
Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

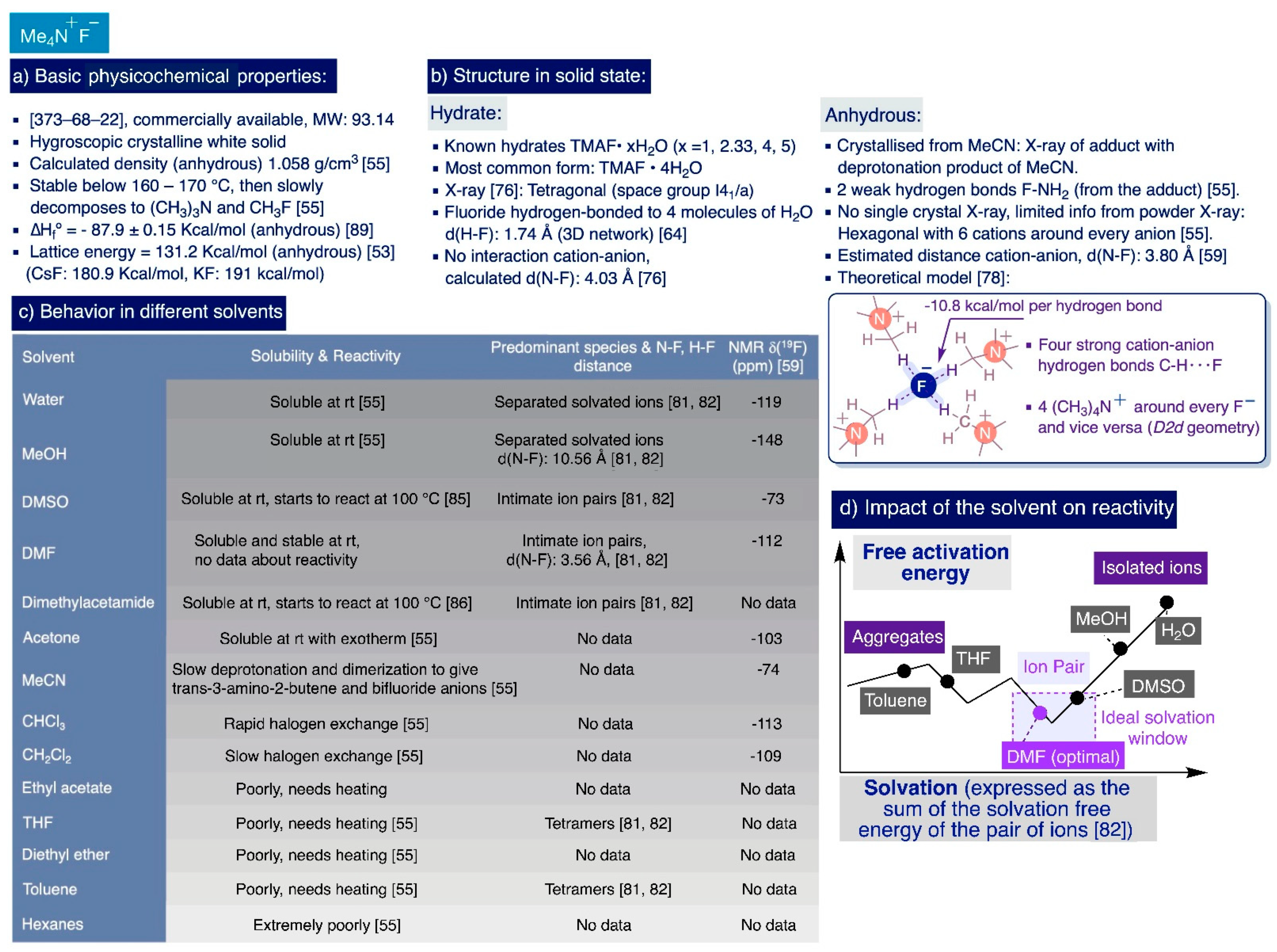
2. TMAF: General Physicochemical Properties and Preparation
2.1. “Nakedness” of the Fluoride Anion

2.2. Physicochemical Properties and Behaviour of TMAF in Different Solvents
2.2.1. Physicochemical Properties and Solid Structure
2.2.2. Solubility of TMAF in Different Solvents

2.2.3. Impact of the Solvent on the Chemical Reactivity of TMAF
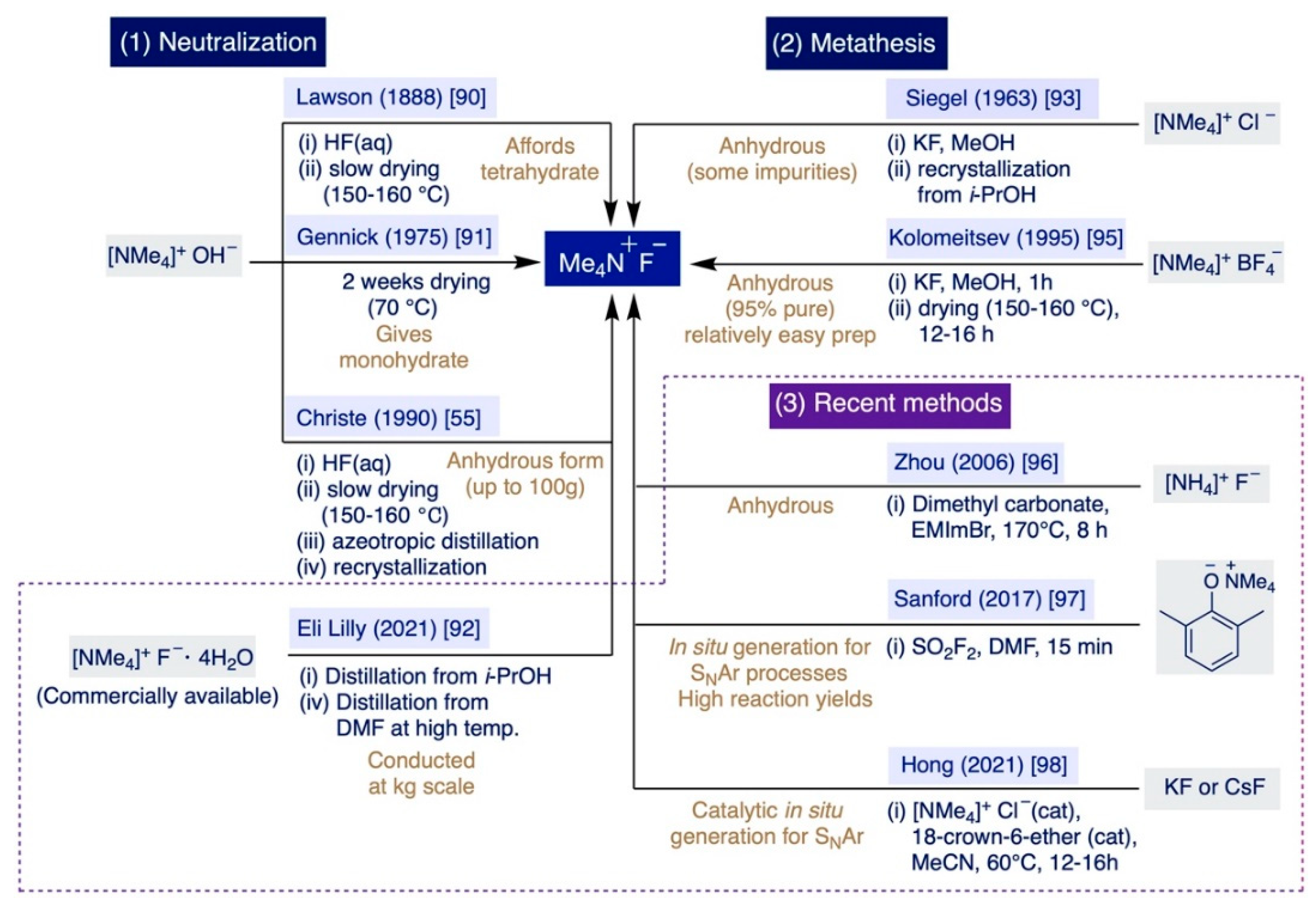
2.3. Preparation of TMAF

3. TMAF as Ionic Source of Fluoride for C-F Bond Formation
3.1. Comparison of TMAF with Other Nucleophilic Sources of Fluoride

3.2. C(sp2)-F Bond Formation with TMAF: Nucleophilic Aromatic Substitution
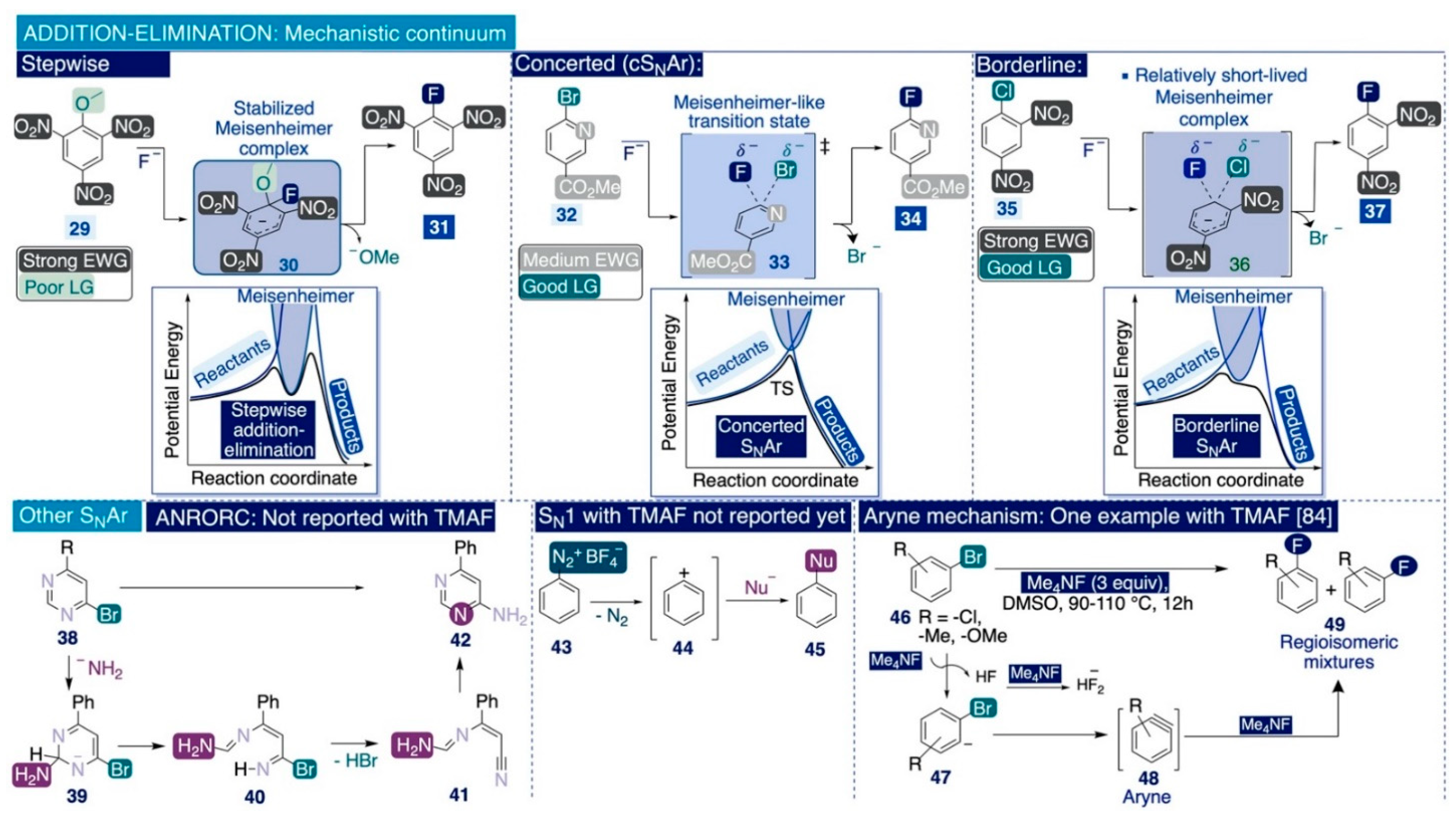
3.2.1. Nucleophilic Aromatic Substitution. General Mechanistic Overview

3.2.2. Seminal Nucleophilic Aromatic Substitution Reactions on Nitro- and Halo-Arenes


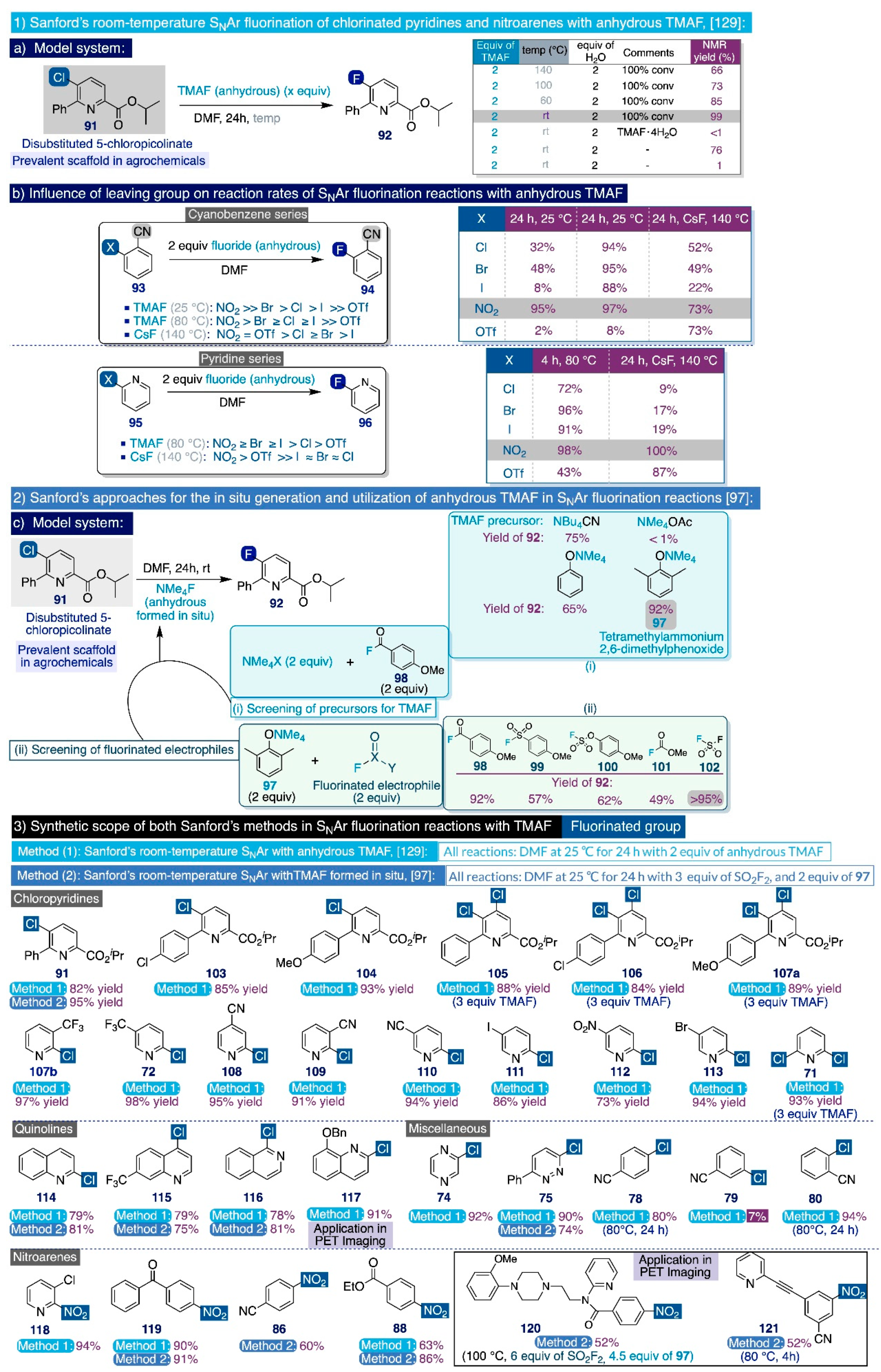
3.2.3. Recent SNAr Fluorination Methodologies with TMAF



3.3. Deoxyfluorination of Phenols and Aldehydes with TMAF
3.3.1. Seminal Examples for the Nucleophilic Deoxyfluorination of Phenolic Compounds
3.3.2. Usage of TMAF for the Deoxyfluorination of Phenols


3.3.3. Usage of TMAF for the Deoxyfluorination of Aldehydes and Ketoesters

3.4. C(sp3)-F Bond Formation with TMAF


3.5. Other Types of C-F Bond Formation with TMAF
3.5.1. Nucleophilic Additions to C=X bonds with TMAF

3.5.2. Nucleophilic Addition to Diaryliodonium Salts with TMAF
4. Use of TMAF as a Base
4.1. TMAF Participation in Classic Acid-Base Transformations

4.2. Novel Uses of TMAF as a Base


5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swallow, S. Fluorine in Pharmaceutical and Medicinal Chemistry. From biophysical Aspects to Clinical Applications, 1st ed.; Gouverneur, V., Müller, K., Eds.; ICP: Oxford, UK, 2012; Volume 6, pp. 141–174. [Google Scholar]
- Ametamey, S.M.; Honer, M.; Shubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Tredwell, M.; Gouverneur, V. 18F Labeling of Arenes. Angew. Chem. Int. Ed. 2012, 51, 11426–11437. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [18F]fluorination: New solutions to old problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [Green Version]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Hiyama, T.; Yamamoto, H. Fluorine-Containing Materials. In Organofluorine Compounds; Yamamoto, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Babudri, F.; Farinola, G.M.; Naso, F.; Ragni, R. Fluorinated organic materials for electronic and optoelectronic applications: The role of fluorine atom. Chem. Commun. 2007, 1003–1022. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in Health Care: Organofluorine Containing Blockbuster Drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2018 FDA drug approvals. Nat. Rev. Drug. Discov. 2019, 18, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug. Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amines Basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry, J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Thiehoff, C.; Rey, Y.P.; Gilmour, R. The Fluorine Gauche Effect: A Brief History. Isr. J. Chem. 2017, 57, 92–100. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Gribble, G.W. Naturally Occurring Organofluorines. In Organofluorines. The Handbook of Environmental Chemistry; (Volume 3 Series: Anthropogenic Compounds), vol 3N; Neilson, A.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 3. [Google Scholar] [CrossRef]
- Gribble, G.W. A recent survey of naturally occurring organohalogen compounds. Environ. Chem. 2015, 12, 396–405. [Google Scholar] [CrossRef]
- Höfler, G.T.; But, A.; Hollmann, F. Haloperoxidases as catalysts in organic synthesis. Org. Biomol. Chem. 2019, 17, 9267–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.; Huang, Q.; Li, M.; Wang, J. Enzyme-catalyzed C–F bond formation and cleavage. Bioresour. Bioprocess. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, D.; Schaffrath, C.; Cobb, S.L.; Hamilton, J.T.G.; Murphy, C.D. Biosynthesis of an organofluorine molecule. Nature 2002, 416, 279. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Hammond, C. Catalytic C(sp3)-F bond formation: Recent achievements and pertaining challenges. Green Chem. 2020, 22, 5195–5209. [Google Scholar] [CrossRef]
