

Tetranuclear Copper Complexes with Bulky Aminoalcohol Ligands as Catalysts for Oxidative Phenoxazinone Synthase-like Coupling of Aminophenol: A Combined Experimental and Theoretical Study

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
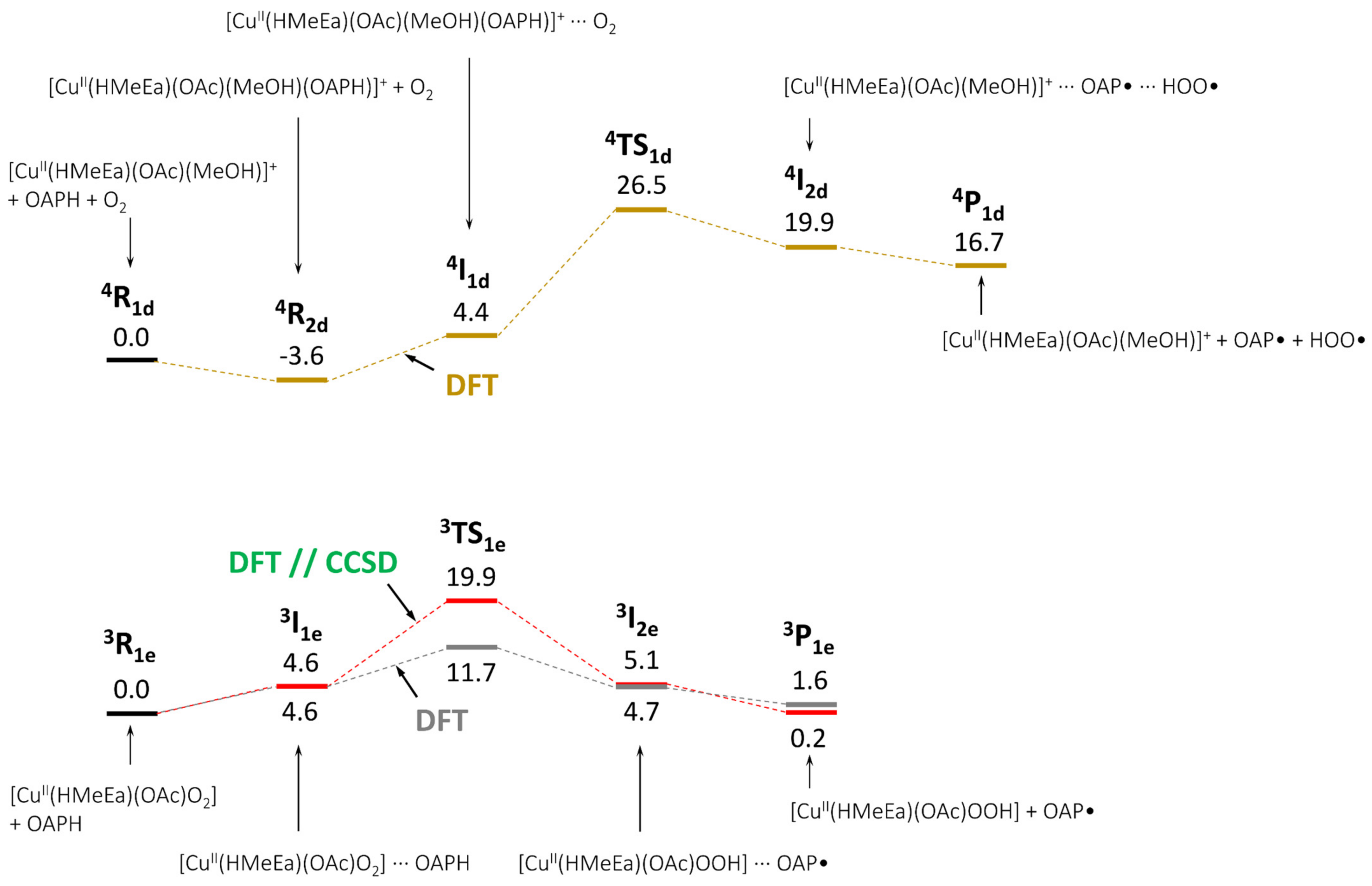{kind=link}
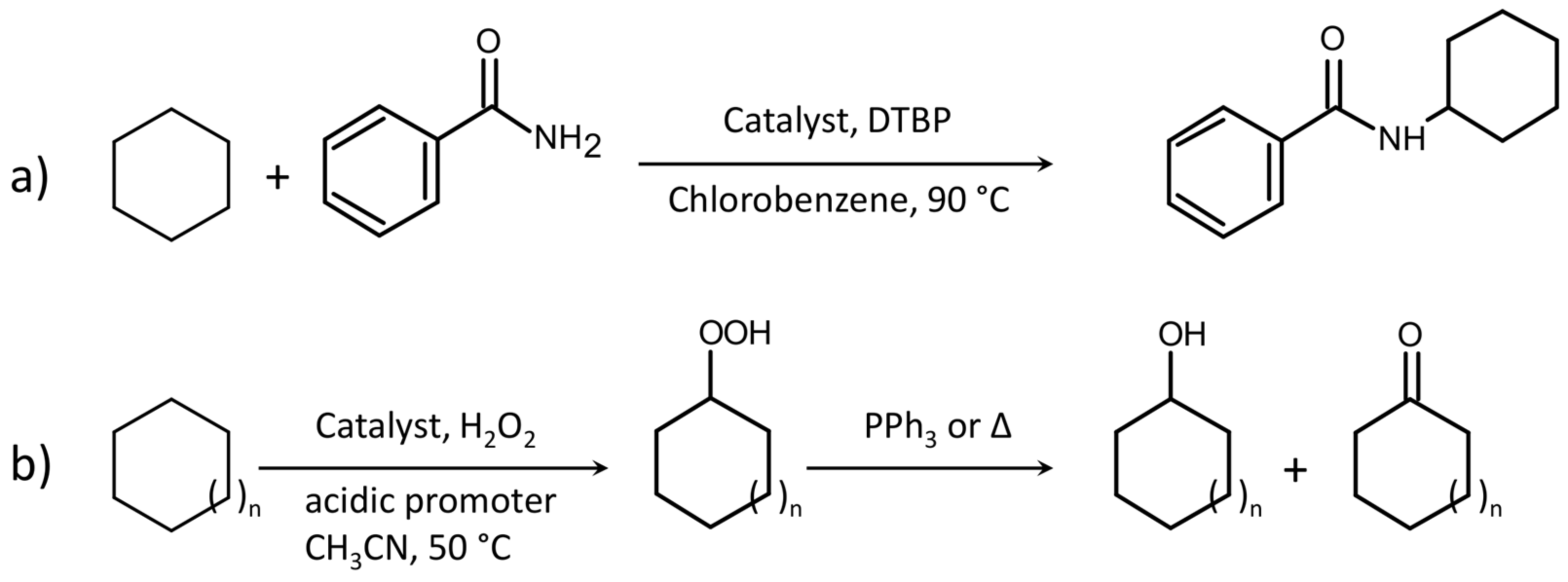{kind=link}
1. Introduction
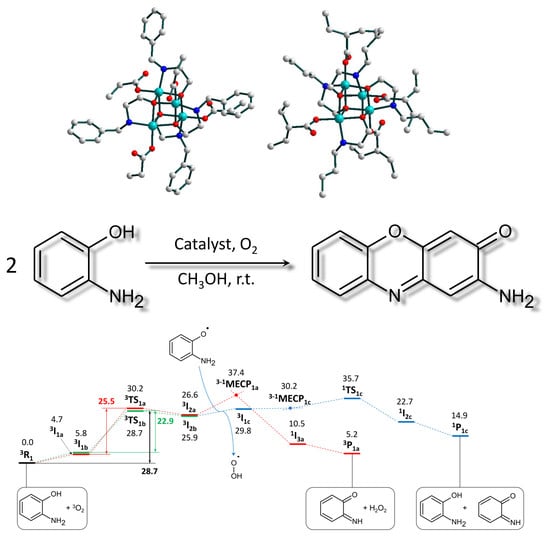
2. Results
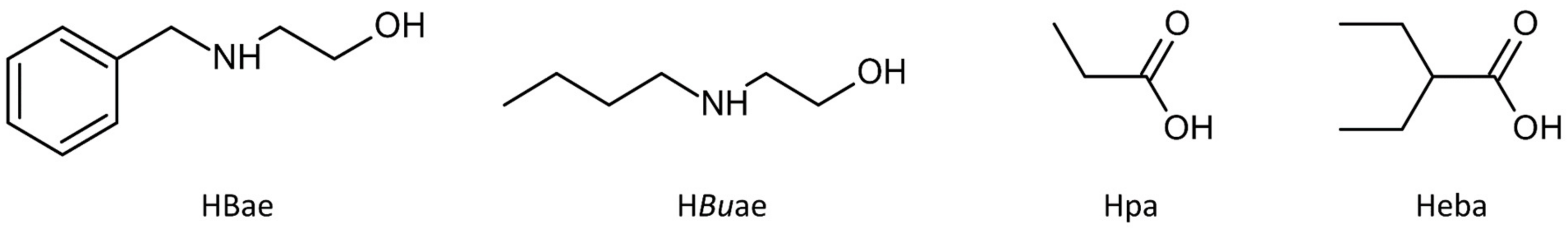
2.1. Synthesis and Spectroscopic Analysis
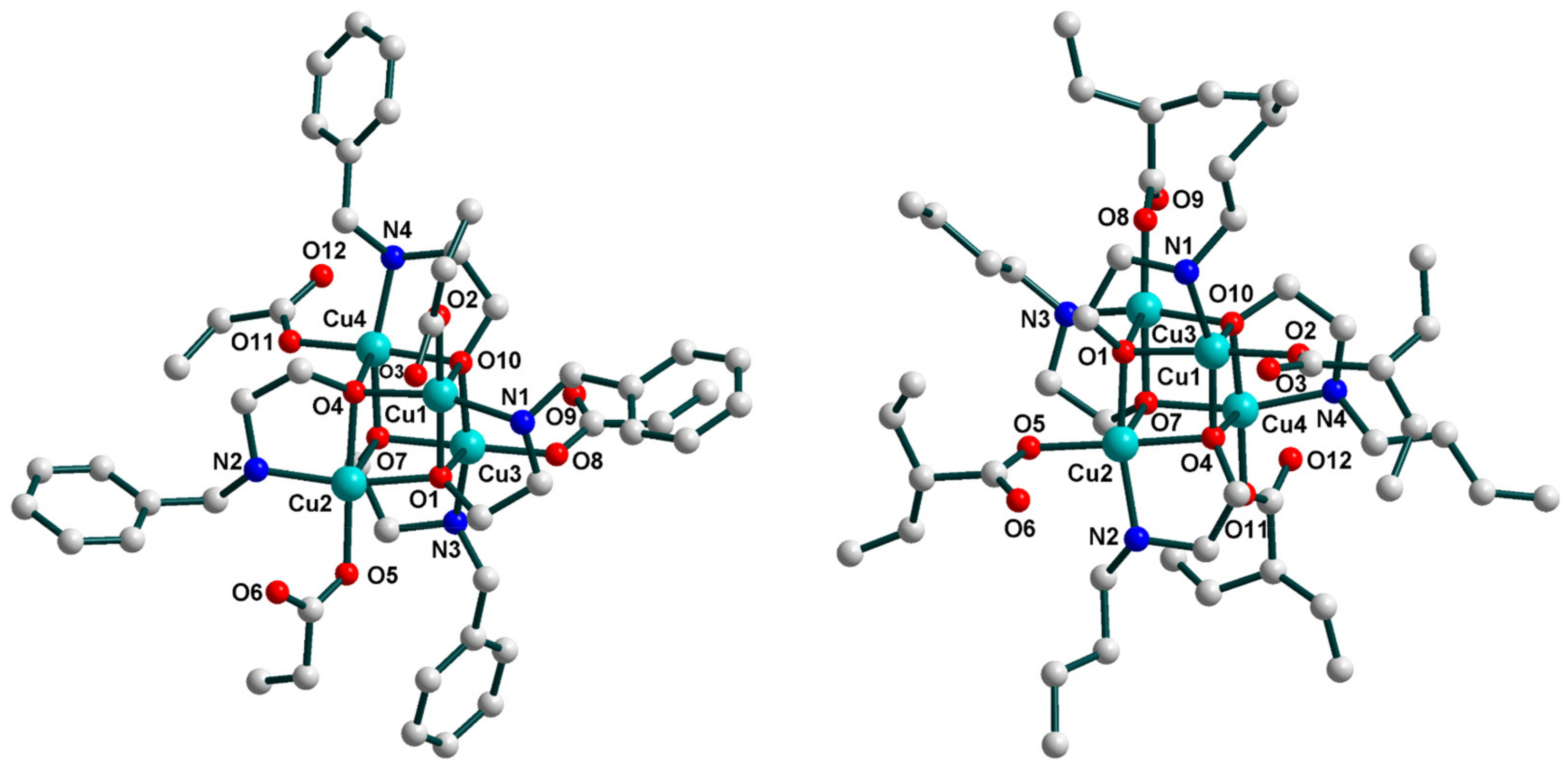
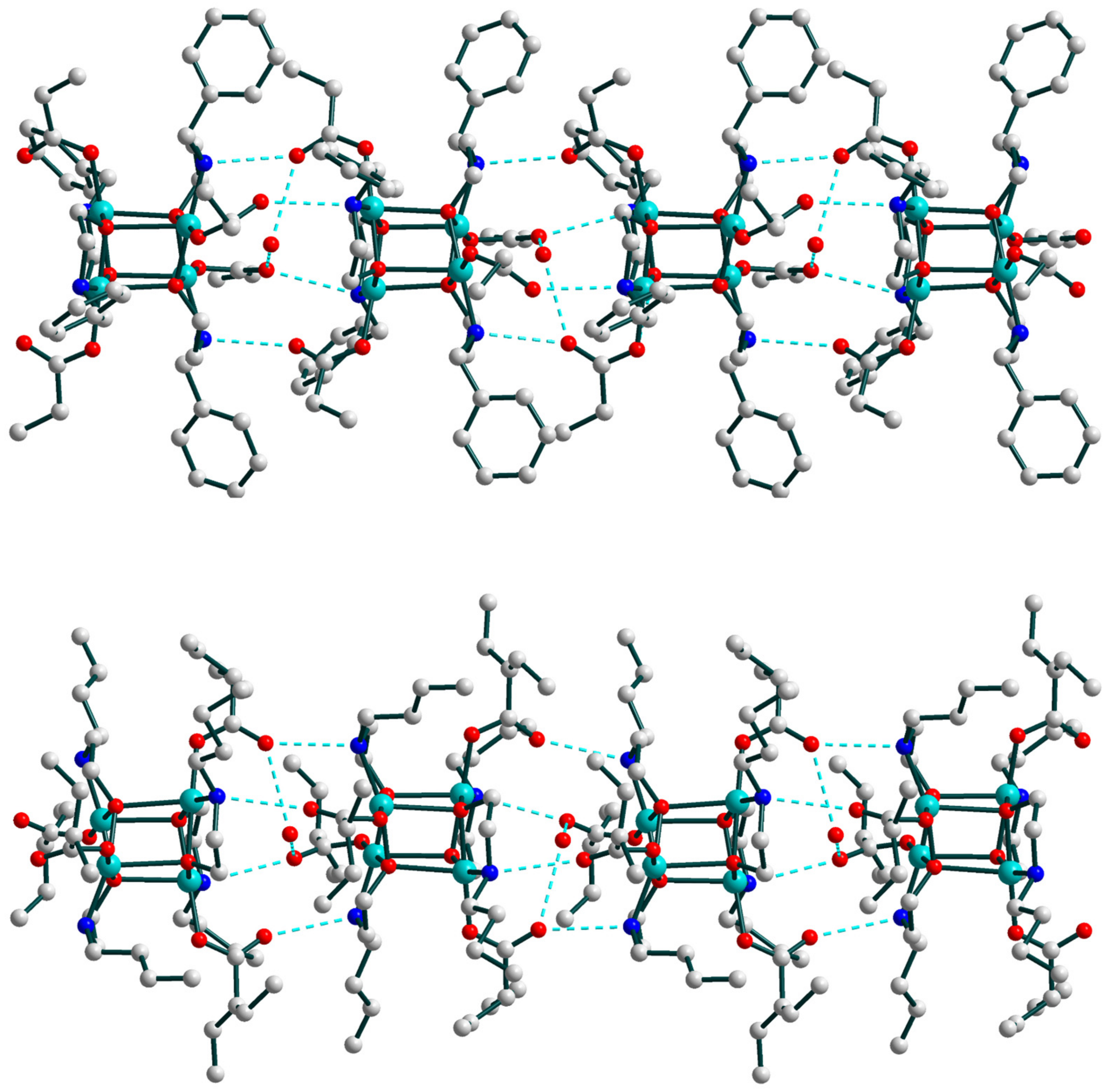
2.2. Crystal Structures
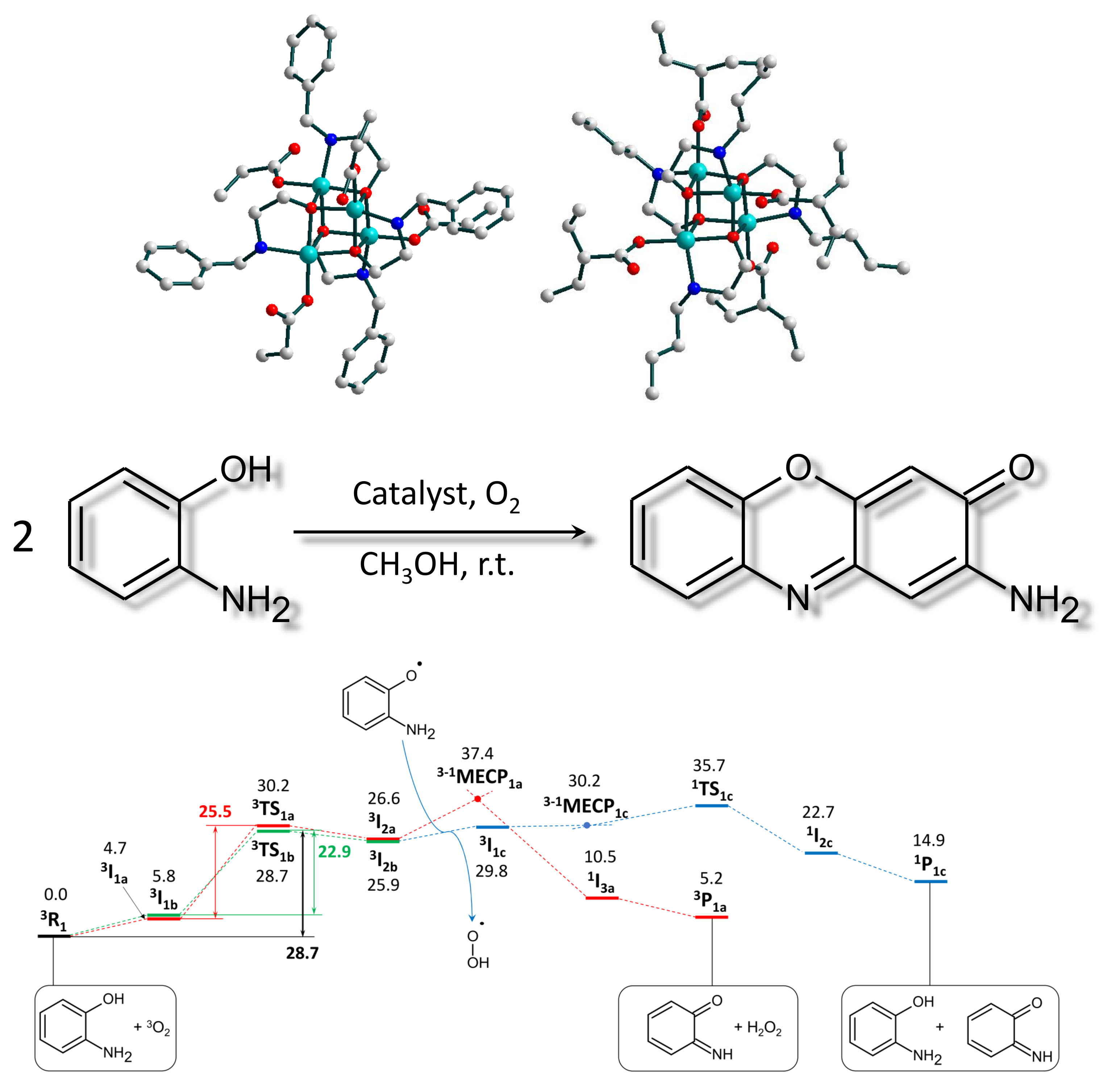
2.3. Catalytic Activity
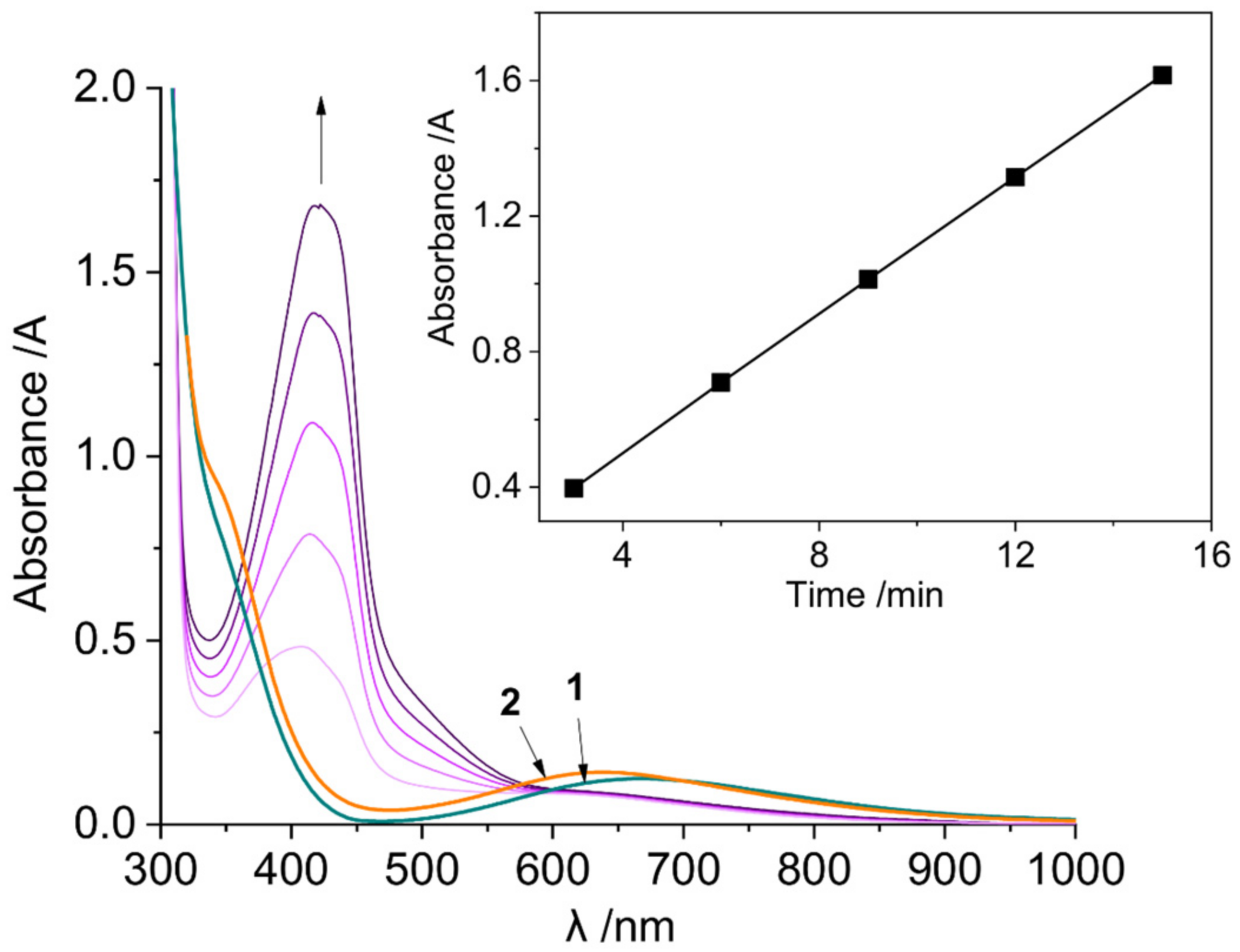
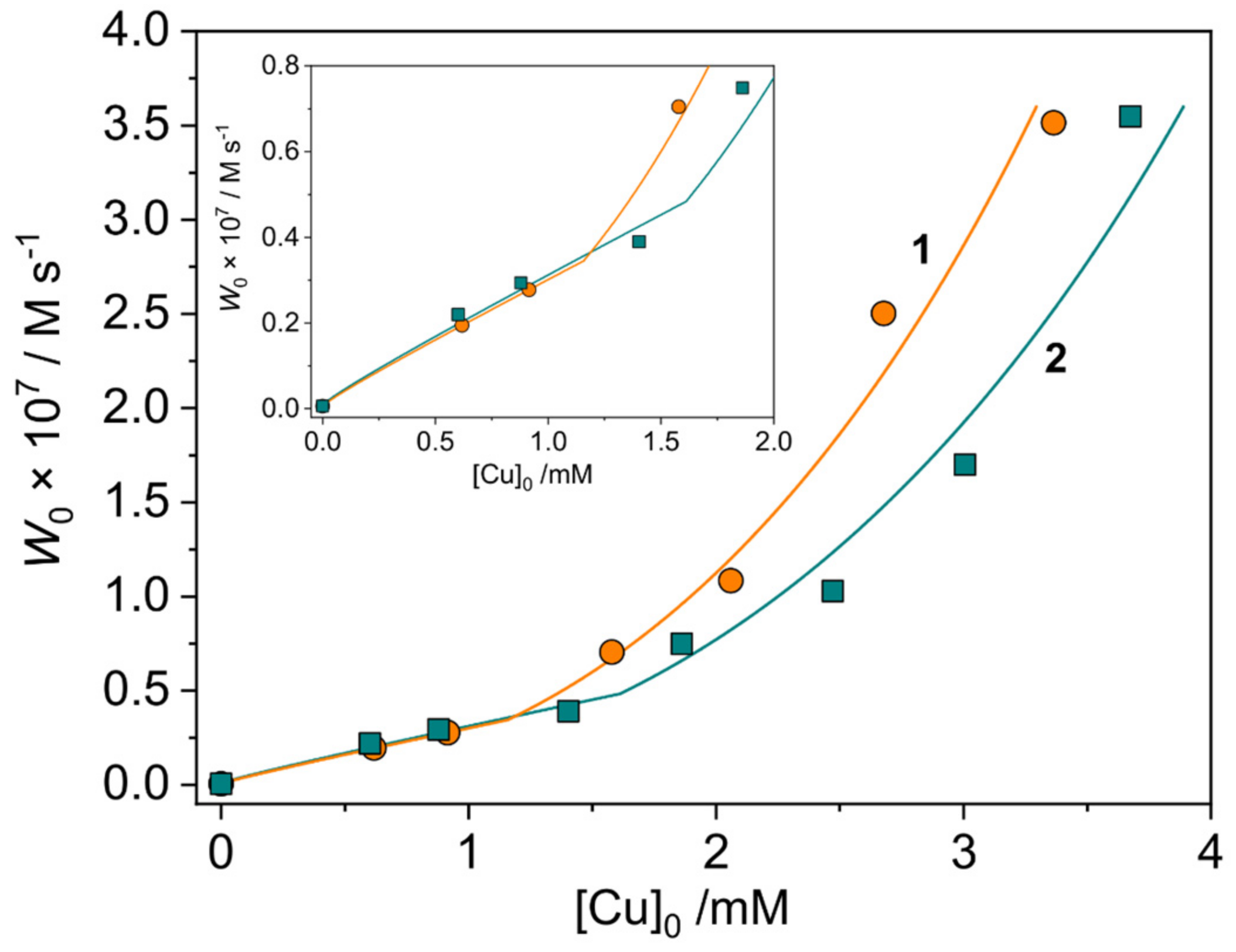
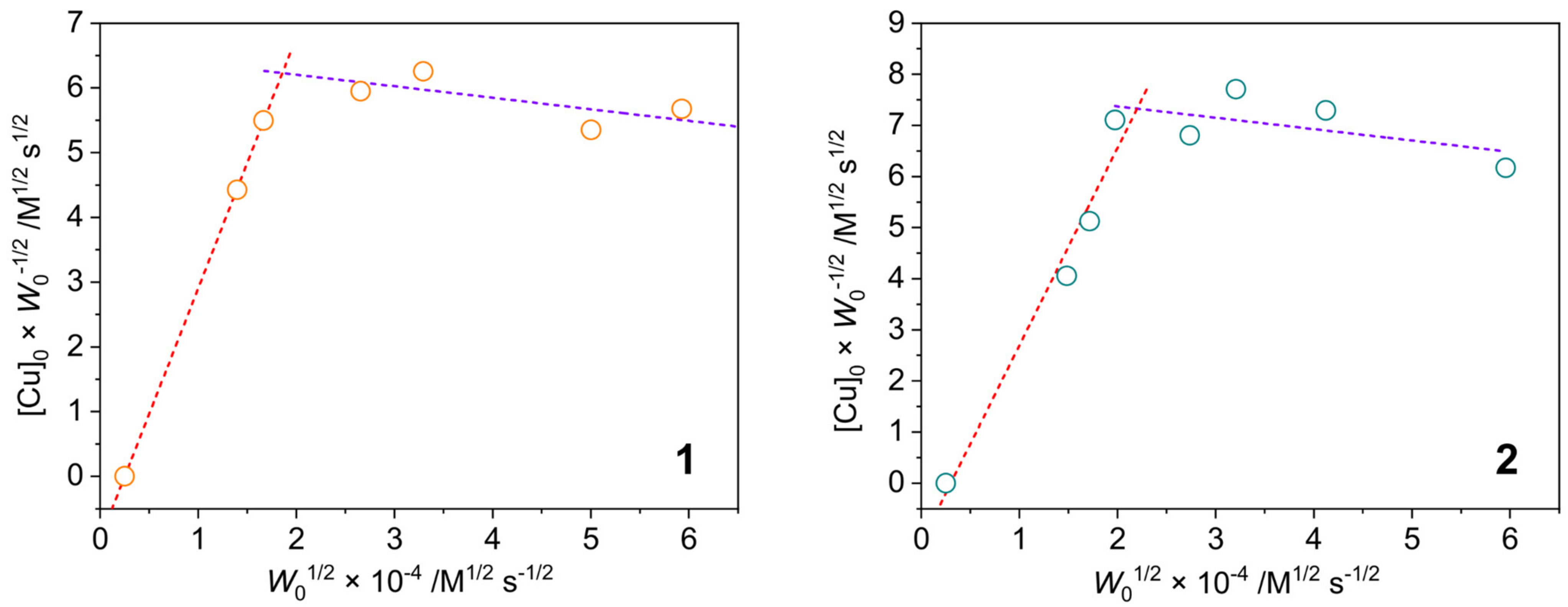
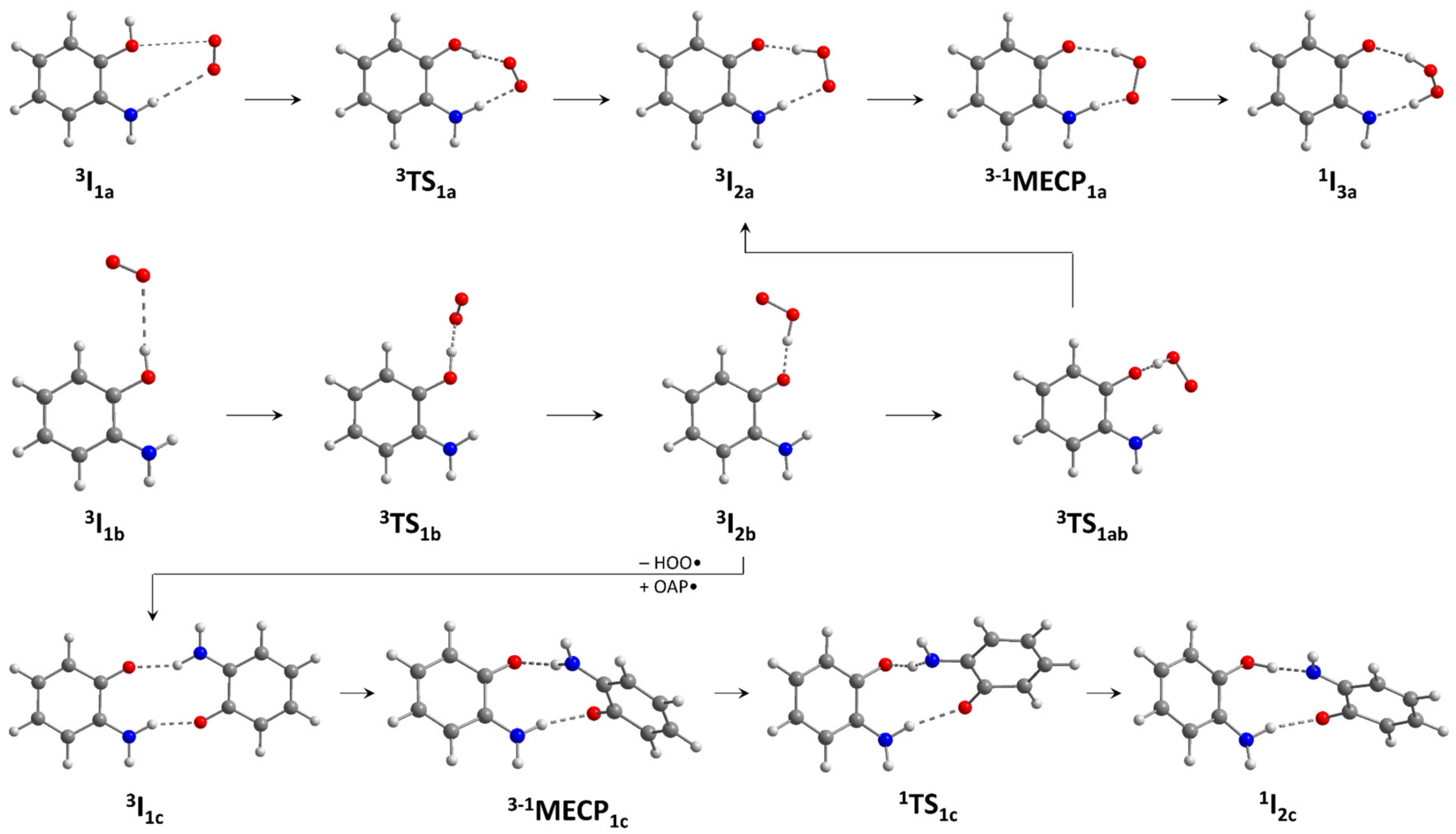
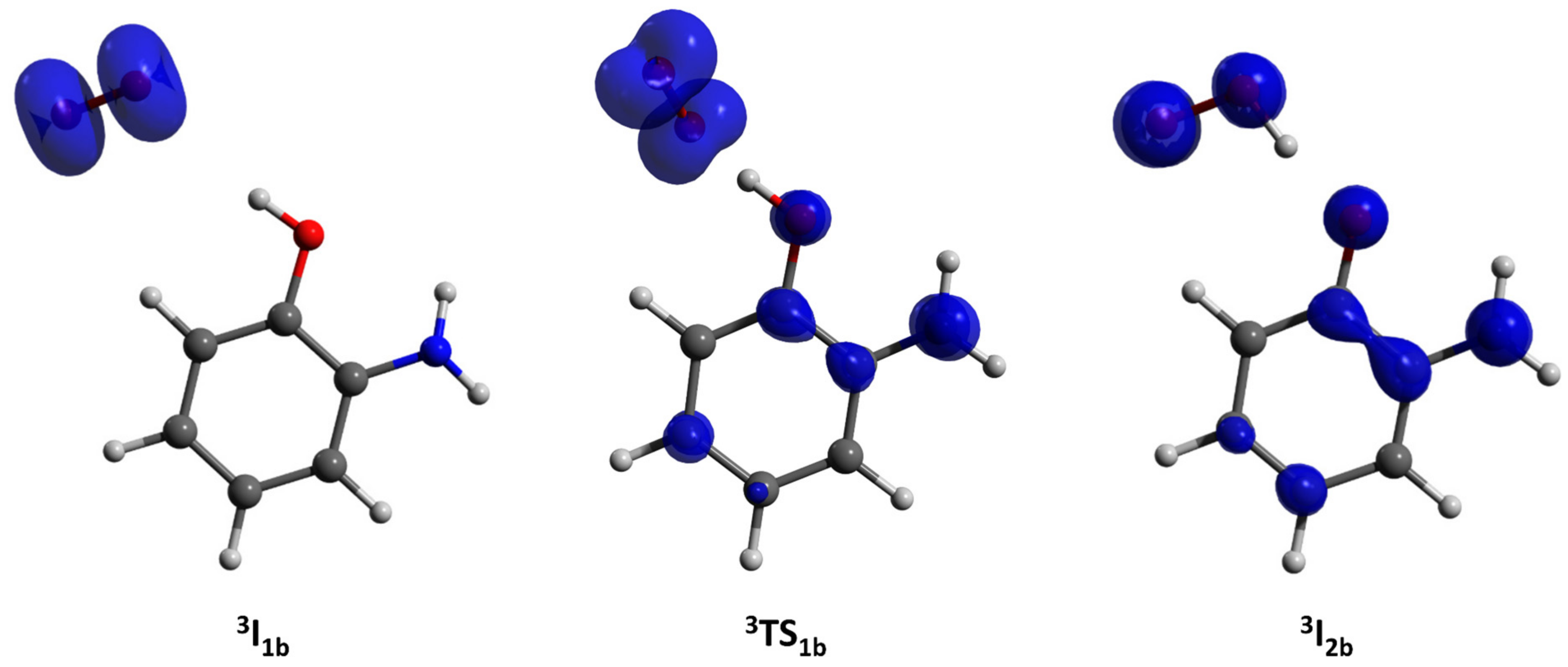
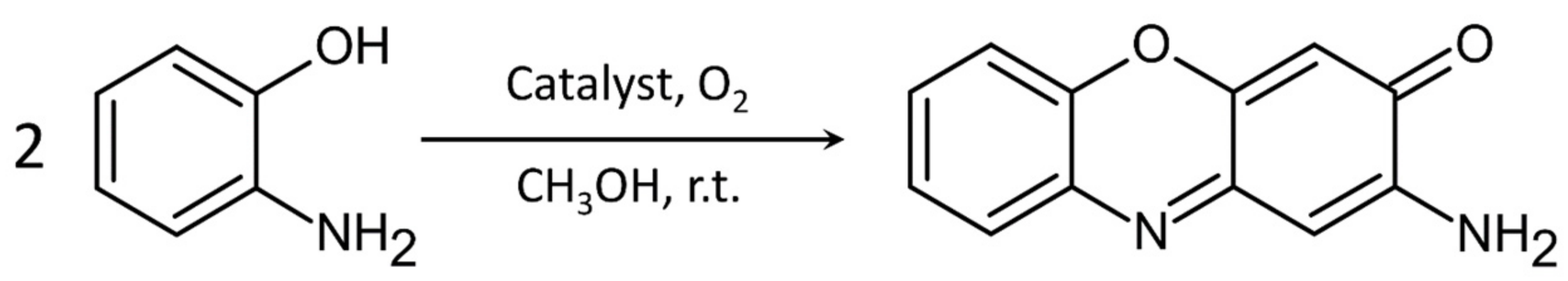
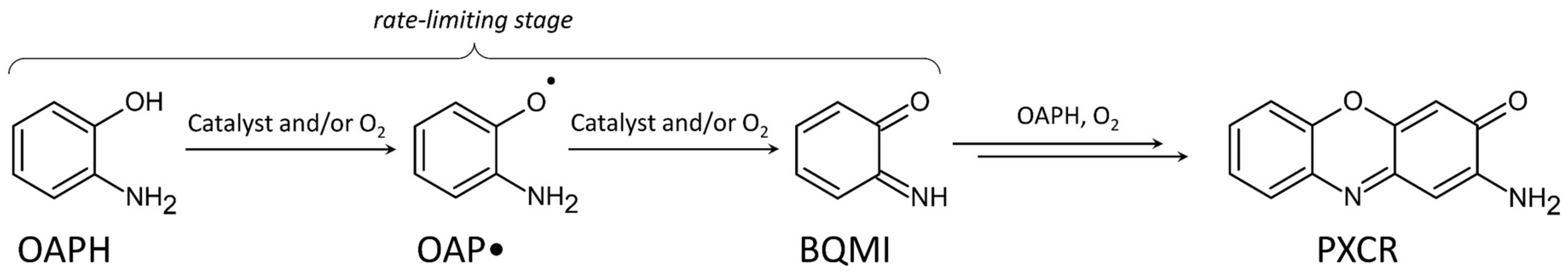
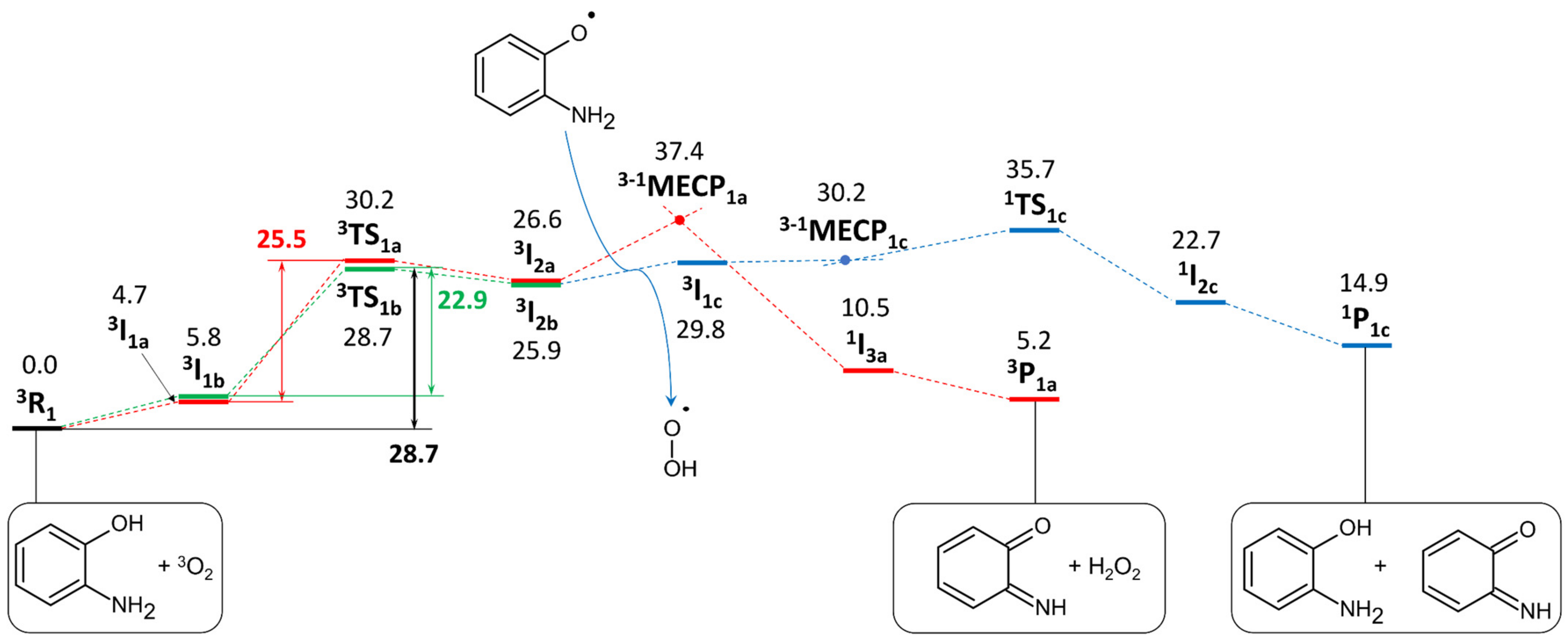
2.3.1. Phenoxazinone Synthase-Like Activity
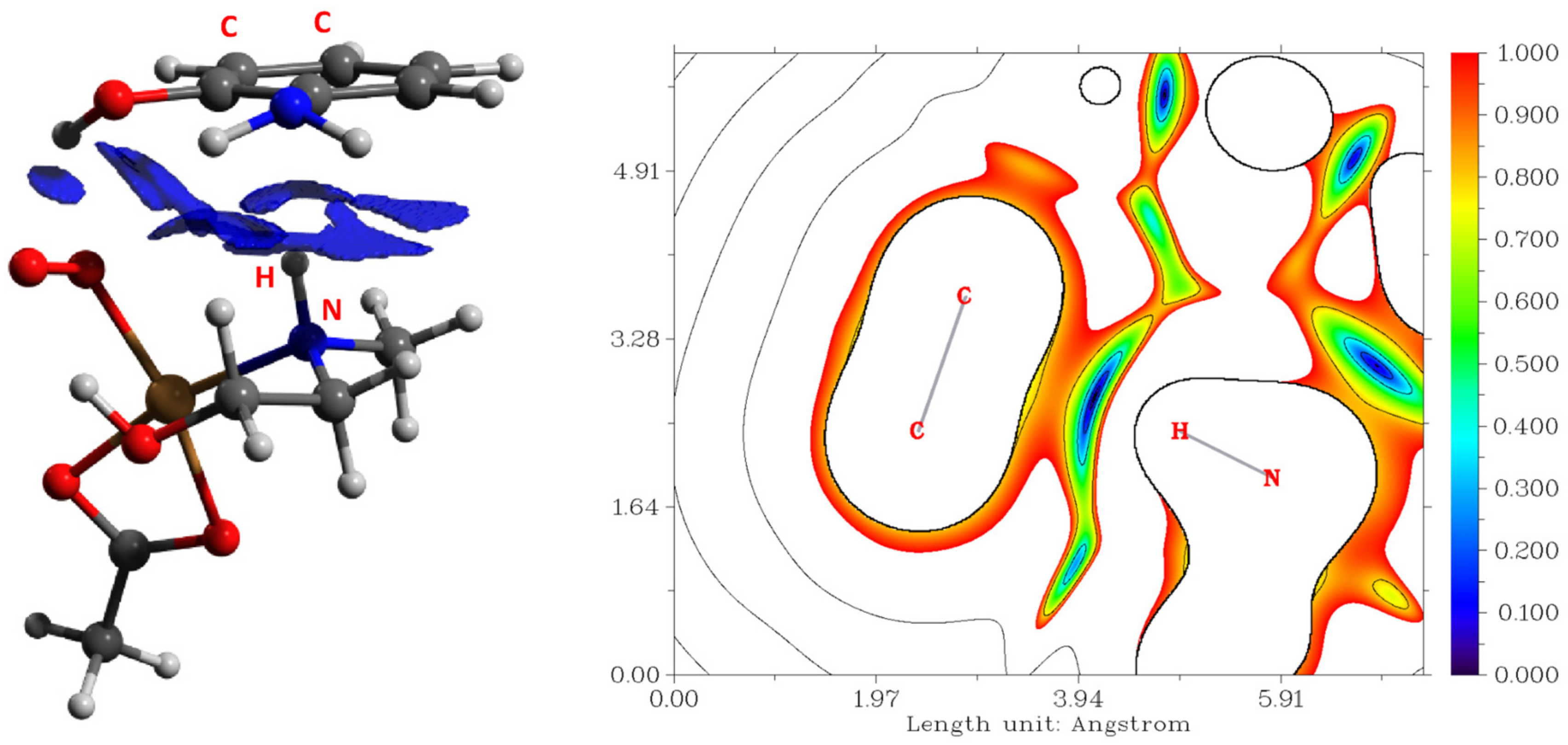
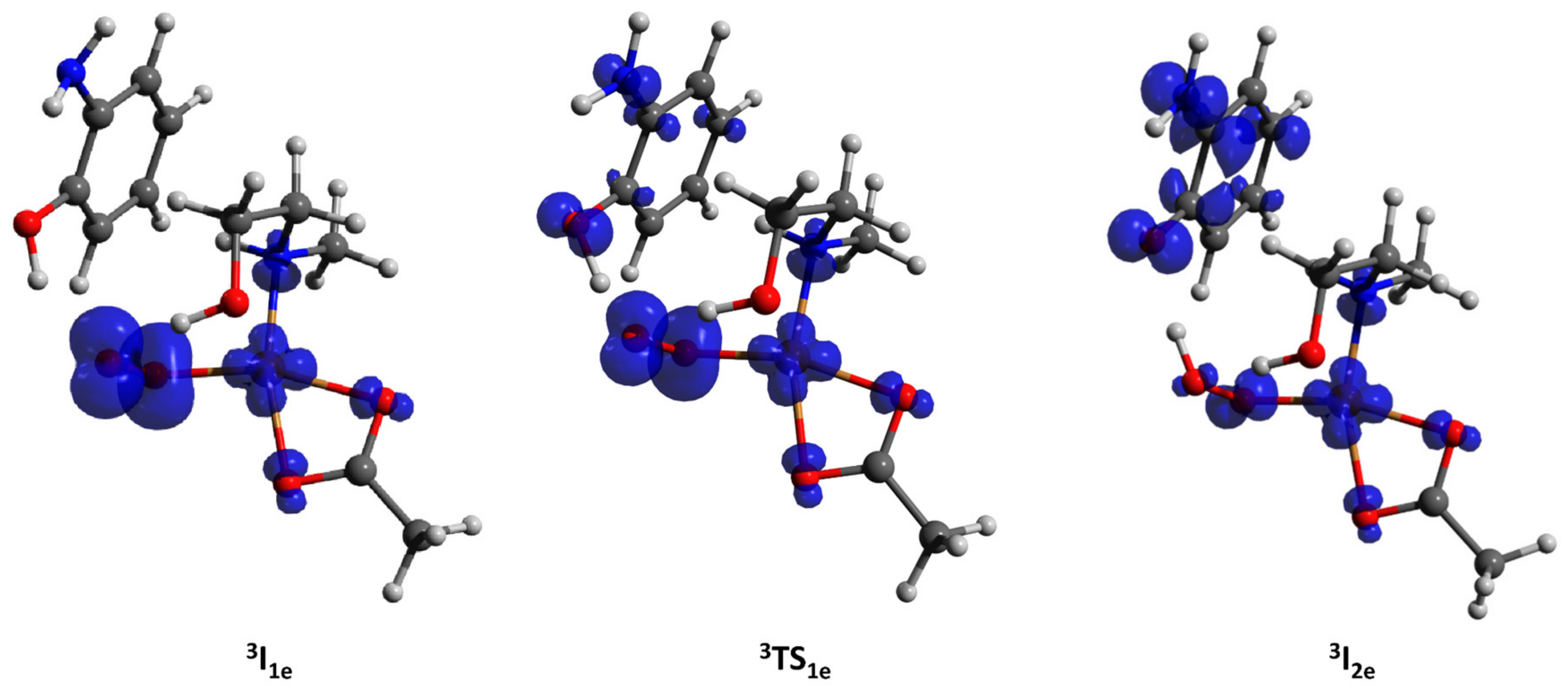
2.3.2. Amidation and Hydroxylation of Cyclohexane
3. Conclusions
4. Experimental Section
4.1. General
4.1.1. Synthesis of [Cu4(pa)4(Bae)4]·H2O (1)
4.1.2. Synthesis of [Cu4(eba)4(Buae)4]·H2O (2)
4.2. Crystallography
4.3. Phenoxazinone Synthase Activity Study
4.4. Catalytic Amidation of Cyclohexane
4.5. Catalytic Hydroxylation of Cyclohexane
4.6. Gas Chromatography
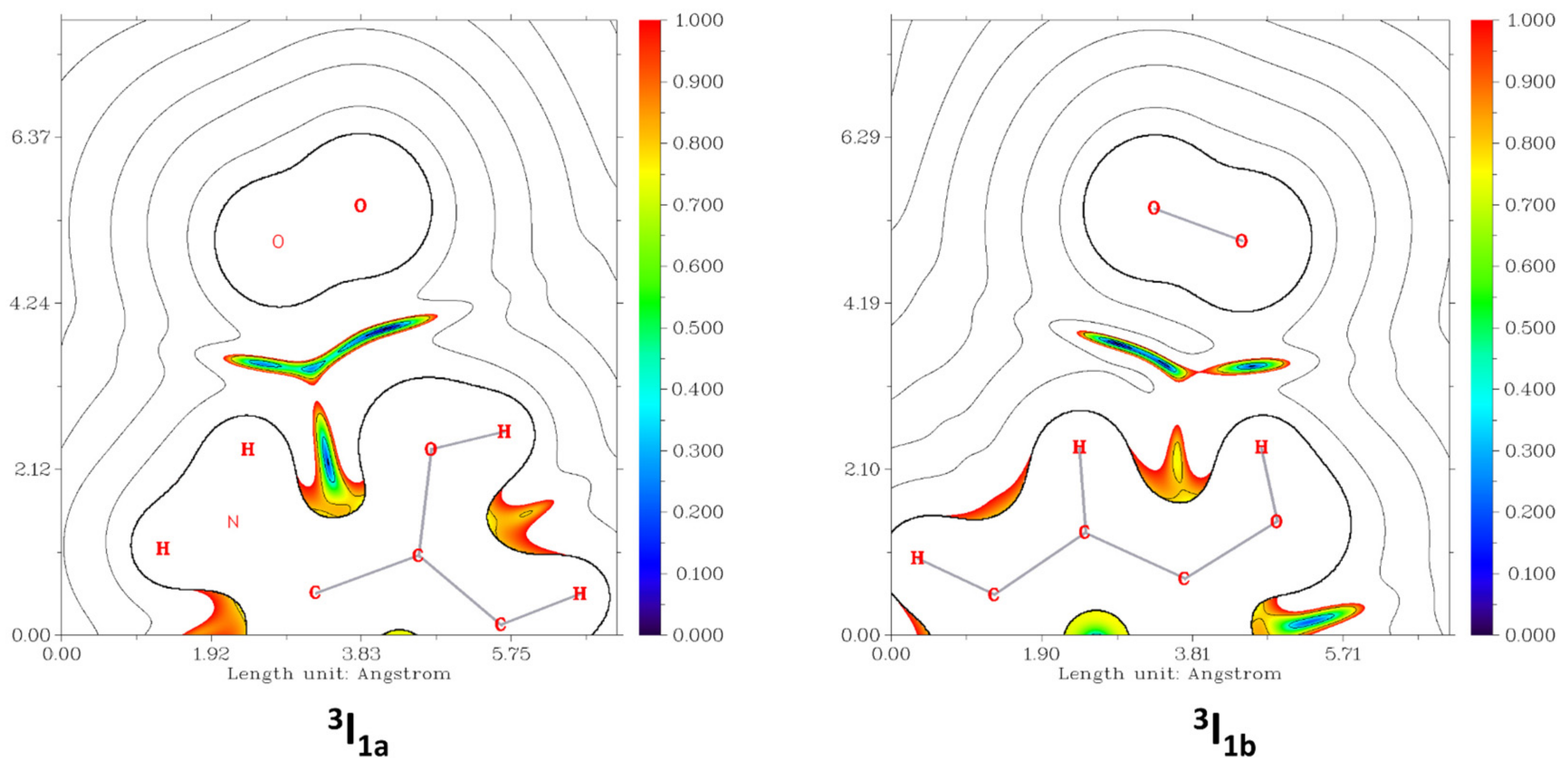
4.7. Theoretical Calculations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Singh, D.; Buratto, W.R.; Torres, J.F.; Murray, L.J. Activation of Dinitrogen by Polynuclear Metal Complexes. Chem. Rev. 2020, 120, 5517–5581. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhao, L. Polynuclear organometallic clusters: Synthesis, structure, and reactivity studies. Chem. Commun. 2020, 56, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C-H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, B.; Yao, Q.F.; Yang, Y.; Xie, J.P.; Yan, N. Recent advances in the synthesis and catalytic applications of ligand-protected, atomically precise metal nanoclusters. Coord. Chem. Rev. 2016, 322, 1–29. [Google Scholar] [CrossRef]
- Buchwalter, P.; Rose, J.; Braunstein, P. Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]
