalanine dehydrogenase specificity towards phenylalanine substrate by site-directed mutagenesis. Arch. Biochem. Biophys. 2017, 635, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Busca, P.; Paradisi, F.; Moynihan, E.; Maguire, A.R.; Engel, P.C. Enantioselective synthesis of non-natural amino acids using phenylalanine dehydrogenases modified by site-directed mutagenesis. Org. Biomol. Chem. 2004, 2, 2684. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, S.; Kuwamori, Y.; Asano, Y. Discrimination of aliphatic substrates by a single amino acid substitution in Bacillus badius and Bacillus sphaericus phenylalanine dehydrogenases. Biosci. Biotechnol. Biochem. 2009, 73, 729–732. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Engel, P.C. Efficient screening for new amino acid dehydrogenase activity: Directed evolution of Bacillus sphaericus phenylalanine dehydrogenase towards activity with an unsaturated non-natural amino acid. J. Biotechnol. 2009, 142, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Takada, H.; Tanizawa, K.; Yoshimura, T.; Esaki, N.; Ohshima, T.; Soda, K. Construction and characterization of chimeric enzyme consisting of an amino-terminal domain of phenylalanine dehydrogenase and a carboxy-terminal domain of leucine dehydrogenase. J. Biochem. 1994, 116, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Kataoka, K.; Jin, Y.; Suzuki, S.; Soda, K. Fragmentary form of thermostable leucine dehydrogenase of Bacillus stearothermophilus: Its construction and reconstitution of active fragmentary enzyme. Biochem. Biophys. Res. Commun. 2001, 280, 1177–1182. [Google Scholar] [CrossRef]
- Bommarius, B.R.; Schürmann, M.; Bommarius, A.S. A novel chimeric amine dehydrogenase shows altered substrate specificity compared to its parent enzymes. Chem. Commun. 2014, 50, 14953–14955. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, M.A.; Engel, P.C. Modular coenzyme specificity: A domain-swopped chimera of glutamate dehydrogenase. Proteins 2009, 77, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.; Sharkey, M.A.; Engel, P.C.; Khan, A.R. Crystal structure of a chimaeric bacterial glutamate dehydrogenase. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 462–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaus, T.; Böhmer, W.; Mutti, F.G. Amine dehydrogenases: Efficient biocatalysts for the reductive amination of carbonyl compounds. Green Chem. 2017, 19, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Pushpanath, A.; Siirola, E.; Bornadel, A.; Woodlock, D.; Schell, U. Understanding and overcoming the limitations of Bacillus badius and Caldalkalibacillus thermarum amine dehydrogenases for biocatalytic reductive amination. ACS Catal. 2017, 7, 3204–3209. [Google Scholar] [CrossRef]
- Wang, D.-H.; Chen, Q.; Yin, S.-N.; Ding, X.-W.; Zheng, Y.-C.; Zhang, Z.; Zhang, Y.-H.; Chen, F.-F.; Xu, J.-H.; Zheng, G.-W. Asymmetric reductive amination of structurally diverse ketones with ammonia using a spectrum-extended amine dehydrogenase. ACS Catal. 2021, 11, 14274–14283. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L., Jr.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]







| EC Number (1.4.1.X) a | Enzyme | Main Amino Acid Substrate | Refs. |
|---|---|---|---|
| 2–4 | GluDH |  | [12,13] |
| 8, 23 | ValDH |  | [14] |
| 9 | LeuDH |  | [14] |
| 19 | TrpDH |  | [8] |
| 20 | PheDH |  | [15] |
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | BsGluDH | M101SI |  | N.D. b | N.D. | 9.405 | 5.68, 2.27 | [13] |
| 2 | G82KI | N.D. | N.D. | 7.28 | 3.45, 4.16 | |||
| 3 | PpGluDH | I170MT |  | 99 | (S), >99 | 0.62181 | 12.33, 290 | [40] |