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorination of C−H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [Green Version]
- Fustero, S.; Sedgwick, D.M.; Román, R.; Barrio, P. Recent advances in the synthesis of functionalized monofluorinated compounds. Chem. Commun. 2018, 54, 9706–9725. [Google Scholar] [CrossRef]
- Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef]
- Pan, Y. The Dark Side of Fluorine. ACS Med. Chem. Lett. 2019, 10, 1016–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.J.; Shu, Y.-Z.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compound. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Ritter, T. Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev. 2015, 115, 612–633. [Google Scholar] [CrossRef] [PubMed]
- See, Y.Y.; Morales-Colón, M.T.; Bland, D.C.; Sanford, M.S. Development of SNAr Nucleophilic Fluorination: A Fruitful Academia-Industry Collaboration. Acc. Chem. Res. 2020, 53, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.S.; Pez, G.P.; Syvret, R.G. Electrophilic NF Fluorinating Agents. Chem. Rev. 1996, 96, 1737–1755. [Google Scholar] [CrossRef]
- Murphy, G.K.; Gulder, T. Hypervalent Iodine Fluorination for Preparing Alkyl Fluorides (Stoichiometrically and Catalytically). In Fluorination. Synthetic Organofluorine Chemistry; Hu, J., Umemoto, T., Eds.; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Rozatian, N.; Ashworth, I.W.; Sandford, G.; Hodgson, D.R.W. A quantitative reactivity scale for electrophilic fluorinating reagents. Chem. Sci. 2018, 9, 8692–8702. [Google Scholar] [CrossRef] [Green Version]
- Sibi, M.P.; Landais, Y. Csp3-F Bond Formation: A Free-Radical Approach. Angew. Chem. Int. Ed. 2013, 52, 3570–3572. [Google Scholar] [CrossRef]
- Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F. Recent advances in radical fluorination. Synthesis 2015, 47, 2554–2569. [Google Scholar] [CrossRef]
- Lantaño, B.; Postigo, A. Radical fluorination reactions by thermal and photoinduced methods. Org. Biomol. Chem. 2017, 15, 9954–9973. [Google Scholar] [CrossRef]
- Koike, T.; Akita, M. Photocatalytic Introduction of Fluorinated Groups. In Science of Synthesis; König, B., Ed.; Thieme Verlagsgruppe: Stuttgart, Germany, 2019; pp. 559–574. [Google Scholar]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of Solvation of Ions. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Lemal, D.M. Perspective on Fluorocarbon Chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Liotta, C.L.; Harris, H.P. Chemistry of Naked Anions. I. Reactions of the 18-Crown-6 Complex of Potassium Fluoride with Organic Substrates in Aprotic Organic Solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Emsley, J. Very Strong Hydrogen Bonding. Chem. Soc. Rev. 1980, 9, 91–124. [Google Scholar] [CrossRef]
- Seppelt, K. Does the Naked Fluoride Ion Exist? Angew. Chem. Int. Ed. 1992, 31, 292–293. [Google Scholar] [CrossRef]
- Christe, K.O.; Jenkins, H.D.B. Quantitative Measure for the “Nakedness” of Fluoride Ion Sources. J. Am. Chem. Soc. 2003, 125, 9457–9461. [Google Scholar] [CrossRef]
- Borrmann, T.; Lork, E.; Mews, R.; Stohrer, W.-D. Fluoride Ion Transfer and Stabilisation of Reactive Ions. J. Fluor. Chem. 2004, 125, 903–916. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Wilson, R.D.; Bau, R.; Feng, J.A. Syntheses, Properties, and Structures of Anhydrous Tetramethylammonium Fluoride and Its 1:1 Adduct with Trans-3-Amino-2-Butenenitrile. J. Am. Chem. Soc. 1990, 112, 7619–7625. [Google Scholar] [CrossRef]
- Harmon, K.M.; Southworth, B.A.; Harmon, J. Hydrogen Bonding Part 46. Thermodynamic and IR Study of Stability and Structure of Tetraethylammonium Fluoride·2.75H2O, Tetraethylammonium Fluoride·2.00H2O, Tetramethylammonium Fluoride·3.00H2O, and Tetraethylammonium Fluoride·5.00H2O. J. Mol. Struct. 1993, 300, 339–349. [Google Scholar] [CrossRef]
- Harmon, K.M.; LaFave, N.J. Hydrogen Bonding Part 66. Further Studies of the Fluoride Ion Assisted Dissolution of 1-Methyl-4,5-Dicarboxyimidazole: Absence of Cation Participation and Stoichiometric Considerations. J. Mol. Struct. 1997, 404, 297–306. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W. Nuclear Magnetic Resonance Spectrum of the Fluoride Anion. J. Fluor. Chem. 1990, 46, 339–342. [Google Scholar] [CrossRef]