- Vaz, M.G.F.; Andruh, M. Molecule-based magnetic materials constructed from paramagnetic organic ligands and two different metal ions. Coord. Chem. Rev. 2021, 427, 213611. [Google Scholar] [CrossRef]
- Aguila, D.; Roubeau, O.; Aromi, G. Designed polynuclear lanthanide complexes for quantum information processing. Dalton Trans. 2021, 50, 12045–12057. [Google Scholar] [CrossRef]
- Sharples, J.W.; Collison, D. The coordination chemistry and magnetism of some 3d-4f and 4f amino-polyalcohol compounds. Coord. Chem. Rev. 2014, 260, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, V.C.C.; Maji, S.; Chen, P.R.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef]
- Dey, S.K.; Mukherjee, A. Catechol oxidase and phenoxazinone synthase: Biomimetic functional models and mechanistic studies. Coord. Chem. Rev. 2016, 310, 80–115. [Google Scholar] [CrossRef]
- Yano, J.; Yachandra, V. Mn4Ca Cluster in Photosynthesis: Where and How Water is Oxidized to Dioxygen. Chem. Rev. 2014, 114, 4175–4205. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S. Polynuclear Cobalt Complexes as Catalysts for Light-DrivenWater Oxidation: A Review of Recent Advances. Catalysts 2018, 8, 602. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Kokozay, V.N.; Pombeiro, A.J.L. Polynuclear Heterometallic Complexes from Metal Powders: The “Direct Synthesis” Approach. Eur. J. Inorg. Chem. 2014, 2014, 4496–4517. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Graiff, C.; Tiripicchio, A.; Pombeiro, A.J.L. Direct synthesis and crystal structure of a new pentanuclear heterotrimetallic Cu/Co/Ni complex with 2-(dimethylamino)ethanol. Discussion of possible “butterfly-like” molecular structure types. CrystEngComm 2011, 13, 5348–5353. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Makhankova, V.G.; Kokozay, V.N.; Skelton, B.W. Direct synthesis and crystal structures of new heteropolynuclear complexes containing aminoalcohol ligands: From heterobi- (Co/Zn) to heterotrimetallic (Cu/Co/Zn) compounds. Inorg. Chim. Acta 2005, 358, 4519–4526. [Google Scholar] [CrossRef]
- Liu, X.; Manzur, C.; Novoa, N.; Celedon, S.; Carrillo, D.; Hamon, J.-R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Jezierska, J.; Nesterova, O.V.; Pombeiro, A.J.L.; Ozarowski, A. An unprecedented octanuclear copper core with C3i symmetry and a paramagnetic ground state. Chem. Commun. 2014, 50, 3431–3434. [Google Scholar] [CrossRef] [Green Version]