| 4 | A167GI | 99 | (S), >99 | 0.5582 | 715.20, 290 | [36] | ||
| 5 | V378AI | 99 | (S), >99 | 0.5313 | 735.98, 320 | |||
| 6 | T121N/L123Y | 99 | (S), >99 | 0.4961 | 598.56, 290 | [30] | ||
| 7 | EcGluDH | K92VI |  | N.D. | N.D. | 1.3 × 10−3 | N.D. | [41] |
| 8 | K92VI/T195SI |  | N.D. | N.D. | N.D. | 90.2, 8.4 | [19] | |
| 9 | AtGluDH | K76LI/T180CI |  | 99.9 | (S), 99.9 | 985.7 | 3.24, 2.52 | [18] |
| 10 | CsGluDH | K89LI/S380AI/A163GI |  | N.D. | N.D. | 2.1 | 17.6 c | [12] |
| 11 | K89LI/A163GI |  | N.D. | N.D. | N.D. | 9.6, 6.6 | ||
| 12 | HsGluDH | K89LI/A163GI/S367AI |  | N.D. | N.D. | N.D. | 19, 11 | [42] |
| 13 |  | N.D. | N.D. | N.D. | 19, 12 | |||
| 14 | EcGluDH | K92AI/A166GI/T195AI/V377AI/S380AI |  | N.D. | N.D. | N.D. | 6.79, 0.24 | [43] |
| 15 | Ec-GAmDH | K116QO/N348MO |  | 97 | (R), >99 | 2.82 | 2.28, 824 | [26] |
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | SaValDH | A124GI |  | N.D. b | N.D. | 1.587 | N.D. | [16] |
| 2 | BcLeuDH | wild type |  | 90.9 | (S), >99 | N.D. | 0.149, 1.890 | [29] |
| 3 | E116VT | 95.7 | (S), >99 | N.D. | 0.168, 0.705 | |||
| 4 | wild type |  | >99 | (S), >99 | N.D. | 94.14, 60.82 | [20] | |
| 5 | E24V/E116VT | >99 | (S), >99 | N.D. | 66.21, 7.92 | |||
| 6 | BsLeuDH | A113GI |  | N.D. | N.D. | N.D. | 18, 4.1 | [17] |
| 7 | L40KI/V294SI |  | N.D. | N.D. | N.D. | 0.57, ND | ||
| 8 | L40DI/V294SI |  | N.D. | N.D. | N.D. | 0.45, 72 |
| Entry No. | Enzyme | Variants a | Substrate | Conversion (%) | Product Configuration and ee (%) | Specific Activity (U/mg) | kcat (s−1), Km (mM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gk-PAmDH | K78SO/N276LO |  (R = -OCH3) | 99 | (R), >99 | 0.30 | N.D. b | [25] |
| 2 | K78SO/N276CO |  | N.D. | N.D. | 9.35 | N.D. | ||
| 3 | K78SO/N276TO |  (R = -CH2OH) | N.D. | N.D. | 4.59 | N.D. | ||
| 4 | Gk-PAmDH-M3 | K78SO/N276LO/V144AI/V309AI/A310GI |  (n = 2) | 99 | (R), >99 | N.D. | 0.62, 4.8 | [70] |
| 5 |  (n = 2; R =-F, -CH3, -OCH3, -OH) | 99, 99, >99, 99 | (R), >99 | N.D. | N.D. | |||
| 6 |  (n = 2; R = -OCH3, -NO2) | 99, 97 | (R), >99 | N.D. | N.D. | |||
| 7 |  (n = 2) | 99 | (R), >99 | N.D. | N.D. | |||
| 8 |  (n = 2; R = -OCH3, -NO2) | 21, 72 | (R), >99 | N.D. | N.D. | |||
| 9 |  | 99 | (R), >99 | N.D. | N.D. | |||
| 10 | Gk-PAmDH-M8 | K78SO/N276LO/V144AI/V309AI/A310GI/S156TI/Q308AI/C79NI/F86MI |  (n = 2) | >99 | (R), >99 | N.D. | 3.10, 11.8 | |
| 11 |  | >99 | (R), >99 | N.D. | N.D. | |||
| 12 |  (n = 1, 2, 3, 4) | >99, >99, 96, 75 | (R), >99 | N.D. | N.D. | |||
| 13 |  | 80 | (R), >99 | N.D. | N.D. | |||
| 14 |  | 88 | (R), >99 | N.D. | N.D. | |||
| 15 |  (n = 2) | 88 | (R), >99 | N.D. | N.D. | |||
| 16 |  (n = 2) | 52 | (R), >99 | N.D. | N.D. | |||
| 17 |  (n = 1, 2, 3; R= -CH2CH3) | 74, 77, 21 | (R), >99 | N.D. | N.D. | |||
| 18 |  (n = 2, 3; R = -CH2CH2CH3) | 31, 7 | (R), >99 | N.D. | N.D. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, F.; Xu, Y.; Nie, Y.; Mu, X. Substrate-Specific Engineering of Amino Acid Dehydrogenase Superfamily for Synthesis of a Variety of Chiral Amines and Amino Acids. Catalysts 2022, 12, 380. https://doi.org/10.3390/catal12040380
Zhou F, Xu Y, Nie Y, Mu X. Substrate-Specific Engineering of Amino Acid Dehydrogenase Superfamily for Synthesis of a Variety of Chiral Amines and Amino Acids. Catalysts. 2022; 12(4):380. https://doi.org/10.3390/catal12040380
Chicago/Turabian StyleZhou, Feng, Yan Xu, Yao Nie, and Xiaoqing Mu. 2022. "Substrate-Specific Engineering of Amino Acid Dehydrogenase Superfamily for Synthesis of a Variety of Chiral Amines and Amino Acids" Catalysts 12, no. 4: 380. https://doi.org/10.3390/catal12040380