- Gerken, M.; Boatz, J.A.; Kornath, A.; Haiges, R.; Schneider, S.; Schroer, T.; Christe, K.O. The 19F NMR Shifts Are Not a Measure for the Nakedness of the Fluoride Anion. J. Fluor. Chem. 2002, 116, 49–58. [Google Scholar] [CrossRef]
- Shodai, Y.; Kohara, S.; Ohishi, Y.; Inaba, M.; Tasaka, A. Anionic Species (FH) x F—In Room-Temperature Molten Fluorides (CH 3) 4 NF· m HF. J. Phys. Chem. A 2004, 108, 1127–1132. [Google Scholar] [CrossRef]
- Harmon, K.M.; Southworth, B.A.; Wilson, K.E.; Keefer, P.K. N,N,N-Trimethyl-1-Adamantylammonium Fluoride, a Completely Anhydrous Quaternary Ammonium Fluoride Salt. J. Org. Chem. 1993, 58, 7294–7295. [Google Scholar] [CrossRef]
- Mahjoub, A.R.; Zhang, X.; Seppelt, K. Reactions of the “Naked” Fluoride Ion: Syntheses and Structures of SeF 62− and BrF 6−. Chem. Eur. J. 1995, 1, 261–265. [Google Scholar] [CrossRef]
- Gnann, R.Z.; Wagner, R.I.; Christe, K.O.; Bau, R.; Olah, G.A.; Wilson, W.W. Naked Fluoride Ion Sources: Synthesis, Characterization, and Coupling Reaction of 1-Methylhexamethylenetetramine Fluoride. J. Am. Chem. Soc. 1997, 119, 112–115. [Google Scholar] [CrossRef]
- Kornath, A.; Neumann, F.; Oberhammer, H. Tetramethylphosphonium Fluoride: “Naked” Fluoride and Phosphorane. Inorg. Chem. 2003, 42, 2894–2901. [Google Scholar] [CrossRef]
- Schwesinger, R.; Link, R.; Wenzl, P.; Kossek, S. Anhydrous Phosphazenium Fluorides as Sources for Extremely Reactive Fluoride Ions in Solution. Chem. Eur. J. 2005, 12, 438–445. [Google Scholar] [CrossRef]
- Ryan, S.J.; Schimler, S.D.; Bland, D.C.; Sanford, M.S. Acyl Azolium Fluorides for Room Temperature Nucleophilic Aromatic Fluorination of Chloro- and Nitroarenes. Org. Lett. 2015, 17, 1866–1869. [Google Scholar] [CrossRef]
- Elias, S.; Karton-Lifshin, N.; Yehezkel, L.; Ashkenazi, N.; Columbus, I.; Zafrani, Y. Synthesis, Characterization, and Reactivity of Thermally Stable Anhydrous Quaternary Ammonium Fluorides. Org. Lett. 2017, 19, 3039–3042. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; DiMagno, S.G. Anhydrous Tetrabutylammonium Fluoride. J. Am. Chem. Soc. 2005, 127, 2050–2051. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.K.; Harrison, R.G.; Richmond, T.G. Cobaltocenium Fluoride: A Novel Source of “Naked” Fluoride Formed by Carbon-Fluorine Bond Activation in a Saturated Perfluorocarbon. J. Am. Chem. Soc. 1994, 116, 11165–11166. [Google Scholar] [CrossRef]
- Grushin, V.V. Generation of “Naked” Fluoride Ions in Unprecedentedly High Concentrations from a Fluoropalladium Complex. Angew. Chem. Int. Ed. 1998, 37, 994–996. [Google Scholar] [CrossRef]
- Chen, Z.; Tonouchi, Y.; Matsumoto, K.; Saimura, M.; Atkin, R.; Nagata, T.; Katahira, M.; Hagiwara, R. Partially Naked Fluoride in Solvate Ionic Liquids. J. Phys. Chem. Lett. 2018, 9, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilcher, A.S.; DeShong, P. Utilization of Tetrabutylammonium Triphenyldifluorosilicate as a Fluoride Source for Silicon−Carbon Bond Cleavage †. J. Org. Chem. 1996, 61, 6901–6905. [Google Scholar] [CrossRef]
- Middleton, W.J. New Fluorinating Reagents. Dialkylaminosulfur Fluorides. J. Org. Chem. 1975, 40, 574–578. [Google Scholar] [CrossRef]
- Behr, J.-B.; Massicot, F. Tetramethylammonium Fluoride. Encycl. Reag. Org. Synth. 2001, 1–5. [Google Scholar] [CrossRef]
- Aue, D.H.; Webb, H.M.; Bowers, M.T. A Thermodynamic Analysis of Solvation Effects on the Basicities of Alkylamines. An Electrostatic Analysis of Substituent Effects. J. Am. Chem. Soc. 1976, 98, 318–329. [Google Scholar] [CrossRef]
- McLean, W.J.; Jeffrey, G.A. Crystal Structure of Tetramethylammonium Fluoride Tetrahydrate. J. Chem. Phys. 1967, 47, 414–417. [Google Scholar] [CrossRef]
- Stäben, D.; Mootz, D. Die Kristallinen Hydrate von Tetramethylammoniumfluorid. Bildung, Struktur, Wasserstoffbrückenbindung [1]/The Crystalline Hydrates of Tetramethylammonium Fluoride. Formation, Structure, Hydrogen Bonding. Z. Naturforsch. B 1993, 48, 1057–1064. [Google Scholar] [CrossRef]
- Harmon, K.M.; Madeira, S.L. Hydrogen Bonding. Part 77. Molecular Orbital Study of C–H⋯F Hydrogen Bonds in Tetramethylammonium Fluoride; Potential Effects on Solid State Structure. J. Mol. Struct. 2001, 560, 179–188. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables; 4th ed.; Monograph 9 (Part I and Part II) (1998); 1963. Available online: https://janaf.nist.gov/ (accessed on 13 January 2022).