- Conceição, N.R.; Nesterova, O.V.; Rajnak, C.; Boca, R.; Pombeiro, A.J.L.; Guedes da Silva, M.F.C.; Nesterov, D.S. New members of the polynuclear manganese family: MnII2MnIII2 single-molecule magnets and MnII3MIII8 antiferromagnetic complexes. Synthesis and magnetostructural correlations. Dalton Trans. 2020, 49, 13970–13985. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Alegria, E.C.B.A.; Jezierska, J. A new member of CuII8 family: Synthesis, structure and magnetic properties of an octanuclear copper complex with N-tert-butyldiethanolamine. Inorg. Chim. Acta 2017, 460, 83–88. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Vranovicova, B.; Boca, R.; Pombeiro, A.J.L. Heterometallic CuIIFeIII and CuIIMnIII alkoxobridged complexes revealing a rare hexanuclear M6(µ-X)7(µ3-X)2 molecular core. Dalton Trans. 2018, 47, 10941–10952. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Nesterov, D.S.; Jezierska, J.; Pombeiro, A.J.L.; Ozarowski, A. Copper(II) Complexes with Bulky N-Substituted Diethanolamines: High-Field Electron Paramagnetic Resonance, Magnetic, and Catalytic Studies in Oxidative Cyclohexane Amidation. Inorg. Chem. 2018, 57, 12384–12397. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Kuznetsov, M.L.; Pombeiro, A.J.L.; Shul’pin, G.B.; Nesterov, D.S. Homogeneous oxidation of C-H bonds with m-CPBA catalysed by a Co/Fe system: Mechanistic insights from the point of view of the oxidant. Catal. Sci. Technol. 2022, 12, 282–299. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Pombeiro, A.J.L.; Nesterov, D.S. Novel H-Bonded Synthons in Copper Supramolecular Frameworks with Aminoethylpiperazine-Based Ligands. Synthesis, Structure and Catalytic Activity. Materials 2020, 13, 5435. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Kasyanova, K.V.; Makhankova, V.G.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Stereospecific sp3 C-H oxidation with m-CPBA: A CoIII Schiff base complex as pre-catalyst vs. its CoIIICdII heterometallic derivative. Appl. Catal. A 2018, 560, 171–184. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Chygorin, E.N.; Kokozay, V.N.; Bon, V.V.; Boca, R.; Kozlov, Y.N.; Shul’pina, L.S.; Jezierska, J.; Ozarowski, A.; Pombeiro, A.J.L.; et al. Heterometallic CoIII4FeIII2 Schiff Base Complex: Structure, Electron Paramagnetic Resonance, and Alkane Oxidation Catalytic Activity. Inorg. Chem. 2012, 51, 9110–9122. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Bondarenko, O.E.; Pombeiro, A.J.L.; Nesterov, D.S. Phenoxazinone synthase-like catalytic activity of novel mono- and tetranuclear copper(ii) complexes with 2-benzylaminoethanol. Dalton Trans. 2020, 49, 4710–4724. [Google Scholar] [CrossRef]
- Freeman, J.C.; Nayar, P.G.; Begley, T.P.; Villafranca, J.J. Stoichiometry and spectroscopic identity of copper centers in phenoxazinone synthase—A new addition to the blue copper oxidase family. Biochemistry 1993, 32, 4826–4830. [Google Scholar] [CrossRef]
- Barry, C.E.; Nayar, P.G.; Begley, T.P. Phenoxazinone synthase-mechanism for the formation of the phenoxazinone chromophore of actinomycin. Biochemistry 1989, 28, 6323–6333. [Google Scholar] [CrossRef]
- Hollstein, U. Actinomycin-chemistry and mechanism of action. Chem. Rev. 1974, 74, 625–652. [Google Scholar] [CrossRef]
- Le Roes-Hill, M.; Goodwin, C.; Burton, S. Phenoxazinone synthase: What’s in a name? Trends Biotechnol. 2009, 27, 248–258. [Google Scholar] [CrossRef]
- Smith, A.W.; Camara-Artigas, A.; Wang, M.T.; Allen, J.P.; Francisco, W.A. Structure of phenoxazinone synthase from Streptomyces antibioticus reveals a new type 2 copper center. Biochemistry 2006, 45, 4378–4387. [Google Scholar] [CrossRef]
- Sagar, S.; Sengupta, S.; Mota, A.J.; Chattopadhyay, S.K.; Ferao, A.E.; Riviere, E.; Lewis, W.; Naskar, S. Cubane-like tetranuclear Cu(II) complexes bearing a Cu4O4 core: Crystal structure, magnetic properties, DFT calculations and phenoxazinone synthase like activity. Dalton Trans. 2017, 46, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, C.; Weyhermuller, T.; Bothe, E.; Rentschier, E.; Chaudhuri, P. A Tetracopper(II)-Tetraradical cuboidal core and its reactivity as a functional model of phenoxazinone synthase. Inorg. Chem. 2007, 46, 9895–9905. [Google Scholar] [CrossRef]
- Podder, N.; Mandal, S. Aerobic oxidation of 2-aminophenol catalysed by a series of mononuclear copper(II) complexes: Phenoxazinone synthase-like activity and mechanistic study. New J. Chem. 2020, 44, 12793–12805. [Google Scholar] [CrossRef]