- Atkins, P.; Overton, T.; Rourke, J.; Weller, M.; Armstrong, F. Inorganic Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2010; p. 94. [Google Scholar]
- Pliego, J.R., Jr.; Piló-Veloso, D. Effects of Ion-Pairing and Hydration on the SNAr Reaction of the F − with p-Chlorobenzonitrile in Aprotic Solvents. Phys. Chem. Chem. Phys. 2008, 10, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Dalessandro, E.V.; Pliego, J.R., Jr. Reactivity and Stability of Ion Pairs, Dimers and Tetramers versus Solvent Polarity: SNAr Fluorination of 2-Bromobenzonitrile with Tetramethylammonium Fluoride. Theor. Chem. Acc. 2020, 139, 27. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W. Reaction of the Fluoride Anion with Acetonitrile. Chloroform and Methylene Chloride. J. Fluor. Chem. 1990, 47, 117–120. [Google Scholar] [CrossRef]
- Grushin, V.V.; Marshall, W.J. Fluorination of Nonactivated Haloarenes via Arynes under Mild Conditions, Resulting from Further Studies toward Ar−F Reductive Elimination from Palladium(II). Organometallics 2008, 27, 4825–4828. [Google Scholar] [CrossRef]
- Bastos, M.M.; Barbosa, J.P.; Pinto, A.C.; Kover, W.B.; Takeuchi, Y.; Boechat, N. Reductive Debromination of 1-Methyl-2,4,5-Tribromoimidazole Mediated by Dry Tetramethylammonium Fluoride in Aprotic Solvents. J. Braz. Chem. Soc. 2001, 12, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.J.; Clark, J.H.; Nightingale, D.J. The Effect of Basicity on Fluorodenitration Reactions Using Tetramethylammonium Salts. Tetrahedron 1999, 55, 7725–7738. [Google Scholar] [CrossRef]
- Yang, Q.; Sheng, M.; Henkelis, J.J.; Tu, S.; Wiensch, E.; Zhang, H.; Zhang, Y.; Tucker, C.; Ejeh, D.E. Explosion Hazards of Sodium Hydride in Dimethyl Sulfoxide, N,N-Dimethylformamide, and N,N-Dimethylacetamide. Org. Process Res. Dev. 2019, 23, 2210–2217. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Sheng, M.; Huang, Y. Potential Safety Hazards Associated with Using N,N-Dimethylformamide in Chemical Reactions. Org. Process Res. Dev. 2020, 24, 1586–1601. [Google Scholar] [CrossRef]
- Wilson, J.W. Standard Enthalpy of Solution and Formation of Tetramethylammonium Fluoride. J. Chem. Thermodyn. 1976, 8, 1107–1108. [Google Scholar] [CrossRef]
- Lawson, A.T.; Collie, N. XLVII.—The action of heat on the salts of tetramethylammonium. J. Chem. Soc. Trans. 1888, 53, 624–636. [Google Scholar] [CrossRef] [Green Version]
- Harmon, K.M.; Gennick, I. Hydrogen Bonding. V. Possible Existence of Strongly Hydrogen-Bonded Water-Fluoride and Water-Hydroxide Complex Anions, (F-.H2O)22- and (OH-.H2O)22-, in Tetramethylammonium Ion Salt Hydrates. Inorg. Chem. 1975, 14, 1840–1845. [Google Scholar] [CrossRef]
- Hawk, M.K.; Ryan, S.J.; Zhang, X.; Huang, P.; Chen, J.; Liu, C.; Chen, J.; Lindsay-Scott, P.J.; Burnett, J.; White, C.; et al. Tetramethylammonium Fluoride Tetrahydrate for SNAr Fluorination of 4-Chlorothiazoles at a Production Scale. Org. Process Res. Dev. 2021, 25, 1167–1175. [Google Scholar] [CrossRef]
- Tunder, R.; Siegel, B. The SF5 Anion. J. Inorg. Nucl. Chem. 1963, 25, 1097–1098. [Google Scholar] [CrossRef]
- Klanberg, F.; Muetterties, E.L. Nuclear Magnetic Resonance Studies on Pentacoordinate Silicon Fluorides. Inorg. Chem. 1968, 7, 155–160. [Google Scholar] [CrossRef]
- Kolomeitsev, A.A.; Seifert, F.U.; Röschenthaler, G.-V. Simple Preparation of Difluorophosphoranes Using Anhydrous Zinc and Tetramethylammonium Fluorides. J. Fluor. Chem. 1995, 71, 47–49. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, T.; Zhou, X. The Synthesis of Quaternary Ammonium Salts from Ammonium Salts and Dialkyl Carbonate. Chem. Commun. 2006, 1864–1865. [Google Scholar] [CrossRef]
- Cismesia, M.A.; Ryan, S.J.; Bland, D.C.; Sanford, M.S. Multiple Approaches to the In Situ Generation of Anhydrous Tetraalkylammonium Fluoride Salts for SNAr Fluorination Reactions. J. Org. Chem. 2017, 82, 5020–5026. [Google Scholar] [CrossRef]
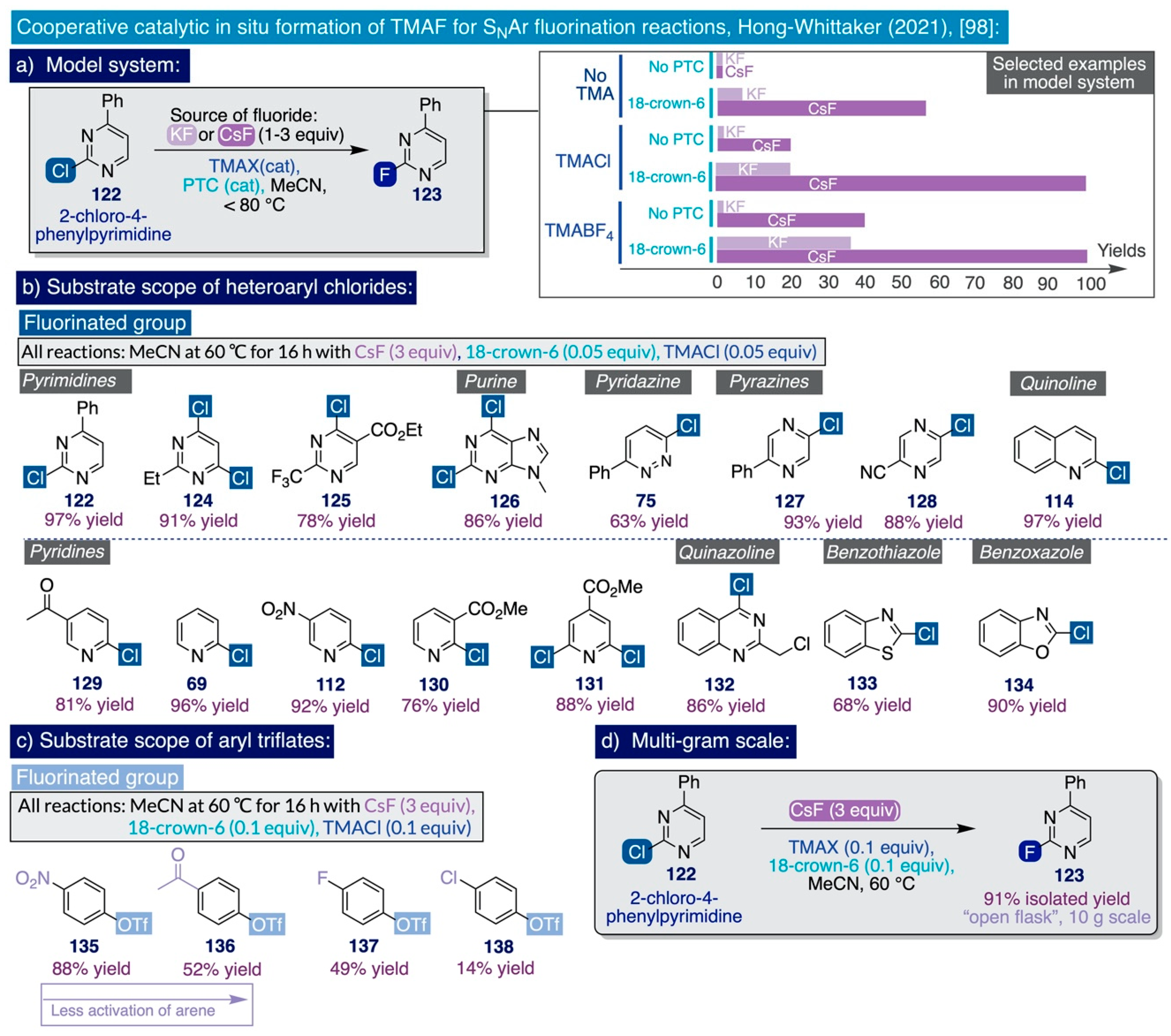
- Hong, C.M.; Whittaker, A.M.; Schultz, D.M. Nucleophilic Fluorination of Heteroaryl Chlorides and Aryl Triflates Enabled by Cooperative Catalysis. J. Org. Chem. 2021, 86, 3999–4006. [Google Scholar] [CrossRef]
- Lee, S.J.; Morales-Colón, M.T.; Brooks, A.F.; Wright, J.S.; Makaravage, K.J.; Scott, P.J.H.; Sanford, M.S. SNAr Radiofluorination with In Situ Generated [18F]Tetramethylammonium Fluoride. J. Org. Chem. 2021, 86, 14121–14130. [Google Scholar] [CrossRef]
- Bloom, S.; Pitts, C.R.; Miller, D.C.; Haselton, N.; Holl, M.G.; Urheim, E.; Lectka, T. A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination. Angew. Chem. Int. Ed. 2012, 51, 10580–10583. [Google Scholar] [CrossRef]
- Guo, R.; Huang, J.; Zhao, X. Organoselenium-Catalyzed Oxidative Allylic Fluorination with Electrophilic N–F Reagent. ACS Catal. 2018, 8, 926–930. [Google Scholar] [CrossRef]
- Sorlin, A.M.; Usman, F.O.; English, C.K.; Nguyen, H.M. Advances in Nucleophilic Allylic Fluorination. ACS Catal. 2020, 10, 11980–12010. [Google Scholar] [CrossRef]
- Terrier, F. Modern Nucleophilic Aromatic Substitution; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–94. [Google Scholar] [CrossRef]
- Lennox, A.J.J. Meisenheimer Complexes in SNAr Reactions: Intermediates or Transition States? Angew. Chem. Int. Ed. 2018, 57, 14686–14688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artamkina, G.A.; Egorov, M.P.; Beletskaya, I.P. Some Aspects of Anionic. Sigma.-Complexes. Chem. Rev. 1982, 82, 427–459. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G.; Uggerud, E. Rate-Determining Factors in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 2010, 75, 2971–2980. [Google Scholar] [CrossRef]
- Cramption, M.R.; Gold, V. 824. Reactions of Aromatic Nitro-Compounds in Alkaline Media. Part IX. Nuclear Magnetic Resonance Spectra of Meisenheimer Complexes. J. Chem. Soc. 1964, 4293–4295. [Google Scholar] [CrossRef]
- Kwan, E.E.; Zeng, Y.; Besser, H.A.; Jacobsen, E.N. Concerted Nucleophilic Aromatic Substitutions. Nat. Chem. 2018, 10, 917–923. [Google Scholar] [CrossRef]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted Nucleophilic Aromatic Substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.N.; Ritter, T. Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res. 2017, 50, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
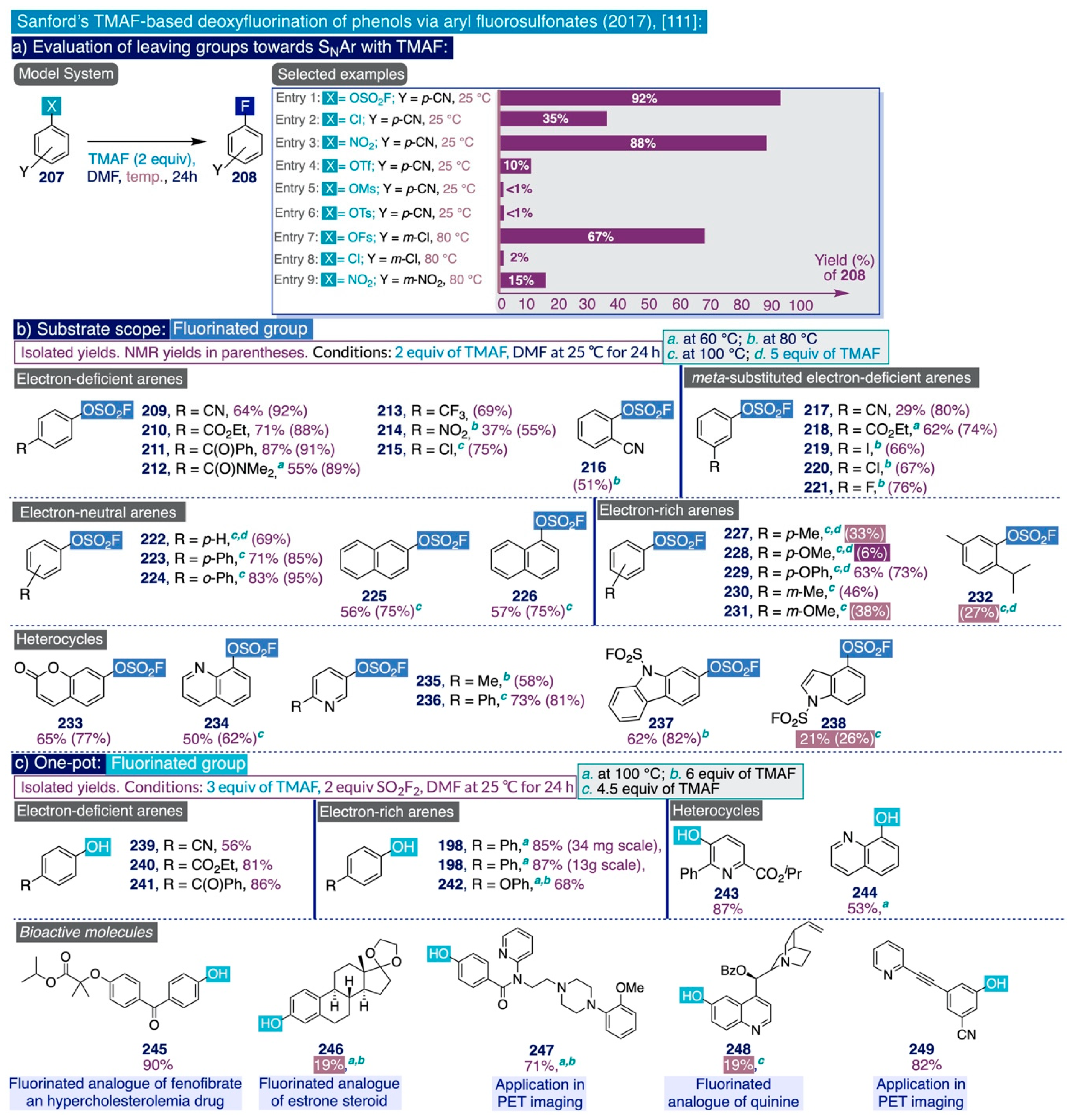
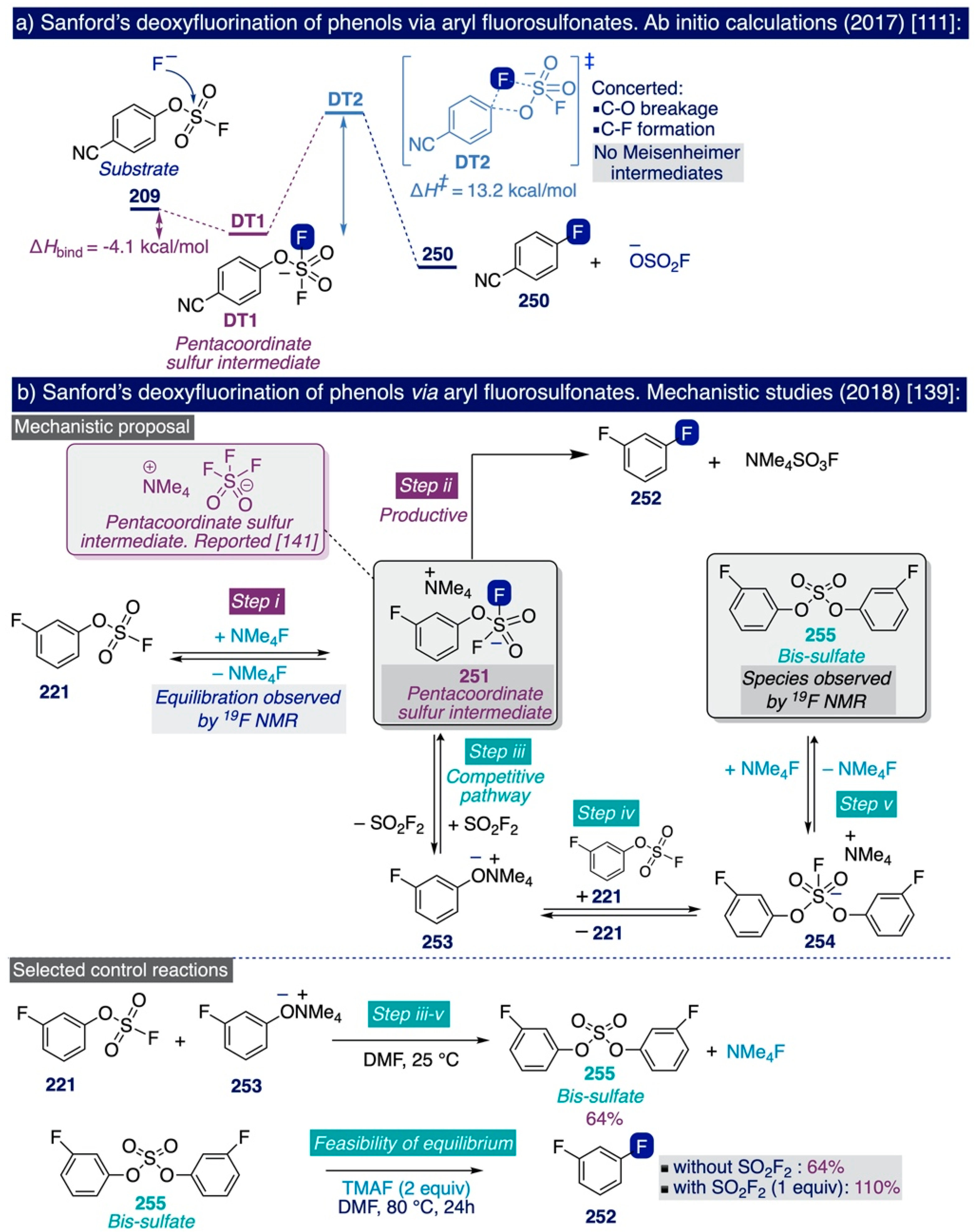
- Schimler, S.D.; Cismesia, M.A.; Hanley, P.S.; Froese, R.D.J.; Jansma, M.J.; Bland, D.C.; Sanford, M.S. Nucleophilic Deoxyfluorination of Phenols via Aryl Fluorosulfonate Intermediates. J. Am. Chem. Soc. 2017, 139, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Ba-Saif, S.; Luthra, A.K.; Williams, A. Concertedness in Acyl Group Transfer in Solution: A Single Transition State in Acetyl Group Transfer between Phenolate Ion Nucleophiles. J. Am. Chem. Soc. 1987, 109, 6362–6368. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- der Plas, H.C.V. The SN(ANRORC) Mechanism: A New Mechanism for Nucleophilic Substitution. Acc. Chem. Res. 1978, 11, 462–468. [Google Scholar] [CrossRef]
- Balz, G.; Schiemann, G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Ber. Dtsch. Chem. Ges. B 1927, 60, 1186–1190. [Google Scholar] [CrossRef]
- Cresswell, A.J.; Davies, S.G.; Roberts, P.M.; Thomson, J.E. Beyond the Balz–Schiemann Reaction: The Utility of Tetrafluoroborates and Boron Trifluoride as Nucleophilic Fluoride Sources. Chem. Rev. 2014, 115, 566–611. [Google Scholar] [CrossRef]
- Gottlieb, H.B. The Replacement of Chlorine by Fluorine in Organic Compounds. J. Am. Chem. Soc. 1936, 58, 532–533. [Google Scholar] [CrossRef]
- Finger, G.C.; Kruse, C.W. Aromatic Fluorine Compounds. VII. Replacement of Aromatic -Cl and -NO2 Groups by -F 1,2. J. Am. Chem. Soc. 1956, 78, 6034–6037. [Google Scholar] [CrossRef]
- Clark, J.H.; Macquarrie, D.J. The Synthesis of Organofluorine Compounds Using Potassium Fluoride-Tetraphenylphosphonium Bromide Systems. Tetrahedron Lett. 1987, 28, 111–114. [Google Scholar] [CrossRef]
- Boechat, N.; Clark, J.H. Fluorodenitrations using tetramethylammonium fluoride. J. Chem. Soc. Chem. Commun. 1993, 921–922. [Google Scholar] [CrossRef]
- Clark, J.H.; Wails, D. Fluorodenitration of Activated Diphenyl Sulphones Using Tetramethylammonium Fluoride. J. Fluor. Chem. 1995, 70, 201–205. [Google Scholar] [CrossRef]
- Adams, D.J.; Clark, J.H.; Nightingale, D.J. Tetramethylammonium Hydrogendifluoride: A Convenient Source of Nucleophilic Fluoride. Synth. Commun. 1998, 28, 4295–4301. [Google Scholar] [CrossRef]
- Adams, D.J.; Clark, J.H.; McFarland, H. The Formation of 4,4′-Difluorobenzophenone from 4,4′-Dinitrodiphenylmethane. J. Fluor. Chem. 1998, 92, 127–129. [Google Scholar] [CrossRef]
- Sun, H.; DiMagno, S.G. Room-Temperature Nucleophilic Aromatic Fluorination: Experimental and Theoretical Studies. Angew. Chem. Int. Ed. 2006, 45, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Eckelbarger, J.D.; Epp, J.B.; Schmitzer, P.R.; Siddall, T.L. 3-Alkenyl-6-halo-4-aminopicolinates and Their Use as Herbicides. U.S. Patent 20120190548A1, 26 July 2012. [Google Scholar]
- Whiteker, G.T.; Arndt, K.E.; Renga, J.M.; Yuanming, Z.; Lowe, C.T.; Siddall, T.L.; Podhorez, D.E.; Roth, G.A.; West, S.P.; Arndt, C. Process for the preparation of 4-amino-5-fluoro-3-halo-6-(substituted)picolinates. U.S. Patent 20120190860, 26 July 2012. [Google Scholar]
- Yerkes, C.N.; Lowe, C.T.; Eckelbarger, J.D.; Epp, J.B.; Guenthenspberger, K.A.; Siddall, T.L.; Schmitzer, P.R. Arylalkyl Esters of 4-amino-6-(substitutedphenyl)picolinates and 6-amino-2-(substitutedphenyl)-4-pyrimidinecarboxylates and Their Use as Selective Herbicides for Crops. U.S. Patent 20150025238A1, 22 January 2015. [Google Scholar]