- Dhara, A.K.; Singh, U.P.; Ghosh, K. Radical pathways and O2 participation in benzyl alcohol oxidation, and catechol and o-amino-phenol oxidase activity studies with novel zinc complexes: An experimental and theoretical investigation. Inorg. Chem. Front. 2016, 3, 1543–1558. [Google Scholar] [CrossRef]
- Kaizer, J.; Csonka, R.; Speier, G. TEMPO-initiated oxidation of 2-aminophenol to 2-aminophenoxazin-3-one. J. Mol. Catal. A 2002, 180, 91–96. [Google Scholar] [CrossRef]
- Khairy, M.; Mahmoud, A.H.; Khalil, K.M.S. Synthesis of highly crystalline LaFeO3 nanospheres for phenoxazinone synthase mimicking activity. RSC Adv. 2021, 11, 17746–17754. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W.T. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Nanayakkara, S.; Tao, Y.W.; Kraka, E. Comment on “Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory”. J. Comput. Chem. 2021, 42, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Rezac, J.; Hobza, P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem. Rev. 2016, 116, 5038–5071. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Abdo, M.; Gerges, S.; El-Khalafy, S. Study of the oxidation of 2-aminophenol by molecular oxygen catalyzed by cobalt(II) phthalocyaninetetrasodiumsulfonate in water. J. Mol. Catal. A 2008, 287, 53–56. [Google Scholar] [CrossRef]
- Simandi, L.I.; Barna, T.; Nemeth, S. Kinetics and mechanism of the cobaloxime(II)-catalysed oxidation of 2-aminophenol by dioxygen. A phenoxazinone synthase model involving free-radical intermediates. J. Chem. Soc. Dalton Trans. 1996, 4, 473–478. [Google Scholar] [CrossRef]
- Szigyarto, I.C.; Simandi, T.M.; Simandi, L.I.; Korecz, L.; Nagy, N. A functional phenoxazinone synthase model based on dioximatomanganese(II)-Kinetics and mechanism of the catalytic oxidation of 2-aminophenols by dioxygen. J. Mol. Catal. A 2006, 251, 270–276. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Najibi, A.; Goerigk, L. DFT-D4 counterparts of leadingmeta-generalized-gradient approximation and hybrid density functionals for energetics and geometries. J. Comput. Chem. 2020, 41, 2562–2572. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mat. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Suzuki, T.; Oshita, H.; Yajima, T.; Tani, F.; Abe, H.; Shimazakix, Y. Formation of the CuII-Phenoxyl Radical by Reaction of O2 with a CuII-Phenolate Complex via the CuI-Phenoxyl Radical. Chem. Eur. J. 2019, 25, 15805–15814. [Google Scholar] [CrossRef]