- Schimler, S.D.; Ryan, S.J.; Bland, D.C.; Anderson, J.E.; Sanford, M.S. Anhydrous Tetramethylammonium Fluoride for Room-Temperature SNAr Fluorination. J. Org. Chem. 2015, 80, 12137–12145. [Google Scholar] [CrossRef] [Green Version]
- Hicken, E.J.; Marmsater, F.P.; Munson, M.C.; Schlachter, S.T.; Robinson, J.E.; Allen, S.; Burgess, L.E.; DeLisle, R.K.; Rizzi, J.P.; Topalov, G.T.; et al. Discovery of a Novel Class of Imidazo[1,2- a ]Pyridines with Potent PDGFR Activity and Oral Bioavailability. ACS Med. Chem. Lett. 2013, 5, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, R.; Ironside, M.D.; Vogt, P.F. Safety Notables: Information from the Literature. Org. Process Res. Dev. 2010, 14, 1480–1484. [Google Scholar] [CrossRef]
- Morales-Colón, M.T.; See, Y.Y.; Lee, S.J.; Scott, P.J.H.; Bland, D.C.; Sanford, M.S. Tetramethylammonium Fluoride Alcohol Adducts for SNAr Fluorination. Org. Lett. 2021, 23, 4493–4498. [Google Scholar] [CrossRef]
- Kim, D.W.; Jeong, H.; Lim, S.T.; Sohn, M. Tetrabutylammonium Tetra(Tert-Butyl Alcohol)-Coordinated Fluoride as a Facile Fluoride Source. Angew. Chem. Int. Ed. 2008, 47, 8404–8406. [Google Scholar] [CrossRef]
- Gupta, H.K.; Pardasani, D.; Mazumder, A.; Purohit, A.K.; Dubey, D.K. Tetrabutylammonium Tetra (Tert-Butyl Alcohol) Coordinated Fluoride-an Efficient Reagent for the Synthesis of Fluorine Derivatives of Phosphorus(V) Compounds. Tetrahedron Lett. 2009, 50, 2697–2699. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Kryachko, E.S.; Vanquickenborne, L.G. General and Theoretical Aspects of Phenols. In The Chemistry of Phenols; Rappoport, Z., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2003. [Google Scholar] [CrossRef]
- Nemoto, H.; Nishiyama, T.; Akai, S. Nucleophilic Deoxyfluorination of Catechols. Org. Lett. 2011, 13, 2714–2717. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sonoda, H.; Fukumura, K.; Nagata, T. 2,2-Difluoro-1,3-Dimethylimidazolidine (DFI). A New Fluorinating Agent. Chem. Commun. 2002, 1618–1619. [Google Scholar] [CrossRef]
- Tang, P.; Wang, W.; Ritter, T. Deoxyfluorination of Phenols. J. Am. Chem. Soc. 2011, 133, 11482–11484. [Google Scholar] [CrossRef] [Green Version]
- Schimler, S.D.; Froese, R.D.J.; Bland, D.C.; Sanford, M.S. Reactions of Arylsulfonate Electrophiles with NMe4F: Mechanistic Insight, Reactivity, and Scope. J. Org. Chem. 2018, 83, 11178–11190. [Google Scholar] [CrossRef]
- Fry, S.E.; Pienta, N.J. Effects of Molten Salts on Reactions. Nucleophilic Aromatic Substitution by Halide Ions in Molten Dodecyltributylphosphonium Salts. J. Am. Chem. Soc. 1985, 107, 6399–6400. [Google Scholar] [CrossRef]
- Hohenstein, C.; Kadzimirsz, D.; Ludwig, R.; Kornath, A. Synthesis and Characterization of Tetramethylammonium Trifluorosulfate. Chem. Eur. J. 2011, 17, 925–929. [Google Scholar] [CrossRef]
- Melvin, P.R.; Ferguson, D.M.; Schimler, S.D.; Bland, D.C.; Sanford, M.S. Room Temperature Deoxyfluorination of Benzaldehydes and α-Ketoesters with Sulfuryl Fluoride and Tetramethylammonium Fluoride. Org. Lett. 2019, 21, 1350–1353. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Melvin, P.R.; Sanford, M.S. Deoxyfluorination of (Hetero)Aryl Aldehydes Using Tetramethylammonium Fluoride and Perfluorobutanesulfonyl Fluoride or Trifluoromethanesulfonic Anhydride. Isr. J. Chem. 2020, 60, 398–401. [Google Scholar] [CrossRef]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H. Bis(2-Methoxyethyl)Aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999, 64, 7048–7054. [Google Scholar] [CrossRef]
- Veryser, C.; Demaerel, J.; Bieliūnas, V.; Gilles, P.; Borggraeve, W.M.D. Ex Situ Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org. Lett. 2017, 19, 5244–5247. [Google Scholar] [CrossRef] [PubMed]
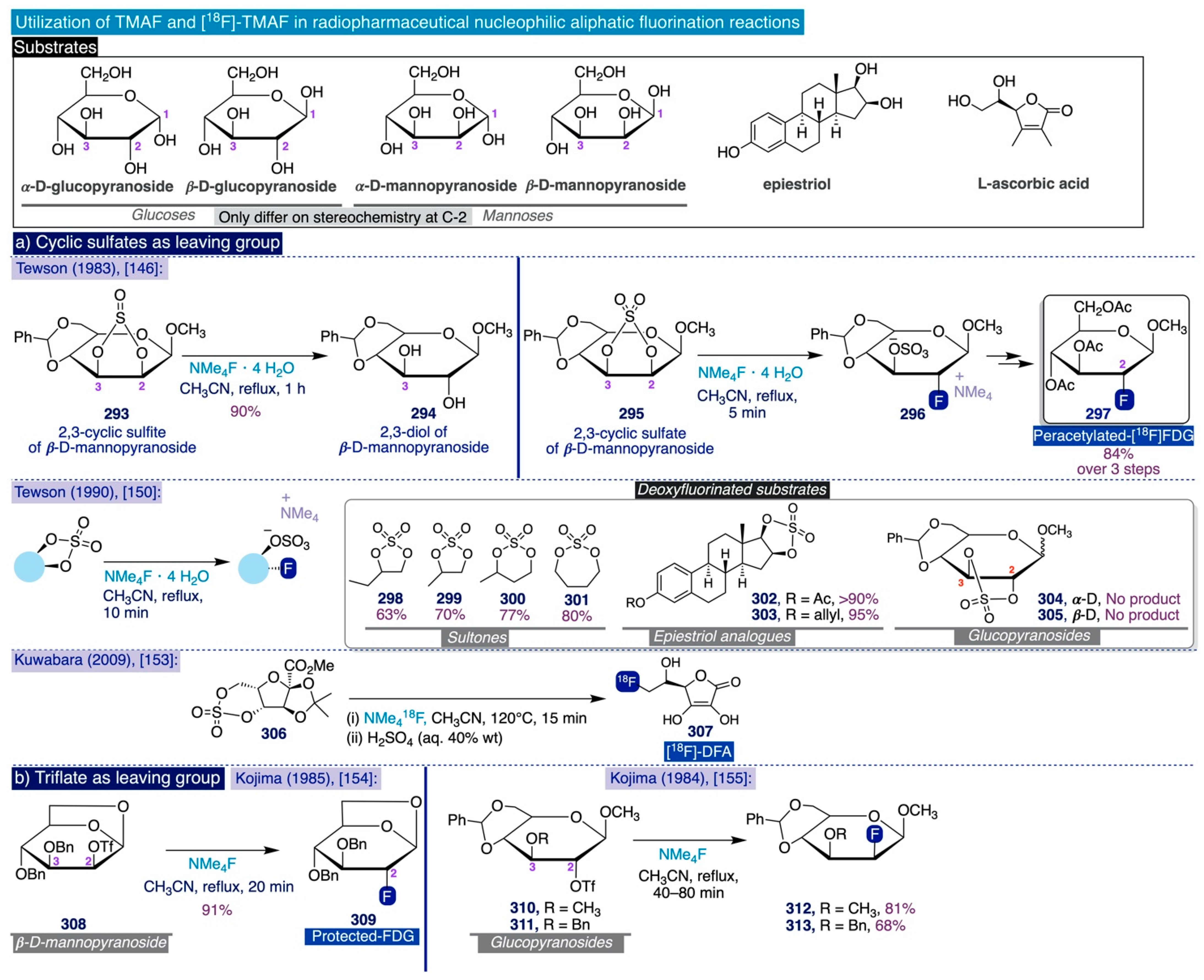
- Tewson, T.J. Cyclic Sulfur Esters as Substrates for Nucleophilic Substitution. A New Synthesis of 2-Deoxy-2-Fluoro-D-Glucose. J. Org. Chem. 1983, 48, 3507–3510. [Google Scholar] [CrossRef]
- Fatangare, A.; Svatoš, A. Applications of 2-Deoxy-2-Fluoro-D-Glucose (FDG) in Plant Imaging: Past, Present, and Future. Front. Plant. Sci. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido, T.; Wan, C.; Casella, V.; Fowler, J.S.; Wolf, A.P.; Reivich, M.; Kuhl, D.E. Labeled 2-deoxy-D-glucose Analogs. 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose. J. Labelled Compd. Radiopharm. 1978, 14, 175–183. [Google Scholar] [CrossRef]
- Gatley, S.J.; Shaughnessy, W.J. Synthesis of 18F-3-Deoxy-3-Fluoro-D-Glucose with Reactor-Produced 18F. Int. J. Appl. Radiat. Isot. 1980, 31, 339–341. [Google Scholar] [CrossRef]
- Berridge, M.S.; Franceschini, M.P.; Rosenfeld, E.; Tewson, T.J. Cyclic Sulfates: Useful Substrates for Selective Nucleophilic Substitution. J. Org. Chem. 1990, 55, 1211–1217. [Google Scholar] [CrossRef]