- Liu, J.Q.; Lorraine, S.C.; Dolinar, B.S.; Hoover, J.M. Aerobic Oxidation Reactivity of Well-Defined Cobalt(II) and Cobalt(III) Aminophenol Complexes. Inorg. Chem. 2022, 61, 6008–6016. [Google Scholar] [CrossRef]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation. J. Am. Chem. Soc. 2013, 135, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Gozzo, F. Radical and non-radical chemistry of the Fenton-like systems in the presence of organic substrates. J. Mol. Catal. A 2001, 171, 1–22. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Lee, Y.M.; Nam, W.; Fukuzumi, S. Amphoteric reactivity of metal-oxygen complexes in oxidation reactions. Coord. Chem. Rev. 2018, 365, 41–59. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.M.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Perdew, J.P.; Tao, J.M.; Staroverov, V.N.; Scuseria, G.E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. J. Chem. Phys. 2004, 120, 6898–6911. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-initio calculation of vibrational absorption and circular-dichroism spectra using density-functional force-fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation-energy of the inhomogeneous electron-gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Bannwarth, C.; Hansen, A.; Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 2018, 148, 13. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sanchez, P.; Yang, W.T. Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theor. Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef]
- Simon, L.; Goodman, J.M. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GGA functionals. Org. Biomol. Chem. 2011, 9, 689–700. [Google Scholar] [CrossRef]
- Murakami, M.; Ishida, N. Beta-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett. 2017, 46, 1692–1700. [Google Scholar] [CrossRef]
- Tran, B.L.; Li, B.J.; Driess, M.; Hartwig, J.F. Copper-Catalyzed Intermolecular Amidation and Imidation of Unactivated Alkanes. J. Am. Chem. Soc. 2014, 136, 2555–2563. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. A Comparative Study of the Catalytic Behaviour of Alkoxy-1,3,5-Triazapentadiene Copper(II) Complexes in Cyclohexane Oxidation. Inorganics 2019, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Gryca, I.; Czerwinska, K.; Machura, B.; Chrobok, A.; Shul’pina, L.S.; Kuznetsov, M.L.; Nesterov, D.S.; Kozlov, Y.N.; Pombeiro, A.J.L.; Varyan, I.A.; et al. High Catalytic Activity of Vanadium Complexes in Alkane Oxidations with Hydrogen Peroxide: An Effect of 8-Hydroxyquinoline Derivatives as Noninnocent Ligands. Inorg. Chem. 2018, 57, 1824–1839. [Google Scholar] [CrossRef]
- Astakhov, G.S.; Bilyachenko, A.N.; Korlyukov, A.A.; Levitsky, M.M.; Shul’pina, L.S.; Bantreil, X.; Lamaty, F.; Vologzhanina, A.V.; Shubina, E.S.; Dorovatovskii, P.V.; et al. High-Cluster Cu9 Cage Silsesquioxanes: Synthesis, Structure, and Catalytic Activity. Inorg. Chem. 2018, 57, 11524–11529. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L. A hydroperoxo-rebound mechanism of alkane oxidation with hydrogen peroxide catalyzed by binuclear manganese(IV) complex in the presence of an acid with involvement of atmospheric dioxygen. Inorg. Chim. Acta 2017, 455, 666–676. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Krogul-Sobczak, A.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Synthesis, crystal structures and catalytic activity of Cu(II) and Mn(III) Schiff base complexes: Influence of additives on the oxidation catalysis of cyclohexane and 1-phenylehanol. J. Mol. Catal. A 2017, 426, 506–515. [Google Scholar] [CrossRef]
- Wierzchowski, P.T.; Zatorski, L.W. Determination of cycle C-6 and C-7 peroxides and hydroperoxides by gas chromatography. Chromatographia 2000, 51, 83–86. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Bilyachenko, A.N.; Nesterov, D.S.; Shul’pina, L.S.; Zubavichus, Y.V.; Pombeiro, A.J.L.; Levitsky, M.M.; Yalymov, A.I.; Shul’pin, G.B. Alkane oxidation with peroxides catalyzed by cage-like copper(II) silsesquioxanes. New J. Chem. 2015, 39, 187–199. [Google Scholar] [CrossRef]
- Kim, H.Y.; Oh, K. Recent advances in the copper-catalyzed aerobic Csp3-H oxidation strategy. Org. Biomol. Chem. 2021, 19, 3569–3583. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Shrestha, B.; Gurung, S.K.; Giri, R. Copper-catalysed cross-coupling: An untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.D.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiri, P.; Aboonajmi, J. A systematic review on silica-, carbon-, and magnetic materials-supported copper species as efficient heterogeneous nanocatalysts in “click” reactions. Beilst. J. Org. Chem. 2020, 16, 551–586. [Google Scholar] [CrossRef] [Green Version]