- Lim, J.L.; Zheng, L.; Berridge, M.S.; Tewson, T.J. The Use of 3-Methoxymethyl-16β, 17β-Epiestriol-O-Cyclic Sulfone as the Precursor in the Synthesis of F-18 16α-Fluoroestradiol. Nucl. Med. Biol. 1996, 23, 911–915. [Google Scholar] [CrossRef]
- Seimbille, Y.; Ali, H.; Lier, J.E. van Synthesis of 2,16α- and 4,16α-Difluoroestradiols and Their 11β-Methoxy Derivatives as Potential Estrogen Receptor-Binding Radiopharmaceuticals. J. Chem. Soc. Perkin Trans. 2002, 1, 657–663. [Google Scholar] [CrossRef]
- Kim, J.; Yamamoto, F.; Kuwabara, Y.; Honda, H.; Mukai, T.; Maeda, M. 6-[18F]Fluoro-Dehydroascorbic Acid:Synthesis and Tissue Distribution in Mice. Radioisotopes 2009, 58, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Haradahira, T.; Maeda, M.; Kai, Y.; Kojima, M. A New, High Yield Synthesis of 2-Deoxy-2-Fluoro- D -Glucose. J. Chem. Soc. Chem. Commun. 1985, 364–365. [Google Scholar] [CrossRef]
- Haradahira, T.; Maeda, M.; Omae, H.; Yano, Y.; Kojima, M. Synthesis of 2-Deoxy-2-Fluoro-D-Mannose Using Fluoride Ion. Chem. Pharm. Bull. 1984, 32, 4758–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
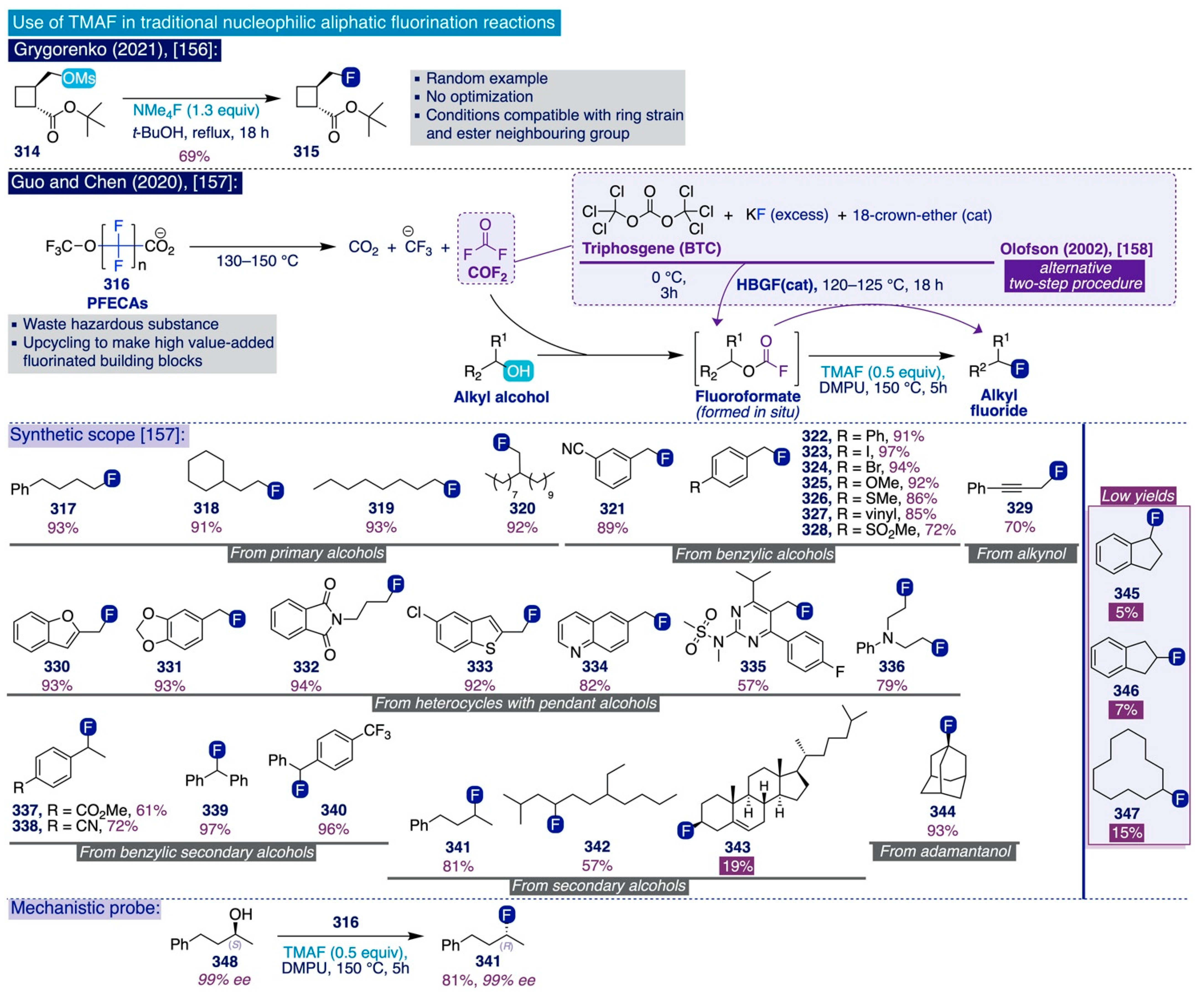
- Demchuk, O.P.; Hryshchuk, O.V.; Vashchenko, B.V.; Trofymchuk, S.A.; Melnykov, K.P.; Skreminskiy, A.; Volochnyuk, D.M.; Grygorenko, O.O. Fluoroalkyl-Containing 1,2-Disubstituted Cyclobutanes: Advanced Building Blocks for Medicinal Chemistry. Eur. J. Org. Chem. 2021, 2021, 87–95. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Su, Z.; Cao, W.; Wu, C.; Chen, Q.-Y. A Series of Deoxyfluorination Reagents Featuring OCF2 Functional Groups. Org. Lett. 2020, 22, 8634–8637. [Google Scholar] [CrossRef] [PubMed]
- Flosser, D.A.; Olofson, R.A. A Useful Conversion of Alcohols to Alkyl Fluorides. Tetrahedron Lett. 2002, 43, 4275–4279. [Google Scholar] [CrossRef]
- Zhang, X.; Gross, U.; Seppelt, K. Fluorocarbonate, [FCO2]−: Preparation and Structure. Angew. Chem. Int. Ed. 1995, 34, 1858–1860. [Google Scholar] [CrossRef]
- Lawlor, L.; Passmore, J. Existence of Cesium Salts of CO2F-, CO2F22- and NO2F2-. Inorg. Chem. 1979, 18, 2923–2924. [Google Scholar] [CrossRef]
- Rüdiger, S.; Seppelt, K. On Reactions of Carbon Disulphide Induced by ‘Naked’ Fluoride Part 2: Reactions with 2-H-Heptafluoropropane, Hexafluoropropene, and Bis (2,2,2-Trifluoroethyl) Amine. J. Fluor. Chem. 1997, 82, 29–32. [Google Scholar] [CrossRef]
- Rüdiger, S.; Seppelt, K. On Reactions of Carbon Disulphide Induced by ‘Naked’ Fluoride Part 1: Reactions with Fluoroaromatics. J Fluor. Chem. 1997, 82, 25–28. [Google Scholar] [CrossRef]
- Kovács, S.; Bayarmagnai, B.; Goossen, L.J. Preparation of Electrophilic Trifluromethylthio Reagents from Nucleophilic Tetramethylammonium Trifluoromethylthiolate. Adv. Synth. Catal. 2017, 359, 250–254. [Google Scholar] [CrossRef]
- Clark, J.H.; Tavener, S.J. The Preparation of Trifluoromethyl Aryl Sulfides Using KF and Thiophosgene. J. Fluor. Chem. 1997, 85, 169–172. [Google Scholar] [CrossRef]
- Dmowski, W.; Haas, A. Trifluoromethanethiolate Ion. Part 2. Nucleophilic Substitution in Pentafluoropyridine. Synthesis and Characteristics of Trifluoromethylthio and Trifluoromethylsulphonyl Derivatives. J. Chem. Soc. Perkin Trans. 1987, 1, 2119–2124. [Google Scholar] [CrossRef]
- Tavener, S.J.; Adams, D.J.; Clark, J.H. Trifluoromethylthiolation of Aromatic Substrates Using Thiophosgene—Fluoride Salt Reagents, and Formation of Byproducts with Multi-Carbon Chains. J Fluor. Chem. 1999, 95, 171–176. [Google Scholar] [CrossRef]
- Tyrra, W.; Naumann, D.; Hoge, B.; Yagupolskii, Y.L. A New Synthesis of Trifluoromethanethiolates—Characterization and Properties of Tetramethylammonium, Cesium and Di(Benzo-15-Crown-5)Cesium Trifluoromethanethiolates. J. Fluor. Chem. 2003, 119, 101–107. [Google Scholar] [CrossRef]
- Yin, G.; Kalvet, I.; Schoenebeck, F. Trifluoromethylthiolation of Aryl Iodides and Bromides Enabled by a Bench-Stable and Easy-To-Recover Dinuclear Palladium(I) Catalyst. Angew. Chem. Int. Ed. 2015, 54, 6809–6813. [Google Scholar] [CrossRef]
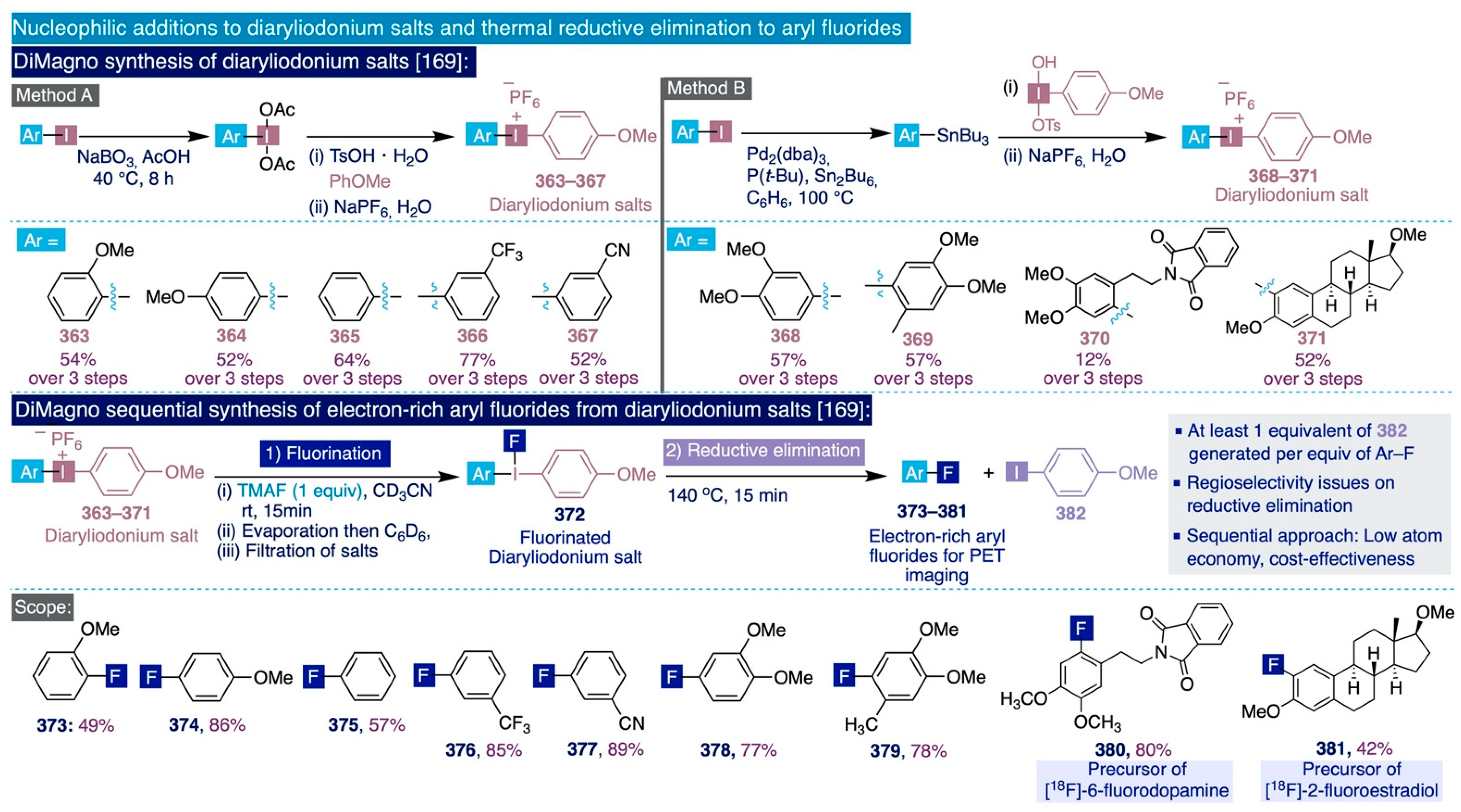
- Wang, B.; Qin, L.; Neumann, K.D.; Uppaluri, S.; Cerny, R.L.; DiMagno, S.G. Improved Arene Fluorination Methodology for I(III) Salts. Org. Lett. 2010, 12, 3352–3355. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, B.; DiMagno, S.G. A Method for Detecting Water in Organic Solvents. Org. Lett. 2008, 10, 4413–4416. [Google Scholar] [CrossRef] [Green Version]
- Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef]
- Clark, J.H. Fluoride Ion as a Base in Organic Synthesis. Chem. Rev. 1980, 80, 429–452. [Google Scholar] [CrossRef]
- Hameed, A.; Alharthy, R.D.; Iqbal, J.; Langer, P. The Role of Naked Fluoride Ion as Base or Catalyst in Organic Synthesis. Tetrahedron 2016, 72, 2763–2812. [Google Scholar] [CrossRef]