- Bruker. APEX2 & SAINT; AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: A review. J. Mol. Catal. A 2002, 189, 39–66. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 18. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscipl. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscipl. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Zheng, J.J.; Xu, X.F.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theor. Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef]
- Asgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jonsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theor. Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Shao, Y.H.; Mei, Y.; Sundholm, D.; Kaila, V.R.I. Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores. J. Chem. Theor. Comput. 2020, 16, 587–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesterova, O.V.; Vassilyeva, O.Y.; Skelton, B.W.; Benko, A.; Pombeiro, A.J.L.; Nesterov, D.S. A novel o-vanillin Fe(III) complex catalytically active in C-H oxidation: Exploring the magnetic exchange interactions and spectroscopic properties with different DFT functionals. Dalton Trans. 2021, 50, 14782–14796. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterova, O.V.; Pombeiro, A.J.L.; Nesterov, D.S. Tetranuclear Copper Complexes with Bulky Aminoalcohol Ligands as Catalysts for Oxidative Phenoxazinone Synthase-like Coupling of Aminophenol: A Combined Experimental and Theoretical Study. Catalysts 2022, 12, 1408. https://doi.org/10.3390/catal12111408
Nesterova OV, Pombeiro AJL, Nesterov DS. Tetranuclear Copper Complexes with Bulky Aminoalcohol Ligands as Catalysts for Oxidative Phenoxazinone Synthase-like Coupling of Aminophenol: A Combined Experimental and Theoretical Study. Catalysts. 2022; 12(11):1408. https://doi.org/10.3390/catal12111408
Chicago/Turabian StyleNesterova, Oksana V., Armando J. L. Pombeiro, and Dmytro S. Nesterov. 2022. "Tetranuclear Copper Complexes with Bulky Aminoalcohol Ligands as Catalysts for Oxidative Phenoxazinone Synthase-like Coupling of Aminophenol: A Combined Experimental and Theoretical Study" Catalysts 12, no. 11: 1408. https://doi.org/10.3390/catal12111408