- Ménand, M.; Dalla, V. TMAF-Catalyzed Conjugate Addition of Oxazolidinone and Thiols. Synlett 2004, 2005, 95–98. [Google Scholar] [CrossRef]
- Cid, M.B.; Duce, S.; Morales, S.; Rodrigo, E.; Ruano, J.L.G. Nitrophenylacetonitriles as Versatile Nucleophiles in Enantioselective Organocatalytic Conjugate Additions. Org. Lett. 2010, 12, 3586–3589. [Google Scholar] [CrossRef] [PubMed]
- Messire, G.; Massicot, F.; Vallée, A.; Vasse, J.; Behr, J. Aza-Henry Reaction with Nitrones, an Under-Explored Transformation. Eur. J. Org. Chem. 2019, 2019, 1659–1668. [Google Scholar] [CrossRef]
- da Silva, V.B.; Campos, B.R.; de Oliveira, J.F.; Decout, J.L.; de Lima, M.D. Medicinal Chemistry of Antischistosomal Drugs: Praziquantel and Oxamniquine. Bioorg. Med. Chem. 2017, 25, 3259–3277. [Google Scholar] [CrossRef] [PubMed]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic Studies on the Michael Addition of Amines and Hydrazines To Nitrostyrenes: Nitroalkane Elimination via a Retro-Aza-Henry-Type Process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-G.; Pu, M.; Kundu, G.; Schoenebeck, F. Selective Methylation of Amides, N-Heterocycles, Thiols, and Alcohols with TMAF. Org. Lett. 2020, 22, 331–334. [Google Scholar] [CrossRef]
- Schönherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
- Scattolin, T.; Pu, M.; Schoenebeck, F. Investigation of (Me4N)SCF3 as a Stable, Solid and Safe Reservoir for S=CF2 as a Surrogate for Thiophosgene. Chem. Eur. J. 2018, 24, 567–571. [Google Scholar] [CrossRef]
- For full details of Schoenebeck’s Mechanistic DFT Studies for the Chemoselective Methylation of Secondary Amides and Alcohols, See pp. S14–S21 in the Supporting Information of That Work.
- Wang, X.; Xu, Y.; Mo, F.; Ji, G.; Qiu, D.; Feng, J.; Ye, Y.; Zhang, S.; Zhang, Y.; Wang, J. Silver-Mediated Trifluoromethylation of Aryldiazonium Salts: Conversion of Amino Group into Trifluoromethyl Group. J. Am. Chem. Soc. 2013, 135, 10330–10333. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; König, B. Photo-Induced Thiolate Catalytic Activation of Inert Caryl-Hetero Bonds for Radical Borylation. Chem 2021, 7, 1653–1665. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, G.F.S.; Denny, W.A.; Santos, J.L.D. Boron in Drug Design: Recent Advances in the Development of New Therapeutic Agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Plescia, J.; Moitessier, N. Design and Discovery of Boronic Acid Drugs. Eur. J. Med. Chem. 2020, 195, 112270. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Wrigstedt, P.; Moslova, K.; Iashin, V.; Mäkkylä, H.; Ghemtio, L.; Heikkinen, S.; Tammela, P.; Perea-Buceta, J.E. Installation of an Aryl Boronic Acid Function into the External Section of N-Aryl-Oxazolidinones: Synthesis and Antimicrobial Evaluation. Eur. J. Med. Chem. 2021, 211, 113002. [Google Scholar] [CrossRef]
- Tian, Y.-M.; Guo, X.-N.; Braunschweig, H.; Radius, U.; Marder, T.B. Photoinduced Borylation for the Synthesis of Organoboron Compounds. Chem. Rev. 2021, 121, 3561–3597. [Google Scholar] [CrossRef] [PubMed]
- Schmalzbauer, M.; Marcon, M.; König, B. Excited State Anions in Organic Transformations. Angew. Chem. Int. Ed. 2021, 60, 6270–6292. [Google Scholar] [CrossRef]
- Wang, S.; Lokesh, N.; Hioe, J.; Gschwind, R.M.; König, B. Photoinitiated Carbonyl-Metathesis: Deoxygenative Reductive Olefination of Aromatic Aldehydes via Photoredox Catalysis. Chem. Sci. 2019, 10, 4580–4587. [Google Scholar] [CrossRef] [Green Version]
- Schmalzbauer, M.; Svejstrup, T.D.; Fricke, F.; Brandt, P.; Johansson, M.J.; Bergonzini, G.; König, B. Redox-Neutral Photocatalytic C−H Carboxylation of Arenes and Styrenes with CO2. Chem 2020, 6, 2658–2672. [Google Scholar] [CrossRef]
- Wang, M.; Shi, Z. Methodologies and Strategies for Selective Borylation of C–Het and C–C Bonds. Chem. Rev. 2020, 120, 7348–7398. [Google Scholar] [CrossRef]
- Jin, S.; Dang, H.T.; Haug, G.C.; He, R.; Nguyen, V.D.; Nguyen, V.T.; Arman, H.D.; Schanze, K.S.; Larionov, O.V. Visible Light-Induced Borylation of C–O, C–N, and C–X Bonds. J. Am. Chem. Soc. 2020, 142, 1603–1613. [Google Scholar] [CrossRef]
- Fernández, I.; Bickelhaupt, F.M. The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Davies, I.W. The Digitization of Organic Synthesis. Nature 2019, 570, 175–181. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iashin, V.; Wirtanen, T.; Perea-Buceta, J.E. Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base. Catalysts 2022, 12, 233. https://doi.org/10.3390/catal12020233
Iashin V, Wirtanen T, Perea-Buceta JE. Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base. Catalysts. 2022; 12(2):233. https://doi.org/10.3390/catal12020233
Chicago/Turabian StyleIashin, Vladimir, Tom Wirtanen, and Jesus E. Perea-Buceta. 2022. "Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base" Catalysts 12, no. 2: 233. https://doi.org/10.3390/catal12020233